
INTRODUCTION
Breast cancer is the most frequently diagnosed cancer among women in industrialized countries [1]. According to immunohistochemical staining of estrogen receptor α (ERα), progesterone receptor (PR) and human epidermal growth factor receptor 2 (HER2), breast cancer is categorized into the following groups; Luminal, subtype A: ERα+/PR+/HER2-; luminal, subtype B: ERα+/PR+/HER2+; HER2 overexpression: ERα-/PR-/HER2+ and triple-negative breast cancer (TNBC): ERα-/PR-/HER2- [2]. TNBC accounts for approximately 15 to 20% of all breast cancers and is often more aggressive with higher rates of recurrence and more frequent distant metastasis than other types of breast cancer [3]. Due to a lack of targeted therapy options available to subgroups that express ERα or with HER2 amplification, cytotoxic chemotherapy is the systemic therapy currently available for TNBC patients [4, 5]. Thus, there is a clear unmet medical need to identify new therapeutic targets for TNBC and understanding the mechanism of their action.
ERs belong to the nuclear receptor protein superfamily of ligand-activated transcription factors [6, 7]. For a long time, only one ER, ERα, was thought to exist [8]. However, in 1996, we reported a second ER, ERβ [9]. Like many other genes, ERβ is expressed as different isoforms, the functions of which need to be studied in order to understand the physiological functions of ERβ. The ERβ protein that shares the domain structure of other nuclear receptors is referred to as the full length wild type ERβ or ERβ1. The most studied human variant of ERβ, ERβ2, is the result of alternative splicing, the last 61 amino acids of ERβ1 being replaced by 26 unique amino acids from an alternative last exon. ERβ2 lacks an intact ligand binding domain and activation function 2 (AF-2). Since ERβ2 has an intact DNA-binding domain and an intact N-terminal domain, including the AF-1 region, it could be involved in gene regulation.
ERα has been shown to generally promote growth of ERα-positive breast cancer, which forms the basis for the use of ER antagonists as first line therapy in this breast cancer subgroup. ERβ1 has been suggested to display anti-proliferative properties, including in breast cancer [10, 11]. The prognostic impact of ERβ2 expression in breast cancer remains controversial. Several studies suggest that high ERβ2 expression is associated with poor outcome in breast cancer [12]. In contrast, some studies have reported that ERβ2 was associated with good clinical outcome or no prognostic value in breast cancer [13–16]. In a recent publication, Andersson et al show that most used ERβ antibodies are not sufficiently specific, providing a potential explanation for the apparent discrepancy of certain published studies [17]. Studies of the molecular mechanism of ERβ2 action have been limited by the lack of cellular models that express ERβ2, alone or in the presence of ERα and/or ERβ1. We have previously demonstrated, using a derivate of the ERα-positive breast cancer cell line MCF-7 engineered to express ERβ2, that ERβ2 heterodimerizes with ERα and inhibits ligand induced ERα transcriptional activity by inducing proteasome-dependent degradation of ERα [18]. However, the possible role of endogenous ERβ2 in breast cancer cells remains to be determined.
HIF-1α is a major determinant of invasion and metastasis in a wide variety of tumor types including breast cancer [19]. Its expression is regulated through modifications at the posttranslational level. In the presence of oxygen, three prolyl hydroxylase (PHD) enzymes, PHD1, PHD2 and PHD3, can hydroxylate two HIF-1α proline residues (p402 and p564) in the oxygen-dependent degradation domain. Hydroxylated HIF-1α is recognized by the tumour suppressor von Hippel-Lindau protein, which targets HIF-1α for degradation [20].
In this study, we identify TNBC breast cancer cell lines that express endogenous ERβ2 but not ERα or ERβ1. We then explore BT549 and MDA-MB-231 cell lines to approach the function of ERβ2 independently of ERα and ERβ1. Our results show that ERβ2, when expressed alone, promotes cell proliferation and invasion. We also investigate ERβ2 dependent global effects on gene expression and suggest a mechanism by which ERβ2 contributes to an invasive phenotype. Whether our results can be translated into an understanding of the molecular mechanism for the invasive nature of TNBC and potentially the identification of targeted therapies remains to be determined.
RESULTS
Triple-negative breast cancer (TNBC) cell lines express high level of ERβ2
We screened the expression of ERβ2 in a panel of ten breast cancer cell lines, including four ER-positive luminal (MCF-7, MDA-MB-175, ZR-751, CAMA-1), two HER2-positive (SK-BR-3 and HCC1569) and four TNBC (Hs578T, MDA-MB-231, BT549 and BT20) cell lines. The highest level of endogenous ERβ2 expression was observed in the BT549, MDA-MB-231 and BT20 TNBC cell lines (Figure 1). The mRNA expression levels of ERβ1 and ERα in these cell lines were undetectable (data not shown). BT549 and MDA-MB-231 cells were used for further functional studies due to the possibility to achieve high transfection efficiency in these two cell lines.
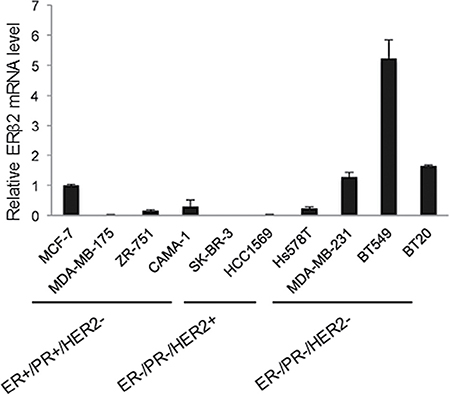
Figure 1: The TNBC cell lines BT549 and MDA-MB-231 express endogenous ERβ2. Analysis of ERβ2 mRNA levels in ER+/PR+/HER2- breast cancer cell lines (MCF-7, MDA-MB-175, ZR-751 and CAMA-1), ER-/PR-/HER2+ breast cancer cell lines (SK-BR-3 and HCC1569) and ER-/PR-/HER2- TNBC cell lines (Hs578T, MDA-MB-231, BT549 and BT20) by qPCR. mRNA levels are normalized to GAPDH, and mRNA levels are presented as means ± SD, relative to the expression level in MCF-7 cells.
