
INTRODUCTION
Prostate cancer (PRCA) is one of the most heritable cancers, with latest estimates of the genetic contribution to total risk near 58% [1]. To date, a total of 202 PRCA risk-associated loci have been reported by genome-wide association studies [2–16], which collectively explain approximately one third of the total familial risk. The majority of these variants does not occur within genic regions and are presumed to be regulatory in nature. Multiple expression quantitative trait loci (eQTL) studies have investigated associations between PRCA susceptibility loci and transcript expression levels of nearby genes [17, 18]. These studies have identified a large number of dysregulated genes that may be relevant to the development and progression of PRCA.
Due to the high testing-dimensionality presented by evaluating transcriptome-wide associations, most eQTL studies of trait-associated genetic variation focus on cis-acting regulation (cis-eQTLs). Thus, tested associations are limited to genes near the variants of interest. However, a growing number of studies have identified trans-eQTLs are also likely to play a major role in disease etiology [19–21]. As transcriptional regulation is highly determined by cell-type, large tissue-specific datasets and sophisticated methods are necessary to discover trans-associations of trait-associated loci. For example, Yao et al. [22, 23] have leveraged a large whole-blood eQTL dataset from the Framingham Heart Study to investigate the role of both cis- and trans-eQTLs among SNPs associated with cardiometabolic traits relevant to cardiovascular disease. In PRCA, Chen et al. [24] applied a Bayesian clustering approach toward investigating the role of trans-associations of reported risk loci in a relatively small set of tumor-adjacent stromal tissue samples. Other approaches, including adaptive false discovery rate estimation [25] and cross-phenotype meta-analysis [26], have focused on trans-eQTL “hotspots”, whereby genetic loci are associated in trans with expression levels of multiple transcripts.
Multiple studies have indicated trans associations are likely mediated by the products of cis-regulated transcripts [22, 27], such as transcription factors and signaling cascade proteins. A plausible strategy to improve discovery of PRCA risk SNP trans-eQTL associations under such a model is leveraging patterns of gene co-expression with cis-regulated genes. Gene co-expression analysis is a powerful data-driven approach for uncovering relevant regulatory networks in high-dimensional expression data. Recent improvements in the construction of sparse undirected graphs using regularized Gaussian graphical models have enabled sparse network inference on large gene expression datasets [28]. In this study, we systematically investigate potential downstream trans-acting dysregulation of protein-coding gene expression by PRCA risk loci using cis-mediator analysis on a large prostate tissue eQTL dataset. We first substantially reduce the search space of trans-eQTL associations by constructing an undirected co-expression network of genes exhibiting at least modest eQTL associations with PRCA risk loci. We then identify cis-eQTL genes as potential mediators of trans-eQTL associations. We then apply a network-driven strategy to determine if neighboring genes in the expression graph exhibit trans-eQTL associations that are mediated by cis-regulatory effects using causal inference analyses. Finally, we interpret putative regulatory targets of dysregulated cis-eQTL genes in the context of PRCA susceptibility.
RESULTS
A total of 3763 expressed transcripts met our transcriptome-wide eQTL screening criteria for inclusion in the co-expression network inference (FDR < 0.2). The estimated undirected graph for this gene subset consisted of 36,728 connections involving 3757 unique transcripts. Of the 3130 candidate cis-eQTL target genes, we identified 86 significant cis-genes associated with 72 unique PRCA risk loci variants (Supplementary Table 1). A total of 1168 neighbor nodes of the significant cis-genes in the expression network met our definition of trans with the corresponding cis-eQTL variant, defining cis-mediator trios eligible for causal inference. Of these, three cis-mediator trios resulted in mediation p-values below the Bonferroni-adjusted significance threshold of 0.05/1168 ≈ 4.3E-05: rs11263762 →HNF1B→SRC, rs11263762→HNF1B→MIA2, and rs10993994→MSMB→NDRG1. A flowchart of these analyses is represented in Figure 1, while complete results for seven trios that exhibited at least suggestive associations (mediation P < 1E-03) are presented in Table 1. Two additional genes corresponded to suggestive cis-mediator relationships with rs11263762 and HNF1B: KIF12 (mediation P = 1.2E-04) and SEMA6A (mediation P = 3.0E-04). As HNF1B encodes the transcription factor HNF-1B, these results highlight multiple putative targets of trans-acting dysregulation via PRCA risk SNP rs11263762.
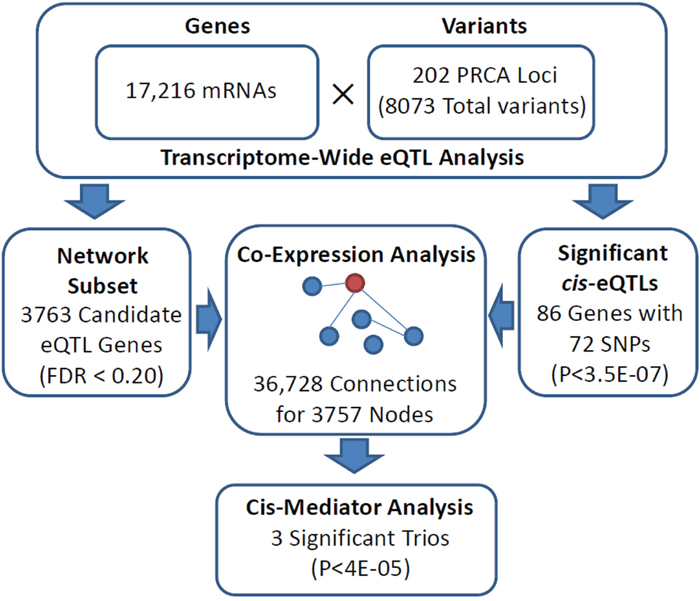
Figure 1: Flowchart indicating analytical stages in identifying significant cis-mediator relationships between PRCA susceptibility loci, proximal cis-genes, and distal trans-genes.
Table 1: Significant and suggestive (mediation P < 1E-05) cis-mediator analysis results from the PRCA susceptibility gene expression network
eQTL Variant |
Cis-Gene |
Trans-Gene |
Mediation Analysis |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
rsID |
Chr:Pos (hg19) |
Allelesa |
Gene |
βC |
PC |
Gene |
BT |
PT |
P |
M |
|
rs11263762 |
17: 36101926 |
G/A |
HNF1B |
0.15 |
4.4E-12 |
SRC |
0.04 |
3.5E-07 |
3.5E-07 |
0.02 |
0.63 |
rs11263762 |
17: 36101926 |
G/A |
HNF1B |
0.15 |
4.4E-12 |
MIA2 |
0.21 |
1.2E-06 |
1.2E-06 |
0.10 |
0.55 |
rs10993994 |
10: 51549496 |
C/T |
MSMB |
-0.32 |
7.4E-38 |
NDRG1 |
-0.10 |
1.0E-05 |
1.0E-05 |
-0.02 |
0.83 |
rs11191385 |
10: 104513049 |
G/T |
AS3MT |
-0.22 |
1.0E-32 |
TMEM121 |
-0.08 |
3.5E-05 |
1.2E-04 |
-0.03 |
0.56 |
rs11263762 |
17: 36101926 |
G/A |
HNF1B |
0.15 |
4.4E-12 |
KIF12 |
0.13 |
1.2E-04 |
1.2E-04 |
0.04 |
0.66 |
rs11263762 |
17: 36101926 |
G/A |
HNF1B |
0.15 |
4.4E-12 |
SEMA6A |
0.14 |
2.4E-10 |
3.0E-04 |
0.07 |
0.50 |
rs6958572 |
7:97789351 |
G/A |
TECPR1 |
0.08 |
3.8E-13 |
UBA5 |
0.02 |
4.1E-04 |
9.9E-04 |
0.01 |
0.58 |
Reference and alternate alleles were defined via frequency in the dataset.
aMajor/Minor as defined by observed minor allele frequency among 471 prostate tissue samples.
