
INTRODUCTION
Cancer-testis antigens are immunogenic proteins normally restricted to expression in the male germline but are frequently activated in various types of cancer [1-4]. Their immunogenicity, however, has become a less defining trait of members of this group, compared to the expression pattern, so the corresponding genes are also referred to as cancer-testis (CT) genes [5]. Despite a wealth of knowledge accumulated about CT genes, their biology in both cancer and in the normal germline is far from being understood. Particularly, areas such as their role as cancer drivers rather than biomarkers, their contribution to the stemness of tumor cells, and their molecular functions have yet to be fully addressed.
BORIS/CTCFL (Brother Of the Regulator of Imprinted Sites / CCCTC-binding Factor Like) [6] is a CT gene that is commonly activated in cancers [7]. BORIS is a paralog of CTCF (CCCTC-Binding Factor) [8], which has been branded a “master weaver of the epigenome” in multicellular organisms [9]. Indeed, CTCF is an inherently versatile and multifunctional DNA-binding protein, using 11 zinc fingers (ZF) to associate with a wide spectrum of target sequences [10]. CTCF has both a cis and trans modes of action: either directly controlling gene expression via enhancer or promoter binding in the first case [8, 11-20] or organizing chromatin domain compartmentalization and interstrand interaction in the second [18, 20-27]. Whilst the DNA binding domains of CTCF and BORIS are nearly identical and show indistinguishable DNA binding specificity in vitro [28-31] there is practically no similarity between the two proteins outside of the DNA binding domain. Thus, BORIS for quite some time was thought to be an alternative form of CTCF, functional only in the germline and in some cancers. A homozygous deletion of CTCF in mice resulted in early embryonic lethality [32], while BORIS knockout mice showed subfertility phenotype with an array of spermatogenic defects including a delay in gametogenesis and a reduction in testis size [28, 31]. Nevertheless, a recent study has revealed that BORIS emergence has likely resulted in the evolution of CTCF target sites CTSes themselves, so that BORIS binding preferentially occurs at clustered CTS, i.e. including two or more consensus DNA motifs in tandem [33]. Furthermore, clustered CTSes are able to bind both BORIS homodimers as well as heterodimers between CTCF and BORIS [33]. That breakthrough has resolved a nagging question on the mode of BORIS interaction with CTCF, firmly indicating that their relationship is mostly cooperative [33] rather than competitive [6, 31, 34] in cell types where both proteins are expressed. First of all, it was shown that BORIS binding is overwhelmingly associated with promoters and enhancers, i.e. substantially more specialized for direct transcriptional regulation compared to CTCF. Second, it was documented that cancer cells expressing high level of BORIS tend to display a characteristic pattern of BORIS binding to chromosomal locations, which is largely independent of the cancer cell origin, but instead recapitulates the binding landscape in male germ cells [33, 35]. Consistent with this interpretation, the ectopic expression of BORIS in MCF7 cells induced notable changes in the expression level of corresponding genes and recapitulated the germ cell pattern of BORIS binding.
K562 cells are chronic myeloid leukemia (CML) cells are dependent on BORIS, which is normally tightly repressed in soma [33], for the proliferation and self-renewal of stemness. Despite the progress in our understanding of BORIS as a broad spectrum transcriptional regulator, it still remains unknown what pathways result in the differentiation of these CML cells upon KO of the BORIS gene. Such knowledge is relevant in the context of anti-cancer differentiation therapy in particular, and for our general understanding of BORIS activation and its contribution to the self-renewal of cancer cells. The analysis of the gene expression in already-differentiated cells is not informative with respect to the identity of the events, downstream of BORIS suppression that triggered the differentiation. Thus, analysis of gene expression should be undertaken shortly after BORIS downregulation. We undertook a gene expression study using RNA-seq in order to identify potential pathways of immediate response to the downregulation of BORIS in K562 cells, which are dependent on BORIS for their proliferation.
RESULTS
BORIS downregulation results in a differential expression of genes in K562 cells
Cell types possessing stemness, such as K562, can enter a differentiated state as a result of two concurrent processes: genes controlling the self-renewal become repressed, and the triggers of specific differentiation pathways are turned on. In case of K562 the two major differentiation pathways are into erythroid and megakaryocytic lineages (Figure 1A). With respect to gene regulation, both repression and activation could be potentially handled by BORIS, as we previously showed that it can act as either an activator or repressor of transcription, dependent on the particular gene [33, 36].
