
INTRODUCTION
Glioblastoma is a highly malignant brain tumor with an extremely dismal prognosis. Standard methods of treatment, which often involve surgical resection, chemotherapy, and radiotherapy, prolong patient survival but only to an extent. With this, survival is often limited to a little over one year following diagnosis [1, 2]. Because of the aggressive nature of this disease and the limited treatment options available, new and further developed anti-tumor modalities of care are desperately needed. One difficulty, notwithstanding all other considerations, related to the development of novel treatment modalities is the unique immune repertoire and tumor microenvironment of glioma [3–5]. As a vastly heterogeneous tumor, glioblastoma (previously referred to with the descriptor of ‘multiforme’ for this very reason) has a tumor microenvironment that is immunosuppressive such that a conventional immune response against the tumor does not ordinarily take place [2–4]. Standard modes of treatment do not modulate the tumor microenvironment in this regard. Fortunately, progressively developed modes of combating these hurdles have come to the forefront such as to enable the active targeting of the tumor as well as its immunosuppressive microenvironment. Oncolytic virotherapy has been developed as a therapeutic modality that utilizes viral infectivity to specifically target the tumor [6–10]. The tropism of the virus can be modified in order to enhance the viral infectivity of neoplastic cells specifically while simultaneously limiting the infection of non-neoplastic tissue. This specific oncolytic activity proves to be a gainful approach for attaining a marked anti-tumor response [9, 11–16]. Clinical trials of such studies as these have demonstrated some success that further supports the advance of these therapies while also encouraging the discovery of other experimental developments that may increase their efficacy [17–22] .
As noted already, the uniquely distinct tumor microenvironment of glioma creates immunosuppressive conditions that ordinarily do not allow for significant anti-tumor immune responsiveness. Immunotherapy strives to overcome this intrinsic tumor immunosuppression and promotes a defined immune response against the tumor through immune system modulations [23–25]. Defined by the presence of regulatory T cells, M2 macrophages, and myeloid-derived suppressor cells (MDSCs), the immunosuppressive glioma microenvironment may prove susceptible to approaches that promote an anti-tumor immune response through T cell activation [23–25]. Immunomodulation of the expression of cell surface receptors such as CTLA4, PD-1, and 4-1BB may be capitalized on to thus yield a T cell immune response in order to achieve an anti-tumor response [26–29]. Several clinical trials have utilized this mode of a novel therapeutic modality, some of which have garnered appreciable success [30–38].
Together, oncolytic virotherapy and immunotherapy have proven to be viable targeting options for the treatment of glioma. Their combination represents a promising treatment modality that has the potential for success in the clinic in ways that have not before been described. This could result in the synergistic destruction of the tumor, thus augmenting the survival benefit for patients, while simultaneously minimizing the risk of systemic side effects and toxicities. Ongoing and upcoming clinical trials will further explore their clinical benefit while studies attempting to augment their efficacy and demonstrate their applicability for the treatment of a disease where new treatment options are desperately needed.
Oncolytic Adenoviral Virotherapy in Glioma
Human adenovirus serotype 5 (Ad5), is one of the most common virus types used in glioma therapy since the biology of this double-stranded DNA virus is well studied and its use has been widely applied for gene therapy approaches [10, 15, 39]. Moreover, Ad5 is comparably easier to genetically modify without affecting its structural stability and host cell infectivity, which allows for redirecting its viral tropism (cell-specific targeting) and for restricting its replication (cell-specific lysis) [40–42]. Due to its well studied unique characteristics, proven safety, and efficacy found during the past few decades, many cancer-specific, targeted oncolytic adenoviruses are currently being tested in clinical trials [17–22, 43–45].
Types of oncolytic adenoviral vehicles
Different modifications of Ad5 tropism have brought current research studies a step closer to a well designed, targeted oncolytic therapy against glioma. Selective tropism, the ability to bind to and enter target cells, is the ultimate goal of effective viral infection for therapeutic purposes [9, 12, 13, 19, 40, 46]. The selective tropism of adenovirus is dictated by the fiber-knob domain [47]. In this context, several changes in this structure have been proposed to achieve targeted interaction in glioma cells. Since most cancerous cells, including much of those involved in glioma, lack the primary adenovirus receptor, CAR (Coxsackievirus and adenovirus receptor), various viral modification approaches are required and thus have been made to achieve efficient infection [11, 48]. Given the presence of surface proteins overexpressed primarily on the surface of glioma cells, it is possible to develop targeted viral vectors by incorporating a binding moiety of these surface proteins onto the adenoviral fiber for both gene therapy and oncolytic virotherapy [49, 50].
Among them, Ad5-pK7, Ad5.RGD, and Ad5/3 represent some of the leading contenders which bind to anionic surface proteins, integrins, and CD80/CD86/ [51, 52]/Desmoglein 2 [53], respectively (Figure 1). These modifications have become important to achieve a high efficacy of transduction to glioma cells. Ad5-pK7 is an insertional modification, which has been created by incorporating seven poly-lysine (pK7) residues onto the fiber-knob domain [50, 54–56]. These residues (pK7) were found to be a motif that binds to over-expressed heparan sulfate proteoglycans (HSPGs) on the surface of glioma cells [57]. Because of the high level of binding affinity to these receptors, this configuration is known to be highly effective and, based on currently available research studies, has the highest infectivity among the tested adenoviral modifications (an average of 70% infectivity in tested glioma cell lines such as U87, U251, and patient derived GBM43) [17, 50, 54–56]. Based on prior work in this area, this Ad5-pK7-based approach is being used in a phase I clinical trial for patients with glioma [17].
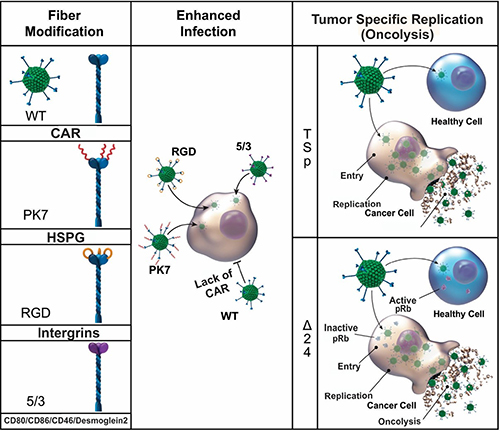
Figure 1: A schematic diagram of infectivity-enhanced fiber modifications and tumor-specific replication of adenovirus. Various viral modification approaches have been constructed to achieve efficient infection and cancer-specific replication. Among the various types of viral modification, the most common approaches for tropism modification (pk7, RGD, and 5/3) and tumor-restricted replication (tumor-specific promoter and delta24) are depicted.
Another approach involves targeting integrins, which are overexpressed on many cells, including glioma cells. Specifically, the integrins αvβ3 and αvb5 have been associated with enhanced tumor angiogenesis and are therefore viable targets for oncolytic virotherapy [58, 59]. Moreover, integrins αvβ3 and αvb5 were found to be over-expressed in glioma cells. Because of this, adenoviral vector Ad5.RGD was constructed by inserting an arginine (R)-glycine (G)-aspartic (D) acid-4C complex at the level of HI-loop in the Ad5 fiber-knob domain. This modification has led to the enhanced infection of cancers that overexpress this set of integrins [39, 46]. Many clinical trials including glioma, ovarian cancer, and other selected gynecologic cancers are being treated using this enhanced infectivity motif incorporated through Ad5RGD viral vehicles [60–66].
A third approach involves a chimeric fiber protein construct termed Ad5/3 [14, 39, 67, 68]. In this particular scenario, the fiber knob domain of Ad5 has been replaced with that of Ad3, allowing for enhanced transduction of cells overexpressing CD 80, CD86, and Desmoglein 2 [49, 50, 52]. Binding activity and replication efficiency of these chimeric constructs is significant, and a relevant oncolytic effect has been observed in murine cancer models, including glioma, pancreatic, ovarian, and prostate cancers [69–73].
All mentioned modifications at the level of the Ad5 fiber domain have become means of achieving more effective infection of glioma cells. By combining the available fiber modifications with specific promoters to drive the expression of replication of essential genes which will be discussed in later section, many have become excellent tools with remarkable oncolytic capabilities for glioma therapies [6–8, 11, 12, 16, 19, 40–42, 46, 49].
Restricted replication, cancer specific oncolysis of Adenovirus
Firstly, an adenovirus releases its progeny through the lysis of the host cell rather than exocytosis. As such, proper control of viral replication restricts host cell lysis to cancerous cells. To achieve tumor-restricted replication, many genetic modification approaches have been applied to the adenoviral vector based on the unique biology of neoplastic cells such as hyperactive promoter/mutations of gene(s) [39, 46, 74, 75] (Figure 1).
One most common approaches is to regulate the expression of adenoviral replication essential gene sequence, called early gene 1 (E1) under a tumor-specific promoter such as Survivin, CXCR4, or midkine. These tumor-specific promoters are known to be hyperactive in tumor cells (‘tumor always on’ status) while having nominal activity in normal cells. For instance, Survivin, a member of the inhibitor of apoptosis (IAP) family, has been shown to have elevated expression in glioma with a correlated association of poorer prognosis and increased rates of recurrence [39, 46, 74, 75]. Therefore, by taking advantage of this tumor-specific transcriptional regulation, replication-mediated oncolysis can be restricted only to cancerous cells.