Quantification of eQTL effect mediation by for significant cis-mediator relationships indicated incomplete attenuation of the trans-eQTL by the mediating cis-genes, ranging from 0.50 to 0.83 (Supplementary Figure 1). However, this is not altogether surprising, as such partial mediation is expected in the presence of measurement error and incorrect selection of the underlying causal regulatory variant due to LD [27]. To further evaluate the robustness of these associations, we conducted permutation testing as similarly conducted in Franzen et al. [29]. Briefly, for each cis-mediator trio, trans-gene expression values were randomly permuted across samples within a given genotype group defined by levels of , holding all other measurements fixed. The mediation analysis p-values from the permuted datasets were compared to the original results and permutation p-values were computed as the proportion of permuted data p-values as or more extreme than the p-value derived from the true data. Under 100,000 permutations and the same significance threshold defined above, all reported associations in Table 1 corresponded to the minimum permutation p-value (1.0E-05) and declared as significant aside from rs11263762→HNF1B→SEMA6A (P = 1.5E-04).
The latest release of Gene Tissue Expression (GTEx) project [30] currently has 87 prostate tissue samples with available genotype data. We queried the constituent trans-eQTL associations that corresponded to the cis-mediator relationships presented in Table 1 in the GTEx Portal to investigate whether consistent associations were observed in an independent dataset (Supplementary Table 2). For genes connected to HNF1B, we allowed any of 19 genotyped positions in LD with rs11263762 to be considered. Of the seven trans-associations reported, four corresponded to trans-eQTLs with p-values < 0.05 (rs4430796→MIA2, P = 6.5E-04; rs11263763→KIF12, P = 0.0044; rs10993994→NDRG1, P = 0.013; rs7405696→SRC, P = 0.038), with GTEx effect estimate directionality consistent with all seven discovery findings. Co-expression scatterplots for each of the four cis-mediator results with GTEx-replicated trans-eQTLs are presented in Figure 2.
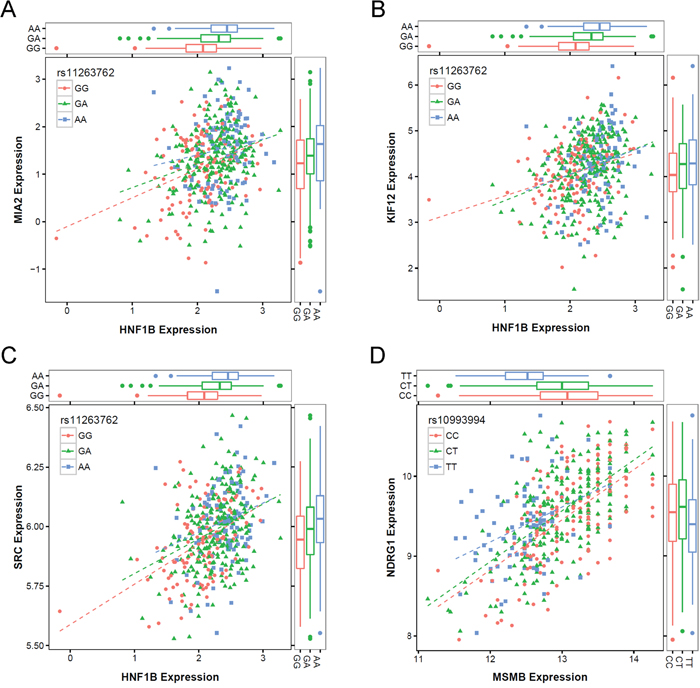
Figure 2: Co-expression scatterplots for four cis-mediator trios with evidence of replication in GTEx, including (A) rs11263762→HNF1B→MIA2, (B) rs11263762→HNF1B →KIF12, (C) rs11263762→HNF1B→SRC, and (D) rs10993994→MSMB→NDRG1. Each sub-figure presents the cis-gene expression on the horizontal axis and the trans-gene expression on the vertical axis, with individual points separated by the cis-eQTL genotype based on color and shape. Boxplots on the margins also summarize the expression distributions by eQTL genotype.
Fine-mapping studies of the PRCA risk locus near HNF1B have indicated potentially multiple variants independently contributing to disease susceptibility for prostate [31] and endometrial cancers [32]. Thus, the peak cis-eQTL association may not completely capture downstream regulatory effects on potential HNF-1B targets. We subsequently relaxed the constraint of investigating trans-associations with peak HNF1B cis-SNP rs11263762 to all 33 potential PRCA risk cis-variants (Supplementary Table 3) for the nine HNF1B-connected gene nodes. Although the peak trans-eQTL SNP association varied for each gene, SNPs corresponding to significant or suggestive eQTLs were all included in a single LD block consisting of 20 SNPs (Supplementary Figure 2). The significance status of the cis-mediator relationship across the nine trans genes remained the same regardless of the selected SNP (peak cis or peak trans) within the LD block.
Previous studies investigating HNF-1B transcription factor targets in PRCA cell lines identified multiple potentially dysregulated genes relevant to PRCA. Hu et al. [33] discovered six putative HNF-1B target genes by leveraging publicly available microarray datasets and conducting HNF-1B transfection studies of multiple prostate cell lines. More recently, Ross-Adams et al. [34] discovered two separate genes (FLRT3 and SLC14A1) that exhibited reduced expression in accordance with HNF1B over-expression in PC3. In our analyses, none of these genes exhibited compelling evidence of trans-eQTL effects with HNF1B cis-eQTLs consistent with HNF-1B regulation in normal prostate epithelium (min. trans-eQTL P = 0.009 across 272 tests; Supplementary Table 4).
HNF1B and MSMB also encode multiple isoforms, and differential isoform expression for these genes has been observed in comparisons of tumor and normal prostate tissues [35]. To explore whether significant eQTL SNPs corresponded to differential isoform usage, we conducted supplementary isoform-ratio QTL analyses using the R package sQTLSeeker [36] and isoform abundances derived from StringTie v1.2.4 [37]. We identified one intronic SNP, rs3110641, to be associated with differential isoform expression (P = 2.0E-04), although this SNP was not a significant trans-eQTL for any HNF1B-connected genes. Similarly, we did not observe any increase in mediation by any one particular HNF1B isoform, although the most highly expressed transcript (ENST00000225893) did account for the largest mediation effects across all significant cis-mediator trios and exhibited the strongest eQTL association with rs11263762 (P = 1.5E-05, Supplementary Figure 3). No alternative isoform transcripts were observed in sufficient quantity for MSMB.