We knocked down BORIS in K562 cells to evaluate the immediate/early response, i.e. genes that change expression within 96 hours after KD induction. Typical downregulation of BORIS mRNA ranged from 2.5 to 4.5 fold. 12 knockdowns (KDs) were subjected to RNA-seq. The best four data sets with a > 4-fold downregulation of BORIS mRNA were taken into the downstream analysis. This corresponded to over 10-fold reduction of BORIS protein level (Figure 1B). As a control, three independent experiments were used, with K562 cells stably infected by the corresponding empty lentivirus vector (EV) and induced by doxycycline. RNA-seq samples showed good correlation between the replicates (based on Pearson correlation) and robust changes in gene expression in response to BORIS KD (Figure 1C). The datasets with lower degrees of BORIS mRNA depletion, while excluded from subsequent analyses, were nevertheless utilized to pinpoint the most robust DE genes (shown in color in Figure 1D).
Chronic myeloid leukemia is commonly triggered by a chromosome 9 to 22 translocation that generates a fusion of the BCR (Breakpoint Cluster Region) and ABL encoding a tyrosine kinase. Incidentally, BORIS does have a strong binding at the BCR promoter region in K562, yet the BCR locus is not differentially expressed in KD. This indicates that the regulation of BCR-ABL1 transcript is unlikely to be responsible for K562 proliferation inhibition upon BORIS KO [33]. Therefore, some alternative and/or downstream pathways must be responsible. In the RNA-seq data, 149 genes were significantly downregulated upon BORIS KD (Cufflinks q-value < 0.05, log2(fold) > 0.4), and 85 were significantly upregulated (Cufflinks q-value < 0.05, log2(fold) < -0.4) (Supplementary Table 1). A more stringent cutoff, i.e. the expression level of higher than 0.5 FPKM and the minimal DE ratio of two-fold resulted in 20 up and 54 down genes, which are discussed below. A subset of these genes (10 upregulated, and 12 downregulated excluding BORIS, in color in Figure 1E) had significant DE even when datasets with lower BORIS downregulation were included in the analysis (BORIS mRNA between 2 and 4 fold, and GAL3ST1 over 1.5-fold downregulated). These genes may be prime candidates for the most responsive genes upon BORIS downregulation.
Upon the comparison of BORIS KD DE genes to the RNA-seq results of KO in [33] it was apparent that the datasets are overlapping only slightly, and the similarly affected genes, were not particularly revealing with respect to the trigger of the differentiation in KO (Figure 1A). Out of three upregulated genes in common, i.e. IL10RA, PASD1, and SLC44A1, only PASD1 may be relevant, as it is a potent regulator of transcription and a CT gene itself [37, 38]. The commonly significantly downregulated genes, i.e. COBL, COL1A2, EFEMP1, LRCH2, MS4A3, MS4A4A, OAF, and SDC2, do not contain regulatory proteins, apart from BORIS itself. This outcome, however, could have been expected, as the majority of gene expression in cells that spontaneously differentiated into megakaryocytic lineage upon stable BORIS downregulation in KO would have been too distant/downstream from the initial phase of the BORIS downregulation, and thus unlikely to correspond to the first-response genes.
One could assume that the genes with BORIS binding in cis, e.g. at the promoters, are most likely to be the direct targets of BORIS regulation. However, it was evident that among DE loci, the genes with BORIS binding in the immediate promoter’s vicinity were approximately evenly represented with ones without any BORIS peaks there. The latter class likely reflects the control of gene expression by BORIS via enhancer activity and/or in general folding of chromatin domains in K562 [33].