Alternatively, adenoviral replications can be restricted to neoplastic cells by taking advantage of the malfunctioning cellular transcriptional machinery: inactivation of retinoblastoma protein (pRb) and p53 in cancer cells [76–78]. Upon the entry into normal cells, the first protein expressed from adenoviral DNA is the early gene 1 (E1) sequence, which produces E1A and E1B proteins that, among other things, bind to pRb and p53, respectively. This interaction continuously turns on cellular transcriptional machinery and allows for the massive production of viral proteins for the generation of its progeny. However, when a deletion of the binding sites for these proteins (24 nucleotides (Δ24) on E1A) or deletion of E1B (dl1152) is made to the adenoviral essential gene sequence, subsequent viral protein production cannot take place unless the function of pRb and p53 have already been impaired [79–82]. Since the majority of neoplastic cells have non-functioning pRb and p53, the cancer-restricted replication of an adenovirus is achieved by utilizing this understanding of the natural interaction between an adenovirus and relevant cellular machinery [79–82].
Additionally, although it is not direct adenoviral modification mediated oncolysis, incorporation of cytotoxic gene(s) such as HSV- thymidine kinase (tk) into a cancer specific adenovirus, called Ad-TK have been used and shown its clinical efficacy in GBM [83–85]. The usages of adenovirus as gene therapeutic oncolytic approaches can be found elsewhere [86–89].
Advantages and disadvantages of adenoviral vector usages for glioma therapies in a clinical setting
There are several well-established advantages of adenoviral vectors that make them ideal for clinical applications. First, they are considered to have no or low pathogenicity. Second, it is easy to produce clinically necessary high titers of viruses: an average 1012 viral particles per milliliter (vp/ml) on each purification when 109~1010 (vp/ml) is used in clinic. Third, the viral DNA is not integrated into the host chromosome such that a therapeutic gene of interest incorporated into the viral DNA is expressed only transiently. And most importantly, there is a high degree of customizability (flexibility for genetic modifications: structural modification and insertion of therapeutic genes–maximal insertion capacity of 7.5Kb) [6, 11, 13, 15, 19, 46, 49].
By taking advantage of these features, many adenoviral vectors have been investigated and demonstrated efficient cancer-specific therapeutic efficacy. Furthermore, this adenovirus-mediated oncolysis has been shown to induce a tumor-specific immune response through the release of tumor antigens [90–94]. Additionally, oncolytic therapy may sensitize cancerous cells in order to inhibit the DNA repair system upon conventional therapies such as chemotherapy and radiotherapy [95–97]. Given these promising findings, many clinical trials utilizing adenoviral agents for patients with gliomas are currently underway. A dose escalation trial (phase I) of ONYX-015 (oncolytic adenovirus with E1B mutation) showed no serious adverse effect upon the treatment (doses up to 1010 pfu) with 6.2 months of median survival [98]. Also, oncolytic adenovirus DNX-2401 (also known as Delta-24-RGD) based clinical trials are now actively recruiting subjects for phase I and II trials [61] (Table 1).
Table 1: Clinical trials of oncolytic adenovirus-based virotherapy in glioma
Trial number |
Type of treatment |
Phase |
Therapeutic agent |
Additional Information |
|---|---|---|---|---|
NCT00805376 |
DNX2401 (Formerly Known as Delta-24-RGD-4C) + TMZ |
Phase I |
Ad.Delta-24-RGD-4C |
Drug: DNX-2401 Procedure: Tumor Removal * Completed |
NCT02197169 |
DNX-2401 With Interferon Gamma (IFN-γ) for Recurrent Glioblastoma or Gliosarcoma Brain Tumors (TARGET-I) |
Phase I |
CRAd DNX-2401 (Formerly Named Delta-24-RGD) |
Drug: Single intratumoral injection of DNX-2401 Drug: Interferon-gamma * In the process of recruiting participants |
NCT03072134 |
Neural Stem Cell Based Virotherapy of Newly Diagnosed Malignant Glioma |
Phase I |
Neural stem cells loaded with Ad5-pK7 |
Biological: Neural stem cells loaded with Ad5-pK7 * In the process of recruiting participants |
NCT02798406 |
Combination Adenovirus + Pembrolizumab to Trigger Immune Virus Effects |
Phase II |
CRAd DNX-2401 |
Biological: DNX-2401 and pembrolizumab * In the process of recruiting participants |
NCT01301430 |
Parvovirus H-1 (ParvOryx) in Patients With Progressive Primary or Recurrent Glioblastoma Multiforme |
Phase I/II |
Parvovirus H-1 |
Drug: H-1PV * Completed |
NCT01582516 |
Trial of a Conditionally Replication-competent Adenovirus (Delta-24-rgd) Administered by Convection Enhanced Delivery in Patients With Recurrent Glioblastoma |
Phase I/II |
Ad.Delta-24-RGD |
Biological: delta-24-RGD adenovirus * Completed |
NCT01956734 |
Virus DNX2401 and Temozolomide in Recurrent Glioblastoma (D24GBM) |
Phase I |
DNX-2401 (Formerly Named Delta-24-RGD) |
Procedure: DNX2401 and Temozolomide * Ongoing trial |
However, there are still some challenges facing adenoviral oncolytic therapy. For instance, the majority of the human population has been pre-exposed to various adenovirus serotypes and it has been shown that therapeutic efficacy of adenovirus based agents could be negatively affected by any pre-existing immunity [99, 100]. Also, due to the nature of adenoviral tropism, this viral vector can be sequestered in liver, causing hepatotoxicity when delivered systemically [101–104]. As a result, many efforts strive to overcome these problems in order to increase infection efficacy and limit biotoxicity [101–104] (ref). For example, modifying the adenoviral capsid protein of the hexon to be less immunogenic and liver tropic can help circumvent unwanted targeting effects [101–104] . Furthermore, limited penetration of oncolytic adenoviruses throughout the depth of the solid tumor mass has been reported which is an inherent problem concerning oncolytic adenoviral therapy for glioma [86, 105]. In this case, it has been found that boosting the immune response against tumor antigen(s) released during oncolytic activity or activating cytotoxic T cells could augment the efficacy of adenoviral oncolytic activity [105–107].
Immunotherapy in Glioma
Immunotherapy has become a rapidly progressing field in the treatment of glioma. Immunotherapeutic efficacy has been shown to be less toxic and more specific in comparison to the other current treatment modalities presently available (resection, chemotherapy, and radiation) [108]. In the context of glioma, immunotherapy has been largely based on glioma vaccination and immune system modulation, which can regulate/suppress the immunosuppressive glioma microenvironment that is defined by the presence of regulatory T cells, M2 macrophages, and myeloid-derived suppressor cells (MDSCs) [109].
Pulsed DC-based therapies
One of the predominant immunotherapies for glioma that is already in clinical trials for patient treatment, is the pulsed dendritic cell (DC) vaccine (Supplementary Table 1). As DCs are the most effective antigen presenting cells (APCs) capable of eliciting a specific cell-mediated anti-tumor immune response, this approach is an attractive strategy to activate anti-tumor immunity for a variety of cancer types. However, due to the lack of in vivo DC-specific delivery vehicles, multi-step ex vivo strategies are more common. In this case, the patient’s autologous DCs are collected via plasmapheresis, stimulated ex vivo, and then pulsed with tumor specific antigen or whole tumor cell lysate. Finally, activated and pulsed DCs are systemically injected back into the patient in order to activate the T cell immune response [23, 109].
Initially, this DC-based tumor vaccine utilized a selected tumor-specific, antigen (TA)-based DC pulsing method. However, as glioma is highly heterogeneous and there being difficulty in identifying a glioma-specific antigen, this approach showed very limited efficacy [110]. Alternatively, it is presently more common to use whole tumor cell lysates pulsed with prepared DCs in the hope that these pulsed DCs will preferentially present tumor-specific antigen(s) not self-antigen(s) that would eventually cause serious autoimmune disease [111]. Therefore, to overcome these current limitations, single-step, in vivo DC delivery vehicles that can facilitate highly efficient multi-epitope TA loading for maximal anti-tumor immunity are needed to achieve efficacious glioma immunotherapy. As expected, side effects of these DC-based immunotherapies have been reported including transient fever, autoimmune vitiligo in melanoma patients, mild headache, and erythema in glioma patients [111].
Regarding the tumor immune response after DC vaccination in clinical trials NCT00612001 and NCT00068510, study authors found that decreased Regulatory T (Treg) cell and Natural Killer (NK) cell populations is correlated with increased survival for study patients [112]. In another study (NCT00846456), authors showed that when DC vaccines are loaded with immunosuppressive glioma stem cells (GSC) mRNA, they can recognize and activate CD8+ T cells and NK cells, consistent with their preclinical results [113]. A different study (NCT00323115) examined the percentage and number of the Tregs post-treatment in which there was an increase in percentage of Tregs but not in their absolute numbers. They also showed that there was an increase in CD8+ memory cells and naive B cells [114].
Immunomodulators
Cytotoxic T Lymphocyte-associated Antigen 4 (CTLA-4)
One of the costimulatory signals for T cell activation involves the binding of B7-1 or B7-2 cofactors to their receptor, CD28 (positive regulator of T cell activation).With higher affinity, these cofactors (B7s) also bind to CTLA-4, a key negative regulator of T cell activation. These higher affinity-based interactions with CTLA-4 cause the inactivation and cell cycle arrest of T cells. Therefore, the balance between positive and negative regulation is essential to generate and maintain the T cell-based immune response [34, 36, 37]. Based on this understanding, anti-CTLA-4 (inhibition of the negative regulator) has been shown to induce tumor antigen-specific T cell immunity (adoptive immunity) and many clinical trials have been developed using this anti-CTLA-4 based T cell immunity induction (Supplementary Table 1).