It is possible due to measurement error or hidden confounders that the estimated undirected graph failed to include relevant trans-gene connections with significantly cis-regulated genes, and true co-expression relationships may have gone undetected. We conducted supplementary sensitivity analyses to identify all trans-eQTL associations for the 72 SNPs that were significant cis-eQTLs based on similar cis-mediator analyses agnostic of the expression network connections, yielding approximately 1.3 million tests. Using the same mediation p-value significance threshold defined above (P < 4.3E-05), only four additional trios were detected (Supplementary Table 5), none of which would have achieved significance by Bonferroni correction under this new multiple testing dimensionality (all P > 1E-07).
DISCUSSION
It is now widely believed that a large proportion of genetic associations with complex traits are regulatory in nature, as trait-associated GWAS findings have been shown to be significantly enriched for eQTLs [38]. While multiple studies have investigated the potential regulatory roles of PRCA susceptibility loci on gene expression, they have largely focused on dysregulation of proximal genes. Thus, association findings only elucidate the initial functional consequences of these variants, which may in turn disrupt downstream molecular pathways. Using an expression network-directed analysis strategy, we identified evidence of three significant cis-mediator trios indicating potential downstream dysregulation of protein coding genes SRC, NDRG1, and MIA2 by PRCA susceptibility loci in normal prostate epithelium, all of which are known to play prominent roles in tumorigenesis.
HNF1B encodes the protein hepatocyte nuclear factor-1 beta (HNF-1B), a transcription factor that plays a critical regulatory role in nephron and pancreas development [39]. HNF-1B and related protein HNF-1A comprise the HNF-1 sub-family of homeodomain-containing transcription factors, and may bind to DNA as homodimers or heterodimers [40]. Although primarily expressed in the liver, these transcription factors are known to regulate tissue-specific gene expression in the epithelia of a variety of organs [41], and may either activate or suppress transcription. Multiple cancer risk SNP associations have been reported at the HNF1B locus [31], although expression studies have produced conflicting results with respect to the regulatory effect of risk alleles on HNF1B in benign prostate tissue. These results have led to differing perspectives on the role of HNF-1B in progression of PRCA. For example, Griziano et al. [42] identified increased normal prostate HNF1B expression levels with the rs4430796-A allele across multiple ethnicities, and HNF1B knockdown in the LNCaP PRCA cell line resulted in reduced colony formation, proliferation, and viability. In contrast, Ross-Adams et al. [34] found no significant eQTL associations between four PRCA risk SNPs in normal prostate tissue samples; however, PRCA risk alleles for rs3760511 and rs11649743 corresponded to elevated HNF1B expression in tumor samples, with additional evidence indicating these variants are associated with reduced promoter methylation. In our analyses, risk-associated alleles exhibited patterns of upregulatory effects on HNF1B expression, suggesting oncogenic properties of HNF1B in the development of PRCA. Similar results were observed in the fine-mapping analysis of the HNF1B locus by Painter et al. [32] in endometrioid cancer, which identified the protective allele of SNP rs11263763 (r2 with rs11263762 = 0.61; HNF1B cis-eQTL P = 4.8E-12) to be associated with reduced HNF1B promoter activity. Recent analysis of HNF1B in breast cancer has also indicated HNF1B overexpression induces transformation and epithelial-to-mesenchymal transition in the NMuMG epithelial cell-line [43], providing further evidence of an oncogenic role of HNF1B in cancers of epithelial origin.
HNF1B corresponded to four out of seven of the reported cis-mediator associations we identified, indicating potential dysregulation of multiple HNF-1B transcription factor targets by PRCA susceptibility variants near HNF1B. SRC encodes the proto-oncogene c-Src, a member of the Src family kinases [44], and Src pathways play a prominent role in PRCA tumorigenesis [45, 46]. SRC expression has also been shown to be directly regulated by HNF-1A (although not HNF-1B) via an alternative tissue-specific HNF-1 promoter in multiple cell-types [47]. MIA2 encodes the melanoma-inhibitory activity 2 (MIA2) protein, which belongs to the MIA gene family, and is similarly transcriptionally regulated by HNF-1A [48]. Although corresponding to tumor suppressive properties in hepatocellular carcinoma [49], MIA2 exhibits protumoral properties in oral squamous cell carcinoma, demonstrating increases in invasion, survival, and angiogenesis [50]. HNF-1A induced expression of MIA2 has also been implicated in pancreatic cancer [51]. Additionally, expression of KIF12 has been shown to be directly regulated by HNF-1B in kidneys [52]. While KIF12 has been implicated as a disease severity modifier of renal cystic disease via HNF-1B-induced transcription [53], its potential role in PRCA etiology is not immediately clear. Eight previously identified PRCA HNF-1B target genes were not replicated in our analyses; however, it is important to note these genes were validated in the correspondent studies based on HNF-1B transfected PC3 PRCA cell-line experiments, not normal prostate epithelial cells.
MSMB encodes the prostate secretory protein 94 (PSP94), which is predominantly expressed in prostate. Reduced or lost MSMB expression is commonly observed in PRCA tumors [54, 55] and is generally associated with poor prognosis and increased risk of recurrence [56, 57], although other studies have produced contrary findings [58]. Suppression of MSMB in prostate epithelial cells also promotes anchorage-independent growth [59]. Multiple genetic association studies [60–62] have replicated the correspondent eQTL variant, rs10993994, with PRCA susceptibility, and the risk-associated T allele has been shown to result in reduced expression of PSP94 [63, 64]. Consequently, PSP94 is widely believed to confer protective effects against PRCA tumorigenesis, although the underlying mechanism is poorly understood. It has been postulated PSP94 may act as a tumor suppressor or limit fungal pathogenic infection of prostate tissue [65]. In our analyses, we identified a trans-assocition of NDRG1 expression with PRCA risk SNP rs10993994 to be mediated by MSMB expression, with the two genes exhibiting positively correlated co-expression patterns. Although the potential biological mechanisms linking MSMB expression to dysregulation of NDRG1 are not immediately clear, it is hypothesized that PSP94 peptides may activate signal transduction pathways relevant to apoptosis via cell surface receptors [66]. NDRG1 encodes the N-myc downstream regulated gene 1 (NDRG1) protein, and NDRG1 gene expression is repressed by N-myc and c-myc [67]. The NDRG1 protein participates in multiple cancer -related pathways, and P53-induced expression of NDRG1 has been shown to suppress cell growth and proliferation [68]. NDRG1 has also been shown to inhibit activation of c-Src by preventing protein-protein interactions between c-Src and EGFR [69]. Specific to PRCA, NDRG1 expression is inversely associated with Gleason score [70], and immunohistochemistry experiments have indicated reduced NDRG1 expression in neoplastic tissue compared to adjacent normal cells [71]. Thus, MSMB may confer protective effects against PRCA via signaling cascades that upregulate expression of NDRG1.
