
INTRODUCTION
Acute right heart failure (RHF) is a common but rarely studied disease that might be caused by rapidly increased right ventricle (RV) afterload during acute pulmonary embolism, hypoxic pulmonary vasoconstriction, or after cardiac transplantation or prolonged cardiopulmonary bypass. In these conditions, acute RHF is a major cause of morbidity and mortality. However, according to the Working Group on Cellular and Molecular Mechanisms of Right Heart Failure of the National Heart, Lung and Blood Institute, there is ‘‘a paucity of basic knowledge at all levels about the RV’s normal and pathological function’’ [1]. Therefore, the molecular mechanisms of RV dysfunction should be investigated.
The protein kinase B, also known as Akt, signaling pathway is a factor in cardiac hypertrophy, remodeling, and cardiomyocyte proliferation in the left ventricle (LV) [2, 3]. Activated Akt protects cardiomyocytes from apoptosis induced by ischemia-reperfusion injury in vivo [4]. Exercise can activate Akt, which was observed after pressure and volume overload in rabbit models [5, 6]. Cardiac-specific constitutively active Akt prevented cardiac dilatation and sudden death in an animal model [7]. However, few studies have been conducted on Akt signaling in the right heart. The RV is embryologically, structurally, and physiologically different from the LV [8, 9]. Akt signaling is an important factor in the development of the RV [10]. Our previous study found that phosphorylated Akt strongly expressed in the threonine 308 site but not in the serine 473 site in the RV compared with the LV, and proline-rich Akt-substrate 40 (PRAS40), an indicator of Akt activity, was significantly increased in its phosphorylated form but total PRAS40 was not increased in the RV compared with the LV [11]. In PAB-induced acute RHF, the effects of Akt signaling are still unknown.
In normal adults, blood flow in the right coronary artery mostly occurs during systole, whereas blood flow in the left coronary artery mostly occurs during diastole [12]. Therefore, pressure overload can predispose the RV to dysfunction and subendocardial ischemia compared with the LV [12, 13]. Studies demonstrated that coronary perfusion occurs primarily during diastole in patients with PH [14] and capillary density was reduced in several PH models [15–17], which caused a mismatch between decreased coronary blood supply and increased oxygen demand in the RV. Prostacyclin therapy in rats with PH was reported to improve both capillary density and survival [15]. Oxygen radical scavengers were also increased in rats with MCT-induced chronic PH [18]. In addition, metabolic shifts from fatty acid oxidation (FAO) to glycolysis were observed in the chronic pressure-loaded RV [19] and in MCT-induced chronic PH [20, 21]. Studies have suggested that reducing FAO and increasing glucose oxidation (GO) are promising treatments in heart failure patients [22]. Trimetazidine (TMZ) is an anti-anginal agent and can attenuate cardiac FAO and shifts metabolism to GO via the Randle cycle by selectively inhibiting long-chain 3-ketoacyl CoA thiolase (3-KAT) [23]. TMZ can relieve mild RV dysfunction caused by long-term PAB [24]. However, in the acute RHF model, whether changes occur in metabolic gene profiles and whether TMZ has some protective effects in right heart function are still unknown.
We developed a PAB-induced acute right heart model evaluated by hemodynamic measurement. We found that Akt activity was increased and peroxisome proliferator-activated receptor gamma coactivator 1-beta (PGC-1β) and peroxisome proliferator-activated receptor alpha (PPARα) were decreased in rats with PAB compared with rats in the sham group. TMZ pre-treatment before PAB can alleviate PAB-induced acute right heart dysfunction and activate PRAS40 as well as upregulate peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), PGC-1β, PPARα, peroxisome proliferator-activated receptor delta (PPARδ) and pyruvate dehydrogenase lipoamide kinase isozyme 4 (PDK4). TMZ pre-treatment might be an alternative for patients with acute RHF caused by rapidly increased afterload such as pulmonary hypertension and pulmonary embolism.
RESULTS
Development of acute right heart failure in rats
Supplementary Figures 1A, 1E, and 1F show that less than 71% constriction degree caused a rapid increase in maximum right ventricular pressure (RVP max), and then a persistent high level lasting at least 30 minutes. More than 70% constriction degree caused a rapid increase, and then a decrease in RVP max. Neither blood pressure (BP) nor heart rate (HR) were changed in the less than 71% constriction degree group but were significantly decreased in the more than 70% constriction degree group compared with the control group (Supplementary Figures 1A, 1B, 1C, and 1D). The positive rate of rise of right ventricle pressure (+dP/dT) decreased, whereas the negative rate of rise right ventricle pressure (–dP/dT) significantly increased compared with control (Supplementary Figures 1G and 1H).
An acute RHF model in rats was established by PAB of more than 70% constriction degree.
Short-term PAB induced increases in pathological markers of the RV that were reversed by TMZ treatment in rats
Figure 1A shows no significant differences in constriction degree between the PAB group and the PAB+TMZ group. Among sham, PAB, and PAB+TMZ groups, the ratio of heart weight to body weight had no significant differences (Figure 1B).
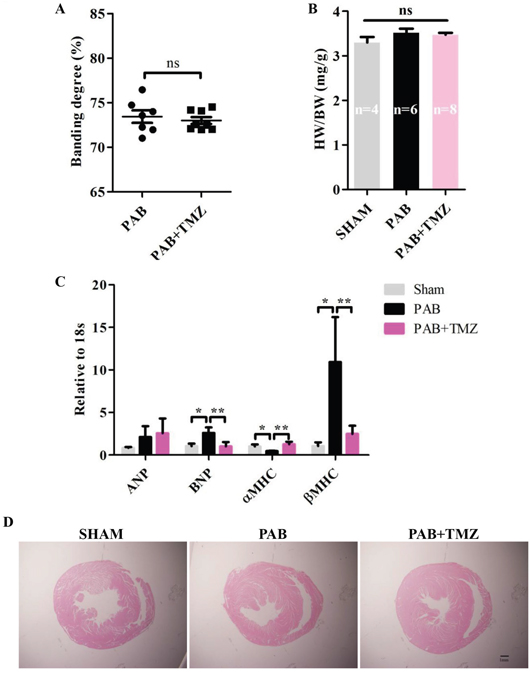
Figure 1: PAB increased mRNA levels of RV pathologic markers and TMZ reversed effects in rats. (A) Statistical analysis of constriction degree (%) in the PAB and PAB+TMZ groups. (B) Statistical analysis of the ratio of heart weight to body weight in the sham, PAB, and PAB+TMZ groups. (C) mRNA levels of ANP, BNP, αMHC, and βMHC in the sham, PAB and PAB+TMZ groups. (D) Representative H&E staining of histological section in the sham, PAB, and PAB+TMZ groups (Scale bar = 1.0 mm). Abbreviations: PAB, pulmonary artery banding; TMZ, trimedazidine; ANP, atrial natriuretic peptide; BNP, brain natriuretic peptide; αMHC, alpha myosin heavy chain; βMHC, beta myosin heavy chain; H&E, hematoxylin and eosin. *P < 0.05, **P < 0.01, n = 4-8 in each group.