This method has been used as both a monotherapy and in combination with vaccines, radiation, chemotherapy, and surgery, demonstrating highly synergistic efficacy [109]. In a recent phase III clinical trial, ipilimumab (commercial anti-CTLA antibody) monotherapy showed an overall response rate of 10.9%. Although significant tumor regression has been observed in many cancer treatment such as melanoma, prostate cancer, malignant mesothelioma, and lung cancer, autoimmune side effects (thyroiditis, colitis, and vitiligo) were observed in certain subsets of patients but all patients recovered after discontinuation of further treatment and steroid therapy [115]. These unwanted effects should be examined more carefully in order to better treat patients in these smaller categories considering that significant anti-tumor effects are observed and thus represent an exciting avenue of anti-glioma treatment.
Programmed death 1(PD1) and programmed death ligand 1(PD-L1)
PD-1 is a member of the CD28/CTLA-4 family, a type I membrane protein in the immunoglobulin superfamily, which has two ligands: PD-L1 and PD-L2. As PD-L1 is highly upregulated in macrophages and DCs, the interaction between PD-L1 and PD-1 on T cells negatively regulates T cell activation. PD-L1 has been shown to be mostly expressed on tumor cells, causing T cell apoptosis or anergy when it binds to PD-1 on T cells [106, 116].
The expression of PD-L1 is regulated mostly by the Akt pathway. Akt is activated through PI(3)K, which, in turn, upregulates the expression of PD-L1. This activation is negatively regulated by PTEN. So when a mutation or loss of PTEN (a common phenomenon of cancer cells) occurs, Akt is constitutively active, yielding high expression of PD-L1 [117].Through this mechanism, cancerous cells accumulate PD-L1 on their surface in order to protect them from the anti-tumor T cell response.
Anti PD-1/PD-L1 antibody-based therapies have seen significant success in clinical trials for different cancer treatments (Supplementary Table 1). A phase I clinical trial with PD-L1 antibody (BMS-936559/MDX-1105) demonstrated observable tumor regression in 6–17% of patients and prolonged disease stabilization in 12–41% patients at 24 weeks post-treatment in melanoma and kidney cancer therapies. In a PD-1 antibody-based clinical therapy for melanoma, non-small cell lung cancer, and renal cell cancer (BMS-936558/MDX-1106/nivolumab), 31%, 17%, and 29% of patients, respectively, showed marked therapeutic response [118]. A common side effect for this treatment involves fatigue albeit to the extent that it did not require suspending treatment and did not negate from the therapeutic efficacy [116]. In some patients, however, there were some serious side effects (pneumonitis and interstitial nephritis) that required adjunct treatment and the arrest of the anti-PD-1 treatment [118, 119] . Other than in this small collection of patients, the treatment was generally durable and, specifically in patients with PD-L1 positive tumor cell surface, an even better response was noted [119]. However, this approach, in recent phase III clinical trial did not show any therapeutic efficacy in patients with the recurrence of glioblastoma multiforme (GBM) and further analysis are needed (unpublished, data were presented at the World Federation of Neuro-Oncology Societies (WFNOS) meeting in 2017).
3.2.3. 4-1BB and 4-1BBL
4-1BB (CD137) is an agonistic receptor, mostly present on activated T cells, natural killer cells (NKs), and antigen-activated regulatory T-cells [120]. This receptor belongs to the tumor necrosis factor (TNF) super family and binds to its ligand 4-1BBL (CD137L) present on APCs [121], such as B cells, DCs, and macrophages [122]. When 4-1BB binds to its ligand, 4-1BBL, it causes the production of costimulatory cytokines such as IFN-γ and IL-2, while also resulting in an increase in the cytotoxicity of the CD8 T-cells through T cell receptor (TCR) signaling [123]. For this reason, 4-1BB-based immunotherapy can elicit both innate and adaptive immune responses against cancerous cells, indicative of a high potency in a clinical setting.
Anti-4-1BB (anti-CD 137) antibody has been used in clinical trials as a single therapy as well as in combination with other check-point blockades or the typical standard of care, related to advanced solid tumors, as well as non-small cell lung cancer, colorectal cancer, head and neck cancer, multiple myeloma, and malignant melanoma. A phase I clinical study of anti-4-1BB (PF-05082566) showed a best overall response of stable disease in 22% (6/27) of patients with advanced cancers. The most common side effects that were reported with this treatment were fatigue, neutropenia, rash, and diarrhea. The most common clinical abnormalities were increased liver function (ALT and AST counts), leukopenia, thrombocytopenia, and hyperbilirubinemia [124].
Advantage and disadvantage of immunotherapeutic approaches in glioma clinical settings
As recognition of the importance of cancer immunotherapy has increased, many pre-clinical (animal models) and clinical investigations for both DC-based vaccines and immunomodulatory-based agents have been explored [23, 24, 27, 32, 108, 109, 124]. Given the rapid progress for this field, all immunotherapies except for anti-4-1BB (anti CD137) introduced in this review have been studied in clinical trials for glioma. Supplementary Table 1 lists the clinical trials for glioblastoma that have been completed or are currently in progress. However, there has been concern that these approaches could change the overall balance of the immune system in such a way that may lead to serious autoimmune side effects. Moreover, many agents of these currently developed check-point blockades cannot pass the BBB which could minimize the therapeutic efficacy for tumors of the CNS [3, 6, 7, 19, 24, 48, 52, 111, 115]. For that reason, it has been suggested that the local delivery of these agents via tumor-specific delivery vehicles around/in neoplastic cells may be immensely beneficial.
Combinatory therapies: oncolytic adenoviral vehicle with local delivery of check point blockade based immunomodulator(s)
As mentioned in previous sections, both oncolytic virotherapy and immunotherapy have their advantages and disadvantages. Nevertheless, both approaches face the inherent hurdle that all glioma therapies face: a highly immunosuppressive microenvironment and guard by the BBB [23, 27, 108, 110]. However, combining these two therapeutic modules can maximize the eradication efficiency of glioma, since oncolytic adenoviruses can be customized to specifically replicate within and destroy tumor cells but also transiently (in significant yet controllable amounts) deliver therapeutic genes such as immunomodulator(s) that have been incorporated into the viral vectors.
Importantly, the accumulated evidence confirms the safety of oncolytic virus usage in clinical studies for the treatment of glioma [1, 6, 9, 11, 13, 19, 46]. As such, the oncolytic viruses and therapeutic immunomodulator(s) described previously can be investigated in conjunction and be expected to boost the efficacy of one another. For instance, therapeutic efficacy of Ad-Flt3L, oncolytic adenovirus expressing fms-like tyrosine kinase ligand (an immunostimulatory cytokine that recruits DCs to tumor sites) in combination with conditionally replicating oncolytic adenovirus (Ad-TK), and Ad-hIL12 have been shown to be more efficient in glioma animal models and are currently under investigation in a phase I clinical trial, Table 1. Both oncolytic adenovirus based therapy expressing IL-12 and 4-1BBL in a melanoma animal model system and oncolytic adenovirus expression of soluble PD-1 in a colon cancer model have demonstrated, albeit not in a glioma model system, highly enhanced combinatory therapeutic efficacy [121, 125].
Although immunological treatments are currently more pursued for clinical cancer therapies, the potential for even better cancer eradication can be achieved with the aid of oncolytic adenoviruses’ therapeutic capabilities. This new combinatorial platform can exploit multiple therapeutic features simultaneously. First, oncolytic viruses will enter cancerous cells and express immunomodulator(s) locally around the glioma. This will change the immunosuppressive microenvironment of glioma to an immuno-vulnerable environment by either enhancing the activation of cytotoxic T cells or removing the immunosuppressive protection of glioma cells. Second, oncolytic adenoviruses will replicate in a tumor-specific manner and lyse tumor cells in a coordinated manner. Third, oncolysis-mediated release of tumor antigen(s) will ensue for which the said antigen(s) will be engulfed by APCs such as DCs and processing/presentation will follow, eliciting an immune response. Fourth, although there are not many supporting mechanistic analyses, it has shown that oncolytic adenoviruses can also inhibit the DNA repair system in cancerous cells upon radiation therapy (radio-sensitization) [9, 10, 13, 95, 97]. It is therefore possible that oncolytic adenovirus-mediated impairment of DNA repair can cause the death of glioma cells in addition to the oncolytic activity [95, 97]. Lastly, after the loss of the immunosuppressive characterization of glioma via the first therapeutic effect, the un-penetrable, residual solid tumor mass can be eradicated completely by activated immune cells which are orchestrated by tumor-specific antigen presenting DCs, as shown in the Figure 2.
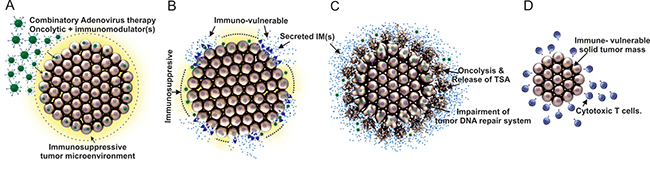
Figure 2: Mechanism of combinatory therapies for the treatment of glioma: oncolytic virotherapy paired with immunotherapy. (A) Entry of oncolytic adenovirus into glioma cells (immunosuppressive microenvironment in yellow color). (B) Release of immunomodulator (IM) from oncolytic adenovirus infected glioma cells (a change of the tumor microenvironment from immunosuppressive to immunovulnerable via activation of cytotoxic T cells or removal of glioma cells’ immunosuppression capability affecting systemic immune repertoire). (C) Specific lysis of cancerous cells, induction of tumor specific antigens (TSA) release, and lastly, via the least known mechanism, impairment of tumor DNA repair system. (D) Eradication of the residual, immunovulnerable solid tumor mass by anti-cancer immune responses such as cytotoxic T cells.
