
Rho GTPases and cellular metabolism in malignant transformation
Our initial efforts to identify novel small molecule inhibitors that block malignant transformation were directed at Rho family GTPase-signaling pathways. There were a number of reasons for this, perhaps foremost being that our laboratory has been studying the small GTPase Cdc42, as well as the closely related proteins Rac, and RhoA, and their signaling partners for a number of years. Signals originating from members of this GTPase family have been shown to be important for a broad array of cellular processes ranging from actin cytoskeletal rearrangements to cell polarity, migration, and cell-cycle progression [1]. However, these GTPases have also been implicated in a variety of diseases and developmental disorders, with a number of lines of evidence linking Rho family members to cancer [2]. For example, their hyper-activation as it occurs either through mutations or the de-regulation of their upstream activators, i.e. guanine nucleotide exchange factors which catalyze the exchange of GDP for GTP on these GTPases (such as members of the Dbl family of oncoproteins), results in cellular transformation [3,4]. Cells expressing constitutively active forms of Rho GTPases have been shown to be capable of growing under conditions of serum deprivation and in the absence of a substratum (i.e. anchorage-independent growth), as well as inducing tumor formation when injected into immuno-compromised mice [5-7]. The over-expression of Rho GTPases has been reported in tumors of the colon, lung, and in advanced stage breast cancers, in testicular germ cell and urinary tract tumors, and in pancreatic cancer [8-14]. Two members of the family, RhoA and RhoC, have been implicated in metastasis [15-18], and the expression of the Rho-GTPase-activating protein (Rho-GAP) DLC1 (for Deleted in Liver Cancer 1) is suppressed in liver cancer tissue and in a number of other cancers [19,20]. Thus, collectively these findings make the Rho GTPases and their regulatory proteins attractive candidates for targets of intervention in human cancer.
A surprising connection between Rho GTPase-induced cellular transformation and cellular metabolic activity
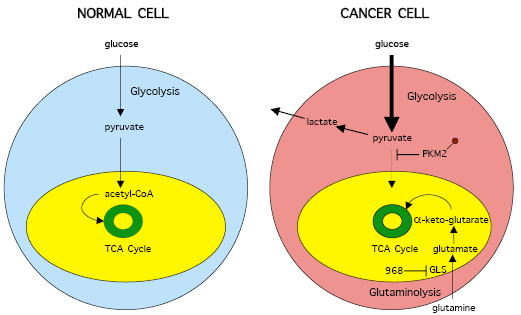
We have recently discovered a new role for Rho GTPases in cancer progression through a previously unappreciated connection to cellular metabolism [21]. In particular, we have found that the hyper-activation of Cdc42 as well as related Rho GTPases (e.g. Rac1, RhoA and RhoC) signals the activation of a mitochondrial enzyme, glutaminase, that plays a key role in glutamine metabolism by hydrolyzing glutamine to glutamate and ammonia. The importance of cellular metabolism in the development of cancer is rooted in the early observations of Warburg that tumor cells exhibit enhanced glycolytic activity (i.e. the “Warburg effect”) [22]. This phenomenon has been receiving a great deal of renewed attention [23-26].
Cancer cells undergo marked changes in metabolic activity in order to sustain their malignant phenotypes (Figure 1). One such set of changes is the up-regulation of the expression of enzymes in the glycolytic pathway, thus accelerating many of the reactions in this pathway. However, importantly, the penultimate step in glycolysis, the conversion of phosphoenolpyruvate to pyruvate, catalyzed by the enzyme pyruvate kinase, is attenuated (rather than accelerated) in cancer cells [24,25]. This occurs as a result of the tyrosine phosphorylation of a specific isoform of pyruvate kinase (M2) that is preferentially expressed in cancer cells, as well as in embryonic cells, but not in differentiated cells [24,25,27]. The net outcome of this attenuation is that pyruvate is generated through a unique enzymatic mechanism that is uncoupled from ATP production and involves the phosphorylation of phosphoglycerate mutase by phosphoenolpyruvate [26]. Pyruvate, when produced through this “alternative glycolytic pathway”, is converted primarily to lactic acid, rather than acetyl-CoA for citrate synthesis, with citrate then normally entering the citric acid cycle. The increased production of lactic acid by cancer cells, as a result of these changes in the glycolytic pathway, was a seminal observation of Warburg’s nearly 80 years ago, and reflects one aspect of the metabolic remodeling that frequently accompanies cellular transformation.
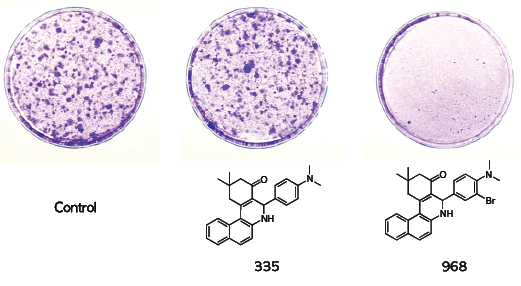
κ
κκκακακκκκκ
á
áéκ
κκ
κ
κ