Depletion of ERβ2 inhibits cellular proliferation and invasion in vitro
We used siRNA to silence ERβ2 expression in BT549 and MDA-MB-231 cells. Successful knockdown of ERβ2 was confirmed at the mRNA level (Figures 2A and S1A). Interestingly, ERβ2 knockdown decreased proliferation of both BT549 (Figure 2B) and MDA-MB-231 cells (Supplementary Figure 1B). Moreover, in invasion assays, ERβ2 knockdown significantly reduced the number of invading cells, from 34.6 ± 9.0 cells per field after control siRNA transfection to 13.4 ± 5.5 cells per field after ERβ2 siRNA transfection of BT549 cells (Figure 2C) (p < 0.01). These results were further validated in the MDA-MB-231 cell line where the number of invading cells per field was 12.7 ± 4.5 in the control siRNA transfected cells and 4.1 ± 3.1 in the ERβ2 siRNA transfected cells (Supplementary Figure 1C) (p < 0.001). The inhibition of invasion was also confirmed with a second set of siRNA targeting ERβ2 (data not shown), supporting that the observed effects are not related to off-target effects.
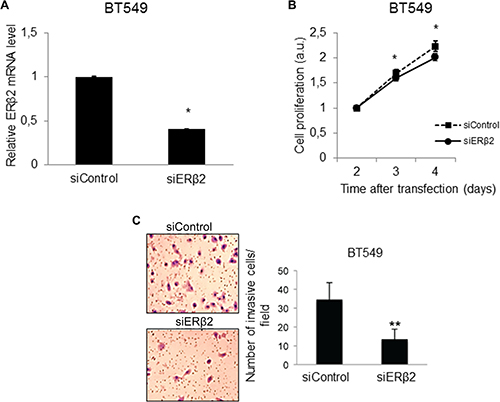
Figure 2: Depletion of ERβ2 inhibits cellular proliferation and invasion. (A) ERβ2 siRNA down-regulates ERβ2 mRNA in BT549 cells. ERβ2 mRNA level was determined by qPCR after transfection with control siRNA or ERβ2 siRNA. Data are normalized to 36B4 and shown as relative fold change compared to control siRNA ± SD. *P < 0.05. (B) ERβ2 depletion reduces proliferation of the BT549 cell line. BT549 cells were transfected with control siRNA or ERβ2 siRNA. WST-1 assays as a measure of cellular proliferation were carried out at the indicated time points after siRNA transfection. Ratio of absorbance to day 1 is calculated. Data are shown as means ± SD. *P < 0.05. The experiment was repeated three times. One representative experiment is shown. (C) ERβ2 depletion reduces invasion of BT549 cell line. BT549 cells were transfected with control siRNA or ERβ2 siRNA, and cell invasion was evaluated by the BD Biocoat growth factor reduced Matrigel invasion chamber assay. Data represent means ± SD. **P < 0.01. Experiment was repeated twice. One representative experiment is shown. A, B, C, p values were calculated by t-test.
ERβ2 overexpression confers a more proliferative and invasive phenotype in vitro
To further support the effects of ERβ2 on cellular proliferation and invasion, we investigated these phenotypes after ERβ2 overexpression. Successful overexpression of ERβ2 was confirmed by Western blot analysis (Figures 3A and S2A). Importantly, ERβ2 overexpression promoted cell proliferation in both investigated TNBC cell lines (Figures 3B and S2B). In addition, cells overexpressing ERβ2 acquired a more invasive phenotype with 9.4 ± 2.3 cells migrating through the chamber for ERβ2 overexpression cells compared to 2.4 ± 1.4 for the control cells for the BT549 cell line (Figure 3C) (p < 0.001). Similarly, overexpression of ERβ2 in MDA-MB-231 cells significantly increased cell invasion with 11.3 ± 5.9 invading cells per field for ERβ2 overexpression cells compared to 2.5 ± 1.8 invading cells for the control cells (Supplementary Figure 2C) (p < 0.001). These results further support the link between ERβ2 levels and cellular proliferation and invasion.
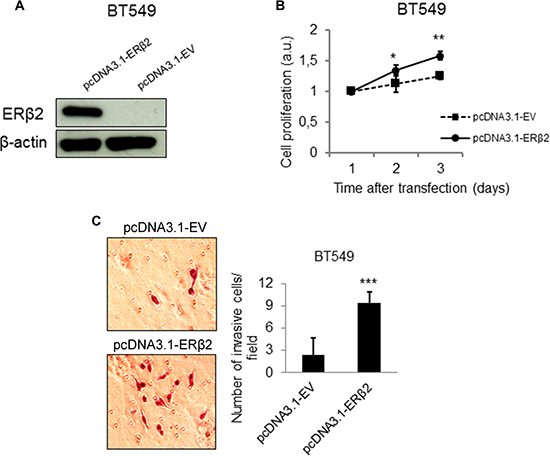
Figure 3: ERβ2 overexpression confers a more proliferative and invasive phenotype in vitro. (A) Western blot analysis showing increased protein level of ERβ2 after transient overexpression of ERβ2 protein. ERβ2 was detected by the PPZ0506 antibody. β-actin was used as a loading control. (B) ERβ2 overexpression promotes cell proliferation in the BT549 cells. WST-1 assays of cell proliferation were carried out at the indicated time points after transfection of ERβ2 or empty vector (EV). Ratio of absorbance to day 1 is calculated. Data are shown as means of relative absorbance ± SD. *P < 0.05, **P < 0.01. Experiments were repeated three times. One representative experiment is shown. (C) ERβ2 overexpression promotes cell invasion in the BT549 cell line. BT549 cells were transfected with ERβ2 or EV, and cell invasion was evaluated by the BD Biocoat growth factor reduced Matrigel invasion chamber assay. Data represent means ± SD. ***P < 0.001. Experiment was repeated twice. One representative experiment is shown. B,C, p values were calculated by t-test.