CONCLUSIONS
This review outlines some of the challenges associated with traditional glioblastoma therapies like surgery and chemotherapy. These tumors are poorly circumscribed and invasive in nature, making traditional modes of treatment vastly less effective [9, 109]. Next, this paper describes some of the newly identified hurdles facing biologically active immunosuppressive agents and virotherapy for the treatment of glioma, such as the immunosuppressive tumor microenvironment and innate immunity to viral agents [6, 7, 9, 13, 22, 23, 32, 108, 109]. Nevertheless, there is encouraging data for immunotherapy and virotherapy in terms of their safety and/or effectiveness for other solid tumors. The benefit of combining these two therapeutic approaches to produce a synergistic anti-tumor effect and greater efficacy while maintaining patient safety is an attractive source of therapeutic consideration and thus warrants further investigation that can be applied for augmented therapeutic efficacy in anti-tumor effects and enhanced patient survival.
Adenoviral vectors have repeatedly been shown to have genomes that can be successfully modified for the generation of targeted, specific oncolytic virotherapy [6, 9, 11–13, 19]. Multiple steps in the viral infection and replication life cycle can be customized to create specific and selective agents for tumor destruction. For example, the cell surface receptor targeted by the viral fiber can be altered to enhance tumor infection, and the transcription of viral proteins can be restricted to cancerous cells, resulting in a viral construct with the ability to easily infect and selectively kill glioma cells [9–11, 13, 19, 46, 49]. However, limited distribution of the virus throughout the tumor and pre-existing systemic immunity and toxicity present hurdles for the efficacy of oncolytic virotherapy that must be overcome in order for the treatment to manifest into a therapy that has pronounced effectiveness [41, 42, 46, 49, 50].
Immunotherapy has been shown to be substantially effective for multiple solid tumor types. This approach is particularly enticing for glioma therapy given the strongly immunosuppressive nature of the tumor environment. There are multiple phases in the adaptive immune response that can be targeted: TA presentation with DCs, T cell checkpoint blockades to reduce the immunosuppression present in the tumor environment, and direct stimulation of activated T cells to name a few. Even these powerful therapies face challenges when utilized for glioma therapy like the restrictive nature of the BBB and systemic autoimmune generation [23, 25, 27, 32, 109, 110].
The ability for these seemingly divergent therapies, one being infection and the other an immune system activator, to complement each other may yield numerous benefits. For example, the autoimmune adverse effects of systemic checkpoint inhibition can be fatal, forcing some patients to discontinue immunomodulatory therapy [110, 111, 122]. But if these agents are only transiently delivered directly/locally to the tumor microenvironment, there should be a significantly reduced risk of systemic side effects. Although there is impairment of the BBB within the tumor, this impairment is not universally present throughout the entire tumor capillary bed. As such, systemic immunotherapies may not penetrate the entire tumor area. However, if the immunotherapeutic protein is actually produced when a viral vector that was directly injected into the tumor intra-operatively infects the tumor cells, themselves, then this is a barrier to therapy that can be overcome. Furthermore, it has been well established that viral cell death can generate an immune response and result in the exposure of tumor antigens to the immune system along with impairment of the DNA repair system in tumor cells such that combining this tumor-specific, immunologic cell death with immune stimulating therapy should be capable of producing a robust immune response [6, 9, 16, 40, 42, 46, 93].
There are still certain limitations that need to be addressed. Modifying the viral genome to carry the gene for a large immune system modulating protein may impair the viruses’ ability to replicate and spread. Additionally, turning the infected tumor cell into a “factory” for therapeutic immune-activating protein production requires that the cell is able to sufficiently generate enough therapeutic protein prior to its lysis. Recruiting the immune system to the tumor could potentially have the unwanted effect of limiting viral spread as well if the immune system begins to recognize and kill viral-infected cells. But if a robust immune response is generated against the tumor, there is potential for significant cerebral edema to occur, which can have catastrophic complications. However, these limitations can be also overcome by current and future pre-clinical and clinical research investigating more efficient and less toxic delivery for such treatment approaches in the treatment of glioblastoma.
The translational studies of oncolytic adenovirus-based therapies have made their way to many pre-clinical/clinical trials that have, by and large, shown pronounced safety and efficacy. Such approaches mechanistically induce a multi-level combination of tumor eradication activities: (1) specific lysis of cancerous cells, (2) induction of tumor specific antigen(s) release, and, (3) lastly, via the least known mechanism, impairment of tumor DNA repair system. However, based on current research in the field of oncolytic adenovirus-based therapy, even with well-crafted modifications to enhance affinity and tumor-restricted replication, there is still a need for additional support with other therapeutic approaches due to (1) the incapability to penetrate a solid mass of glioma and (2) inhibition of additional therapeutic immune activation/boost against tumor antigen(s) released via oncolytic activity by the immunosuppressive activity of glioma [6, 9, 13, 19, 95, 97].
As a rapidly progressing field in cancer therapy, immunotherapy is one of the most expanding and well-funded research areas at this time [23, 109]. DC-based vaccination and the use of immunomodulators (T cell check point blockades) aim to manipulate the immune system by either boosting anti-tumor immunity or extinguishing the immunosuppressive tumor microenvironment [23, 32, 93, 109, 110]. Both of these approaches have shown clinical efficacy, albeit with side effects such as autoimmunity [23, 24, 109]. However, in the case of current ex vivo DC-based tumor vaccine approaches, these applications are highly labor intensive and only allowed to be performed in good manufacturing practice (GMP) facilities [109, 111]. For future, optimal use of this application in a more logistically feasible manner, it is essential to generate single-step, in vivo DC-specific delivery vehicles that can facilitate highly efficient multi-epitope tumor antigen loading for maximal anti-tumor immunity. Additionally, problems resultant of the generation of systemic autoimmunity and the hindrance of delivery beyond the BBB are still faced. Specifically in immunomodulatory approaches, most immunomodulators in use cannot be transferred across the BBB and, in high concentration, have shown to be poorly tolerated by patients and are linked to the advent of autoimmunity as well as various other side effects [23, 109].
In light of this fact, the activation of the immune system and, specifically, local immune response activation are necessary to achieve better therapeutic effect in glioma treatment. Future endeavors to develop efficient and clinically relevant immunotherapies will require alleviation of these concerns in the context of both glioma abrogation and patient safety. Therefore, multi-level therapy such as combination of immunomodulators and viral vector approaches should be considered as a subject of further research investigations. In this approach, the infection of neoplastic cells with oncolytic viruses carrying immunomodulator(s), in addition to operating in an oncolytic capacity, introduces the immunomodulatory(s) to become locally expressed by the cancerous cells, thus allowing for a change of the tumor microenvironment from immunosuppressive to immune-vulnerable via activation of cytotoxic T cells or removal of glioma cells’ immunosuppressive capability affecting the systemic immune repertoire, followed by the oncolytic activity-mediated tumor eradication processes.
In conjunction with immunomodulatory approaches, it is expected that these therapies can boost the efficacy of one another. Future studies will be necessary to evaluate the effects of this combination in the hope that their conjunction will result in more pronounced anti-glioma effects that are pertinent to patient safety at the same time. Therefore, in the heart of this review, we propose the marriage of two sets of demonstrably efficacious and well-studied anti-tumor therapies for the treatment of glioma: oncolytic virotherapy paired with immunotherapy.
CONFLICTS OF INTEREST
None declared.
FUNDING
This work is supported by NIH grants R01CA122930, R35CA197725, R01NS077388 and R01NS87990, R01NS093903.
REFERENCES
1. Assi H, Candolfi M, Baker G, Mineharu Y, Lowenstein PR, Castro MG. Gene therapy for brain tumors: basic developments and clinical implementation. Neurosci Lett. 2012; 527:71–7.
2. Grossman SA, Ye X, Piantadosi S, Desideri S, Nabors LB, Rosenfeld M, Fisher J, Consortium NC. Survival of patients with newly diagnosed glioblastoma treated with radiation and temozolomide in research studies in the United States. Clin Cancer Res. 2010; 16:2443–9. https://doi.org/10.1158/1078–0432.CCR-09-3106.
3. Wainwright DA, Dey M, Chang A, Lesniak MS. Targeting Tregs in Malignant Brain Cancer: Overcoming IDO. Front Immunol. 2013; 4:116. https://doi.org/10.3389/fimmu.2013.00116.
4. Wei J, Wu A, Kong LY, Wang Y, Fuller G, Fokt I, Melillo G, Priebe W, Heimberger AB. Hypoxia potentiates glioma-mediated immunosuppression. PLoS One. 2011; 6:e16195. https://doi.org/10.1371/journal.pone.0016195.
5. Xu S, Shao QQ, Sun JT, Yang N, Xie Q, Wang DH, Huang QB, Huang B, Wang XY, Li XG, Qu X. Synergy between the ectoenzymes CD39 and CD73 contributes to adenosinergic immunosuppression in human malignant gliomas. Neuro Oncol. 2013; 15:1160–72. https://doi.org/10.1093/neuonc/not067.
6. Choi JW, Lee JS, Kim SW, Yun CO. Evolution of oncolytic adenovirus for cancer treatment. Adv Drug Deliv Rev. 2012; 64:720–9.
7. Galanis E. Therapeutic potential of oncolytic measles virus: promises and challenges. Clin Pharmacol Ther. 2010; 88:620–5. https://doi.org/10.1038/clpt.2010.211.