There are a number of limitations to our study that warrant mention. First, our network inference is based solely on mRNA expression, and only captures a fraction of the biomolecular intermediaries of causal pathways impacted by dysregulation of the cis-associated genes. The relatively small sample size in comparison to the large gene count may also have limited our ability to estimate the graphical network structure, and smaller partial correlations may have gone undetected. Second, other types of RNA beyond protein-coding transcripts are also known to possess regulatory effects, including long non-coding RNAs and miRNAs. For example, HNF-1B was recently identified to regulate the miR-200 cluster in renal cells [72]. Integrative analyses of the co-expression patterns between the variety of RNA species may provide a more complete perspective on the impact of PRCA susceptibility genetics on the prostate transcriptome, although these likely necessitate larger datasets. Third, rigorous replication and lab validation of our findings will be necessary to verify the regulatory associations we have identified.
By integrating gene co-expression patterns and causal mediation analyses in the evaluation of transcriptional dysregulation by PRCA risk loci, we identified multiple plausible downstream effects mediated by PRCA risk genes MSMB and HNF1B. Our work provides the foundation for novel hypotheses for further investigation into the functional genetics of PRCA susceptibility and tumor progression.
MATERIALS AND METHODS
Study samples
All analyses were conducted on a normal prostate tissue eQTL dataset comprised of 471 samples that passed strict quality control criteria (dbGap accession phs000985.v1.p1), previously detailed elsewhere [17, 73]. Briefly, normal prostate tissue was acquired from an archive collection of fresh frozen material obtained from patients with either radical prostatectomy or cystoprostatectomy, which was reviewed to identify samples that met the following criteria: absence of prostate tumor, Gleason score was <7 for the presenting tumor, absence of high-grade prostatic intraepithelial neoplasia and benign prostatic hyperplasia, normal prostatic epithelial glands representing ≥ 40% of all cells, lymphocytic population representing ≤ 2% of all cells, and the normal epithelium was from the posterior region of the prostate. Informed consent was obtained from all subjects and the study was approved by the Mayo Clinic Institutional Review Board.
Genotyping and imputation
DNA was extracted using the Puregene tissue extraction protocol per the manufacturer’s recommendations and DNA quality was assessed by examining 260/280 ratio and DNA yield. Samples were genotyped using Illumina Infinium 2.5M bead arrays based on the manufacturer’s protocol (Illumina, San Diego, CA, USA). Standard quality control analyses were performed to identify poor-quality samples or SNPs. Untyped SNPs as well as missing genotypes for typed SNPs were imputed using SHAPEIT [74] and IMPUTE2 [75] with reference files from the 1000 Genomes Phase I integrated variant set.
RNA sequencing and expression quantification
RNA was extracted using the QIAGEN miRNeasy Mini Kit and the QIAcube instrument in accordance with the manufacturer’s instructions, and RNA quality was assessed by evaluating the RNA integrity number (>7) and the 260/280 ratio. RNA libraries were prepared using the TruSeq RNA Sample Prep Kit v2 (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions. Paired-end sequencing was performed on an Illumina HiSeq 2000 using TruSeq SBS sequencing kit version 3 and HCS v2.0.12 data collection software. A minimum of 50 million total reads per sample was required for analysis; samples with insufficient reads were re-sequenced and resultant BAM files were merged.
RNA-seq data were analyzed with the use of the MAP-R-Seq pipeline [76]. Paired-end reads were aligned by TopHat 2.0.6 [77] against the hg19 genome build using the bowtie1 aligner option. RSeQC [78] was used to calculate various quality control metrics to identify problematic samples. Such metrics include: the genomic distance between paired-end reads, the sequencing depth for predicted alternate splicing events, the rate of duplicate reads, and the evenness of each sample’s gene body coverage. Gene counts were quantified for 102,279 RNA features based on the ENSEMBL GRCh37.75 gene definition file, of which 17,216 were identified as protein coding genes from the biotype annotation field and declared to be expressed based on a median gene read count ≥ 10.
To remove potential biases such as GC content and differences in sequencing depth, gene read counts were normalized using conditional quantile normalization [79]. To account for latent sources of non-genetic variation in gene expression, we applied principal components analysis (PCA) to the complete normalized gene expression matrix, identifying 13 PCs for inclusion as covariates in the eQTL analysis, each accounting for ≥1% of the total variation.
eQTL analyses
For the 202 previously reported PRCA risk SNPs (Supplementary Table 1), we declared all observed and high-quality imputed variants (allelic ) within 200kb and with linkage disequilibrium (LD) r2>0.5 eligible for eQTL analysis as candidate risk variants, resulting in a total of 8073 variants of interest. All eQTL association analyses were conducted using the MatrixEQTL R package [80], adjusting for PE, PL, and the top 13 expression PCs. Associations were defined as cis if the eQTL SNP was within 1 Mb of the transcript. To avoid spurious associations due to long-range LD patterns, we declared all transcripts at least 10 Mb from a cis-gene to be eligible as cis-mediated trans-genes.
Network inference
To identify a large subset of potentially relevant PRCA susceptibility genes for network inference, we conducted an initial transcriptome-wide eQTL screening for expressed transcripts with all tag PRCA risk SNPs under liberal selection criteria. The rationale for this strategy was the notion that risk SNP associations would propagate through relevant co-expression networks. The smallest eQTL association p-value per transcript across all tested SNPs was Bonferroni-adjusted for the number of original risk loci (i.e., 202), and all transcripts corresponding to a FDR < 0.20 were selected for network analysis. This permissive significance threshold accommodated efficient dimensionality reduction while limiting the potential exclusion of false negative results. We estimated an undirected graph using the Meinshausen-Buhlmann method as part of the huge R package [81]. Default settings were used for all regularization parameters.
Cis-Mediators and causal inference
A cis-mediator causal relationship is comprised of the eQTL variant, denoted , the cis-regulated transcript (or cis-gene), denoted , and the trans-regulated transcript (or trans-gene), denoted . Thus, the eQTL variant, cis-gene, and trans-gene comprise a candidate cis-mediator trio , where the causal relationship can be characterized as with arrows indicating causal direction (Figure 3A). The peak cis-eQTL SNP per gene was defined according to the smallest eQTL association p-value. All peak cis-eQTL associations with a p-value below a Bonferroni-adjusted threshold for all cis-eQTL association tests (P < 0.05/144,628 = 3.5E-07) were considered to be significant. All mRNAs connected to significant cis-genes in the expression network and in trans with the corresponding peak cis-eQTL variant were declared to be eligible cis-mediator trios (Figure 3B).
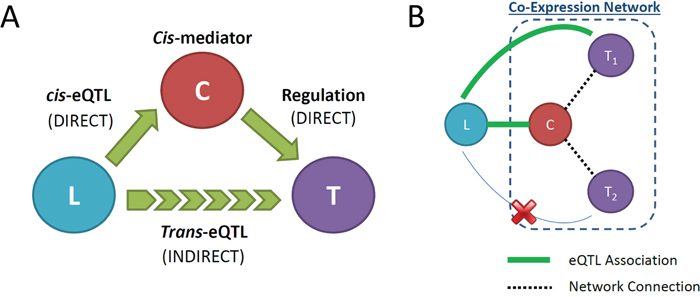
Figure 3: (A) Diagram indicating the causal relationships among the cis-eQTL variant, L, the cis-eQTL gene, C, and the trans-eQTL gene, T, that define the cis-mediator causal pathway. The trans-eQTL association is an indirect relationship mediated by the cis-regulated gene C. (B) Illustration of co-expression network strategy for identifying potential cis-mediated trans-eQTLs. Solid green lines indicate significant eQTL association, while dotted black lines indicate co-expression network connections between node genes. Here, locus L demonstrates a shared eQTL association with gene C and network-connected gene T1(but not T2), which can be further investigated using causal mediation analysis.
