
INTRODUCTION
Childhood acute lymphoblastic leukemia (cALL) is the most frequent cancer in children between 1 and 14 years old and accounts for ≈25% of all pediatric tumours [1]. Precursor B cell cALL (pre-B cALL) is the predominant form of cALL accounting for 85% of cALL patients. Several studies have described expression signatures for classifying molecularly-defined ALL subtypes and improving outcome prediction [2–9]. A new class of molecule, long non-coding RNAs (lncRNAs), play regulatory roles in various processes including pluripotency and tumorigenesis [10, 11]. Recent studies have highlighted their involvement in leukemia initiation and progression. Indeed, the BALR-2 and BALR-6 lncRNAs were found to be involved in cell survival or glucocorticoid response in both human and mouse B cells [12, 13]. Furthermore, we and others have demonstrated that lncRNA transcription profiles can discriminate pre-B cALL subtypes accurately [2, 12, 14, 15] or can act as prognostic biomarkers [2]. Although the importance of lncRNAs in tumor biology is clear, little is known about their precise function. Here we studied the impact of three lncRNAs whose transcription is repressed in a cohort of pre-B cALL samples [14, 15]. We found that they significantly affected proliferation, migration, response to cytotoxic drugs, and DNA damage response when overexpressed in pre-B leukemic cells. These findings shed light on lncRNA function in leukemia and point the way towards new biomarkers and therapeutic targets.
RESULTS
Overexpression of lncRNAs in pre-B cALL increases apoptosis in response to genotoxic stress
We studied the impact of three lncRNAs, RP11-624C23.1, RP11-203E8, and RP11-446E9, which were downregulated in our pre-B cALL cohort (Supplementary Figure 1 and 2 [14, 15]) by overexpressing them in the Reh pre-B cALL cell line (Supplementary Figure 3). We assessed their impact on apoptosis in response to camptothecin (CPT; a DNA-damaging agent), doxorubicine (DOX; an anthracycline), and prednisolone (a corticosteroid). All three lncRNAs significantly increased apoptosis after CPT exposure: > 2-fold increase in RP11-624C23.1-overexpressing cells and a > 3-fold increase for those overexpressing RP11-203E8 or RP11-446E9 (Figure 1A). However, no effect was observed upon either DOX or prednisolone treatment (Supplementary Figure 6). This increased sensitivity to CPT disappeared in rescue experiments, where cell lines overexpressing these lncRNAs are stably transfected with shRNAs targeting those (Figure 1A and Supplementary Figure 4). The increased sensitivity to CPT upon RP11-624C23.1, RP11-203E8, or RP11-446E9 overexpression was also seen in another pre-B cALL cell line, NALM-6 (Supplementary Figure 7), indicating it is not cell-line specific.
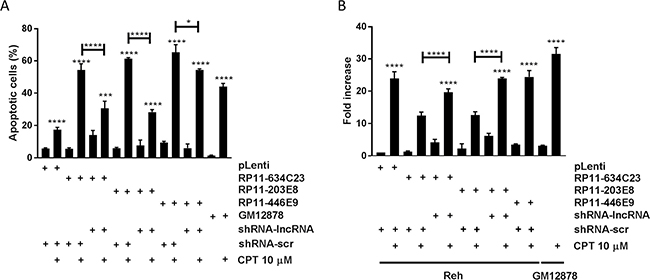
Figure 1: The lncRNAs RP11-624C23.1 and RP11-203E8 are involved in camptothecin resistance and DNA damage response. Reh-pLenti, Reh-RP11-624C23.1, Reh-RP11-203E8, Reh-RP11-446E9, Reh-RP11-624C23.1-sh2, Reh-RP11-203E8-sh2, and Reh-RP11-446E9-sh2 were treated with camptothecin (CPT) 10 μM for six hours. Apoptosis (A) and γ-H2A.X (B) were then measured. *, *** and ****P ≤ 0.05, ≤ 0.001 and ≤ 0.0001, respectively. The GM12878 non-leukemic lymphoid cell line was used as a normal cell control.