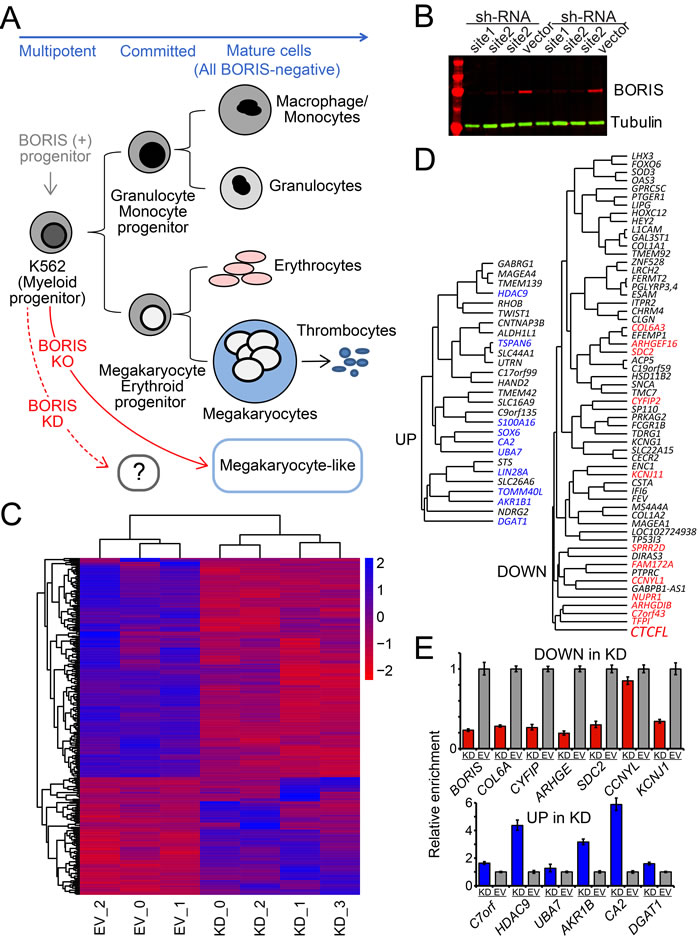
Figure 1: KD of BORIS leads to a limited but robust response in K562 transcriptome. A. The differentiation pathways for K562 cells. K562 cells have properties of pluripotent precursors of normal hematopoiesis, and express both erythroid and megakaryocytic specific markers. The further differentiation of K562 along hematopoietic lineages could be triggered by specific treatments. This differentiation results in proliferation arrest, changed morphology and adhesion properties, as well as gain of protein markers specific for the terminal differentiation [146]. The KO of all transcribed copies of BORIS in K562 results in cells possessing a megakaryocyte-like morphology with a substantial number of corresponding markers activated [33]. As no intermediate steps were available for transcriptome analysis upon KO, the rapid downregulation of BORIS (KD) was analyzed in this work. B. Downregulation of BORIS protein upon knockdown. Immunoblotting shows reproducible depletion of BORIS protein over 10-fold (quantified by LI-COR) within 96 hours after shRNA induction, in two independent experiments with site 1 and site 2 shRNAs (see Methods). Anti-BORIS mAbs were as previously described [33], anti alpha-tubulin antibodies were used as loading control for quantification. C. A heatmap of the hierarchical clustering of differentially expressed genes based on mRNA-seq analysis. Only significant DE genes are shown. Blue - upregulated genes, red - downregulated genes. Both rows and columns are clustered based on their distance similarity. The color gradient corresponds to log2 of fold expression difference for each gene. EV- are control samples, KD_0 and KD_2 are shRNA-1 KD, KD_1 and KD_3 are shRNA-2 KD. D. The subset of hierarchical clustering of differentially expressed genes based on mRNA-seq analysis filtered for expression levels and fold change in expression. Shown in color are the robust DE genes, i.e. ones that displayed significant change even when datasets with lower than 4-fold downregulation of BORIS mRNA were included (only if the downregulation of GAL3ST1, a known direct downstream target of BORIS [28], was at least 1.5 fold). E. RT-qPCR validation of differentially expressed genes upon BORIS KD. Both downregulated and upregulated DE genes were normalized to the K562 cell line stably infected with the empty vector (EV) lentivirus.
Genes upregulated upon BORIS KD
It seemed likely that the DE genes involved in the cell cycle would have been the primary candidates for how BORIS controls the cascade that eventually results in cell differentiation. Nevertheless, the STS gene, with the strongest relative upregulation , encodes a steroid sulfatase, which is involved in hormone metabolism. However, the upregulated transcription was limited to a longer transcript or a minor isoform originating from an alternative promoter, which might indicate that it represents a regulatory transcription, possibly germline-related. Indeed, the corresponding RNAP II binding in K562 was limited to the immediate vicinity of BORIS peak at this site (not shown). This transcription could also be related to the fact that STS is a gene that escapes X-chromosome inactivation [39]. Among other genes that were clustered with STS (Figure 1D) were several genes with ubiquitous expression in the normal tissues and documented overexpression in cancers: SLC26A6, TOMM40L, AKR1B1, NDRG2 and DGAT1 (based on TCGA data). All of them have non-zero expression in K562 and have BORIS binding in their promoters, which likely results in their upregulation upon BORIS KD, as exemplified in Figure 2A, with DGAT1. However, these genes do not seem to be obviously linked to K562 differentiation. Nevertheless, sampling of protein expression, e.g. AKR1B1 (Figure 2B), by immunoblotting showed that protein upregulation could be readily detected.