8. Hardcastle J, Kurozumi K, Chiocca EA, Kaur B. Oncolytic viruses driven by tumor-specific promoters. Curr Cancer Drug Targets. 2007; 7:181–9.
9. Haseley A, Alvarez-Breckenridge C, Chaudhury AR, Kaur B. Advances in oncolytic virus therapy for glioma. Recent Pat CNS Drug Discov. 2009; 4:1–13.
10. Kay MA. State-of-the-art gene-based therapies: the road ahead. Nat Rev Genet. 2011; 12:316–28.
11. Arnberg N. Adenovirus receptors: implications for targeting of viral vectors. Trends Pharmacol Sci. 2012; 33:442–8. https://doi.org/10.1016/j.tips.2012.04.005.
12. Chen RF, Lee CY. Adenoviruses types, cell receptors and local innate cytokines in adenovirus infection. Int Rev Immunol. 2014; 33:45–53.
13. Hedley SJ, Chen J, Mountz JD, Li J, Curiel DT, Korokhov N, Kovesdi I. Targeted and shielded adenovectors for cancer therapy. Cancer Immunol Immunother. 2006; 55:1412–9.
14. Kawakami Y, Li H, Lam JT, Krasnykh V, Curiel DT, Blackwell JL. Substitution of the adenovirus serotype 5 knob with a serotype 3 knob enhances multiple steps in virus replication. Cancer Res. 2003; 63:1262–9.
15. Kim JW, Morshed RA, Kane JR, Auffinger B, Qiao J, Lesniak MS. Viral Vector Production: Adenovirus. Methods Mol Biol. 2016; 1382:115–30. https://doi.org/10.1007/978-1-4939-3271-9_9.
16. Liu TC, Kirn D. Gene therapy progress and prospects cancer: oncolytic viruses. Gene Ther. 2008; 15:877–84.
17. Kim JW, Auffinger B, Spencer DA, Miska J, Chang AL, Kane JR, Young JS, Kanojia D, Qiao J, Mann JF, Zhang L, Wu M, Ahmed AU, et al. Single dose GLP toxicity and biodistribution study of a conditionally replicative adenovirus vector, CRAd-S-pk7, administered by intracerebral injection to Syrian hamsters. J Transl Med. 2016; 14:134. https://doi.org/10.1186/s12967-016-0895-8.
18. Sterman DH, Alley E, Stevenson JP, Friedberg J, Metzger S, Recio A, Moon EK, Haas AR, Vachani A, Katz SI, Sun J, Heitjan D, Hwang WT, et al. Pilot And Feasibility Trial Of Immuno-Gene Therapy Of Malignant Mesothelioma Using Intrapleural Delivery Of Adenovirus- Interferon-Alpha Combined With Chemotherapy. Clin Cancer Res. 2016. https://doi.org/10.1158/1078-0432.ccr-15-2133.
19. Buijs PR, Verhagen JH, van Eijck CH, van den Hoogen BG. Oncolytic viruses: From bench to bedside with a focus on safety. Hum Vaccin Immunother. 2015; 11:1573–84. https://doi.org/10.1080/21645515.2015.1037058.
20. Freytag SO, Stricker H, Lu M, Elshaikh M, Aref I, Pradhan D, Levin K, Kim JH, Peabody J, Siddiqui F, Barton K, Pegg J, Zhang Y, et al. Prospective randomized phase 2 trial of intensity modulated radiation therapy with or without oncolytic adenovirus-mediated cytotoxic gene therapy in intermediate-risk prostate cancer. Int J Radiat Oncol Biol Phys. 2014; 89:268–76. https://doi.org/10.1016/j.ijrobp.2014.02.034.
21. Hemminki O, Parviainen S, Juhila J, Turkki R, Linder N, Lundin J, Kankainen M, Ristimaki A, Koski A, Liikanen I, Oksanen M, Nettelbeck DM, Kairemo K, et al. Immunological data from cancer patients treated with Ad5/3-E2F-Delta24-GMCSF suggests utility for tumor immunotherapy. Oncotarget. 2015; 6:4467–81. https://doi.org/10.18632/oncotarget.2901.
22. Ye W, Liu R, Pan C, Jiang W, Zhang L, Guan Z, Wu J, Ying X, Li L, Li S, Tan W, Zeng M, Kang T, et al. Multicenter randomized phase 2 clinical trial of a recombinant human endostatin adenovirus in patients with advanced head and neck carcinoma. Mol Ther. 2014; 22:1221–9. https://doi.org/10.1038/mt.2014.53.
23. Binder DC, Davis AA, Wainwright DA. Immunotherapy for cancer in the central nervous system: Current and future directions. Oncoimmunology. 2016; 5:e1082027. https://doi.org/10.1080/2162402x.2015.1082027.
24. Lieberman NA, Moyes KW, Crane CA. Developing immunotherapeutic strategies to target brain tumors. Expert Rev Anticancer Ther. 2016. https://doi.org/10.1080/14737140.2016.1192470.
25. Lim M, Weller M, Chiocca EA. Current State of Immune-Based Therapies for Glioblastoma. Am Soc Clin Oncol Educ Book. 2016; 35:e132–9. https://doi.org/10.14694/edbk_159084.
26. Wainwright DA, Chang AL, Dey M, Balyasnikova IV, Kim CK, Tobias A, Cheng Y, Kim JW, Qiao J, Zhang L, Han Y, Lesniak MS. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin Cancer Res. 2014; 20:5290–301. https://doi.org/10.1158/1078-0432.ccr-14-0514.
27. Marrone KA, Ying W, Naidoo J. Immune-related Adverse Events from Immune Checkpoint Inhibitors. Clin Pharmacol Ther. 2016. https://doi.org/10.1002/cpt.394.
28. McNamara MJ, Hilgart-Martiszus I, Barragan Echenique DM, Linch SN, Kasiewicz MJ, Redmond WL. Interferon-gamma production by peripheral lymphocytes predicts survival of tumor-bearing mice receiving dual PD-1/CTLA-4 blockade. Cancer Immunol Res. 2016. https://doi.org/10.1158/2326-6066.cir-16-0022.
29. Planchard D, Yokoi T, McCleod MJ, Fischer JR, Kim YC, Ballas M, Shi K, Soria JC. A Phase III Study of Durvalumab (MEDI4736) With or Without Tremelimumab for Previously Treated Patients With Advanced NSCLC: Rationale and Protocol Design of the ARCTIC Study. Clin Lung Cancer. 2016; 17:232–6.e1. https://doi.org/10.1016/j.cllc.2016.03.003.
30. Armand P, Nagler A, Weller EA, Devine SM, Avigan DE, Chen YB, Kaminski MS, Holland HK, Winter JN, Mason JR, Fay JW, Rizzieri DA, Hosing CM, et al. Disabling immune tolerance by programmed death-1 blockade with pidilizumab after autologous hematopoietic stem-cell transplantation for diffuse large B-cell lymphoma: results of an international phase II trial. J Clin Oncol. 2013; 31:4199–206. https://doi.org/10.1200/jco.2012.48.3685.
31. Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, Stankevich E, Pons A, Salay TM, McMiller TL, Gilson MM, Wang C, Selby M, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010; 28:3167–75. https://doi.org/10.1200/jco.2009.26.7609.
32. Godwin JL, Zibelman M, Plimack ER, Geynisman DM. Immune checkpoint blockade as a novel immunotherapeutic strategy for renal cell carcinoma: a review of clinical trials. Discov Med. 2014; 18:341–50.
33. Hodi FS, Lawrence D, Lezcano C, Wu X, Zhou J, Sasada T, Zeng W, Giobbie-Hurder A, Atkins MB, Ibrahim N, Friedlander P, Flaherty KT, Murphy GF, et al. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol Res. 2014; 2:632–42. https://doi.org/10.1158/2326-6066.cir-14-0053.
34. Kwek SS, Dao V, Roy R, Hou Y, Alajajian D, Simko JP, Small EJ, Fong L. Diversity of antigen-specific responses induced in vivo with CTLA-4 blockade in prostate cancer patients. J Immunol. 2012; 189:3759–66. https://doi.org/10.4049/jimmunol.1201529.
35. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, Ferrucci PF, Hill A, Wagstaff J, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015; 373:23–34. https://doi.org/10.1056/NEJMoa1504030.
36. Ribas A, Benz MR, Allen-Auerbach MS, Radu C, Chmielowski B, Seja E, Williams JL, Gomez-Navarro J, McCarthy T, Czernin J. Imaging of CTLA4 blockade-induced cell replication with (18)F-FLT PET in patients with advanced melanoma treated with tremelimumab. J Nucl Med. 2010; 51:340–6. https://doi.org/10.2967/jnumed.109.070946.
37. Santegoets SJ, Stam AG, Lougheed SM, Gall H, Scholten PE, Reijm M, Jooss K, Sacks N, Hege K, Lowy I, Cuillerot JM, von Blomberg BM, Scheper RJ, et al. T cell profiling reveals high CD4+CTLA-4 + T cell frequency as dominant predictor for survival after prostate GVAX/ipilimumab treatment. Cancer Immunol Immunother. 2013; 62:245–56. https://doi.org/10.1007/s00262-012-1330-5.
38. Westin JR, Chu F, Zhang M, Fayad LE, Kwak LW, Fowler N, Romaguera J, Hagemeister F, Fanale M, Samaniego F, Feng L, Baladandayuthapani V, Wang Z, et al. Safety and activity of PD1 blockade by pidilizumab in combination with rituximab in patients with relapsed follicular lymphoma: a single group, open-label, phase 2 trial. Lancet Oncol. 2014; 15:69–77. https://doi.org/10.1016/s1470-2045(13)70551-5.