Causal inference was conducted for all eligible trios to investigate whether trans-gene associations with cis-SNPs were mediated by the corresponding cis-gene expression. We employed model-based causal inference analysis using the cit R package [82, 83]. The mediation analysis methods in cit correspond to a conservative omnibus intersection-union test for four constituent association relationships that comprise the causal mediation relationship , returning the maximum p-value across these tests. We additionally quantified mediation by calculating the proportion of the trans-eQTL effect estimate βˆT attenuated by the additional adjustment of the corresponding cis-gene expression values as a covariate. For cis-gene adjusted trans-eQTL effect , we define the estimated mediated proportion as . All mediation tests with p-values below a Bonferroni-adjusted -level of 0.05 were reported as significant cis-mediated trans-eQTLs.
Abbreviations
eQTL, expression quantitative trait locus; FDR, false discovery rate; GTEx, Gene Tissue Expression project; GWAS, genome-wide association study; LD, linkage disequilibrium; PCA, principal components analysis; PRCA, prostate cancer; SNP, single-nucleotide polymorphism.
Author contributions
NBL conceived and conducted the primary analyses as well as wrote the manuscript; SKD, ZF, ML, SR, SB, AW, AAN, DO, and JD contributed to data preparation and intermediate analyses. JC, LW, DJS and SNT contributed to original study design and data collection. DJS and SNT also provided guidance on interpretation of results. All co-authors contributed to the final manuscript.
CONFLICTS OF INTEREST
The authors do not declare any conflicts of interest.
FUNDING
This work was supported by a Department of Defense Grant (W81XWH-11-1-0261) and a grant from the National Institutes of Health (CA151254 to SNT). SNP genotyping and RNA-seq was partially supported by National Institute of Health (R01CA157881 to LW).
REFERENCES
1. Hjelmborg JB, Scheike T, Holst K, Skytthe A, Penney KL, Graff RE, Pukkala E, Christensen K, Adami HO, Holm NV, Nuttall E, Hansen S, Hartman M, et al. The heritability of prostate cancer in the Nordic Twin Study of Cancer. Cancer Epidemiol Biomarkers Prev. 2014; 23:2303–10.
2. Gudmundsson J, Sulem P, Manolescu A, Amundadottir LT, Gudbjartsson D, Helgason A, Rafnar T, Bergthorsson JT, Agnarsson BA, Baker A, Sigurdsson A, Benediktsdottir KR, Jakobsdottir M, et al. Genome-wide association study identifies a second prostate cancer susceptibility variant at 8q24. Nat Genet. 2007; 39:631–37.
3. Yeager M, Orr N, Hayes RB, Jacobs KB, Kraft P, Wacholder S, Minichiello MJ, Fearnhead P, Yu K, Chatterjee N, Wang Z, Welch R, Staats BJ, et al. Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet. 2007; 39:645–49.
4. Salinas CA, Kwon E, Carlson CS, Koopmeiners JS, Feng Z, Karyadi DM, Ostrander EA, Stanford JL. Multiple independent genetic variants in the 8q24 region are associated with prostate cancer risk. Cancer Epidemiol Biomarkers Prev. 2008; 17:1203–13.
5. Schaid DJ, McDonnell SK, Riska SM, Carlson EE, Thibodeau SN. Estimation of genotype relative risks from pedigree data by retrospective likelihoods. Genet Epidemiol. 2010; 34:287–98.
6. Kote-Jarai Z, Olama AA, Giles GG, Severi G, Schleutker J, Weischer M, Campa D, Riboli E, Key T, Gronberg H, Hunter DJ, Kraft P, Thun MJ, et al, and UK Genetic Prostate Cancer Study Collaborators/British Association of Urological Surgeons’ Section of Oncology, and UK ProtecT Study Collaborators, The Australian Prostate Cancer BioResource, and PRACTICAL Consortium. Seven prostate cancer susceptibility loci identified by a multi-stage genome-wide association study. Nat Genet. 2011; 43:785–91.
7. Cheng I, Chen GK, Nakagawa H, He J, Wan P, Laurie CC, Shen J, Sheng X, Pooler LC, Crenshaw AT, Mirel DB, Takahashi A, Kubo M, et al. Evaluating genetic risk for prostate cancer among Japanese and Latinos. Cancer Epidemiol Biomarkers Prev. 2012; 21:2048–58.
8. Akamatsu S, Takata R, Haiman CA, Takahashi A, Inoue T, Kubo M, Furihata M, Kamatani N, Inazawa J, Chen GK, Le Marchand L, Kolonel LN, Katoh T, et al. Common variants at 11q12, 10q26 and 3p11.2 are associated with prostate cancer susceptibility in Japanese. Nat Genet. 2012; 44:426-9, S1. https://doi.org/10.1038/ng.1104..
9. Kote-Jarai Z, Saunders EJ, Leongamornlert DA, Tymrakiewicz M, Dadaev T, Jugurnauth-Little S, Ross-Adams H, Al Olama AA, Benlloch S, Halim S, Russell R, Dunning AM, Luccarini C, et al, and PRACTICAL Consortium. Fine-mapping identifies multiple prostate cancer risk loci at 5p15, one of which associates with TERT expression. Hum Mol Genet. 2013; 22:2520–28. https://doi.org/10.1093/Hmg/Ddt086
10. Amin Al Olama A, Kote-Jarai Z, Schumacher FR, Wiklund F, Berndt SI, Benlloch S, Giles GG, Severi G, Neal DE, Hamdy FC, Donovan JL, Hunter DJ, Henderson BE, et al, and UK Genetic Prostate Cancer Study Collaborators/British Association of Urological Surgeons’ Section of Oncology, and UK ProtecT Study Collaborators, and Australian Prostate Cancer Bioresource, and PRACTICAL Consortium. A meta-analysis of genome-wide association studies to identify prostate cancer susceptibility loci associated with aggressive and non-aggressive disease. Hum Mol Genet. 2013; 22:408–15. https://doi.org/10.1093/Hmg/Dds425
11. Hazelett DJ, Rhie SK, Gaddis M, Yan C, Lakeland DL, Coetzee SG, Henderson BE, Noushmehr H, Cozen W, Kote-Jarai Z, Eeles RA, Easton DF, Haiman CA, et al, and Ellipse/GAME-ON consortium, and Practical consortium. Comprehensive functional annotation of 77 prostate cancer risk loci. PLoS Genet. 2014; 10:e1004102.