Functionally, one gene did catch our attention: LIN28A [40] is a potent pluripotency factor [41, 42] and a marker of cancer stem cells [43-45]. Thus, it could link BORIS to powerful pathways of mRNA and miRNA regulation, as LIN28A is an RNA-binding protein [40]. Nevertheless, LIN28A does not appear to be regulated by BORIS in cis, as the closest strong BORIS binding peak [33] is 80 kb downstream, and there is no BORIS/CTCF-anchored chromatin domains enclosing it [46].
Another set of genes clustered together (Figure 1D) included four genes, UBA7, CA2, SOX6 and a rather uninformative S100A16. CA2 has a strong BORIS&CTCF binding site in the promoter and encodes a carbonic anhydrase. It is widely expressed in many tissues, including erythroid lineage [47, 48]. Thus, its activation in BORIS KD K562 (Figure 2B) could be a sign of commitment to the differentiation. The two remaining genes are more insightful. In the corresponding KO mice, the UBA7 homolog, Ube1L, was shown to be directly involved in the regulation of the transplantation proficiency, i.e. proliferation, of hematopoietic progenitors [49]. Incidentally, the UBA7 gene does not have BORIS bound at the promoter in K562 cells, indicating that the BORIS-mediated regulation is exerted indirectly, possibly by rearranging 3D chromosomal structure like CTCF [33]. Indeed, while the UBA7-containing region is involved in multiple interactions as shown in ChIA-PET (Chromatin Interaction Analysis by Paired-End Tag/Sequencing) data [46], there is a UBA7 locus-specific CTCF loop, with one anchor enriched by BORIS, while the other is occupied by CTCF only, with corresponding strong RNAP II-specific interactions present only in BORIS-positive K562, but not in MCF7 (Supplementary Figure S1).
Another gene clustering with UBA7 is the transcription factor SOX6 (Figure 1D). With concentrated extracts, the expression of protein itself is detectable in BORIS KD (Figure 2C). The SOX6 gene is large, spanning almost 500 kb, with strong BORIS binding sites both upstream and downstream of the locus. The main promoter region of SOX6 is not occupied by either BORIS or CTCF in K562 (Figure 2D). This could reflect the fact that the transcriptional control by BORIS and CTCF at the SOX6 locus is mediated by these proteins via a larger chromatin domain. The CTCF ChIA-PET data [46] indicates that the SOX6 locus loop is fully enclosed between CTCF sites regardless of BORIS presence (Figure 2D) . Nevertheless, the regulatory potential of BORIS might be exerted through anchor sites in cooperation with CTCF (Figure 2D). Indeed, the RNAP II ChIA-PET data [46] indicate that there is a lot of RNA polymerase-dependent interactions within SOX6, many of them anchored at BORIS bound sites, in BORIS-positive K562, but not in MCF7 (Figure 2D). SOX6 overexpression is known to induce differentiation in several cell types [50-54].
Among other genes in the DE upregulated group, which could be considered informative, is TWIST1. Its overexpression has been directly implicated in both CML [55] and AML [56] biology and in epithelial-mesenchymal transition [57]. It provides an additional argument that the highest amplitude effect upon BORIS KD could involve a larger chromatin domain control, rather than a direct promoter regulation. Indeed, the locus identified in RNA-seq as corresponding to TWIST1 (chr7.14) in fact overlapped with another upregulated gene HDAC9 [58]. Both genes are virtually silent in K562 in the presence of BORIS. Incidentally, co-regulation of Hdac9 and Twist1 genes has been reported in mice and was attributed to having a common enhancer within the Hdac9 intron [59].