ERβ2 effects on global gene expression profiles
To approach the molecular mechanism responsible for the effects of ERβ2 on cellular phenotypes, we determined changes in gene expression profiles for BT549 cells upon ERβ2 knockdown. We identified 662 genes, applying a false discovery rate of less or equal to 0.1, as upregulated (fold change equal or greater than 1.5, p < 0.05) while the expression of 263 genes was repressed (fold change equal or less than 1.5, p < 0.05) upon ERβ2 knockdown. Network analysis revealed the top three ranked networks regulated by inhibiting endogenous ERβ2 in BT549 cells as cell morphology, DNA replication and repair, and cell death and survival (Table 1). Molecular and cellular functional classification analysis shows how alterations of gene expression were predicted to disrupt various molecular and cellular functions. The top 5 highlighted molecular and cellular functions after ERβ2 knockdown were cell cycle, cell death and survival, morphology, development and organization (Table 2).
Table 1: Changed networks after knockdown of ERβ2 in BT549 cells
Top Networks |
||
|---|---|---|
ID |
Associated Network Functions |
Score |
1 |
Cell Morphology, DNA Replication, Recombination and Repair, Developmental Disorder |
32 |
2 |
Cancer, Reproductive System Disease, Cell Dearth and Survival |
24 |
3 |
Cell Morphology, Dermatological Diseases and Conditions, Developmental Disorder |
24 |
Table 2: Changed molecular functions after knockdown of ERβ2 in BT549 cells
Name |
p-value |
#Molecules |
|---|---|---|
Cell Cycle |
1.94E-03–4.01E-02 |
5 |
Cell Death and Survival |
1.94E-03–4.46E-02 |
5 |
Cell Morphology |
1.94E-03–4.57E-02 |
8 |
Cellular Development and Organization |
1.94E-03–3.63-02 |
7 |
Cellular Development |
1.94E-03–3.79-02 |
4 |
Validation of global gene expression profiling data by qPCR
qPCR analysis was performed to confirm changed expression of 5 genes (PHD3, RECK, TGFB3, E2F2, GAB1) identified as being regulated by ERβ2 in the global gene expression profiling analysis. These represent genes well known to be involved in cell growth, cell death, apoptosis, cell migration and invasion [21–26]. As shown in Figure 4A, the qPCR data confirmed the data from the microarray assay of BT549 cells. In addition, qPCR analysis of the expression of these genes was performed in the MDA-MB-231 cell line. Importantly, changed expression of 4 of the 5 genes was also observed in the MDA-MB-231 cell line upon ERβ2 knockdown, the exception being RECK which was not regulated in the MDA-MB-231 cell line upon ERβ2 knockdown (Figure 4B).
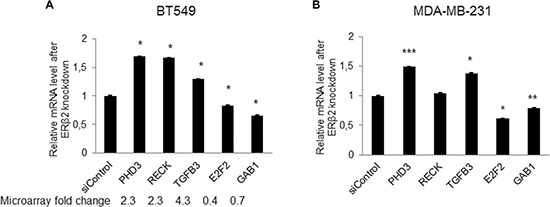
Figure 4: Validation of gene expression profiling data by qPCR. (A) Real-time PCR analysis for a subset of ERβ2 regulated genes identified by microarray analysis in BT549 cells. mRNA levels are normalized to 36B4. Data represent means ± SD. *P < 0.05. Fold change derived from microarray analysis is presented as numbers below the bars. (B) Real-time PCR analysis of selected genes in MDA-MB-231 cells. mRNA levels are normalized to 36B4. Data represent means ± SD. *P < 0.05, **P < 0.01, ***P < 0.001. A, B, p values were calculated by t-test relative to control siRNA-treated cells.
ERβ2 promotes cell invasion potentially through repression of PHD3 with associated up-regulation of HIF-1α
PHD3, one of the most regulated genes upon ERβ2 knockdown is known to play a critical role in suppressing the growth of diverse tumor types. Figure 5A shows that PHD3 mRNA and protein levels are increased by ERβ2 knockdown in the BT549 cell line. Importantly, depletion of PHD3 results in increased cell invasiveness (Figure 5B). These findings suggest a link between ERβ2, PHD3 and cell invasion. Furthermore, in support of the link between ERβ2 and PHD3 expression, PHD3 mRNA and protein levels are reduced by overexpression of ERβ2 (Figure 5C). As PHD3 has been shown to promote degradation of HIF-1α [27] and HIF-1α has been shown to play key roles in many crucial aspects of breast cancer biology, including invasion and metastasis [28], we hypothesized that HIF-1α could be a mediator of the invasive effects of ERβ2 in this system. In support of this hypothesis, knockdown of ERβ2 decreased whereas overexpression of ERβ2 increased HIF-1α protein levels in BT549 cells as assayed by Western blot analysis and ELISA assays (Figure 5D). Knockdown and overexpression of ERβ2 also regulated the mRNA levels of HIF-1α with small fold changes (~1.2-fold) in BT549 cells (Figure 5E). In summary, our data suggest that ERβ2 decreased PHD3 gene expression and increased HIF-1α protein levels, thus contributing to the proliferative and invasive phenotype of TNBC cell lines.