12. Al Olama AA, Kote-Jarai Z, Berndt SI, Conti DV, Schumacher F, Han Y, Benlloch S, Hazelett DJ, Wang Z, Saunders E, Leongamornlert D, Lindstrom S, Jugurnauth-Little S, et al, and Breast and Prostate Cancer Cohort Consortium (BPC3), and PRACTICAL (Prostate Cancer Association Group to Investigate Cancer-Associated Alterations in the Genome) Consortium, and COGS (Collaborative Oncological Gene-environment Study) Consortium, and GAME-ON/ELLIPSE Consortium. A meta-analysis of 87,040 individuals identifies 23 new susceptibility loci for prostate cancer. Nat Genet. 2014; 46:1103–09.
13. Yang Z, Hu S, Cheng J, Xu J, Shi W, Zhu B, Zhang Y, Yao Z, Pan H, Zhang Y. Prevalence and risk of cancer of incidental uptake in prostate identified by fluorine-18 fluorodeoxyglucose positron emission tomography/computed tomography. Clin Imaging. 2014; 38:470–74.
14. Hoffmann TJ, Van Den Eeden SK, Sakoda LC, Jorgenson E, Habel LA, Graff RE, Passarelli MN, Cario CL, Emami NC, Chao CR, Ghai NR, Shan J, Ranatunga DK, et al. A large multiethnic genome-wide association study of prostate cancer identifies novel risk variants and substantial ethnic differences. Cancer Discov. 2015; 5:878–91.
15. Teerlink CC, Leongamornlert D, Dadaev T, Thomas A, Farnham J, Stephenson RA, Riska S, McDonnell SK, Schaid DJ, Catalona WJ, Zheng SL, Cooney KA, Ray AM, et al, and PRACTICAL consortium, and International Consortium for Prostate Cancer Genetics. Genome-wide association of familial prostate cancer cases identifies evidence for a rare segregating haplotype at 8q24.21. Hum Genet. 2016; 135:923–38.
16. Amin Al Olama A, Dadaev T, Hazelett DJ, Li Q, Leongamornlert D, Saunders EJ, Stephens S, Cieza-Borrella C, Whitmore I, Benlloch Garcia S, Giles GG, Southey MC, Fitzgerald L, et al, and PRACTICAL Consortium, and COGS-CRUK GWAS-ELLIPSE (Part of GAME-ON) Initiative, and Australian Prostate Cancer BioResource, and UK Genetic Prostate Cancer Study Collaborators, and UK ProtecT Study Collaborators. Multiple novel prostate cancer susceptibility signals identified by fine-mapping of known risk loci among Europeans. Hum Mol Genet. 2015; 24:5589–602.
17. Thibodeau SN, French AJ, McDonnell SK, Cheville J, Middha S, Tillmans L, Riska S, Baheti S, Larson MC, Fogarty Z, Zhang Y, Larson N, Nair A, et al. Identification of candidate genes for prostate cancer-risk SNPs utilizing a normal prostate tissue eQTL data set. Nat Commun. 2015; 6:8653.
18. Whitington T, Gao P, Song W, Ross-Adams H, Lamb AD, Yang Y, Svezia I, Klevebring D, Mills IG, Karlsson R, Halim S, Dunning MJ, Egevad L, et al. Gene regulatory mechanisms underpinning prostate cancer susceptibility. Nat Genet. 2016; 48:387–97.
19. Kirsten H, Al-Hasani H, Holdt L, Gross A, Beutner F, Krohn K, Horn K, Ahnert P, Burkhardt R, Reiche K, Hackermüller J, Löffler M, Teupser D, et al. Dissecting the genetics of the human transcriptome identifies novel trait-related trans-eQTLs and corroborates the regulatory relevance of non-protein coding loci. Hum Mol Genet. 2015; 24:4746–63.
20. Westra HJ, Peters MJ, Esko T, Yaghootkar H, Schurmann C, Kettunen J, Christiansen MW, Fairfax BP, Schramm K, Powell JE, Zhernakova A, Zhernakova DV, Veldink JH, et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet. 2013; 45:1238–43.
21. Joehanes R, Zhang X, Huan T, Yao C, Ying SX, Nguyen QT, Demirkale CY, Feolo ML, Sharopova NR, Sturcke A, Schäffer AA, Heard-Costa N, Chen H, et al. Integrated genome-wide analysis of expression quantitative trait loci aids interpretation of genomic association studies. Genome Biol. 2017; 18:16.
22. Yao C, Joehanes R, Johnson AD, Huan T, Liu C, Freedman JE, Munson PJ, Hill DE, Vidal M, Levy D. Dynamic Role of trans Regulation of Gene Expression in Relation to Complex Traits. Am J Hum Genet. 2017; 100:571–80.
23. Yao C, Chen BH, Joehanes R, Otlu B, Zhang X, Liu C, Huan T, Tastan O, Cupples LA, Meigs JB, Fox CS, Freedman JE, Courchesne P, et al. Integromic analysis of genetic variation and gene expression identifies networks for cardiovascular disease phenotypes. Circulation. 2015; 131:536–49.
24. Chen X, McClelland M, Jia Z, Rahmatpanah FB, Sawyers A, Trent J, Duggan D, Mercola D. The identification of trans-associations between prostate cancer GWAS SNPs and RNA expression differences in tumor-adjacent stroma. Oncotarget. 2015; 6:1865–73. https://doi.org/10.18632/oncotarget.2763
25. Kang HM, Ye C, Eskin E. Accurate discovery of expression quantitative trait loci under confounding from spurious and genuine regulatory hotspots. Genetics. 2008; 180:1909–25.
26. Brynedal B, Choi J, Raj T, Bjornson R, Stranger BE, Neale BM, Voight BF, Cotsapas C. Large-Scale trans-eQTLs Affect Hundreds of Transcripts and Mediate Patterns of Transcriptional Co-regulation. Am J Hum Genet. 2017; 100:581–91.
27. Pierce BL, Tong L, Chen LS, Rahaman R, Argos M, Jasmine F, Roy S, Paul-Brutus R, Westra HJ, Franke L, Esko T, Zaman R, Islam T, et al. Mediation analysis demonstrates that trans-eQTLs are often explained by cis-mediation: a genome-wide analysis among 1,800 South Asians. PLoS Genet. 2014; 10:e1004818.
28. Wang YX, Huang H. Review on statistical methods for gene network reconstruction using expression data. J Theor Biol. 2014; 362:53–61.
29. Franzén O, Ermel R, Cohain A, Akers NK, Di Narzo A, Talukdar HA, Foroughi-Asl H, Giambartolomei C, Fullard JF, Sukhavasi K, Köks S, Gan LM, Giannarelli C, et al. Cardiometabolic risk loci share downstream cis- and trans-gene regulation across tissues and diseases. Science. 2016; 353:827–30.
30. Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, Hasz R, Walters G, Garcia F, Young N, Foster B, Moser M, Karasik E, et al, and GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013; 45:580–85. https://doi.org/10.1038/Ng.2653
31. Berndt SI, Sampson J, Yeager M, Jacobs KB, Wang Z, Hutchinson A, Chung C, Orr N, Wacholder S, Chatterjee N, Yu K, Kraft P, Feigelson HS, et al. Large-scale fine mapping of the HNF1B locus and prostate cancer risk. Hum Mol Genet. 2011; 20:3322–29.