Compared with the sham group, brain natriuretic peptide (BNP) and beta myosin heavy chain (βMHC) increased in the PAB group 30 minutes after banding, but atria natriuretic peptide (ANP) did not increase (Figure 1C). Conversely, alpha myosin heavy chain (αMHC) decreased significantly in the PAB group compared with the sham group (Figure 1C). TMZ treatment decreased BNP and βMHC levels and increased αMHC levels but had no effect on ANP levels compared with the PAB group (Figure 1C).
Furthermore, compared with the sham group, the RV dilated in the PAB group, and TMZ treatment did not change RV size (Figure 1D).
Short-term PAB caused RHF indicated by pathological markers, and TMZ alleviated the damage of RHF caused by PAB.
Short-term PAB caused acute RV failure that was improved by TMZ treatment in rats
Figures 2A and 2B show that both systolic blood pressure (SBP) and diastolic blood pressure (DBP) decreased in the PAB group compared with both the sham group and baseline 5 seconds after banding. Baseline SBP was not different among the sham, PAB, and PAB+TMZ groups, whereas DBP was lower in the PAB group than in the sham group at baseline. In the PAB+TMZ group, both SBP and DBP decreased compared with the sham group and baseline 5 seconds after banding, but increased compared with the PAB group (Figure 2A and 2B). Heart rate (HR) decreased in the PAB group compared with baseline 20 seconds after banding and compared with the sham group 45 seconds after banding (Figure 2C). In the PAB+TMZ group, HR decreased compared with baseline 10 seconds after banding and had no significant differences from the sham group. Although there were no significant differences in HR between the PAB+TMZ group and the PAB group, the decline in HR in the PAB+TMZ group was not as much as in the PAB group compared with the sham group (Figure 2C). The RVP max was increased in the PAB group at 5 seconds, 10 seconds, and 15seconds after banding compared with baseline and 5 seconds after banding compared with the sham group (Figure 2D).
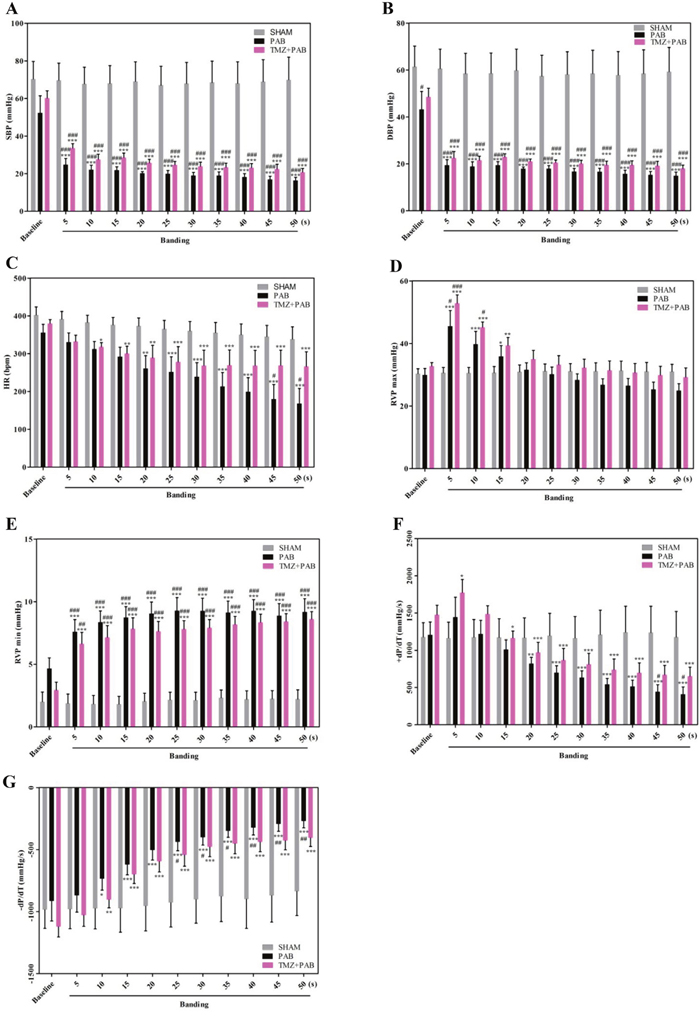
Figure 2: Comparison of hemodynamic parameters among the sham, PAB, and PAB+TMZ groups in rat. (A-G) Statistical analysis of hemodynamic parameters (SBP, DBP, HR, RVP max, RVP min, +dP/dT, and –dP/dT). Abbreviations: PAB, pulmonary artery banding; TMZ, trimedazidine; SBP, systolic blood pressure; DBP, diastolic blood pressure; HR, heart rate; RVP max, maximum right ventricle pressure; RVP min, minim right ventricle pressure. *P<0.05, **P < 0.01, compared with baseline; #P < 0.05, compared with sham group; n = 4-8 in each group.
Figure 2D shows that after an abrupt increase in RVP max after banding, RVP max gradually decreased. Compared with the sham group and baseline, RVP max increased in the PAB and PAB+TMZ groups at 5 seconds after banding. The RVP max was still higher in the PAB+TMZ group but not in the PAB group compared with the sham group 10 seconds after banding. No significant differences in RVP max occurred among the sham, PAB, and PAB+TMZ groups and in the PAB+TMZ and PAB groups compared with baseline at and after 20 seconds of banding, but the RVP max of the PAB+TMZ group was still higher than that of the PAB group. The minimum RVP (RVP min) was increased in the PAB group and the PAB+TMZ group 5 seconds after banding compared with both baseline and the sham group (Figure 2E). Consistent with the RVP trends, +dP/dT increased rapidly after banding, and then decreased gradually (Figure 2F). Beginning 20 seconds after banding, +dP/dT decreased in the PAB and PAB+TMZ groups compared with baseline (Figure 2F). At 45 seconds and 50 seconds after banding, +dP/dT decreased in the PAB group but not in the PAB+TMZ group compared with the sham group (Figure 2F). −dP/dT increased in the PAB group and the PAB+TMZ group 10 seconds after banding compared with baseline. Compared with the sham group, −dP/dT increased significantly in the PAB group but not in the PAB+TMZ group 25 seconds after banding (Figure 2G). The increased levels of −dP/dT and the decreased levels of +dP/dT were higher in the PAB group than in the PAB+TMZ group (Figure 2F and 2G).
Figures 3A, 3B, and 3C show that ejection fraction (EF) of the RV decreased in the PAB group compared with baseline and the sham group, and RV intradiameter in diastole (RVID; d) and RV anterior wall thickness in diastole (RVAW; d) had no significant differences between the PAB group and baseline as well as the PAB group and the sham group. However, RVID; d increased more in post-PAB compared with pre-PAB in the PAB and PAB+TMZ groups than in the sham group. The decline of EF was significantly lower in the PAB+TMZ group after banding compared with the PAB group.