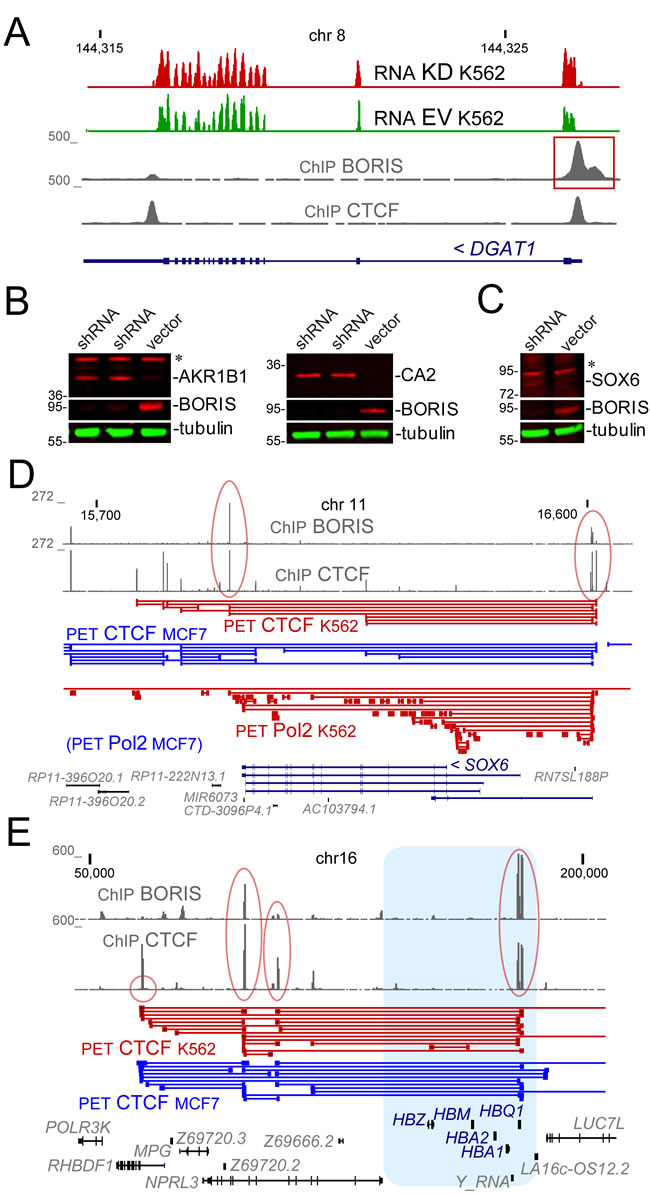
Figure 2: The examples of genomic loci and proteins that were upregulated upon BORIS KD. A. The genomic structure of DGAT1 locus. The RNA signal (TopHat alignment) shows the comparison of KD and EV control. The ChIP signal [33] shows the sites for CTCF and BORIS binding. The BORIS peak at the transcription start site is boxed in red. Numbers on the top show chromosomal coordinates (in kb), and numbers on the left indicate the range of data-view signal scaling (in Genome Browser arbitrary units). B. The upregulation of AKR1B1 and CA2 proteins upon BORIS knockdown. Immunoblotting shows the depletion of BORIS in two independent experiments and the appearance of the corresponding upregulation of AKR1B1 and CA2 protein in the whole cell extracts 96 hours after shRNA induction. Alpha-tubulin is a loading control. Molecular mass markers are shown on the left (x1000). C. The upregulation of SOX6 protein in chromatin fraction upon BORIS knockdown. Immunoblotting shows the depletion of BORIS in the whole cell extracts and the appearance of the corresponding SOX6 protein 96 hours after shRNA induction. Molecular mass markers are indicated on the left (x1000). D. The SOX6 spanning region in human genome with the corresponding CTCF and RNAP II (Pol2) ChIA-PET data [46]. The BORIS and CTCF peaks corresponding to two major anchor sites for the locus are circled in red. Numbers on the top and on the left are as in A.. E. The globin locus. The hemoglobin genes are shown in blue. The ChIP signal pileup shows the two anchor sites for the globin locus (circled in red), with one on the right including a BORIS and CTCF cluster site.