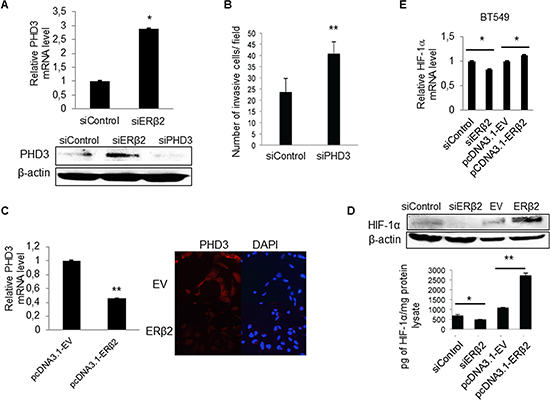
Figure 5: ERβ2 modulates levels of PHD3 and HIF-1α in TNBC cells. (A) ERβ2 knock-down increases PHD3 mRNA and protein levels. BT549 cells were transfected with control siRNA or ERβ2 siRNA. RNA was collected after 48 h while protein was collected after 72 h. PHD3 mRNA levels were determined by a qPCR assay and was normalized to 36B4, *P < 0.05 (top panel) and PHD3 protein levels were determined by Western blot analysis. β-actin was used as a loading control (bottom panel). (B) PHD3 knockdown promotes cell invasiveness for the BT549 cell line. BT549 cells were transfected with control siRNA or PHD3 siRNA. Data represent means ± SD. **P < 0.01. (C) BT549 cells were transfected with ERβ2 containing plasmid or a control EV. After 24 h, RNA was collected; qPCR was used to determine the PHD3 mRNA level, that was normalized to 36B4 (bar chart). Data represent means ± SD. **P < 0.01. BT549 cells were seeded onto microscope cover slide after ERβ2 overexpression; 24 h after plating cells, PHD3 protein level was detected by immunofluorescence. DAPI was used for nuclear staining (right figure). (D) BT549 cells were transfected with control siRNA or ERβ2 siRNA, and with empty vector or ERβ2 containing plasmid. Protein lysates were collected and HIF-1α levels were evaluated by both Western blot analysis (top panel) and HIF-1α ELISA (bottom graph). *P < 0.05, **P < 0.01. p values were calculated by t-test. (E) qPCR analysis of HIF-1α mRNA level after overexpression or knockdown of ERβ2. BT549 cells were transfected with control siRNA or ERβ2 siRNA and with EV or ERβ2 containing plasmid. mRNA levels were normalized to 36B4. Data represent means ± SD. *P < 0.05. A,B,C,D,E, p values were calculated by t-test, relative to control siRNA or empty vector transfected cells.
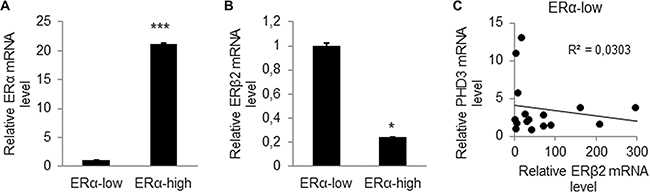
Figure 6: ERβ2 expression is high in breast tumor samples with low ERα expression and negatively correlates with PHD3 expression. (A) qPCR analysis of ERα mRNA level of 50 human breast cancer tissues determined ERα-low expression (n=20) and ERα-high expression (n = 30) breast tumors. Data represent means ± SD. ***P < 0.001. (B) qPCR analysis of ERβ2 mRNA level in ERα-low expression tumor samples compared to ERα-high expression breast tumor samples. Data represent means ± SD. *P < 0.05. (C) Negative correlation of PHD3 mRNA expression to ERβ2 mRNA expression in ERα-low expression breast tumors. *P < 0.05.
ERβ2 expression is high in breast tumor samples with low ERα expression and negatively correlates with PHD3 expression
To determine the clinical relevance of ERβ2 expression in breast cancer, we analysed ERβ2 expression in 50 human breast tumor samples. The tumor samples were divided into ERα-low expression (n = 20) and ERα-high expression (n = 30) based on ERα mRNA levels (p < 0.001) (Figure 6A). We observed that ERα-low expression tumors, that are generally more aggressive, express higher level of ERβ2 as compared to ERα-high expression tumors (Figure 6B) (p < 0.05). This is consistent with the high expression of ERβ2 that we report for TNBC cell lines. We further investigated the relationship between ERβ2 and PHD3 mRNA expression in the clinical samples. Our analysis showed a weak negative correlation between ERβ2 and PHD3 expression in ERα-low expression tumor samples (Figure 6C), which was not observed in ERα-high expression tumor samples (data not shown). These results support our findings in TNBC cell lines that knockdown of ERβ2 increases PHD3 expression. We did not find a correlation between ERβ2 and HIF-1α mRNA expression in clinical samples (data not shown). This may be due to the fact that the effect of ERβ2 on HIF-1α mRNA expression in cell lines is very modest (Figure 5E).
DISCUSSION
The function of the human ERβ variant ERβ2 remains elusive. In this study we report that several TNBC cell lines express endogenous ERβ2 without expression of ERα and ERβ1. To our knowledge this is the first study that addresses the molecular mechanism and cellular phenotypes conferred by endogenous expression of ERβ2 alone, without the influence of ERα and ERβ1. We demonstrate that ERβ2 depletion significantly inhibited proliferation and invasion of TNBC cells. This was confirmed with a second set of siRNA (data not shown), supporting that this does not correspond to off-target effects. Moreover, we observed opposite cellular phenotypes and molecular changes resulting from overexpression of ERβ2, further supporting that the observed effects are related to ERβ2.
We used gene expression profiling to start to elucidate the molecular mechanisms associated with the proliferative and invasive properties of ERβ2 in the BT549 cell line and identified changes in pathways and molecular classes associated with cancer, cell cycle, cell survival and cell death compatible with a role of ERβ2 in cellular proliferation and invasion. To our knowledge, this study is the first to assay regulation of global gene expression by endogenous ERβ2.
In this study we propose that the proliferative and invasive phenotype associated with ERβ2 expression could be mediated via repression of PHD3 and subsequent up-regulation of HIF-1α. PHD3 is known to regulate HIF-1α by posttranslational modification. However, we also observe minor effects on HIF-1α mRNA levels upon modulation of ERβ2 levels. Indeed, studies also show that HIF-1α is additionally subject to transcriptional regulation [29]. It promotes cell proliferation, migration and invasion in various tumor cells [30–33]. PHD3 has been shown to exert anti-proliferative effects including inhibition of tumor growth in pancreatic cancer and renal cell carcinoma [34, 35]. High PHD3 expression is correlated with good clinical prognosis markers for breast cancer such as lower tumor grade, smaller tumor size and lower proliferation [5]. In a recent study, down-regulation of PHD3 expression was reported to occur during epithelial to mesenchymal transition (EMT) [36]. It remains unclear if ERβ2 regulates PHD3 directly or indirectly. In support of an indirect mechanism of ERβ2 regulation of PHD3, we were unable to detect binding of ERβ2 to the promoter region of the PHD3 gene using a ChIP assay (data not shown). Furthermore, our analysis revealed a weak negative correlation between ERβ2 and PHD3 mRNA expression in ERα-low expression breast tumors, consistent with the notion that ERβ2 downregulates PHD3. The weak correlation is presumably due to the small sample size and cell type heterogeneity of the tumor samples. Our data suggest that ERβ2 regulates PHD3 indirectly, which may also account for the poor correlation. Moreover, the lack of a correlation between ERβ2 and PHD3 expression in ERα-high expression tumor samples may be due to the low or even undetectable expression levels of ERβ2 in these tumor samples. Our data are consistent with Peurala et al. who found that PHD3 is mainly expressed in tumors with better prognosis and their preliminary data suggested that PHD3 negativity associates with triple negative tumors [6].