RP11-624C23.1 and RP11-203E8 modulate DNA damage response
The increased sensitivity to CPT prompted us to assess whether these lncRNAs modulate DNA damage response (DDR). To address this question, we quantified phosphorylated histone H2A.X (γ-H2A.X) levels, a well-known DNA damage marker, after CPT treatment. We observed that γ-H2A.X levels decrease to that of control in cells overexpressing RP11-624C23.1 or RP11-203E8, but not RP11-446E9, and that γ-H2A.X levels are restored in rescue experiments (Figure 1B). Taken together with the increased sensitivity to CPT, these results suggest that RP11-624C23.1 and RP11-203E8 modulate the DDR pathway.
RP11-446E9 is involved in cell proliferation and migration
We observed that cell proliferation was decreased upon RP11-446E9 overexpression, while it was partially restored (> 50%) in rescue experiments (Figure 2A). Furthermore, transwell migration using SDF1 as chemoattractant led to a RP11-446E9-specific decrease in cell migration (Figure 2B). Once again, this effect was reversed in rescue experiments, demonstrating specificity (Figure 2B and Supplementary Figure 4). No effect was seen on cell proliferation or migration for RP11-624C23.1 or RP11-203E8 (data not shown).
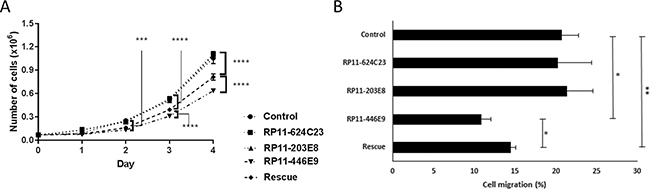
Figure 2: The lncRNA RP11-446E9 regulates cell proliferation and migration. Four-day proliferation tests (A) and migration assays (B) were performed on Reh-pLenti (Control), Reh-RP11-624C23.1 (RP11-624C23), Reh-RP11-203E8 (RP11-203E8), Reh-RP11-446E9 (RP11-446E9), and Reh-RP11-446E9-sh1 (Rescue) cell lines. *, **, *** and ****P ≤ 0.05, ≤ 0.01, ≤ 0.0005 and ≤ 0.0001 respectively.
Taken together, our results indicate that these three lncRNAs play important roles in the regulation of cancer phenotypes such as cell proliferation, migration, and treatment and DNA damage response.
DISCUSSION
While our knowledge on lncRNAs’ role in cancer has progressed in recent years, only a few have been functionally characterized. Examples of these include HOTAIR, which interacts with Polycomb-repressive complex 2 and LSD1 to promote cancer invasiveness [16] and, more recently, BALR-2, which is implicated in prednisolone treatment resistance by inhibiting AP-1 activation [12]. Here we showed that three novel lncRNAs downregulated in pre-B cALL, RP11-624C23.1, RP11-203E8, and RP11-446E9, modulate DDR and migration.
We have shown that both RP11-624C23.1 and RP11-203E8 overexpression increased apoptosis and decreased H2A.X phosphorylation upon CPT treatment. Interestingly, the phenotypes affected and the magnitudes of said effects were very similar for both RP11-624C23.1 and RP11-203E8 overexpression, possibly indicating that they act in the same molecular pathway. Interestingly, these two lncRNAs flank the ADAM28 gene, although whether this is related to their function is unclear. However, we see no effect of these lncRNAs overexpression on neighboring genes ADAM28, LYN, or RPS (Supplementary Figure 5), suggesting that these lncRNAs role do not involve neighboring genes’ modulation. These results strongly suggest that these lncRNAs are involved in DDR, possibly acting on DNA damage checkpoints or directly on the DNA repair mechanisms. While further work is required to fully investigate these hypotheses, there are other reports of non-coding RNAs that play roles in DDR and genotoxic stress response [17–20]. These results indicate that silencing RP11-624C23.1 or RP11-203E8 could provide a selective advantage to leukemic cells by increasing resistance to genotoxic stress, possibly by modulating the DDR pathway.
While overexpression of RP11-446E9 also increases apoptosis in response to CPT treatment, it had no impact on H2A.X phosphorylation. This shows that RP11-446E9 modulates DNA damage-triggered cell death but does not act in the DDR pathway. Furthermore, we have shown that both cell migration and proliferation decrease when RP11-446E9 expression is restored, suggesting a negative role on cell cycle control and cell motility. More work is required to investigate this thoroughly.