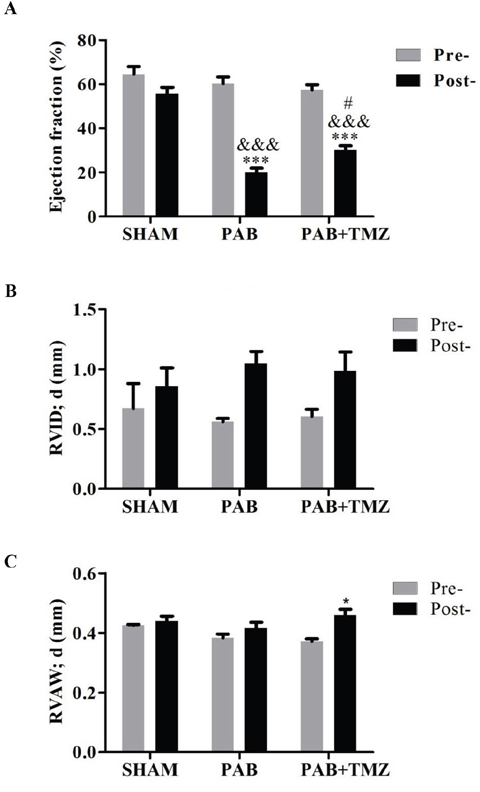
Figure 3: Comparison of right ventricular ejection fraction (EF), RVAWd, and RVIDd among the sham, PAB, and PAB+TMZ groups 24 hours after operation in rats. (A) Right ventricular ejection fraction; (B) RVID; d; (C) RVAW; d. Abbreviations: PAB, pulmonary artery banding; TMZ, trimedazidine; RVID; d, right ventricle internal diameter in diastole; RVAW; d, right ventricle anterior wall thickness in diastole; Pre-, pre-PAB; Post-, 24 hours after PAB. *P < 0.05, ***P < 0.001, Post- versus Pre-; &&&P < 0.001, PAB, PAB+TMZ versus sham; #P < 0.05, PAB+TMZ versus PAB; n = 3-5 in each group.
According to hemodynamic parameters, TMZ treatment partially improved PAB-induced acute right heart dysfunction.
PGC-1β and PPARα were downregulated by short-term PAB, and TMZ treatment upregulated PGC-1α, PGC-1β, PPARα, PPARδ, and PDK4 in rats
Figure 4A shows no significant differences in PDK4, glucose transporter 1 (GLUT1) and glucose transporter 4 (GLUT4) between the PAB group and baseline as well as the PAB and sham groups. PGC-1β and PPARα were decreased in the PAB group compared with the sham group but not PGC-1α, PPARδ, and peroxisome proliferator-activated receptor gamma (PPARγ) (Figure 4B). Compared with the PAB group, besides PGC-1β and PPARα, TMZ increased PDK4, PGC-1α, and PPARδ (Figure 4A and 4B).
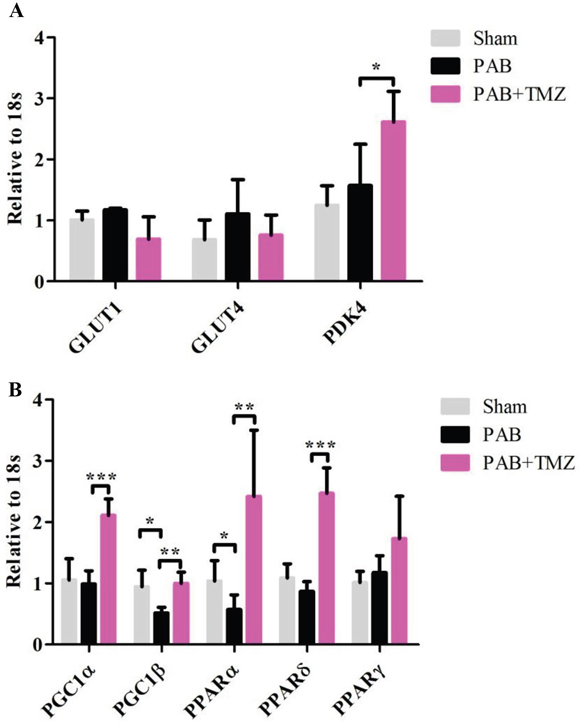
Figure 4: Metabolic gene profile changed after PAB and PAB+TMZ treatment. (A) Glucose metabolic gene profile (GLUT1, GLUT4, and PDK4) in the sham, PAB, and PAB + TMZ groups. (B) Mitochondrial metabolic gene profile (PGC1α and PGC1β) and lipid metabolic gene profile (PPARα, PPARδ, and PPARγ) in the sham, PAB, and PAB + TMZ groups. Abbreviations: PAB, pulmonary artery banding; TMZ, trimedazidine; GLUT1, glucose transporter 1; GLUT4, glucose transporter 4; PDK4, pyruvate dehydrogenase lipoamide kinase isozyme 4; PGC-1α, peroxisome proliferator-activated receptor gamma coactivator 1-alpha; PGC-1β, peroxisome proliferator-activated receptor gamma coactivator 1-beta; PPARα, peroxisome proliferator-activated receptor alpha; PPARδ, peroxisome proliferator-activated receptor delta; PPARγ, peroxisome proliferator-activated receptor gamma. *P < 0.05, **P < 0.01, ***P < 0.001, n = 4-8 in each group.
TMZ treatment increased PRAS40 activity and decreased Akt activity induced by short-term PBA in rats
Figure 5A shows that total Akt was decreased in the PAB group compared with the sham group. Correspondingly, levels of phosphorylated Akt at the serine 473 site was increased but not at the threonine site in the PAB group compared with the sham group (Figures 5A, 5B, 5C, and 5D). Levels of phosphatase and tensin homolog (PTEN), transforming growth factor-beta (TGF-β), PRAS40, and Glycogen synthase kinase-3 beta (GSK-3β) were not different between the PAB and sham groups (Figures 5A and 5E-5J). TMZ treatment increased PRAS40 activity, indicated by increased phosphorylated PRAS40, and inhibited Akt activity, indicated by decreased phosphorylated Akt at the serine 473 site (Figures 5A-5C and 5H). TMZ treatment had no effects on PTEN, GSK-3β, total PRAS40, and phosphorylated Akt at the therine 308 site. Compared with the sham and PAB groups, TMZ treatment increased TGF-β level (Figure 5A and 5G).
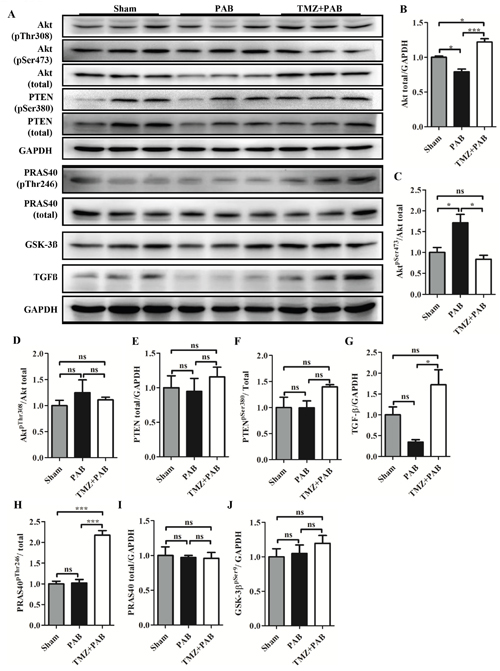
Figure 5: Detection of Akt signaling pathway by Western blot in each group. (A) Representative WB images. (B-J) Statistical analysis of (A). Abbreviations: PAB, pulmonary artery banding; TMZ, trimedazidine; Akt, protein kinase B; PTEN, phosphatase and tensin homolog; TGF-β, transforming growth factor-beta; PRAS40, proline-rich Akt-substrate 40; GSK-3β, glycogen synthase kinase-3 beta; GAPDH, glyceraldehyde 3-phosphate dehydrogenase. *P<0.05, **P<0.01, ***P<0.001, n=4-8 in each group.