In this study we focus on ERβ2 regulation of PDH3. However, our analysis also identified additional ERβ2 target genes that could contribute to effects of ERβ2 on proliferation and invasion. Genes known to be involved in positive regulation of cell proliferation such as E2F2 [30] and GAB1 [29], were decreased upon knockdown of ERβ2 in both analysed TNBC cell lines. Furthermore, the regulators of cell cycle progression, cyclins E and A (CCNE2/CCNA2) were down regulated 1.3 and 1.4 fold, respectively, with a concomitant increase in the expression of p21WAF1/CIP1 (CDKN1A) of 1.5 fold (data not shown). Knockdown of ERβ2 also downregulated gene expression of the Wilms tumor-1 (WT-1) transcription factor known to play an important role in cellular development, cell survival and angiogenesis [32, 33]. Finally, genes involved in inhibition of cell migration and invasion such as RECK [26, 27] and TGFB3 [25] were induced by knockdown of ERβ2 in BT549 cells.
In this study, we screened the expression of ERβ2 in a panel of ten breast cancer cell lines. These cell lines can be classified into three different breast cancer subtypes according to the expression of ER, PR and HER2 receptor: ER-positive (MCF-7, MDA-MB-175, ZR-751 and CAMA-1), HER2-overexpressed (SK-BR-3 and HCC1569) and triple-negative subtypes (Hs578T, MDA-MB-231, BT549 and BT20). Gene expression profiling has further classified breast cancer into five subtypes: luminal A, luminal B, HER2-enriched, basal-like and normal-like [37]. The cell lines used in our study represent different breast cancer subtypes. For example, MCF-7 and ZR-751 cell lines are representative of luminal A and luminal B, respectively. SK-BR-3 closely resembles HER2-enriched, while the TNBC cell lines such as BT549, MDA-MB-231 and Hs578T resemble basal-like tumors. Clinically, the basal-like tumors are characterized by poorer survival outcomes and higher relapse rates than other subtypes of breast cancer [38]. Our results show that the highest level of endogenous ERβ2 expression was observed in the TNBC cell lines, indicating a role of ERβ2 in promotion of an aggressive phenotype. In line with this, our data also showed an increased expression of ERβ2 in ERα-low expression tumors compared with ERα-high expression tumors. TNBC lacks effective specific targeted therapy leading to poor survival. It is possible that a fraction of TNBC expresses ERβ2 and that this class of TNBC would benefit from therapeutic strategies targeting ERβ2. We were unable to detect endogenous ERβ2 protein in BT549 and MDA-MB-231 cells, that have the highest levels of ERβ2 mRNA expression, by Western blotting using the only specific ERβ antibody (PPZ0506) [17]. It is possible that the levels of ERβ2 are still not very high in these cell lines or that the antibody may not provide sufficient sensitivity.
Studies of ERβ2 are limited by the lack of this ERβ2 isoform in rodents. Thus it will be important to generate mouse models that express ERβ2 in different tissues. Furthermore, although ERβ2 does not bind tested ER ligands, it is possible that compounds that inhibit its function can be identified, thus providing tools to further explore the function of ERβ2 as well as be used therapeutic agents, for example in breast cancers that express ERβ2.
In summary, we identify TNBC breast cancer cell lines that express endogenous ERβ2 but not ERα or ERβ1 and show that ERβ2, when expressed alone in these cell lines, promotes cell proliferation and invasion and suggest a mechanism for these effects. Whether our results can be translated into an understanding of the molecular mechanism for the invasive nature of TNBC and potentially the identification of targeted therapies remains to be determined.
MATERIALS AND METHODS
Cell culture
Cell lines were obtained from the American Type Culture Collection (ATCC) and maintained at 37°C and 5% CO2. MCF-7, CAMA-1, SK-BR-3, Hs578T and BT20 cells were grown in DMEM medium (Gibco by Life Technologies, Carlsbad, CA) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin (Gibco); ZR-751, HCC1569, MDA-MB-231 and BT549 cells were grown in RPMI 1640 medium (Gibco) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin (Gibco); MDA-MB-175 cells were grown in L-15 medium (Gibco) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin (Gibco).
RNA isolation and cDNA synthesis
Total RNA was extracted using the RNeasy Plus Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. cDNA was synthesized using SuperScript™ VILO™ MasterMix according to the standard protocol (Invitrogen, Carlsbad, CA). In brief, 1 μg of RNA was mixed with 4 μl of SuperScript VILO MasterMix and DEPC treated water to a total volume of 20 μl. The reaction was gently mixed and incubated at 25°C for 10 mins, at 42°C for 60 mins and at 85°C for 5 mins. cDNA was stored at -20°C until further use.