In conclusion, we have functionally characterized three lncRNAs specifically repressed in pre-B cALL: RP11-624C23.1, RP11-203E8, and RP11-446E9. Restoring their expression in a pre-B cALL cell line promoted tumor suppressor-like phenotypes: apoptosis induction in response to DNA damaging agents and a reduction in cell proliferation and migration. Interestingly, RP11-624C23.1 and RP11-203E8 exhibited identical phenotypes, suggesting that they may be involved in a similar pathway. Our results suggest that lncRNAs play key roles in pre-B cALL disease progression and treatment resistance. While it has been shown that lncRNA expression can accurately discriminate pre-B cALL disease subtype and thus be used as a prognostic tool, further work to dissect their exact modes of action is required and could lead to novel, unexplored therapeutic strategies to overcome treatment resistance by targeting the biological pathways associated with their action.
MATERIALS AND METHODS
Childhood ALL sample cohort and transcriptome profiling
Our cohort, transcriptome sequencing and analysis were described in Ouimet et al. [14]. Our study cohort consisted of 56 pre-B cALL patients (28 females and 28 males) with a mean age at diagnosis of 6.1 ± 3.6 years. All subjects were French-Canadians of European descent diagnosed in the Division of Hematology-Oncology at the Sainte-Justine Hospital (Montreal, Canada) and part of the Quebec childhood ALL cohort (QcALL) [21]. CD10+CD19+ cells isolated from human cord blood were used as controls. Total RNA was extracted from white blood cell pellets obtained from bone marrow and peripheral blood at diagnosis using the mirVana Isolation kit (Ambion) according to manufacturer’s protocol. Following a DNAse treatment to remove possible contamination by genomic DNA, ribosomal RNAs were removed using the RiboMinus Eukaryote kit (Invitrogen). cDNA libraries were prepared using the SOLiD Total RNA-seq kit based on manufacturer’s protocol and sequenced on the Life Technologies SOLiD 4/5500 System (paired-end: 50 × 35 bp and 75 × 35 bp). Reads were aligned to the human genome (hg19) using the Lifescope Genomic Analysis Software (Applied Biosystems; Whole Transcriptome Analysis pipeline, default parameters). Expression levels by gene were determined with the HTseq-count software [22]. using gene models from Ensembl75 combined with (non-overlapping) lncRNA transcripts provided in Casero et al. [23]. The identification of differentially expressed transcripts relative to HCB controls was done using the Generalized Linear Model implemented in the edgeR package [24]. The Sainte-Justine Institutional Review Board approved the research protocol, and informed consent was obtained from all participating individuals and/or their parents.
lncRNA overexpression constructs
cDNA for RP11-624C23.1, RP11-203E8, and RP11-446E9 were amplified from CD19+ primary cells then cloned into pLenti-CMV-Puro-DEST using the Gateway system (ThermoFisher, Waltham, MA, USA). Short hairpin RNA for RP11-624C23.1 (5′-AAGGGAUACACUACUGUUAUGGG-3′), RP11-203E8 (5′-GUCUCAUGUAGGCUGAAUACUUG-3′) and RP11-446E9 (5′-CUAGCCAGCUGGACUGCA AUUUC-3′) were designed (uptime Life Technologies, Carlsbad, CA, USA) then cloned into psiLVRH1H (GeneCopeia, Rockville, MD, USA). A list of PCR primers used can be found in Supplementary Table 1.
Cell culture and lentiviral infections
The Reh pre-B cALL and GM12878 B-lymphoblast cell lines (American Type Culture Collection, MD, USA) were cultured in RPMI 1640 (Wisent Bio Products, QC, Canada) supplemented with fetal calf serum (Wisent Bio Products) and 100 units/mL penicillin and streptomycin (Wisent Bio Products) at 37°C in a humidified atmosphere at 5% CO2. Cells infected with lentiviral plasmids pLenti-CMV-Puro-DEST or psiLVRH1H were cultured by adding puromycin (1 μg/mL) or hygromycin (400 μg/mL) (Sigma-Aldrich, MI, USA), respectively, to the cell culture medium. 600 ng of the lentiviral particles generated using these constructs were used to infect 1 × 106 cells in 1mL RPMI 1640 supplemented with 10% fetal calf serum and 8 μg/mL polybrene (Sigma-Aldrich). After 24 h, cells are washed with PBS and cultured in 1mL RPMI supplemented with 10% fetal calf serum. Cells expressing the constructs were selected by addition of the appropriate drug, as detailed above.