Pathways upregulated upon BORIS KD
When assessing the group of differentially upregulated genes as a whole, without focusing on the individual top scoring genes, one can pinpoint some indicative pathways upregulated by BORIS KD (Supplementary Table S2), despite the relatively small number of DE genes in our case. The most telling, notwithstanding the low expression, is a set of hemoglobin genes, HBA1, HBG1, HBG2, HBA2, HBZ, and HBE1. It is an additional indication of some BORIS involvement in blocking of a differentiation pathway, erythroid in this case, in K562. It also appears likely that BORIS is involved in the control of the whole hemoglobin locus (Figure 2E), as it was established for CTCF [60-62]. Incidentally, both ZBTB7A/POK, which has a defining role during erythroid differentiation and in cancer [63, 64], and BCL11A involved in hemoglobin switching [65, 66] both have strong BORIS binding upstream of their TSS [33 ]and were also moderately upregulated in the present study. At the same time, genes that are required for hemoglobin assembly, such as AHSP, were unaffected by BORIS KD.
Among the upregulated pathways, there were also several prominent transcription factors and cofactors. These factors are controlling substantial networks of downstream genes and involved into morphogenetic and/or differentiation processes: GDF, as well as HAND2 and FOXA2, both prominent early mesoderm genes. HAND2 has a BORIS site at its promoter and is known to be aberrantly methylated in CLL [67], which may indicate an activity of BORIS counteracting aberrant hyper methylation, similar to TP53TG1. While HAND2 was shown to be a player in differentiation and trans-differentiation [68], it shows low expression in K562 and a relatively minor increase. GDF2/BMP9, encoding a TGF-beta superfamily member [69], and FOX2A, on the other hand, do not have BORIS binding at its promoter, and are upregulated less than twofold.
The group of signaling-related and cytokine-related genes also included genes that either did not (OSGIN1 and HYAL1) or did (CA2, SLC26A6, CHRD, RHOB and APBB2) have BORIS binding in the immediate promoter vicinity. Some of these genes could negatively regulate cell growth, such as OSGIN1 [70]. Published data on HYAL1 are somewhat contradictory with respect to its effect on the proliferation of cancer cells [71, 72]. RHOB is probably most interesting in this group. It encodes a small GTPase with a direct link to apoptosis regulation [73]. Furthermore, the mechanism of its involvement is likely mediated by the binding to TP53 isoforms and thus connects to the very core of the ability of cancer cells to proliferate [74].
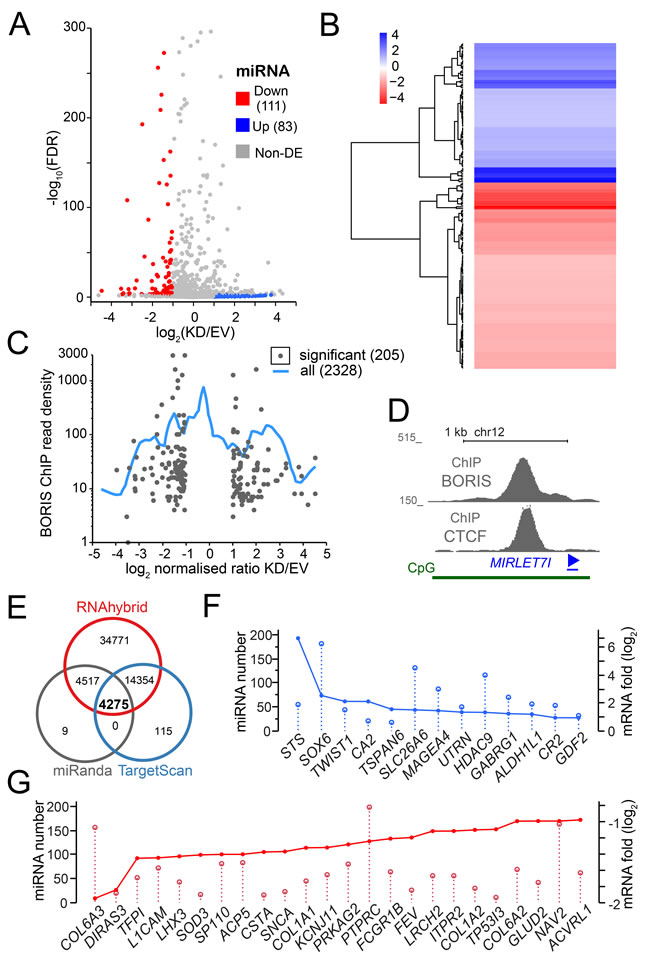
Figure 3: The RNA-seq analysis of miRNA upon BORIS depletion. A. A volcano plot of all differentially expressed miRNA. Each dot corresponds to a potentially differentially expressed miRNA, with colored corresponding to significant DE MiRs and grey to ones under significance threshold, as described in Methods. B. A heatmap of the hierarchical clustering of differentially expressed miRNA based on RNA-seq analysis. Blue - upregulated genes, red - downregulated genes. C. A plot representing the relationship between the differential expression of miRNA and the read density from BORIS ChIP-seq [33], i.e. number of sequencing tags per 2 kb window centered at a given MiR-encoding genomic sequence. The blue smoothing line represents the whole dataset of expressed MiRs converted into genomic coordinates, the dots correspond only to significant DE MiRs. D. MIRLET7I locus structure, with ChIP [33] and ChIA-PET [46] data combined. E. Target analysis of DE miRNA. Vent diagram of potential miRNA regulated targets shows the overlap of all three methods used, with corresponding numbers of target sequences. E. The overlap plot of a sample of upregulated mRNA with the corresponding miRNAs expressed in K562. F. The overlap plot of some significantly downregulated mRNA with the corresponding K562-expressed miRNAs. G. The overlap plot of significantly upregulated mRNA with the corresponding K562-expressed miRNAs.