32. Painter JN, O’Mara TA, Batra J, Cheng T, Lose FA, Dennis J, Michailidou K, Tyrer JP, Ahmed S, Ferguson K, Healey CS, Kaufmann S, Hillman KM, et al, and National Study of Endometrial Cancer Genetics Group (NSECG), and CHIBCHA Consortium, and Australian National Endometrial Cancer Study Group (ANECS), and RENDOCAS, and Australian Ovarian Cancer Study (AOCS), and GENICA Network. Fine-mapping of the HNF1B multicancer locus identifies candidate variants that mediate endometrial cancer risk. Hum Mol Genet. 2015; 24:1478–92.
33. Hu YL, Zhong D, Pang F, Ning QY, Zhang YY, Li G, Wu JZ, Mo ZN. HNF1b is involved in prostate cancer risk via modulating androgenic hormone effects and coordination with other genes. Genet Mol Res. 2013; 12:1327–35.
34. Ross-Adams H, Ball S, Lawrenson K, Halim S, Russell R, Wells C, Strand SH, Ørntoft TF, Larson M, Armasu S, Massie CE, Asim M, Mortensen MM, et al. HNF1B variants associate with promoter methylation and regulate gene networks activated in prostate and ovarian cancer. Oncotarget. 2016; 7:74734–46. https://doi.org/10.18632/oncotarget.12543
35. Harries LW, Perry JR, McCullagh P, Crundwell M. Alterations in LMTK2, MSMB and HNF1B gene expression are associated with the development of prostate cancer. BMC Cancer. 2010; 10:315.
36. Monlong J, Calvo M, Ferreira PG, Guigó R. Identification of genetic variants associated with alternative splicing using sQTLseekeR. Nat Commun. 2014; 5:4698.
37. Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015; 33:290–95.
38. Nicolae DL, Gamazon E, Zhang W, Duan S, Dolan ME, Cox NJ. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 2010; 6:e1000888.
39. Maestro MA, Cardalda C, Boj SF, Luco RF, Servitja JM, Ferrer J. Distinct roles of HNF1beta, HNF1alpha, and HNF4alpha in regulating pancreas development, beta-cell function and growth. Endocr Dev. 2007; 12:33–45.
40. Haumaitre C, Reber M, Cereghini S. Functions of HNF1 family members in differentiation of the visceral endoderm cell lineage. J Biol Chem. 2003; 278:40933–42.
41. Igarashi P, Shao X, McNally BT, Hiesberger T. Roles of HNF-1beta in kidney development and congenital cystic diseases. Kidney Int. 2005; 68:1944–47.
42. Grisanzio C, Werner L, Takeda D, Awoyemi BC, Pomerantz MM, Yamada H, Sooriakumaran P, Robinson BD, Leung R, Schinzel AC, Mills I, Ross-Adams H, Neal DE, et al. Genetic and functional analyses implicate the NUDT11, HNF1B, and SLC22A3 genes in prostate cancer pathogenesis. Proc Natl Acad Sci USA. 2012; 109:11252–57.
43. Matsui A, Fujimoto J, Ishikawa K, Ito E, Goshima N, Watanabe S, Semba K. Hepatocyte nuclear factor 1 beta induces transformation and epithelial-to-mesenchymal transition. FEBS Lett. 2016; 590:1211–21.
44. Thomas SM, Brugge JS. Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol. 1997; 13:513–609.
45. Varkaris A, Katsiampoura AD, Araujo JC, Gallick GE, Corn PG. Src signaling pathways in prostate cancer. Cancer Metastasis Rev. 2014; 33:595–606.
46. Vlaeminck-Guillem V, Gillet G, Rimokh R. SRC: marker or actor in prostate cancer aggressiveness. Front Oncol. 2014; 4:222.
47. Bonham K, Ritchie SA, Dehm SM, Snyder K, Boyd FM. An alternative, human SRC promoter and its regulation by hepatic nuclear factor-1alpha. J Biol Chem. 2000; 275:37604–11.
48. Bosserhoff AK, Moser M, Schölmerich J, Buettner R, Hellerbrand C. Specific expression and regulation of the new melanoma inhibitory activity-related gene MIA2 in hepatocytes. J Biol Chem. 2003; 278:15225–31.
49. Hellerbrand C, Amann T, Schlegel J, Wild P, Bataille F, Spruss T, Hartmann A, Bosserhoff AK. The novel gene MIA2 acts as a tumour suppressor in hepatocellular carcinoma. Gut. 2008; 57:243–51.
50. Kurihara M, Kirita T, Sasahira T, Ohmori H, Matsushima S, Yamamoto K, Bosserhoff AK, Kuniyasu H. Protumoral roles of melanoma inhibitory activity 2 in oral squamous cell carcinoma. Br J Cancer. 2013; 108:1460–69.
51. Kong B, Wu W, Valkovska N, Jäger C, Hong X, Nitsche U, Friess H, Esposito I, Erkan M, Kleeff J, Michalski CW. A common genetic variation of melanoma inhibitory activity-2 labels a subtype of pancreatic adenocarcinoma with high endoplasmic reticulum stress levels. Sci Rep. 2015; 5:8109.
52. Gong Y, Ma Z, Patel V, Fischer E, Hiesberger T, Pontoglio M, Igarashi P. HNF-1beta regulates transcription of the PKD modifier gene Kif12. J Am Soc Nephrol. 2009; 20:41–47.
53. Mrug M, Zhou J, Yang C, Aronow BJ, Cui X, Schoeb TR, Siegal GP, Yoder BK, Guay-Woodford LM. Genetic and Informatic Analyses Implicate Kif12 as a Candidate Gene within the Mpkd2 Locus That Modulates Renal Cystic Disease Severity in the Cys1cpk Mouse. PLoS One. 2015; 10:e0135678.
54. Tsurusaki T, Koji T, Sakai H, Kanetake H, Nakane PK, Saito Y. Cellular expression of beta-microseminoprotein (beta-MSP) mRNA and its protein in untreated prostate cancer. Prostate. 1998; 35:109–16.
55. Imasato Y, Xuan JW, Sakai H, Izawa JI, Saito Y, Chin JL, Moussa M. PSP94 expression after androgen deprivation therapy: a comparative study with prostate specific antigen in benign prostate and prostate cancer. J Urol. 2000; 164:1819–24.
56. Hyakutake H, Sakai H, Yogi Y, Tsuda R, Minami Y, Yushita Y, Kanetake H, Nakazono I, Saito Y. Beta-microseminoprotein immunoreactivity as a new prognostic indicator of prostatic carcinoma. Prostate. 1993; 22:347–55.
57. Bjartell AS, Al-Ahmadie H, Serio AM, Eastham JA, Eggener SE, Fine SW, Udby L, Gerald WL, Vickers AJ, Lilja H, Reuter VE, Scardino PT. Association of cysteine-rich secretory protein 3 and beta-microseminoprotein with outcome after radical prostatectomy. Clin Cancer Res. 2007; 13:4130–38.