qPCR
Real-time PCR was performed with the SYBR Green I dye master mix (Applied Biosystems, Foster City, CA). qPCR reactions were analysed using a 7500 Fast Real-Time PCR System (Applied Biosystems) applying the following conditions: 95°C for 20 sec, followed by 40 cycles at 95°C for 3 sec and 60°C for 30 sec. Primer sequences for analysed genes were: ERβ1/ERβ2 forward 5′-TCCATGCGCCTGGCTAAC-3′, ERβ1 reverse 5′-CAGATGTTCCATGCCCTTGTTA-3′, ERβ2 reverse 5′-CCATCGTTGCTTCAGGCAA-3′, PHD3 forward 5′-GCCGGCTGGGCAAATACTA-3′, reverse 5′-CCGGATAGCAAGCCACCAT-3′, TGFB3 forward 5′-TACTATGCCAACTTCTGCTCAG-3′, reverse 5′-AACTTACCATCCCTTTCCTC-3′, GAB1 forward 5′-CCTGTTGCTCATCAACTGTCAAAGC-3′, reverse 5′-CTACACTCGATGTCCCAGATGGG-3′, E2F2 forward 5′-TGAGGACAAGGCCAACAAGAG-3′, reverse 5′-TTGCCAACAGCACGGATATC-3′, RECK forward 5′-AACAGGCCAACAGAACTTTTCAG-3′, reverse 5′-CATGTCATTCATGGCTCCTTGA-3′, ERα forward 5′-GCTACGAAGTGGGAATGATGAAAG-3′, reverse 5′-TCTGGCGCTTGTGTTTCAAC-3′. mRNA expression levels were normalized to acidic ribosomal phosphoprotein P0 (36B4) mRNA[39] reference gene forward 5′-GTGTTCGACAATGGCAGCAT-3′, reverse 5′-GACACCCTCCAGGAAGCGA-3′ or to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) reference gene forward 5′-GACCCCTTCATTGACCTCAACT-3′, reverse 5′-GAATTTGCCATGGGTGGAAT-3′.
Plasmids and siRNA transfection
The ERβ2 cDNA was cloned into the pcDNA3.1 vector with a FLAG tag at the N-terminus. MDA-MB-231 or BT549 cells at a confluency of about 70-90 % were transfected with 2.5 μg of either pcDNA3.1-ERβ2 or pcDNA3.1-empty vector (pcDNA3.1-EV) plasmid using 5 μl/well of Lipofectamine®2000 according to manufacturer’s instructions (Invitrogen, Carlsbad, CA). For siRNA transfection, BT549 and MDA-MB-231 cells were transfected at about 30-50 % confluency with two different sets of siRNA targeting ERβ2 mRNA (Thermo Scientific and Dharmacon) or control siRNA (Sigma) and siRNA targeting PHD3 mRNA with the sequence GUACUUUGAUGCUGAAGAAUU (Sigma) using 10 μl/well of INTERFERinTM siRNA transfection reagent (Polyplus, Illkirch, France) according to manufacturer’s instructions.
Western blot and ELISA assay
Protein lysates were extracted using RIPA buffer (Thermo Fisher, Waltham, MA) with protease inhibitor cocktail (Roche, Basel, Switzerland). Thirty μg of protein was boiled with 5x Laemli buffer (0.5 M Tris-HCl, 10% SDS, 30% glycerol, 0.25% Bromphenol blue, 20% β-mercaptoethanol) for 10 mins. Protein samples were separated by SDS-PAGE, and electrophoretically transferred to nitrocellulose membranes (GE Healthcare, Danderyd, Sweden). After milk blocking, blots were incubated over night at 4°C with the following primary antibodies: PPZ0506 [17] for ERβ2 (R&D systems, Abingdon, UK), PHD3 (Novus biological, Littleton, CO), HIF-1α (Pharmingen, San Diego, CA) and β-actin (Sigma Aldrich, Dorset, UK). Secondary antibodies were anti-mouse and anti-rabbit IgG (GE Healthcare, Danderyd, Sweden).
For ELISA analysis, thirty μg of protein was used. HIF-1α ELISA was performed using Human/Mouse Total HIF-1 alpha DuoSet IC ELISA kit (R&D systems, Abingdon, UK) according to manufacturer’s instructions.
Immunofluorescence
100000 cells were seeded on microscope cover glass (Menzel Glaser, Germany) 24 h after ERβ2 or control plasmid transfection. The next day, immunofluorescence analysis was performed as described previously [40]. PHD3 (Novus biological, Littleton, CO) antibody was used. DAPI (4′, 6-diamidino-2-phenylindole) (Sigma Aldrich, Dorset, UK) was used at 0.5 μg/mL for nuclear staining. The coverslips were then mounted on a microscope slide SuperFrost®Plus (Menzel Glaser, Germany) with fluorescence mounting solution (Dako, Carpinteria, CA).
Cell proliferation assay
3000 cells per well were seeded in 96-well plates 24 h after ERβ2 or control plasmid transfection and 48 h after ERβ2 siRNA or control siRNA transfection. Cell proliferation was determined after 1, 2, 3 or 4 days using the WST-1 kit (Roche, Basel, Switzerland) according to the instructions of the manufacturer. The absorbance was measured at 450 nm using NanoQuant spectrophotometer (Tecan, Männedorf, Switzerland).
Invasion assay
Twenty-four hours after transfection of control siRNA or ERβ2 siRNA and pcDNA3.1-empty vector or pcDNA3.1-ERβ2, approximately 2.5 × 104 cells were seeded in the upper chambers of 24-well BD Biocoat growth factor-reduced Matrigel Invasion Chambers (Becton Dickinson, Bedford, MA) with media without FBS and incubated at 37°C for 24 h. Medium containing 10% FBS was added to the lower chamber. Non-invading cells in the upper chamber were removed by scraping with cotton sticks and washed with PBS. Cells that migrated to the lower chamber were fixed and stained using Gurr for microscopy (VWR, Radnor, PA) and counted under a microscope Zeiss Axiovert S100 under magnification ×20.
Gene expression microarray analysis
Total RNA from three biological replicates were hybridized to Affymetrix Human Gene 1.1 ST arrays, which contain probes for 33299 gene sequences. Experimental steps such as probe synthesis, hybridization and scanning were done according to the Affymetrix protocol (www.affymetrix.com). Pre-processing for background correction/normalization was performed in the Affymetrix Expression Console using the Robust Multichip Average (RMA) method [41]. Two-tailed Student’s t-test was used to derive p-values, and the false discovery rates were estimated using the q-value. A cut-off fold change of at least 1.5 and p value < 0.05 were used to define differentially regulated genes. The q-value, representing the false discovery rate, was less or equal to 0.1. The microarray raw data are deposited in GEO (accession number GSE57379).
To derive genes as input for pathway analysis the following cut-offs were applied; fold change of ≥ 2, p value < 0.05 and q value ≤ 0.1. The IPA software (www.ingenuity.com) was used for this analysis. The entire IPA core analysis was performed based on information in the Ingenuity Pathway Knowledge Base (IPKB), which derives information from known relationships of molecules, functions and interactions of genes published in the literature.