Genes downregulated upon BORIS KD
As was mentioned, one could expect to find potential genes involved in the maintenance of self-renewal of the K562 cells among the genes that are downregulated upon BORIS KD. Indeed, among genes with highest DE (Figure 1D) several genes did stand out. Among the genes that tightly clustered with BORIS or formed the next cluster (Figure 1D), only TFPI and PTPRC were assigned to a biological process pathway (Supplementary Table S3). The rest, i.e. C7orf43, ARHGDIB, NUPR1, GABPB1-AS1, CCNYL1, FAM172A, DIRAS3, and SPRR2D provide only indirect hints on the potential BORIS facilitation of K562 proliferation and stemness self-renewal. The highest individual fold change in this group was observed for FAM172A. which remains a poorly characterized gene [75-77] The CCNYL1 gene is more informative, with the second highest fold downregulation in BORIS KD and BORIS binding in its promoter. It is a CDK6 cyclin, which is normally specific for germ cells [78].
In general, at present we could only infer some relevance of most genes in the set to BORIS biology in K562. The KD of NUPR1 was shown to inhibit both cell proliferation and migration and resulted in a cell cycle arrest in more than one cancer types [79, 80]. NUPR1 is a chromatin-bound regulatory protein [81] that interfaces with both DNA methylation and histone acetylation genome-wide [82, 83]. The DIRAS3 gene encoding a member of the ras superfamily presents a special case with respect to BORIS-mediated regulation, as the expression of this gene is tightly controlled epigenetically; it is expressed monoallelically due to maternal imprinting [84]. SPRR2D is a part of a cluster of several small proline-reach proteins encoding genes on chr1(q21.3), which was identified as epidermal differentiation complex [85]. The cluster is enclosed into a chromatin loop with one of the CTCF anchors occupied by BORIS, according to ChIA-PET data [46]. CYFIP2, which has a very strong BORIS binding around the TSS, not just in K562 but as well in all other BORIS-positive cells analyzed by ChIP in [33], is directly linked to the actin metabolism and the fragile X syndrome via interaction with FMR1 protein [86]. There are also genes that are known to directly control transcription and chromatin structure: FEV, CECR2, ZNF528, HEY2, FOXO6 and CECR2.