The autophagy and the apoptosis pathways were not altered by short-term PAB and TMZ treatment in rats
Figures 6A-6F show no significant differences in LC3 II, p62, Bcl-2/Bax, and caspase-3 between the PAB group and the sham groups, and TMZ treatment also had no effects.
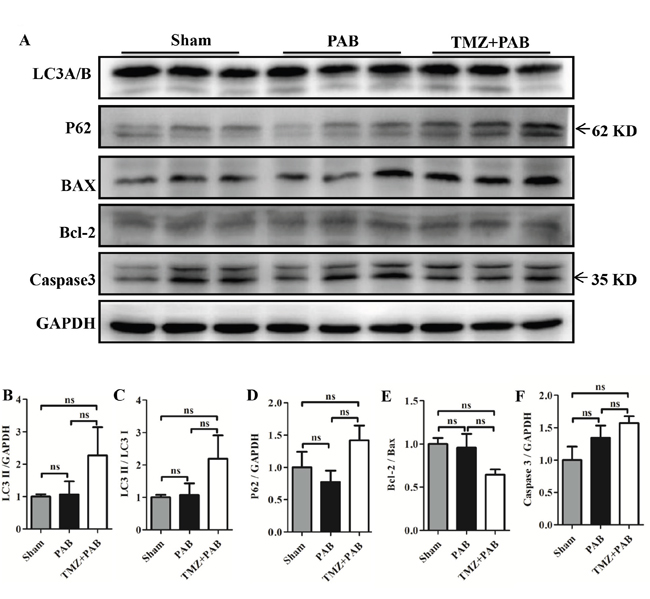
Figure 6: Detection of autophagy and apoptosis pathway by Western blot in each group. (A) Representative WB images. (B-F) Statistical analysis of (A). Abbreviations: PAB, pulmonary artery banding; TMZ, trimedazidine; LC3, microtubule-associated protein 1 light chain 3; P62, ubiquitin-binding protein 62; BAX, bcl-2 associated X protein; Bcl-2, B-cell lymphoma 2;caspase-3, cysteine-aspartic proteases 3; GAPDH, glyceraldehyde 3-phosphate dehydrogenase. *P < 0.05, **P < 0.01, *** P< 0.001, n = 4-8 in each group.
DISCUSSION
Acute RHF has a high mortality but receives little attention from clinicians and basic scientists [25, 26]. Both effective treatment and the molecular mechanisms of RHF are still obscured [27]. In this study, we successfully developed an RHF model in rats and confirmed the protective effects of TMZ in RHF.
Acute right heart failure model
Experimental animal models for RHF, such as monocrotaline (MCT), PAB, and pulmonary lobectomy-induced models take a long time to develop right heart hypertrophy, and then RHF [28–32]. In addition, the MCT model always yields systemic adverse effects that are important confounding factors in the study of RHF [28]. Pulmonary lobectomy not only disrupts right heart function but also disrupts lung function [31]. PAB allows study of the right heart without comorbidities [32]. However, previous experimental PAB models utilized the same gauge regardless of variation in pulmonary artery diameter and did not mimic acute RHF characterized by rapidly increased, and then decreased, right ventricular pressure (RVP) and decreased blood pressure (BP) [33–35]. We analyzed data from rats and found that pulmonary artery diameter correlates with body weight and heart weight, which means a fixed gauge cannot develop a fixed constriction degree in a PAB model [36–38] (Supplementary Figure 2). We measured the pulmonary artery before PAB and selected a corresponding gauge according to the pulmonary artery diameter in each rat and developed a nearly identical constriction degree. Furthermore, we tried different constriction degrees and found rats with approximately 75% constriction degree of the pulmonary artery presented hemodynamic changes in acute RHF (transient soaring, and then lower RVP with decreased BP). The pathological markers BNP and βMHC increased, and αMHC decreased in PAB rats but not in the sham group (Figure 1). The acute RHF in rats was well developed.
Effects of TMZ on acute right heart dysfunction
In the normal RV, coronary perfusion mainly occurs during systole, which means it is blocked by increased afterload caused by PH or pulmonary embolism [12, 14]. Hence, a chronic oxygen demand–supply mismatch might be an underlying mechanism in the transition to failure. Capillary density was reported to be reduced in several PH models, which supports the notion mentioned above [15–17]. Normally, the main substrate for the adult heart is fatty acids that metabolize to glucose and lactose under stress [39]. Fatty acid oxidation (FAO) is reduced in PH patients with severe hypertrophy [19], which appears to be an adaptive mechanism because TMZ alleviates PAB-induced chronic mild RV dysfunction [24]. TMZ also has beneficial effects in MTC-induced RVH [40]. In this study, in the acute RHF model, TMZ had protective effects in right heart function. TMZ treatment before PAB decreased BNP and βMHC levels and increased αMHC levels (Figure 1C). The hemodynamic parameters BP, HR, RVP, and dP/dT improved in the PAB+TMZ group compared with the PAB group (Figure 2). Echocardiographic evaluation 24 hours after operation indicated a higher ejection fraction of the RV in the PAB+TMZ group than in the PAB group (Figure 3). TMZ can alleviate PAB-induced acute RHF evaluated by right catheter and echocardiography.
Underlying mechanism of TMZ improving acute right heart dysfunction
TMZ inhibits the long-chain fatty acid oxidation and increases glucose oxidation in patients with ischemic heart disease [23] and in patients with idiopathic dilated cardiomyopathy [41]. TMZ increases net O2 consumption, reduces formation of oxygen free radicals, and enhances cardiac mitochondrial function in MCT-induced RVH in rats [42]. Our study showed that PGC-1β and PPAR-α were decreased in the PAB-induced acute RHF model compared with the sham group. TMZ treatment before PAB increased not only PGC-1β and PPAR-α but also PGC-1α and PPAR-δ (Figure 4). PGC-1α and PGC-1β are expressed in brown fat, heart, kidney, skeletal muscle, and brain and induce the mitochondrial function [43, 44]. PGC-1 is reduced in animal models of heart disease, accompanied by a switch of substrate use from fatty acids to glucose [45, 46], and PGC-1α is a potent regulator of gluconeogenesis via induction of phosphoenol pyruvate carboxykinase and glucose-6-phosphatase and increased tricarboxylic acid cycle flux [47–50]. PPARα and PPARδ also promote glucose metabolism, increase cardiac levels of endogenous antioxidants, promote mitochondrial biogenesis, and inhibit cardiomyocyte apoptosis, and their deficiency impairs cardiac function in animal models [51, 52]. In addition, the mRNA of PDK4 was also reported to be increased along with PGC-1α after exercise in muscle tissue [53, 54] and downregulated in cardiac tissue during heart failure [55]. In this study, we found PDK4 had no increase in the PAB-induced acute RHF model but increased by TMZ treatment, even though it was reported to be increased in both the PH and PAB models of adaptive RV remodeling [56]. Collectively, TMZ treatment alleviated acute right heart dysfunction, possibly by increasing some metabolic gene profiles associated with mitochondrial biogenesis and glucose oxidation.