58. Girvan AR, Chang P, van Huizen I, Moussa M, Xuan JW, Stitt L, Chin JL, Yamasaki Y, Izawa JI. Increased intratumoral expression of prostate secretory protein of 94 amino acids predicts for worse disease recurrence and progression after radical prostatectomy in patients with prostate cancer. Urology. 2005; 65:719–23.
59. Pomerantz MM, Shrestha Y, Flavin RJ, Regan MM, Penney KL, Mucci LA, Stampfer MJ, Hunter DJ, Chanock SJ, Schafer EJ, Chan JA, Tabernero J, Baselga J, et al. Analysis of the 10q11 cancer risk locus implicates MSMB and NCOA4 in human prostate tumorigenesis. PLoS Genet. 2010; 6:e1001204.
60. Kote-Jarai Z, Easton DF, Stanford JL, Ostrander EA, Schleutker J, Ingles SA, Schaid D, Thibodeau S, Dörk T, Neal D, Donovan J, Hamdy F, Cox A, et al, and PRACTICAL Consortium. Multiple novel prostate cancer predisposition loci confirmed by an international study: the PRACTICAL Consortium. Cancer Epidemiol Biomarkers Prev. 2008; 17:2052–61.
61. Eeles RA, Kote-Jarai Z, Giles GG, Olama AA, Guy M, Jugurnauth SK, Mulholland S, Leongamornlert DA, Edwards SM, Morrison J, Field HI, Southey MC, Severi G, et al, and UK Genetic Prostate Cancer Study Collaborators, and British Association of Urological Surgeons’ Section of Oncology, and UK ProtecT Study Collaborators. Multiple newly identified loci associated with prostate cancer susceptibility. Nat Genet. 2008; 40:316–21.
62. Chang BL, Cramer SD, Wiklund F, Isaacs SD, Stevens VL, Sun J, Smith S, Pruett K, Romero LM, Wiley KE, Kim ST, Zhu Y, Zhang Z, et al. Fine mapping association study and functional analysis implicate a SNP in MSMB at 10q11 as a causal variant for prostate cancer risk. Hum Mol Genet. 2009; 18:1368–75.
63. Whitaker HC, Kote-Jarai Z, Ross-Adams H, Warren AY, Burge J, George A, Bancroft E, Jhavar S, Leongamornlert D, Tymrakiewicz M, Saunders E, Page E, Mitra A, et al, and IMPACT Study Steering Committee, and IMPACT Study Collaborators, and UK GPCS Collaborators. The rs10993994 risk allele for prostate cancer results in clinically relevant changes in microseminoprotein-beta expression in tissue and urine. PLoS One. 2010; 5:e13363.
64. FitzGerald LM, Zhang X, Kolb S, Kwon EM, Liew YC, Hurtado-Coll A, Knudsen BS, Ostrander EA, Stanford JL. Investigation of the relationship between prostate cancer and MSMB and NCOA4 genetic variants and protein expression. Hum Mutat. 2013; 34:149–56.
65. Sutcliffe S, De Marzo AM, Sfanos KS, Laurence M. MSMB variation and prostate cancer risk: clues towards a possible fungal etiology. Prostate. 2014; 74:569–78.
66. Shukeir N, Arakelian A, Kadhim S, Garde S, Rabbani SA. Prostate secretory protein PSP-94 decreases tumor growth and hypercalcemia of malignancy in a syngenic in vivo model of prostate cancer. Cancer Res. 2003; 63:2072–78.
67. Shimono A, Okuda T, Kondoh H. N-myc-dependent repression of ndr1, a gene identified by direct subtraction of whole mouse embryo cDNAs between wild type and N-myc mutant. Mech Dev. 1999; 83:39–52.
68. Ellen TP, Ke Q, Zhang P, Costa M. NDRG1, a growth and cancer related gene: regulation of gene expression and function in normal and disease states. Carcinogenesis. 2008; 29:2–8.
69. Liu W, Yue F, Zheng M, Merlot A, Bae DH, Huang M, Lane D, Jansson P, Lui GY, Richardson V, Sahni S, Kalinowski D, Kovacevic Z, Richardson DR. The proto-oncogene c-Src and its downstream signaling pathways are inhibited by the metastasis suppressor, NDRG1. Oncotarget. 2015; 6:8851–74. https://doi.org/10.18632/oncotarget.3316
70. Song Y, Oda Y, Hori M, Kuroiwa K, Ono M, Hosoi F, Basaki Y, Tokunaga S, Kuwano M, Naito S, Tsuneyoshi M. N-myc downstream regulated gene-1/Cap43 may play an important role in malignant progression of prostate cancer, in its close association with E-cadherin. Hum Pathol. 2010; 41:214–22.
71. Bandyopadhyay S, Pai SK, Gross SC, Hirota S, Hosobe S, Miura K, Saito K, Commes T, Hayashi S, Watabe M, Watabe K. The Drg-1 gene suppresses tumor metastasis in prostate cancer. Cancer Res. 2003; 63:1731–36.
72. Hajarnis SS, Patel V, Aboudehen K, Attanasio M, Cobo-Stark P, Pontoglio M, Igarashi P. Transcription Factor Hepatocyte Nuclear Factor-1β (HNF-1β) Regulates MicroRNA-200 Expression through a Long Noncoding RNA. J Biol Chem. 2015; 290:24793–805.
73. Larson NB, McDonnell S, French AJ, Fogarty Z, Cheville J, Middha S, Riska S, Baheti S, Nair AA, Wang L, Schaid DJ, Thibodeau SN. Comprehensively evaluating cis-regulatory variation in the human prostate transcriptome by using gene-level allele-specific expression. Am J Hum Genet. 2015; 96:869–82.
74. Delaneau O, Zagury JF, Marchini J. Improved whole-chromosome phasing for disease and population genetic studies. Nat Methods. 2013; 10:5–6.
75. Marchini J, Howie B. Genotype imputation for genome-wide association studies. Nat Rev Genet. 2010; 11:499–511.
76. Kalari KR, Nair AA, Bhavsar JD, O’Brien DR, Davila JI, Bockol MA, Nie J, Tang X, Baheti S, Doughty JB, Middha S, Sicotte H, Thompson AE, et al. MAP-RSeq: Mayo Analysis Pipeline for RNA sequencing. BMC Bioinformatics. 2014; 15:224.
77. Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013; 14:R36.
78. Wang L, Wang S, Li W. RSeQC: quality control of RNA-seq experiments. Bioinformatics. 2012; 28:2184–85.
79. Hansen KD, Irizarry RA, Wu Z. Removing technical variability in RNA-seq data using conditional quantile normalization. Biostatistics. 2012; 13:204–16.
80. Shabalin AA. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics. 2012; 28:1353–58.
81. Zhao T, Liu H, Roeder K, Lafferty J, Wasserman L. The huge Package for High-dimensional Undirected Graph Estimation in R. J Mach Learn Res. 2012; 13:1059–62.
82. Millstein J, Zhang B, Zhu J, Schadt EE. Disentangling molecular relationships with a causal inference test. BMC Genet. 2009; 10:23.
83. Millstein J, Chen GK, Breton CV. cit: hypothesis testing software for mediation analysis in genomic applications. Bioinformatics. 2016; 32:2364–65.