IPA analysis identified pathways that were most significantly changed upon ERβ2 knock down. Associated network functions and molecular functions are two of the categories used for enrichment testing within IPA. Biological functions were ranked according to the significance of that function to the network. Fisher’s exact test was used to derive the p-values in IPA.
Human breast tumor samples
The tumor samples in this study have been previously described [18]. The studies were approved by the ethical committee of the Karolinska Institute.
Statistics
Student’s t-test was used to determine statistically significant differences and p < 0.05 was considered to be significant unless otherwise specified.
Abbreviations
ER, estrogen receptor; ERβ2, estrogen receptor beta isoform 2; ERβ1, estrogen receptor beta isoform 1; ERα, estrogen receptor alpha; PHD3, prolyl hydroxylase 3; HIF-1α, hypoxia inducible factor 1α; TGFB3, transforming growth factor beta 3; E2F2, E2F transcription factor 2; GAB1, GRB2-associated-binding protein 1; RECK, Reversion-inducing cysteine-rich protein with Kazal motifs; AF-2, activation function 2; RMA, robust multichip average; IPKB; ingenuity pathway knowledge base; TNBC, triple-negative breast cancer; EMT, epithelial-to-mesenchymal transition; PR, progesterone receptor; HER2, human epidermal growth factor receptor 2; EV, empty vector
Author contributions
LB and LX designed and supervised the study, performed experiments, analyzed and interpreted data and wrote manuscript. CZ, LAH and KDW conceived and supervised project, critically revised manuscript for scientific content, have given final approval of the version to be published. JAG edited the manuscript. All authors read and approved the final manuscript.
FUNDING
This study was supported by the Swedish Cancer Society, the Center for Biosciences and a grant from the Robert A. Welch Foundation (E-0004, J-ÅG). We thank the Bioinformatic and Expression Analysis core facility at the Karolinska Institute (BEA, www.bea.ki.se) for performing the Affymetrix assays.
CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
REFERENCES
1. Bouchardy C, Rapiti E, Fioretta G, Laissue P, Neyroud-Caspar I, Schafer P, Kurtz J, Sappino AP, Vlastos G. Undertreatment strongly decreases prognosis of breast cancer in elderly women. J Clin Oncol. 2003; 21:3580–3587.
2. Haldosen LA, Zhao C, Dahlman-Wright K. Estrogen receptor beta in breast cancer. Mol Cell Endocrinol. 2014; 382:665–672.
3. Elias AD. Triple-negative breast cancer: a short review. Am J Clin Oncol. 2010; 33:637–645.
4. Pal SK, Childs BH, Pegram M. Triple negative breast cancer: unmet medical needs. Breast Cancer Res Treat. 2011; 125:627–636.
5. Peurala E, Koivunen P, Bloigu R, Haapasaari KM, Jukkola-Vuorinen A. Expressions of individual PHDs associate with good prognostic factors and increased proliferation in breast cancer patients. Breast Cancer Res Treat. 2012; 133:179–188.
6. Bhat RA, Harnish DC, Stevis PE, Lyttle CR, Komm BS. A novel human estrogen receptor beta: identification and functional analysis of additional N-terminal amino acids. J Steroid Biochem Mol Biol. 1998; 67:233–240.
7. Ogawa S, Inoue S, Watanabe T, Orimo A, Hosoi T, Ouchi Y, Muramatsu M. Molecular cloning and characterization of human estrogen receptor betacx: a potential inhibitor ofestrogen action in human. Nucleic Acids Res. 1998; 26:3505–3512.
8. Green S, Walter P, Greene G, Krust A, Goffin C, Jensen E, Scrace G, Waterfield M, Chambon P. Cloning of the human oestrogen receptor cDNA. J Steroid Biochem. 1986; 24:77–83.
9. Kuiper GG, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson JA. Cloning of a novel receptor expressed in rat prostate and ovary. Proc Natl Acad Sci USA. 1996; 93:5925–5930.
10. Cotrim CZ, Fabris V, Doria ML, Lindberg K, Gustafsson JA, Amado F, Lanari C, Helguero LA. Estrogen receptor beta growth-inhibitory effects are repressed through activation of MAPK, PI3K signalling in mammary epithelial and breast cancer cells. Oncogene. 2013; 32:2390–2402.
11. Strom A, Hartman J, Foster JS, Kietz S, Wimalasena J, Gustafsson JA. Estrogen receptor beta inhibits 17beta-estradiol-stimulated proliferation of the breast cancer cell line T47D. Proc Natl Acad Sci USA. 2004; 101:1566–1571.
12. Baek JM, Chae BJ, Song BJ, Jung SS. The potential role of estrogen receptor beta2 in breast cancer. Int J Surg. 2015; 14:17–22.
13. Palmieri C, Lam EW, Mansi J, MacDonald C, Shousha S, Madden P, Omoto Y, Sunters A, Warner M, Gustafsson JA, Coombes RC. The expression of ER beta cx in human breast cancer and the relationship to endocrine therapy and survival. Clin Cancer Res. 2004; 10:2421–2428.
14. Honma N, Saji S, Kurabayashi R, Aida J, Arai T, Horii R, Akiyama F, Iwase T, Harada N, Younes M, Toi M, Takubo K, Sakamoto G. Oestrogen receptor-beta1 but not oestrogen receptor-betacx is of prognostic value in apocrine carcinoma of the breast. APMIS. 2008; 116:923–930.
15. Esslimani-Sahla M, Simony-Lafontaine J, Kramar A, Lavaill R, Mollevi C, Warner M, Gustafsson JA, Rochefort H. Estrogen receptor beta (ER beta) level but not its ER beta cx variant helps to predict tamoxifen resistance in breast cancer. Clin Cancer Res. 2004; 10:5769–5776.
16. Saji S, Hirose M, Toi M. Clinical significance of estrogen receptor beta in breast cancer. Cancer Chemother Pharmacol. 2005; 56:21–26.
17. Andersson S, Sundberg M, Pristovsek N, Ibrahim A, Jonsson P, Katona B, Clausson CM, Zieba A, Ramström M, Söderberg O, Williams C, Asplund A. Insufficient antibody validation challenges oestrogen receptor beta research. Nat Commun. 2017; 8:15840.