39. Thomas CE, Ehrhardt A, Kay MA. Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet. 2003; 4:346–58.
40. Ramirez M, Garcia-Castro J, Alemany R. Oncolytic virotherapy for neuroblastoma. Discov Med. 2010; 10:387–93.
41. Volpers C, Kochanek S. Adenoviral vectors for gene transfer and therapy. J Gene Med. 2004; 6:S164–71.
42. Wold WS, Toth K. Adenovirus vectors for gene therapy, vaccination and cancer gene therapy. Curr Gene Ther. 2013; 13:421–33.
43. Balint JP, Gabitzsch ES, Rice A, Latchman Y, Xu Y, Messerschmidt GL, Chaudhry A, Morse MA, Jones FR. Extended evaluation of a phase 1/2 trial on dosing, safety, immunogenicity, and overall survival after immunizations with an advanced-generation Ad5 [E1-, E2b-]-CEA(6D) vaccine in late-stage colorectal cancer. Cancer Immunol Immunother. 2015; 64:977–87. https://doi.org/10.1007/s00262-015-1706-4.
44. Dreno B, Urosevic-Maiwald M, Kim Y, Guitart J, Duvic M, Dereure O, Khammari A, Knol AC, Derbij A, Lusky M, Didillon I, Santoni AM, Acres B, et al. TG1042 (Adenovirus-interferon-gamma) in primary cutaneous B-cell lymphomas: a phase II clinical trial. PLoS One. 2014; 9:e83670. https://doi.org/10.1371/journal.pone.0083670.
45. Rojas-Martinez A, Manzanera AG, Sukin SW, Esteban-Maria J, Gonzalez-Guerrero JF, Gomez-Guerra L, Garza-Guajardo R, Flores-Gutierrez JP, Elizondo Riojas G, Delgado-Enciso I, Ortiz-Lopez R, Aguilar LK, Butler EB, et al. Intraprostatic distribution and long-term follow-up after AdV-tk immunotherapy as neoadjuvant to surgery in patients with prostate cancer. Cancer Gene Ther. 2013; 20:642–9. https://doi.org/10.1038/cgt.2013.56.
46. Nandi S, Lesniak MS. Adenoviral virotherapy for malignant brain tumors. Expert Opin Biol Ther. 2009; 9:737–47. https://doi.org/10.1517/14712590902988451.
47. Fields BN, Knipe DM, Howley PM. Fields virology. (United States: Philadelphia : Wolters Kluwer Health/Lippincott Williams & Wilkins, c2013).
48. Kim JW, Glasgow JN, Nakayama M, Ak F, Ugai H, Curiel DT. An adenovirus vector incorporating carbohydrate binding domains utilizes glycans for gene transfer. PLoS One. 2013; 8:e55533. https://doi.org/10.1371/journal.pone.0055533.
49. Mizuguchi H, Hayakawa T. Targeted adenovirus vectors. Hum Gene Ther. 2004; 15:1034–44. https://doi.org/10.1089/hum.2004.15.1034.
50. Zheng S, Ulasov IV, Han Y, Tyler MA, Zhu ZB, Lesniak MS. Fiber-knob modifications enhance adenoviral tropism and gene transfer in malignant glioma. J Gene Med. 2007; 9:151–60. https://doi.org/10.1002/jgm.1008.
51. Ulasov IV, Tyler MA, Zheng S, Han Y, Lesniak MS. CD46 represents a target for adenoviral gene therapy of malignant glioma. Hum Gene Ther. 2006; 17:556–64. https://doi.org/10.1089/hum.2006.17.556.
52. Ulasov IV, Rivera AA, Han Y, Curiel DT, Zhu ZB, Lesniak MS. Targeting adenovirus to CD80 and CD86 receptors increases gene transfer efficiency to malignant glioma cells. J Neurosurg. 2007; 107:617–27. https://doi.org/10.3171/jns-07/09/0617.
53. Wang H, Li Z, Yumul R, Lara S, Hemminki A, Fender P, Lieber A. Multimerization of adenovirus serotype 3 fiber knob domains is required for efficient binding of virus to desmoglein 2 and subsequent opening of epithelial junctions. J Virol. 2011; 85:6390–402. https://doi.org/10.1128/jvi.00514-11.
54. Rein DT, Breidenbach M, Wu H, Han T, Haviv YS, Wang M, Kirby TO, Kawakami Y, Dall P, Alvarez RD, Curiel DT. Gene transfer to cervical cancer with fiber-modified adenoviruses. Int J Cancer. 2004; 111:698–704. https://doi.org/10.1002/ijc.20295.
55. Wu H, Han T, Lam JT, Leath CA, Dmitriev I, Kashentseva E, Barnes MN, Alvarez RD, Curiel DT. Preclinical evaluation of a class of infectivity-enhanced adenoviral vectors in ovarian cancer gene therapy. Gene Ther. 2004; 11:874–8. https://doi.org/10.1038/sj.gt.3302249.
56. Wu H, Seki T, Dmitriev I, Uil T, Kashentseva E, Han T, Curiel DT. Double modification of adenovirus fiber with RGD and polylysine motifs improves coxsackievirus-adenovirus receptor-independent gene transfer efficiency. Hum Gene Ther. 2002; 13:1647–53. https://doi.org/10.1089/10430340260201734.
57. Wickham TJ, Roelvink PW, Brough DE, Kovesdi I. Adenovirus targeted to heparan-containing receptors increases its gene delivery efficiency to multiple cell types. Nat Biotechnol. 1996; 14:1570–3. https://doi.org/10.1038/nbt1196-1570.
58. Dong W, van Ginkel JW, Au KY, Alemany R, Meulenberg JJ, van Beusechem VW. ORCA-010, a novel potency-enhanced oncolytic adenovirus, exerts strong antitumor activity in preclinical models. Hum Gene Ther. 2014; 25:897–904. https://doi.org/10.1089/hum.2013.229.
59. Krause A, Whu WZ, Qiu J, Wafadari D, Hackett NR, Sharma A, Crystal RG, Worgall S. RGD capsid modification enhances mucosal protective immunity of a non-human primate adenovirus vector expressing Pseudomonas aeruginosa OprF. Clin Exp Immunol. 2013; 173:230–41. https://doi.org/10.1111/cei.12101.
60. Lang FF, Conrad C, Gomez-Manzano C, Tufaro F, Yung WKA, Sawaya R, Weinberg J, Prabhu S, Fuller G, Aldape K, Fueyo J. First-in-human phase i clinical trial of oncolytic delta-24-rgd (dnx-2401) with biological endpoints: implications for viro- immunotherapy. Neuro-Oncology. 2014; 16:iii39–iii. https://doi.org/10.1093/neuonc/nou208.61.
61. Lang FF, Conrad C, Gomez-Manzano C, Tufaro F, Sawaya R, Weinberg J, Prabhu S, Fuller G, Aldape K, Fueyo J. Nt-18Phase I Clinical Trial Of Oncolytic Virus Delta-24-Rgd (Dnx-2401) With Biological Endpoints: Implications For Viro-Immunotherapy. Neuro-Oncology. 2014; 16:v162-v. https://doi.org/10.1093/neuonc/nou265.17.
62. Kim KH, Dmitriev I, O’Malley JP, Wang M, Saddekni S, You Z, Preuss MA, Harris RD, Aurigemma R, Siegal GP, Zinn KR, Curiel DT, Alvarez RD. A phase I clinical trial of Ad5.SSTR/TK.RGD, a novel infectivity-enhanced bicistronic adenovirus, in patients with recurrent gynecologic cancer. Clin Cancer Res. 2012; 18:3440–51. https://doi.org/10.1158/1078-0432.ccr-11-2852.
63. Kimball KJ, Preuss MA, Barnes MN, Wang M, Siegal GP, Wan W, Kuo H, Saddekni S, Stockard CR, Grizzle WE, Harris RD, Aurigemma R, Curiel DT, et al. A phase I study of a tropism-modified conditionally replicative adenovirus for recurrent malignant gynecologic diseases. Clin Cancer Res. 2010; 16:5277–87. https://doi.org/10.1158/1078-0432.ccr-10-0791.
64. Nokisalmi P, Pesonen S, Escutenaire S, Sarkioja M, Raki M, Cerullo V, Laasonen L, Alemany R, Rojas J, Cascallo M, Guse K, Rajecki M, Kangasniemi L, et al. Oncolytic adenovirus ICOVIR-7 in patients with advanced and refractory solid tumors. Clin Cancer Res. 2010; 16:3035–43. https://doi.org/10.1158/1078-0432.ccr-09-3167.
65. Paoloni MC, Tandle A, Mazcko C, Hanna E, Kachala S, Leblanc A, Newman S, Vail D, Henry C, Thamm D, Sorenmo K, Hajitou A, Pasqualini R, et al. Launching a novel preclinical infrastructure: comparative oncology trials consortium directed therapeutic targeting of TNFalpha to cancer vasculature. PLoS One. 2009; 4:e4972. https://doi.org/10.1371/journal.pone.0004972.
66. Pesonen S, Diaconu I, Cerullo V, Escutenaire S, Raki M, Kangasniemi L, Nokisalmi P, Dotti G, Guse K, Laasonen L, Partanen K, Karli E, Haavisto E, et al. Integrin targeted oncolytic adenoviruses Ad5-D24-RGD and Ad5-RGD-D24-GMCSF for treatment of patients with advanced chemotherapy refractory solid tumors. Int J Cancer. 2012; 130:1937–47. https://doi.org/10.1002/ijc.26216.