Our previous study showed that high phosphorylated level of Akt was present in the threonine 308 site but not in the serine 473 site in RV compared with LV, and PRAS40, a promotor of Akt activity, was increased in its phosphorylated form but total PRAS40 was not increased in RV compared with LV [11]. In this study, we found that phosphorylated Akt at serine 473 site was increased accompanied by decreased total Akt in the PAB group (Figures 5A-5D). TMZ treatment reversed the changes in Akt and increased phosphorylated PRAS40, a downstream activator of Akt and promotor of mTORC1 activity (Figures 5A, 5H, and 5I) [57]. PRAS40 treatment was reported to improve the metabolic profile and prevent the development of diabetic cardiomyopathy in obese mice [58]. Additionally, a previous study showed PRAS40 preferentially promotes protective mTORC2 signaling in diseased myocardium, and PRAS40 phosphorylation stimulates physiological hypertrophy both in vitro and in vivo [59]. The beneficial effects of TMZ in rats with acute RHF were possibly induced by increased PRAS40 activity. However, we need further study to confirm that TMZ directly activates PRAS40.
Apoptosis and autophagy have been reported to be factors in the pathological process of RHF [60–63]. However, we have not found any changes among the sham, PAB, and PAB+TMZ groups (Figure 6). The possible reason is that our observations were too early.
Limitations
The limitations of our study are as follows: (1) We did not detect reactive oxygen species (ROS) that possibly contribute to the pathophysiological process of PAB-induced RHF. (2) We have not observed longer effects of TMZ in RHF that might be useful for investigating the mechanisms of RHF induced by PAB. (3) Further study is needed to prove that TMZ directly activates PRAS40 signaling.
MATERIALS AND METHODS
Animal model
Male Sprague Dawley rats (200g-250g) were fed a standard diet. In the PAB+TMZ group, TMZ (3.58 mg/kg/d) was administered by gavage for three days before PAB. All experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication no. 85-23, revised in 1996) and the regulations on mouse welfare and ethics of Gansu Provincial Hospital. All procedures were approved by the Ethics Committee of Gansu Provincial Hospital (Lanzhou, China).
Right ventricular catheterization and pulmonary artery banding
Open-chest RV catheterization was performed under general anesthesia in all animals (sodium pentobarbital, 50 mg/kg, i.p.). Before the procedure, the rats were intubated (16-gauge Teflon tube) and attached to a mechanical ventilator (Micro-Ventilator, UNO, Zevenaar, the Netherlands; ventilator settings: breathing frequency, 80 breaths per minute; pressures, 9/0 cmH2O; inspiratory/expiratory ratio, 1:1). The right ventricle was approached via a middle thoracotomy to the second rib and the sternum retracted by use of a chest retractor. RV pressures were recorded by use of a high-fidelity catheter tip transducer (Mikro-Tip SPR671, Millar Instruments, Houston, TX). Analyses were performed when steady state was reached over an interval of at least 10 seconds and averaged.
After right ventricular catheterization was established, fine-tip 45° angled forceps were used to gently separate the thymus and fat tissue from the pulmonary artery trunk. After identification of the pulmonary artery trunk, a Vernier caliper was used to measure the diameter of pulmonary artery (Supplementary Figure 2F). A vascular clip was then used to clip the pulmonary artery at the main pulmonary artery level, placing a small piece of a selected gauge blunt needle parallel to the main pulmonary artery to yield a residual cavity the same diameter of the gauge in lengths of the short shaft of the ellipse. Two knots were tied around the pulmonary artery and vascular clip to prevent clip and gauge slippage. In sham control rats, the entire procedure is identical, except for the clip of the pulmonary artery. After 1 minute, rats were sacrificed and hearts were harvested for further use. If the heart function was to be assessed by echocardiography after 24 hours, the chest retractor was removed and the rib cage was closed by use of a 6.0 prolene suture with an interrupted suture pattern and the skin was closed using a 6.0 prolene suture with a continuous suture pattern. To assess the effects of different banding degrees on right heart function, we used different gauges to produce different constrictions of pulmonary arteries. The success rate for establishing an acute RHF model is 86.8% (data not show).
Calculation of constriction degree of pulmonary artery
The pulmonary artery was oval shaped after banding and the diameter of the gauge was the same length as the short shaft of the elliptic pulmonary artery. We assumed that the circumference of the pulmonary artery did not change before and after banding. According to the diameter of the pulmonary artery before banding and the length of the short shaft of the pulmonary artery with oval shaped deformation after banding, we could obtain the area of the pulmonary artery before and after banding, and then the constriction degree, indicated as area stenosis degree of the pulmonary artery, was calculated according the formula, banding degree (%) = 1− (π[r−b]/[2+b]b)/ r2 (r, radius of pulmonary artery; b, the length of the short shaft of the elliptic pulmonary artery after banding) (Supplementary Figure 3).
Echocardiography of right heart
Rats were anesthetized with isoflurane and subjected to echocardiography as described by Brittain at al. [64] and Cheng et al. [65]. A Vevo 770 (Visual Sonics), equipped with a 30-MHz transducer, was used for noninvasive transthoracic echocardiography. Two-dimensional guided M-mode tracings were recorded. The parasternal long axis M mode view was used to obtain RV chamber dimension, ejection fraction, and RV wall thickness. Echocardiography was performed without knowledge of the animal group.
Western blotting analysis
Heart lysates of rats were prepared in lysis buffer (20 mM Tris, 150 mM NaCl, 10% glycerol, 20 mM glycerophosphate, 1% NP40, 5 mM EDTA, 0.5 mM EGTA, 1 mM Na3VO4, 0.5 mM PMSF, 1 mM benzamidine, 1 mM DTT, 50 mM NaF, 4 μM leupeptin, pH = 8.0). Samples were resolved by 10% SDS-PAGE and transferred to PVDF membranes (Millipore). Membranes were blocked with 5% non-fat milk in TBST (50 mM Tris, 150 mM NaCl, 0.5 mM Tween-20, pH = 7.5), and then incubated with primary antibodies overnight. Antibodies used in this study were purchased from Cell Signaling Technology (CST; Danvers, MA, USA), Bioworld: total Akt (CST #4691), phospho-Akt (Ser308) (CST #4060), phospho-Akt (Thr473) (CST #13038), phospho-GSK3β (Ser9) (CST #5558), LC3A/B (CST #12741), PRAS40 (CST #2691), phospho-PRAS40 (Thr246) (CST #13175), PTEN (CST #9188), phospho-PTEN (Ser380) (CST #9551), Bax (CST2772s), Bcl2 (CST3498s), GAPDH (#AP0063), anti-rabbit IgG, (HRP-linked antibody) (CST #7074). Image J software (NIH) was used to perform densitometric analysis (http://rsb.info.nih.gov/ij/).