18. Zhao C, Matthews J, Tujague M, Wan J, Strom A, Toresson G, Lam EW, Cheng G, Gustafsson JA, Dahlman-Wright K. Estrogen receptor beta2 negatively regulates the transactivation of estrogen receptor alpha in human breast cancer cells. Cancer Res. 2007; 67:3955–3962.
19. Cai FF, Xu C, Pan X, Cai L, Lin XY, Chen S, Biskup E. Prognostic value of plasma levels of HIF-1α and PGC-1a in breast cancer. Oncotarget. 2016; 7:77793–806. https://doi.org/10.18632/oncotarget.12796.
20. Semenza GL. Expression of hypoxia-inducible factor 1: mechanisms and consequences. Biochem Pharmacol. 2000; 59:47–53.
21. Zhao H, Jiang Y, Cao Q, Hou Y, Wang C. Role of integrin switch and transforming growth factor Beta 3 in hypoxia-induced invasion inhibition of human extravillous trophoblast cells. Biol Reprod. 2012; 87:47.
22. Takahashi C, Sheng Z, Horan TP, Kitayama H, Maki M, Hitomi K, Kitaura Y, Takai S, Sasahara RM, Horimoto A, Ikawa Y, Ratzkin BJ, Arakawa T, Noda M. Regulation of matrix metalloproteinase-9 and inhibition of tumor invasion by the membrane-anchored glycoprotein RECK. Proc Natl Acad Sci USA. 1998; 95:13221–13226.
23. Silveira Correa TC, Massaro RR, Brohem CA, Taboga SR, Lamers ML, Santos MF, Maria-Engler SS. RECK-mediated inhibition of glioma migration and invasion. J Cell Biochem. 2010; 110:52–61.
24. Zhang Y, Cheng JC, Huang HF, Leung PC. Homeobox A7 stimulates breast cancer cell proliferation by up-regulating estrogen receptor-alpha. Biochem Biophys Res Commun. 2013; 440:652–657.
25. Sang H, Li T, Li H, Liu J. Down-regulation of Gab1 inhibits cell proliferation and migration in hilar cholangiocarcinoma. PLoS One. 2013; 8:e81347.
26. Wu N, Xiao L, Zhao X, Zhao J, Wang J, Wang F, Cao S, Lin X. miR-125b regulates the proliferation of glioblastoma stem cells by targeting E2F2. FEBS Lett. 2012; 586:3831–3839.
27. Ke Q, Costa M. Hypoxia-inducible factor-1 (HIF-1). Mol Pharmacol. 2006; 70:1469–1480.
28. Liu ZJ, Semenza GL, Zhang HF. Hypoxia-inducible factor 1 and breast cancer metastasis. J Zhejiang Univ Sci B. 2015; 16:32–43.
29. Kuschel A, Simon P, Tug S. Functional regulation of HIF-1alpha under normoxia--is there more than post-translational regulation? J Cell Physiol. 2012; 227:514–524.
30. Shen SH, Kwan AL, Chen YY, Wang ZX. Effect of silencing HIF-1alpha on proliferation, invasion and migration of glioblastoma U87 cells. Neurol Sci. 2013; 34:365–371.
31. Stoeltzing O, McCarty MF, Wey JS, Fan F, Liu W, Belcheva A, Bucana CD, Semenza GL, Ellis LM. Role of hypoxia-inducible factor 1alpha in gastric cancer cell growth, angiogenesis, and vessel maturation. J Natl Cancer Inst. 2004; 96:946–956.
32. Gao L, Wu GJ, Liu B, Shen MZ, Pan TJ, Yu CG, Wang QH, Ru Y, Liu XP, Niu TS, Wang GD, Wei M, Li RX, et al. Up-regulation of pVHL along with down-regulation of HIF-1alpha by NDRG2 expression attenuates proliferation and invasion in renal cancer cells. PLoS One. 2013; 8:e84127.
33. Zou J, Li P, Lu F, Liu N, Dai J, Ye J, Qu X, Sun X, Ma D, Park J, Ji C. Notch1 is required for hypoxia-induced proliferation, invasion and chemoresistance of T-cell acute lymphoblastic leukemia cells. J Hematol Oncol. 2013; 6:3.
34. Su Y, Loos M, Giese N, Hines OJ, Diebold I, Gorlach A, Metzen E, Pastorekova S, Friess H, Buchler P. PHD3 regulates differentiation, tumour growth and angiogenesis in pancreatic cancer. Br J Cancer. 2010; 103:1571–1579.
35. Tanaka T, Torigoe T, Hirohashi Y, Sato E, Honma I, Kitamura H, Masumori N, Tsukamoto T, Sato N. Hypoxia-inducible factor (HIF)-independent expression mechanism and novel function of HIF prolyl hydroxylase-3 in renal cell carcinoma. J Cancer Res Clin Oncol. 2014; 140:503–513.
36. Place TL, Nauseef JT, Peterson MK, Henry MD, Mezhir JJ, Domann FE. Prolyl-4-hydroxylase 3 (PHD3) expression is downregulated during epithelial-to-mesenchymal transition. PLoS One. 2013; 8:e83021.
37. Holliday DL, Speirs V. Choosing the right cell line for breast cancer research. Breast Cancer Res. 2011; 13:215.
38. Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. 2016; 13:674–90.
39. Ceelen L, De Craene J, De Spiegelaere W. Evaluation of normalization strategies used in real-time quantitative PCR experiments in HepaRG cell line studies. Clin Chem. 2014; 60:451–454.
40. Dey P, Jonsson P, Hartman J, Williams C, Strom A, Gustafsson JA. Estrogen receptors beta1 and beta2 have opposing roles in regulating proliferation and bone metastasis genes in the prostate cancer cell line PC3. Mol Endocrinol. 2012; 26:1991–2003.
41. Dahlman-Wright K, Qiao Y, Jonsson P, Gustafsson JA, Williams C, Zhao C. Interplay between AP-1 and estrogen receptor alpha in regulating gene expression and proliferation networks in breast cancer cells. Carcinogenesis. 2012; 33:1684–1691.