Total RNA purification and reverse transcription
The RNeasy Mini Kit (QIAGEN, Toronto, ON, Canada) was used to extract total RNA as per manufacturer’s instructions. First-strand cDNA synthesis was done from 1 μg total RNA using the QuantiTect Reverse Transcription Kit (QIAGEN, Mississauga, ON, Canada). QPCR was done using SYBR Green PCR Master Mix (Life technologies). The cycling conditions were as follows: 5 min at 95°C, followed by 35 cycles of 95°C (5 sec), 50°C (30 sec) and 72°C (32 sec). Relative expression levels were normalized to GAPDH. QPCR primers used are listed in Supplementary Table 2.
Cell-based functional assays
Apoptosis
Apoptosis was measured using 1 × 106 cells with the FITC AnnexinV/Dead Cell apoptosis kit (ThermoFisher) on a FACS Canto II platform (BD Biosciences, CA, USA) 6 hours after 10 μM camptothecin (Sigma-Aldrich) treatment.
Proliferation
Cells were inoculated in triplicate at 0.9 × 104 cells/mL in 150 μL in a 96-well plate and were counted daily over 4 days with a Z1 Coulter particle counter (Beckman-Coulter, QC, Canada).
ϒ-H2A.X assessment
Cells were seeded in 6-well plates at 0.5 × 106 cells/mL per well, treated with 10μM camptothecin, and cultured for 6 h. Cells were fixed with 1% formaldehyde for 15 min and permeabilized overnight at −20°C using 95% ethanol. Cells were hydrated 5 min with TBS + 0.05% Tween and incubated at 4°C overnight with anti-phospho-H2A.X(S139) (613402; BioLegend). Cells were washed twice with TBS + 0.05% Tween and incubated 90 min in the dark with Alexa Fluor 488 goat anti-mouse IgG (A11001; ThermoFisher). Finally, cells were washed twice with TBS + 0.05% Tween, incubated 1 h with 50 μg/mL propidium iodide (Life Technologies, ON, Canada) supplemented with 1 μg/mL RNase I, and analyzed by flow cytometry as described above.
Migration assay
Polycarbonate filters with a pore size of 8 μm (BD Biosciences, Bedford, MA, USA) were used as previously described [25]. Briefly, 5 × 104 cells were seeded in the upper chamber of the insert and placed in a six-well plate filled with serum-free RPMI-1640 medium supplemented with 30 nM SDF-1 (Sigma-Aldrich) in the lower chamber. After 1 h, cells in the culture medium in the lower chamber were counted with a Z1 Coulter particle counter (Beckman-Coulter).
Statistical analysis
Statistical analyses were done using GraphPad Prism 5.0 (GraphPad Software, CA, USA). The two-sided Student’s p-value used to call statistical significance was p ≤ 0.05 for all experiments.
Abbreviations
cALL: childhood acute lymphoblastic leukemia; lncRNA: long non-coding RNA; shRNA: short hairpin RNA; CPT: camptothecin; DDR: DNA damage response; QcALL: Quebec childhood ALL cohort; qPCR: quantitative polymerase chain reaction.
Author contributions
RG designed and performed all experiments, analyzed the data, and wrote the first draft. SD participated in experimental design and analysis and co-wrote the manuscript. MC and PSO performed bioinformatics analyses. MO assisted RG and provided critical feedback for the manuscript. CR assisted RG and generated RNA sequencing libraries. DS is the principal investigator of the research and participated in the design and interpretation of the results as well as in the writing of the paper.