Quantitative real-time PCR for metabolism relative genes and fetal genes
Total RNA was extracted from RVs by use of TRIzol reagent (Invitrogen, Carlsbad, CA), according to the manufacturer’s protocol. One microgram of total RNA from each specimen was reverse transcribed to cDNA by use of SuperScript Reverse Transcriptase and random hexamers as primers (Invitrogen). Quantitative real-time PCR (qRT-PCR) was performed with an ABI StepOnePlus instrument (Applied Biosystems, Foster City, CA) utilizing a 1×ITaQ SYBR Green Supermix Kit (Bio-Bad, Reinach, Switzerland) and 300 nmol/L as forward and reverse primers in a total volume of 20 μL. The mRNA level was based on the critical threshold (Ct) value. Primer sequences for quantitative real-time PCR are showed in Table 1, and 18S-rRNA was used as internal control.
Table 1: Primer sequences of metabolism genes in rat
Primer |
Forward |
Reverse |
|---|---|---|
GLUT1 |
GCTGTGGCTGGCTTCTCTAA |
CCGGAAGCGATCTCATCGAA |
GLUT4 |
GGCAATGCCAAATTGCTCCA |
GGCACAGTTAAGGTCCCCTC |
PDK4 |
ACAATTCACGGAATGCCCCT |
TACTTGGCGTAGAGACGGGA |
PPARα |
GAGTAGCCTGGGCTGCTTTT |
CTGATCACCAGCAGAGGTCC |
PPARγ |
TACCACGGTTGATTTCTC |
TCTACTTTGATCGCACTTT |
PPARδ |
CATCCGTTCTCTACCCAGCC |
AATTCTGAGCCCGGAGTTGG |
PGC-1α |
TGGAGTGACATAGAGTGTGCTG |
TATGTTCGCGGGCTCATTGT |
PGC-1β |
GGCCACACCTGTCTATGCTT |
AGGCTTGTTGACATCCCGTT |
ANP |
GAGCAAATCCCGTATACAGTGC |
ATCTTCTACCGGCATCTTCTCC |
BNP |
GCTGCTGGAGCTGATAAGAGAA |
GTTCTTTTGTAGGGCCTTGGTC |
α-MHC |
ACATCAGTCAGCAGAACA |
TTCCTCTAGCCTCTCACT |
β-MHC |
GCTGTTATTGCTGCCATT |
TTATCATTCCGAACTGTC |
Abbreviation: GLUT1, glucose transporter 1; GLUT4, glucose transporter 4; PPARα, peroxisome proliferator-activated receptor α; PPARγ, peroxisome proliferator-activated receptor γ; PPARδ, peroxisome proliferator-activated receptor δ; PGC-1α, peroxisome proliferator-activated receptor gamma coactivator 1α; PDK4, Pyruvate dehydrogenase lipoamide kinase isozyme.
Statistics
Results of calculations were presented as means ± SEM. Differences in means between two groups were evaluated by application of unpaired two-tailed Student’s t tests and those among multiple groups with one-way ANOVA followed by Bonferroni post hoc tests. Repeated-measures ANOVA were used to measure blood pressure, heart rate, and right ventricular pressure at multiple time points. All statistics was performed by GraphPad Prism 4.0 software (GraphPad, San Diego, CA, USA). P values of <0.05 were considered statistically significant.
CONCLUSIONS
We successfully developed an acute RHF model by PAB at approximately 75% constriction degree. TMZ, an accelerator of energy metabolism, can alleviate PAB-induced acute RHF, possibly by increasing PGC-1α, PGC-1β, PPAR-α, PPAR-δ and PDK4 levels as well as increasing PRAS40 activity. Therefore, TMZ might be an alternative drug for patients with acute RHF. Also, these findings might provide new insights for therapeutic strategies that utilize PRAS40 directed against afterload-induced acute RHF.
Author contributions
Y.S.C. and J.Y.S. conceived and designed experiments and obtained funding. J.Y.S. performed animal studies, Western blotting, and immunostaining, S.T.S. extracted RNA and conducted real-time PCR. H.L.F and A.Q.W. performed echocardiography of rats. Y.S.C., X.L., and Y.X. analyzed all data and performed statistical analyses. Y.S.C. and M.Z. wrote, formatted, and submitted the manuscripts. X.L.L. previewed the manuscript. Y.S.C, X.L.L., and M.Z. oversaw and coordinated the study. All authors participated in data discussions and approved the submitted version.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (81460072), CAS “Light of West China” Program, the Natural Science Foundation of Shanghai (13ZR1436500), Shanghai Municipal Commission of Health and Family Planning (2014ZYJB0502, ZY3-LCPT-2-1003) and Pudong New Area Municipal Commission of Health and Family Planning (PW2013E-1).
CONFLICTS OF INTEREST
The authors declare no competing financial interests.
REFERENCES
1. Voelkel NF, Quaife RA, Leinwand LA, Barst RJ, McGoon MD, Meldrum DR, Dupuis J, Long CS, Rubin LJ, Smart FW, Suzuki YJ, Gladwin M, Denholm EM, Gail DB, and National Heart, Lung, and Blood Institute Working Group on Cellular and Molecular Mechanisms of Right Heart Failure. Right ventricular function and failure: report of a National Heart, Lung, and Blood Institute working group on cellular and molecular mechanisms of right heart failure. Circulation. 2006; 114:1883–91.
2. Altomare DA, Khaled AR. Homeostasis and the importance for a balance between AKT/mTOR activity and intracellular signaling. Curr Med Chem. 2012; 19:3748–62.
3. Sussman MA, Völkers M, Fischer K, Bailey B, Cottage CT, Din S, Gude N, Avitabile D, Alvarez R, Sundararaman B, Quijada P, Mason M, Konstandin MH, et al. Myocardial AKT: the omnipresent nexus. Physiol Rev. 2011; 91:1023–70.
4. Fujio Y, Nguyen T, Wencker D, Kitsis RN, Walsh K. Akt promotes survival of cardiomyocytes in vitro and protects against ischemia-reperfusion injury in mouse heart. Circulation. 2000; 101:660–67.
5. Fujita S, Rasmussen BB, Cadenas JG, Drummond MJ, Glynn EL, Sattler FR, Volpi E. Aerobic exercise overcomes the age-related insulin resistance of muscle protein metabolism by improving endothelial function and Akt/mammalian target of rapamycin signaling. Diabetes. 2007; 56:1615–22.
6. Miyamoto T, Takeishi Y, Takahashi H, Shishido T, Arimoto T, Tomoike H, Kubota I. Activation of distinct signal transduction pathways in hypertrophied hearts by pressure and volume overload. Basic Res Cardiol. 2004; 99:328–37.
7. Matsui T, Li L, Wu JC, Cook SA, Nagoshi T, Picard MH, Liao R, Rosenzweig A. Phenotypic spectrum caused by transgenic overexpression of activated Akt in the heart. J Biol Chem. 2002; 277:22896–901.
8. Haddad F, Doyle R, Murphy DJ, Hunt SA. Right ventricular function in cardiovascular disease, part II: pathophysiology, clinical importance, and management of right ventricular failure. Circulation. 2008; 117:1717–31.
9. Zaffran S, Kelly RG, Meilhac SM, Buckingham ME, Brown NA. Right ventricular myocardium derives from the anterior heart field. Circ Res. 2004; 95:261–68.