67. Kanerva A, Mikheeva GV, Krasnykh V, Coolidge CJ, Lam JT, Mahasreshti PJ, Barker SD, Straughn M, Barnes MN, Alvarez RD, Hemminki A, Curiel DT. Targeting adenovirus to the serotype 3 receptor increases gene transfer efficiency to ovarian cancer cells. Clin Cancer Res. 2002; 8:275–80.
68. Takayama K, Reynolds PN, Short JJ, Kawakami Y, Adachi Y, Glasgow JN, Rots MG, Krasnykh V, Douglas JT, Curiel DT. A mosaic adenovirus possessing serotype Ad5 and serotype Ad3 knobs exhibits expanded tropism. Virology. 2003; 309:282–93.
69. Koski A, Kangasniemi L, Escutenaire S, Pesonen S, Cerullo V, Diaconu I, Nokisalmi P, Raki M, Rajecki M, Guse K, Ranki T, Oksanen M, Holm SL, et al. Treatment of cancer patients with a serotype 5/3 chimeric oncolytic adenovirus expressing GMCSF. Mol Ther. 2010; 18:1874–84. https://doi.org/10.1038/mt.2010.161.
70. Hemminki O, Bauerschmitz G, Hemmi S, Lavilla-Alonso S, Diaconu I, Guse K, Koski A, Desmond RA, Lappalainen M, Kanerva A, Cerullo V, Pesonen S, Hemminki A. Oncolytic adenovirus based on serotype 3. Cancer Gene Ther. 2011; 18:288–96. https://doi.org/10.1038/cgt.2010.79.
71. Lopez MV, Rivera AA, Viale DL, Benedetti L, Cuneo N, Kimball KJ, Wang M, Douglas JT, Zhu ZB, Bravo AI, Gidekel M, Alvarez RD, Curiel DT, et al. A tumor-stroma targeted oncolytic adenovirus replicated in human ovary cancer samples and inhibited growth of disseminated solid tumors in mice. Mol Ther. 2012; 20:2222–33. https://doi.org/10.1038/mt.2012.147.
72. Oneal MJ, Trujillo MA, Davydova J, McDonough S, Yamamoto M, Morris JC 3rd. Characterization of infectivity-enhanced conditionally replicating adenovectors for prostate cancer radiovirotherapy. Hum Gene Ther. 2012; 23:951–9. https://doi.org/10.1089/hum.2012.047.
73. Raki M, Sarkioja M, Escutenaire S, Kangasniemi L, Haavisto E, Kanerva A, Cerullo V, Joensuu T, Oksanen M, Pesonen S, Hemminki A. Switching the fiber knob of oncolytic adenoviruses to avoid neutralizing antibodies in human cancer patients. J Gene Med. 2011; 13:253–61. https://doi.org/10.1002/jgm.1565.
74. Ulasov IV, Tyler MA, Rivera AA, Nettlebeck DM, Douglas JT, Lesniak MS. Evaluation of E1A double mutant oncolytic adenovectors in anti-glioma gene therapy. J Med Virol. 2008; 80:1595–603. https://doi.org/10.1002/jmv.21264.
75. Ulasov IV, Tyler MA, Zhu ZB, Han Y, He TC, Lesniak MS. Oncolytic adenoviral vectors which employ the survivin promoter induce glioma oncolysis via a process of beclin-dependent autophagy. Int J Oncol. 2009; 34:729–42.
76. Harada JN, Berk AJ. p53-Independent and -dependent requirements for E1B-55K in adenovirus type 5 replication. J Virol. 1999; 73:5333–44.
77. Rothmann T, Hengstermann A, Whitaker NJ, Scheffner M, zur Hausen H. Replication of ONYX-015, a potential anticancer adenovirus, is independent of p53 status in tumor cells. J Virol. 1998; 72:9470–8.
78. Wang HG, Draetta G, Moran E. E1A induces phosphorylation of the retinoblastoma protein independently of direct physical association between the E1A and retinoblastoma products. Mol Cell Biol. 1991; 11:4253–65.
79. Fueyo J, Gomez-Manzano C, Alemany R, Lee PS, McDonnell TJ, Mitlianga P, Shi YX, Levin VA, Yung WK, Kyritsis AP. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene. 2000; 19:2–12. https://doi.org/10.1038/sj.onc.1203251.
80. Ranki T, Kanerva A, Ristimaki A, Hakkarainen T, Sarkioja M, Kangasniemi L, Raki M, Laakkonen P, Goodison S, Hemminki A. A heparan sulfate-targeted conditionally replicative adenovirus, Ad5.pk7-Delta24, for the treatment of advanced breast cancer. Gene Ther. 2007; 14:58–67. https://doi.org/10.1038/sj.gt.3302830.
81. Rivera AA, Wang M, Suzuki K, Uil TG, Krasnykh V, Curiel DT, Nettelbeck DM. Mode of transgene expression after fusion to early or late viral genes of a conditionally replicating adenovirus via an optimized internal ribosome entry site in vitro and in vivo. Virology. 2004; 320:121–34. https://doi.org/10.1016/j.virol.2003.11.028.
82. van Beusechem VW, Mastenbroek DC, van den Doel PB, Lamfers ML, Grill J, Wurdinger T, Haisma HJ, Pinedo HM, Gerritsen WR. Conditionally replicative adenovirus expressing a targeting adapter molecule exhibits enhanced oncolytic potency on CAR-deficient tumors. Gene Ther. 2003; 10:1982–91. https://doi.org/10.1038/sj.gt.3302103.
83. Hulou MM, Cho CF, Chiocca EA, Bjerkvig R. Experimental therapies: gene therapies and oncolytic viruses. Handb Clin Neurol. 2016; 134:183–97. https://doi.org/10.1016/b978-0-12-802997-8.00011-6.
84. Castro MG, Candolfi M, Wilson TJ, Calinescu A, Paran C, Kamran N, Koschmann C, Moreno-Ayala MA, Assi H, Lowenstein PR. Adenoviral Vector-Mediated Gene Therapy for Gliomas: Coming of Age. Expert opinion on biological therapy. 2014; 14:1241–57. https://doi.org/10.1517/14712598.2014.915307.
85. Ghulam Muhammad AKM, Xiong W, Puntel M, Farrokhi C, Kroeger KM, Salem A, Lacayo L, Pechnick RN, Kelson KR, Palmer D, Ng P, Liu C, Lowenstein PR, et al. Safety Profile of Gutless Adenovirus Vectors Delivered into the Normal Brain Parenchyma: Implications for a Glioma Phase 1 Clinical Trial. Human Gene Therapy Methods. 2012; 23:271–84. https://doi.org/10.1089/hgtb.2012.060.
86. Kane JR, Miska J, Young JS, Kanojia D, Kim JW, Lesniak MS. Sui generis: gene therapy and delivery systems for the treatment of glioblastoma. Neuro Oncol. 2015; 17:ii24-ii36. https://doi.org/10.1093/neuonc/nou355.
87. Douglas J. Adenoviral vectors for gene therapy. Molecular Biotechnology. 2007; 36:71–80. https://doi.org/10.1007/s12033-007-0021-5.
88. Spencer DA, Young JS, Kanojia D, Kim JW, Polster SP, Murphy JP, Lesniak MS. Unlocking the promise of oncolytic virotherapy in glioma: combination with chemotherapy to enhance efficacy. Ther Deliv. 2015; 6:453–68. https://doi.org/10.4155/tde.14.123.
89. Young JS, Kim JW, Ahmed AU, Lesniak MS. Therapeutic cell carriers: a potential road to cure glioma. Expert Rev Neurother. 2014; 14:651–60. https://doi.org/10.1586/14737175.2014.917964.
90. Kim KH, Dmitriev IP, Saddekni S, Kashentseva EA, Harris RD, Aurigemma R, Bae S, Singh KP, Siegal GP, Curiel DT, Alvarez RD. A phase I clinical trial of Ad5/3-Delta24, a novel serotype-chimeric, infectivity-enhanced, conditionally-replicative adenovirus (CRAd), in patients with recurrent ovarian cancer. Gynecol Oncol. 2013; 130:518–24. https://doi.org/10.1016/j.ygyno.2013.06.003.
91. Rodriguez-Rocha H, Gomez-Gutierrez JG, Garcia-Garcia A, Rao XM, Chen L, McMasters KM, Zhou HS. Adenoviruses induce autophagy to promote virus replication and oncolysis. Virology. 2011; 416:9–15. https://doi.org/10.1016/j.virol.2011.04.017.
92. Guo ZS, Liu Z, Bartlett DL. Oncolytic Immunotherapy: Dying the Right Way is a Key to Eliciting Potent Antitumor Immunity. Front Oncol. 2014; 4:74. https://doi.org/10.3389/fonc.2014.00074.
93. Kim Y, Clements DR, Sterea AM, Jang HW, Gujar SA, Lee PW. Dendritic Cells in Oncolytic Virus-Based Anti-Cancer Therapy. Viruses. 2015; 7:6506–25. https://doi.org/10.3390/v7122953.
94. Ranki T, Pesonen S, Hemminki A, Partanen K, Kairemo K, Alanko T, Lundin J, Linder N, Turkki R, Ristimaki A, Jager E, Karbach J, Wahle C, et al. Phase I study with ONCOS-102 for the treatment of solid tumors - an evaluation of clinical response and exploratory analyses of immune markers. J Immunother Cancer. 2016; 4:17. https://doi.org/10.1186/s40425-016-0121-5.