ACKNOWLEDGMENTS AND FUNDING
This study was supported by research funds provided by the Canadian Cancer Society Research Institute (CCSRI). DS holds the François-Karl-Viau Research Chair in Pediatric Oncogenomics.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Gilliland DG, Jordan CT, Felix CA. The molecular basis of leukemia. Hematology Am Soc Hematol Educ Program. 2004; 2004:80–97. https://doi.org/10.1182/asheducation-2004.1.80.
2. Yeoh EJ, Ross ME, Shurtleff SA, Williams WK, Patel D, Mahfouz R, Behm FG, Raimondi SC, Relling MV, Patel A, Cheng C, Campana D, Wilkins D, et al. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell. 2002; 1:133–43.
3. Ross ME, Zhou X, Song G, Shurtleff SA, Girtman K, Williams WK, Liu HC, Mahfouz R, Raimondi SC, Lenny N, Patel A, Downing JR. Classification of pediatric acute lymphoblastic leukemia by gene expression profiling. Blood. 2003; 102:2951–9. https://doi.org/10.1182/blood-2003-01-0338.
4. Andersson A, Eden P, Lindgren D, Nilsson J, Lassen C, Heldrup J, Fontes M, Borg A, Mitelman F, Johansson B, Hoglund M, Fioretos T. Gene expression profiling of leukemic cell lines reveals conserved molecular signatures among subtypes with specific genetic aberrations. Leukemia. 2005; 19:1042–50. https://doi.org/10.1038/sj.leu.2403749.
5. Harvey RC, Mullighan CG, Wang X, Dobbin KK, Davidson GS, Bedrick EJ, Chen IM, Atlas SR, Kang H, Ar K, Wilson CS, Wharton W, Murphy M, et al. Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling: correlation with genome-wide DNA copy number alterations, clinical characteristics, and outcome. Blood. 2010; 116:4874–84. https://doi.org/10.1182/blood-2009-08-239681.
6. Bhojwani D, Kang H, Menezes RX, Yang W, Sather H, Moskowitz NP, Min DJ, Potter JW, Harvey R, Hunger SP, Seibel N, Raetz EA, Pieters R, et al. Gene expression signatures predictive of early response and outcome in high-risk childhood acute lymphoblastic leukemia: A Children’s Oncology Group Study [corrected]. J Clin Oncol. 2008; 26:4376–84. https://doi.org/10.1200/JCO.2007.14.4519.
7. Silveira VS, Scrideli CA, Moreno DA, Yunes JA, Queiroz RG, Toledo SC, Lee ML, Petrilli AS, Brandalise SR, Tone LG. Gene expression pattern contributing to prognostic factors in childhood acute lymphoblastic leukemia. Leuk Lymphoma. 2013; 54:310–4. https://doi.org/10.3109/10428194.2012.710330.
8. Chen IM, Harvey RC, Mullighan CG, Gastier-Foster J, Wharton W, Kang H, Borowitz MJ, Camitta BM, Carroll AJ, Devidas M, Pullen DJ, Payne-Turner D, Tasian SK, et al. Outcome modeling with CRLF2, IKZF1, JAK, and minimal residual disease in pediatric acute lymphoblastic leukemia: a Children’s Oncology Group study. Blood. 2012; 119:3512–22. https://doi.org/10.1182/blood-2011-11-394221.
9. Haferlach T, Kohlmann A, Wieczorek L, Basso G, Kronnie GT, Bene MC, De Vos J, Hernandez JM, Hofmann WK, Mills KI, Gilkes A, Chiaretti S, Shurtleff SA, et al. Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: report from the International Microarray Innovations in Leukemia Study Group. J Clin Oncol. 2010; 28:2529–37. https://doi.org/10.1200/JCO.2009.23.4732.
10. Morlando M, Ballarino M, Fatica A. Long Non-Coding RNAs: New Players in Hematopoiesis and Leukemia. Front Med (Lausanne). 2015; 2:23. https://doi.org/10.3389/fmed.2015.00023.
11. Iyer MK, Niknafs YS, Malik R, Singhal U, Sahu A, Hosono Y, Barrette TR, Prensner JR, Evans JR, Zhao S, Poliakov A, Cao X, Dhanasekaran SM, et al. The landscape of long noncoding RNAs in the human transcriptome. Nat Genet. 2015; 47:199–208. https://doi.org/10.1038/ng.3192.