10. Luo W, Zhao X, Jin H, Tao L, Zhu J, Wang H, Hemmings BA, Yang Z. Akt1 signaling coordinates BMP signaling and β-catenin activity to regulate second heart field progenitor development. Development. 2015; 142:732–42.
11. Song J, Shen S, Zhang M, Wang K, Zhang Y, Li X, Wang N, Cao Y. Differences in Akt signaling and metabolism gene expression in the right heart, intraventricular septum and left heart of rodents. Int J Clin Exp Pathol. 2015; 8:12915–21.
12. Zong P, Tune JD, Downey HF. Mechanisms of oxygen demand/supply balance in the right ventricle. Exp Biol Med (Maywood). 2005; 230:507–19.
13. Friedberg MK, Redington AN. Right versus left ventricular failure: differences, similarities, and interactions. Circulation. 2014; 129:1033–44.
14. Wong YY, Ruiter G, Lubberink M, Raijmakers PG, Knaapen P, Marcus JT, Boonstra A, Lammertsma AA, Westerhof N, van der Laarse WJ, Vonk-Noordegraaf A. Right ventricular failure in idiopathic pulmonary arterial hypertension is associated with inefficient myocardial oxygen utilization. Circ Heart Fail. 2011; 4:700–06.
15. van Albada ME, Berger RM, Niggebrugge M, van Veghel R, Cromme-Dijkhuis AH, Schoemaker RG. Prostacyclin therapy increases right ventricular capillarisation in a model for flow-associated pulmonary hypertension. Eur J Pharmacol. 2006; 549:107–16.
16. de Man FS, Handoko ML, van Ballegoij JJ, Schalij I, Bogaards SJ, Postmus PE, van der Velden J, Westerhof N, Paulus WJ, Vonk-Noordegraaf A. Bisoprolol delays progression towards right heart failure in experimental pulmonary hypertension. Circ Heart Fail. 2012; 5:97–105.
17. Bogaard HJ, Natarajan R, Henderson SC, Long CS, Kraskauskas D, Smithson L, Ockaili R, McCord JM, Voelkel NF. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation. 2009; 120:1951–60.
18. Redout EM, Wagner MJ, Zuidwijk MJ, Boer C, Musters RJ, van Hardeveld C, Paulus WJ, Simonides WS. Right-ventricular failure is associated with increased mitochondrial complex II activity and production of reactive oxygen species. Cardiovasc Res. 2007; 75:770–81.
19. Kim Y, Goto H, Kobayashi K, Sawada Y, Miyake Y, Fujiwara G, Chiba H, Okada T, Nishimura T. Detection of impaired fatty acid metabolism in right ventricular hypertrophy: assessment by I-123 beta-methyl iodophenyl pentadecanoic acid (BMIPP) myocardial single-photon emission computed tomography. Ann Nucl Med. 1997; 11:207–12.
20. Buermans HP, Redout EM, Schiel AE, Musters RJ, Zuidwijk M, Eijk PP, van Hardeveld C, Kasanmoentalib S, Visser FC, Ylstra B, Simonides WS. Microarray analysis reveals pivotal divergent mRNA expression profiles early in the development of either compensated ventricular hypertrophy or heart failure. Physiol Genomics. 2005; 21:314–23.
21. Gomez-Arroyo J, Mizuno S, Szczepanek K, Van Tassell B, Natarajan R, dos Remedios CG, Drake JI, Farkas L, Kraskauskas D, Wijesinghe DS, Chalfant CE, Bigbee J, Abbate A, et al. Metabolic gene remodeling and mitochondrial dysfunction in failing right ventricular hypertrophy secondary to pulmonary arterial hypertension. Circ Heart Fail. 2013; 6:136–44.
22. Lopatin YM, Rosano GM, Fragasso G, Lopaschuk GD, Seferovic PM, Gowdak LH, Vinereanu D, Hamid MA, Jourdain P, Ponikowski P. Rationale and benefits of trimetazidine by acting on cardiac metabolism in heart failure. Int J Cardiol. 2016; 203:909–15.
23. Kantor PF, Lucien A, Kozak R, Lopaschuk GD. The antianginal drug trimetazidine shifts cardiac energy metabolism from fatty acid oxidation to glucose oxidation by inhibiting mitochondrial long-chain 3-ketoacyl coenzyme A thiolase. Circ Res. 2000; 86:580–88.
24. Fang YH, Piao L, Hong Z, Toth PT, Marsboom G, Bache-Wiig P, Rehman J, Archer SL. Therapeutic inhibition of fatty acid oxidation in right ventricular hypertrophy: exploiting Randle’s cycle. J Mol Med (Berl). 2012; 90:31–43.
25. Ansari-Ramandi MM, Ansari-Ramandi S, Naderi N. Acute Right Heart Failure in a Patient with Right Heart Thrombus and Pulmonary Thromboembolism. J Clin Diagn Res. 2016; 10:OD03–04.
26. Rich JD, Gosev I, Patel CB, Joseph S, Katz JN, Eckman PM, Lee S, Sundareswaran K, Kilic A, Bethea B, Soleimani B, Lima B, Uriel N, Kiernan M, and Evolving Mechanical Support Research Group (EMERG) Investigators. The incidence, risk factors, and outcomes associated with late right-sided heart failure in patients supported with an axial-flow left ventricular assist device. J Heart Lung Transplant. 2017; 36:50–58
27. Harjola VP, Mebazaa A, Čelutkienė J, Bettex D, Bueno H, Chioncel O, Crespo-Leiro MG, Falk V, Filippatos G, Gibbs S, Leite-Moreira A, Lassus J, Masip J, et al. Contemporary management of acute right ventricular failure: a statement from the Heart Failure Association and the Working Group on Pulmonary Circulation and Right Ventricular Function of the European Society of Cardiology. Eur J Heart Fail. 2016; 18:226–41.
28. Gomez-Arroyo JG, Farkas L, Alhussaini AA, Farkas D, Kraskauskas D, Voelkel NF, Bogaard HJ. The monocrotaline model of pulmonary hypertension in perspective. Am J Physiol Lung Cell Mol Physiol. 2012; 302:L363–69.
29. Gomes AC, Falcão-Pires I, Pires AL, Brás-Silva C, Leite-Moreira AF. Rodent models of heart failure: an updated review. Heart Fail Rev. 2013; 18:219–49.
30. Haidara MA, Assiri AS, Yassin HZ, Ammar HI, Obradovic MM, Isenovic ER. Heart Failure Models: Traditional and Novel Therapy. Curr Vasc Pharmacol. 2015; 13:658–69.
31. Le Cras TD, Fernandez LG, Pastura PA, Laubach VE. Vascular growth and remodeling in compensatory lung growth following right lobectomy. J Appl Physiol. 2005; 98: 1140-48.
32. Hirata M, Ousaka D, Arai S, Okuyama M, Tarui S, Kobayashi J, Kasahara S, Sano S. Novel Model of Pulmonary Artery Banding Leading to Right Heart Failure in Rats. BioMed Res Int. 2015; 2015:753210.