95. Brestovitsky A, Nebenzahl-Sharon K, Kechker P, Sharf R, Kleinberger T. The Adenovirus E4orf4 Protein Provides a Novel Mechanism for Inhibition of the DNA Damage Response. PLoS Pathog. 2016; 12:e1005420. https://doi.org/10.1371/journal.ppat.1005420.
96. Nokisalmi P, Rajecki M, Pesonen S, Escutenaire S, Soliymani R, Tenhunen M, Ahtiainen L, Hemminki A. Radiation-induced upregulation of gene expression from adenoviral vectors mediated by DNA damage repair and regulation. Int J Radiat Oncol Biol Phys. 2012; 83:376–84. https://doi.org/10.1016/j.ijrobp.2011.06.1973.
97. Tobias AL, Thaci B, Auffinger B, Rincon E, Balyasnikova IV, Kim CK, Han Y, Zhang L, Aboody KS, Ahmed AU, Lesniak MS. The timing of neural stem cell-based virotherapy is critical for optimal therapeutic efficacy when applied with radiation and chemotherapy for the treatment of glioblastoma. Stem Cells Transl Med. 2013; 2:655–66. https://doi.org/10.5966/sctm.2013-0039.
98. Chiocca EA, Abbed KM, Tatter S, Louis DN, Hochberg FH, Barker F, Kracher J, Grossman SA, Fisher JD, Carson K, Rosenblum M, Mikkelsen T, Olson J, et al. A phase I open-label, dose-escalation, multi-institutional trial of injection with an E1B-Attenuated adenovirus, ONYX-015, into the peritumoral region of recurrent malignant gliomas, in the adjuvant setting. Mol Ther. 2004; 10:958–66. https://doi.org/10.1016/j.ymthe.2004.07.021.
99. Yu B, Dong J, Wang C, Zhan Y, Zhang H, Wu J, Kong W, Yu X. Characteristics of neutralizing antibodies to adenovirus capsid proteins in human and animal sera. Virology. 2013; 437:118–23. https://doi.org/10.1016/j.virol.2012.12.014.
100. Lopez-Gordo E, Podgorski, II, Downes N, Alemany R. Circumventing antivector immunity: potential use of nonhuman adenoviral vectors. Hum Gene Ther. 2014; 25:285–300. https://doi.org/10.1089/hum.2013.228.
101. Ma J, Duffy MR, Deng L, Dakin RS, Uil T, Custers J, Kelly SM, McVey JH, Nicklin SA, Baker AH. Manipulating adenovirus hexon hypervariable loops dictates immune neutralisation and coagulation factor X-dependent cell interaction in vitro and in vivo. PLoS Pathog. 2015; 11:e1004673. https://doi.org/10.1371/journal.ppat.1004673.
102. Lopez-Gordo E, Denby L, Nicklin SA, Baker AH. The importance of coagulation factors binding to adenovirus: historical perspectives and implications for gene delivery. Expert Opin Drug Deliv. 2014; 11:1795–813. https://doi.org/10.1517/17425247.2014.938637.
103. Short JJ, Rivera AA, Wu H, Walter MR, Yamamoto M, Mathis JM, Curiel DT. Substitution of adenovirus serotype 3 hexon onto a serotype 5 oncolytic adenovirus reduces factor X binding, decreases liver tropism, and improves antitumor efficacy. Mol Cancer Ther. 2010; 9:2536–44. https://doi.org/10.1158/1535-7163.mct-10-0332.
104. Kaliberov SA, Kaliberova LN, Hong Lu Z, Preuss MA, Barnes JA, Stockard CR, Grizzle WE, Arbeit JM, Curiel DT. Retargeting of gene expression using endothelium specific hexon modified adenoviral vector. Virology. 2013; 447:312–25. https://doi.org/10.1016/j.virol.2013.09.020.
105. Chiocca EA, Rabkin SD. Oncolytic Viruses and Their Application to Cancer Immunotherapy. Cancer immunology research. 2014; 2:295–300. https://doi.org/10.1158/2326-6066.CIR-14-0015.
106. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, Lennon VA, Celis E, Chen L. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002; 8:793–800. https://doi.org/10.1038/nm730.
107. Prestwich RJ, Harrington KJ, Pandha HS, Vile RG, Melcher AA, Errington F. Oncolytic viruses: a novel form of immunotherapy. Expert review of anticancer therapy. 2008; 8:1581–8. https://doi.org/10.1586/14737140.8.10.1581.
108. Heimberger AB, Sampson JH. Immunotherapy coming of age: what will it take to make it standard of care for glioblastoma? Neuro Oncol. 2011; 13:3–13. https://doi.org/10.1093/neuonc/noq169.
109. Bloch O. Immunotherapy for malignant gliomas. Cancer Treat Res. 2015; 163:143–58. https://doi.org/10.1007/978-3-319-12048-5_9.
110. Van Gool SW. Brain Tumor Immunotherapy: What have We Learned so Far? Front Oncol. 2015; 5:98. https://doi.org/10.3389/fonc.2015.00098.
111. Yamanaka R, Homma J, Yajima N, Tsuchiya N, Sano M, Kobayashi T, Yoshida S, Abe T, Narita M, Takahashi M, Tanaka R. Clinical evaluation of dendritic cell vaccination for patients with recurrent glioma: results of a clinical phase I/II trial. Clin Cancer Res. 2005; 11:4160–7. https://doi.org/10.1158/1078-0432.ccr-05-0120.
112. Prins RM, Wang X, Soto H, Young E, Lisiero DN, Fong B, Everson R, Yong WH, Lai A, Li G, Cloughesy TF, Liau LM. Comparison of glioma-associated antigen peptide-loaded versus autologous tumor lysate-loaded dendritic cell vaccination in malignant glioma patients. J Immunother. 2013; 36:152–7. https://doi.org/10.1097/CJI.0b013e3182811ae4.
113. Vik-Mo EO, Nyakas M, Mikkelsen BV, Moe MC, Due-Tonnesen P, Suso EM, Saeboe-Larssen S, Sandberg C, Brinchmann JE, Helseth E, Rasmussen AM, Lote K, Aamdal S, et al. Therapeutic vaccination against autologous cancer stem cells with mRNA-transfected dendritic cells in patients with glioblastoma. Cancer Immunol Immunother. 2013; 62:1499–509. https://doi.org/10.1007/s00262-013-1453-3.
114. Fadul CE, Fisher JL, Hampton TH, Lallana EC, Li Z, Gui J, Szczepiorkowski ZM, Tosteson TD, Rhodes CH, Wishart HA, Lewis LD, Ernstoff MS. Immune response in patients with newly diagnosed glioblastoma multiforme treated with intranodal autologous tumor lysate-dendritic cell vaccination after radiation chemotherapy. J Immunother. 2011; 34:382–9. https://doi.org/10.1097/CJI.0b013e318215e300.
115. Phan GQ, Yang JC, Sherry RM, Hwu P, Topalian SL, Schwartzentruber DJ, Restifo NP, Haworth LR, Seipp CA, Freezer LJ, Morton KE, Mavroukakis SA, Duray PH, et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc Natl Acad Sci U S A. 2003; 100:8372–7. https://doi.org/10.1073/pnas.1533209100.
116. Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016; 8:328rv4. https://doi.org/10.1126/scitranslmed.aad7118.
117. Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, Cachola KE, Murray JC, Tihan T, Jensen MC, Mischel PS, Stokoe D, Pieper RO. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007; 13:84–8. https://doi.org/10.1038/nm1517.
118. Dolan DE, Gupta S. PD-1 pathway inhibitors: changing the landscape of cancer immunotherapy. Cancer Control. 2014; 21:231–7.
119. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012; 366:2443–54. https://doi.org/10.1056/NEJMoa1200690.
120. Schoenbrunn A, Frentsch M, Kohler S, Keye J, Dooms H, Moewes B, Dong J, Loddenkemper C, Sieper J, Wu P, Romagnani C, Matzmohr N, Thiel A. A converse 4-1BB and CD40 ligand expression pattern delineates activated regulatory T cells (Treg) and conventional T cells enabling direct isolation of alloantigen-reactive natural Foxp3+ Treg. J Immunol. 2012; 189:5985–94. https://doi.org/10.4049/jimmunol.1201090.
121. Huang JH, Zhang SN, Choi KJ, Choi IK, Kim JH, Lee MG, Kim H, Yun CO. Therapeutic and tumor-specific immunity induced by combination of dendritic cells and oncolytic adenovirus expressing IL-12 and 4-1BBL. Mol Ther. 2010; 18:264–74. https://doi.org/10.1038/mt.2009.205.
122. Wang C, Lin GH, McPherson AJ, Watts TH. Immune regulation by 4-1BB and 4-1BBL: complexities and challenges. Immunol Rev. 2009; 229:192–215. https://doi.org/10.1111/j.1600-065X.2009.00765.x.
123. Wen T, Bukczynski J, Watts TH. 4-1BB ligand-mediated costimulation of human T cells induces CD4 and CD8 T cell expansion, cytokine production, and the development of cytolytic effector function. J Immunol. 2002; 168:4897–906.
124. Ascierto PA, Simeone E, Sznol M, Fu YX, Melero I. Clinical experiences with anti-CD137 and anti-PD1 therapeutic antibodies. Semin Oncol. 2010; 37:508–16. https://doi.org/10.1053/j.seminoncol.2010.09.008.
125. Shin SP, Seo HH, Shin JH, Park HB, Lim DP, Eom HS, Bae YS, Kim IH, Choi K, Lee SJ. Adenovirus expressing both thymidine kinase and soluble PD1 enhances antitumor immunity by strengthening CD8 T-cell response. Mol Ther. 2013; 21:688–95. https://doi.org/10.1038/mt.2012.252.