12. Fernando TR, Rodriguez-Malave NI, Waters EV, Yan W, Casero D, Basso G, Pigazzi M, Rao DS. LncRNA Expression Discriminates Karyotype and Predicts Survival in B-Lymphoblastic Leukemia. Mol Cancer Res. 2015; 13:839–51. https://doi.org/10.1158/1541-7786.MCR-15-0006-T.
13. Rodriguez-Malave NI, Fernando TR, Patel PC, Contreras JR, Palanichamy JK, Tran TM, Anguiano J, Davoren MJ, Alberti MO, Pioli KT, Sandoval S, Crooks GM, Rao DS. BALR-6 regulates cell growth and cell survival in B-lymphoblastic leukemia. Mol Cancer. 2015; 14:214. https://doi.org/10.1186/s12943-015-0485-z.
14. Ouimet M, Drouin S, Lajoie M, Caron M, St-Onge P, Gioia R, Richer C, Sinnett D. A childhood acute lymphoblastic leukemia-specific lncRNA implicated in prednisolone resistance, cell proliferation, and migration. Oncotarget. 2017; 8:7477–88. https://doi.org/10.18632/oncotarget.13936.
15. Lajoie M, Drouin S, Caron M, St-Onge P, Ouimet M, Gioia R, Lafond MH, Vidal R, Richer C, Oualkacha K, Droit A, Sinnett D. Specific expression of novel long non-coding RNAs in high-hyperdiploid childhood acute lymphoblastic leukemia. PLoS One. 2017; 12:e0174124. https://doi.org/10.1371/journal.pone.0174124.
16. Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, Chang HY. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010; 329:689–93. https://doi.org/10.1126/science.1192002.
17. Bertozzi D, Iurlaro R, Sordet O, Marinello J, Zaffaroni N, Capranico G. Characterization of novel antisense HIF-1alpha transcripts in human cancers. Cell Cycle. 2011; 10:3189–97. https://doi.org/10.4161/cc.10.18.17183.
18. Kotake Y, Nakagawa T, Kitagawa K, Suzuki S, Liu N, Kitagawa M, Xiong Y. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15(INK4B) tumor suppressor gene. Oncogene. 2011; 30:1956–62. https://doi.org/10.1038/onc.2010.568.
19. Hung T, Wang Y, Lin MF, Koegel AK, Kotake Y, Grant GD, Horlings HM, Shah N, Umbricht C, Wang P, Wang Y, Kong B, Langerod A, et al. Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat Genet. 2011; 43:621–9. https://doi.org/10.1038/ng.848.
20. Lal A, Pan Y, Navarro F, Dykxhoorn DM, Moreau L, Meire E, Bentwich Z, Lieberman J, Chowdhury D. miR-24-mediated downregulation of H2AX suppresses DNA repair in terminally differentiated blood cells. Nat Struct Mol Biol. 2009; 16:492–8. https://doi.org/10.1038/nsmb.1589.
21. Healy J, Belanger H, Beaulieu P, Lariviere M, Labuda D, Sinnett D. Promoter SNPs in G1/S checkpoint regulators and their impact on the susceptibility to childhood leukemia. Blood. 2007; 109:683–92. https://doi.org/10.1182/blood-2006-02-003236.
22. Anders S, Pyl PT, Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2014:btu638.
23. Casero D, Sandoval S, Seet CS, Scholes J, Zhu Y, Ha VL, Luong A, Parekh C, Crooks GM. Long non-coding RNA profiling of human lymphoid progenitor cells reveals transcriptional divergence of B cell and T cell lineages. Nat Immunol. 2015; 16:1282–91. https://doi.org/10.1038/ni.3299.
24. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015; 43:e47. https://doi.org/10.1093/nar/gkv007.
25. Ramesh R, Mhashilkar AM, Tanaka F, Saito Y, Branch CD, Sieger K, Mumm JB, Stewart AL, Boquoi A, Dumoutier L, Grimm EA, Renauld JC, Kotenko S, et al. Melanoma differentiation-associated gene 7/interleukin (IL)-24 is a novel ligand that regulates angiogenesis via the IL-22 receptor. Cancer Res. 2003; 63:5105–13.