33. Aguero J, Ishikawa K, Hadri L, Santos-Gallego C, Fish K, Hammoudi N, Chaanine A, Torquato S, Naim C, Ibanez B, Pereda D, García-Alvarez A, Fuster V, et al. Characterization of right ventricular remodeling and failure in a chronic pulmonary hypertension model. Am J Physiol Heart Circ Physiol. 2014; 307:H1204–15.
34. Azakie A, Fineman J, He Y. Differential responses of the right ventricle to abnormal loading conditions in vivo: possible pathophysiologic mechanisms. J Thorac Cardiovasc Surg. 2013; 145:1335–44.
35. Borgdorff MA, Bartelds B, Dickinson MG, Steendijk P, de Vroomen M, Berger RM. Distinct loading conditions reveal various patterns of right ventricular adaptation. Am J Physiol Heart Circ Physiol. 2013; 305:H354–64.
36. Kay JM. Comparative morphologic features of the pulmonary vasculature in mammals. Am Rev Respir Dis. 1983; 128:S53–57.
37. Jones EL. Quantitative histological study of the medial thickness of the pulmonary trunk and muscular pulmonary arteries in the vervet monkey. J Pathol. 1969; 99:181–91.
38. Ferencz C. Pulmonary arterial design in mammals. Morphologic variation and physiologic constancy. Johns Hopkins Med J. 1969; 125:207–24.
39. Bartelds B, Knoester H, Smid GB, Takens J, Visser GH, Penninga L, van der Leij FR, Beaufort-Krol GC, Zijlstra WG, Heymans HS, Kuipers JR. Perinatal changes in myocardial metabolism in lambs. Circulation. 2000; 102:926–31.
40. Guarnieri C, Muscari C. Beneficial effects of trimetazidine on mitochondrial function and superoxide production in the cardiac muscle. Cardiovasc Drugs Ther. 1990; 4:814–15.
41. Tuunanen H, Engblom E, Naum A, Någren K, Scheinin M, Hesse B, Juhani Airaksinen KE, Nuutila P, Iozzo P, Ukkonen H, Opie LH, Knuuti J. Trimetazidine, a metabolic modulator, has cardiac and extracardiac benefits in idiopathic dilated cardiomyopathy. Circulation. 2008; 118:1250–58.
42. Guarnieri C, Muscari C. Beneficial effects of trimetazidine on mitochondrial function and superoxide production in the cardiac muscle of monocrotaline-treated rats. Biochem Pharmacol. 1988; 37:4685–88.
43. Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998; 92:829–39.
44. Lin J, Puigserver P, Donovan J, Tarr P, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1beta (PGC-1beta ), a novel PGC-1-related transcription coactivator associated with host cell factor. J Biol Chem. 2002; 277:1645–48.
45. Huss JM, Kelly DP. Nuclear receptor signaling and cardiac energetics. Circ Res. 2004; 95:568–78.
46. Huss JM, Kelly DP. Mitochondrial energy metabolism in heart failure: a question of balance. J Clin Invest. 2005; 115:547–55.
47. Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J, Adelmant G, Stafford J, Kahn CR, Granner DK, Newgard CB, Spiegelman BM. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001; 413:131–38.
48. Handschin C, Lin J, Rhee J, Peyer AK, Chin S, Wu PH, Meyer UA, Spiegelman BM. Nutritional regulation of hepatic heme biosynthesis and porphyria through PGC-1alpha. Cell. 2005; 122:505–15.
49. Burgess SC, Leone TC, Wende AR, Croce MA, Chen Z, Sherry AD, Malloy CR, Finck BN. Diminished hepatic gluconeogenesis via defects in tricarboxylic acid cycle flux in peroxisome proliferator-activated receptor gamma coactivator-1alpha (PGC-1alpha)-deficient mice. J Biol Chem. 2006; 281:19000–08.
50. Koo SH, Satoh H, Herzig S, Lee CH, Hedrick S, Kulkarni R, Evans RM, Olefsky J, Montminy M. PGC-1 promotes insulin resistance in liver through PPAR-alpha-dependent induction of TRB-3. Nat Med. 2004; 10:530–34.
51. Palomer X, Barroso E, Zarei M, Botteri G, Vázquez-Carrera M. PPARβ/δ and lipid metabolism in the heart. Biochim Biophys Acta. 2016; 1861:1569–78.
52. Pol CJ, Lieu M, Drosatos K. PPARs: Protectors or Opponents of Myocardial Function? PPAR Res. 2015; 2015:835985.
53. Pilegaard H, Ordway GA, Saltin B, Neufer PD. Transcriptional regulation of gene expression in human skeletal muscle during recovery from exercise. Am J Physiol Endocrinol Metab. 2000; 279:E806–14.
54. Wang L, Sahlin K. The effect of continuous and interval exercise on PGC-1α and PDK4 mRNA in type I and type II fibres of human skeletal muscle. Acta Physiol (Oxf). 2012; 204:525–32.
55. Razeghi P, Young ME, Ying J, Depre C, Uray IP, Kolesar J, Shipley GL, Moravec CS, Davies PJ, Frazier OH, Taegtmeyer H. Downregulation of metabolic gene expression in failing human heart before and after mechanical unloading. Cardiology. 2002; 97:203–09.
56. Piao L, Marsboom G, Archer SL. Mitochondrial metabolic adaptation in right ventricular hypertrophy and failure. J Mol Med (Berl). 2010; 88:1011–20.
57. Völkers M, Sussman M. mTOR/PRAS40 interaction: hypertrophy or proliferation. Cell Cycle. 2013; 12:3579–80.
58. Völkers M, Doroudgar S, Nguyen N, Konstandin MH, Quijada P, Din S, Ornelas L, Thuerauf DJ, Gude N, Friedrich K, Herzig S, Glembotski CC, Sussman MA. PRAS40 prevents development of diabetic cardiomyopathy and improves hepatic insulin sensitivity in obesity. EMBO Mol Med. 2014; 6:57–65.
59. Völkers M, Toko H, Doroudgar S, Din S, Quijada P, Joyo AY, Ornelas L, Joyo E, Thuerauf DJ, Konstandin MH, Gude N, Glembotski CC, Sussman MA. Pathological hypertrophy amelioration by PRAS40-mediated inhibition of mTORC1. Proc Natl Acad Sci USA. 2013; 110:12661–66.
60. Bogaard HJ, Abe K, Vonk Noordegraaf A, Voelkel NF. The right ventricle under pressure: cellular and molecular mechanisms of right-heart failure in pulmonary hypertension. Chest. 2009; 135:794–804.
61. Suzuki YJ, Ibrahim YF, Shults NV. Apoptosis-based therapy to treat pulmonary arterial hypertension. J Rare Dis Res Treat. 2016; 1:17–24.
62. Ryan JJ, Huston J, Kutty S, Hatton ND, Bowman L, Tian L, Herr JE, Johri AM, Archer SL. Right ventricular adaptation and failure in pulmonary arterial hypertension. Can J Cardiol. 2015; 31:391–406.
63. Vonk-Noordegraaf A, Haddad F, Chin KM, Forfia PR, Kawut SM, Lumens J, Naeije R, Newman J, Oudiz RJ, Provencher S, Torbicki A, Voelkel NF, Hassoun PM. Right heart adaptation to pulmonary arterial hypertension: physiology and pathobiology. J Am Coll Cardiol. 2013; 62:D22–33.