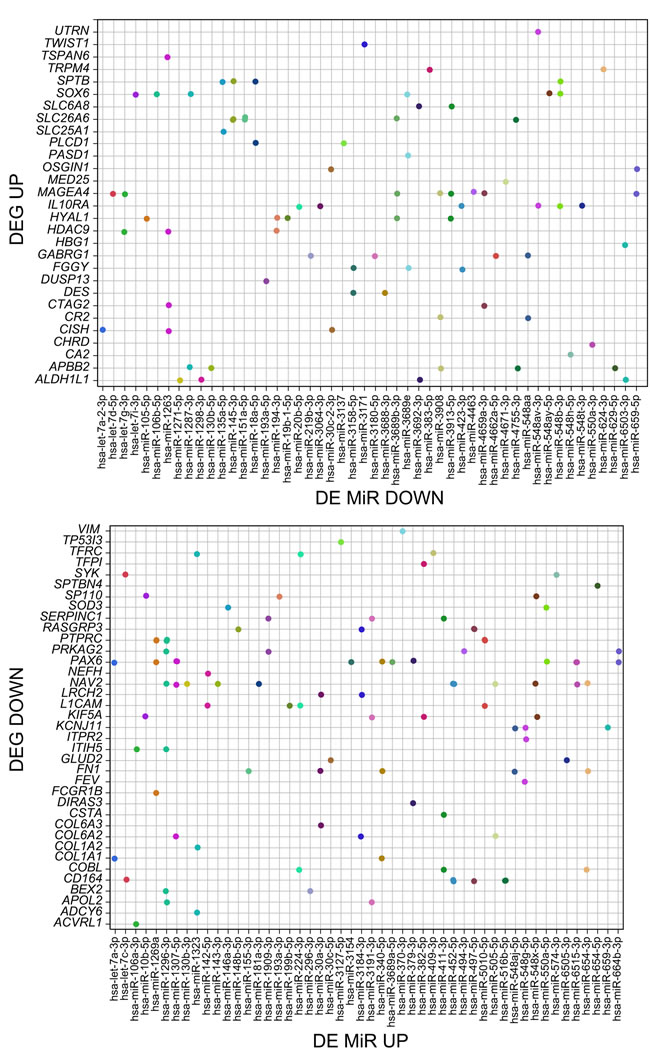
Figure 4: The correspondence between the significantly differentially expressed mRNAs and MiRs upon BORIS KD. The dots represent the individual significant DE MiRs regulating a given gene, but expressed in a reciprocal fashion, as indicated in panel titles.
Pathways downregulated upon BORIS KD
GO analysis showed that 42 of downregulated genes fell into clusters corresponding to defined biological pathways. Due to the small number of downregulated genes, pathway discovery was not particularly informative with respect to the role of BORIS in K562 self-renewal or regarding triggering the differentiation upon its downregulation. Nevertheless, it was noteworthy that the largest group of downregulated genes was the group (12 genes in total) related to blood coagulation (L1CAM, ESAM, ITPR2, KIF5A, FN1, SERPINC1, SYK, TFPI, CD9, COL1A1, COL1A2, DOCK11) (Supplementary Table S3). The coordinated downregulation of this group by BORIS could indicate that one way BORIS interjects itself in the control of myeloid cancer cells expression program, is by controlling the expression of megakaryocytic pathway genes. Incidentally, some other hematopoiesis-related genes and pathways were also evidently regulated by BORIS, which could reflect a broad spectrum of K562 cells’ dependency on BORIS [33]: L1CAM, PTPRC, SYK, PGLYRP4, PGLYRP3, PTPRC, and SYK (Supplementary Table S3).
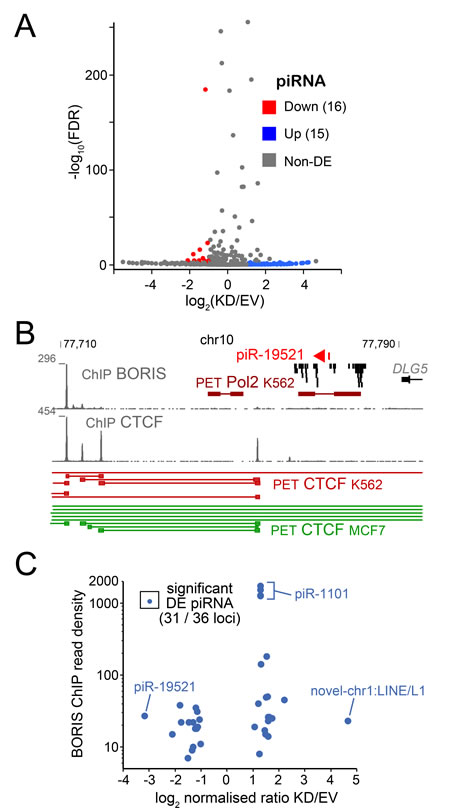
Figure 5: The RNA-seq analysis of piRNA. A. Volcano plot of K562-expressed piRNA shows the relationship between fold change of expression and p-value. B. The structure of pIR19821 locus, with BORIS and CTCF ChIP data in K562 combined with CTCF ChIA-PET data from K562 (BORIS-positive) and MCF7 (BORIS-negative) [46]. C. The differential expression of piRNAs in relation to BORIS ChIP (read density) from [33], with some piRNA examples indicated.
Multiple miRNAs change their expression upon BORIS KD
As was shown above, BORIS displayed a multifaceted involvement in the regulation of downstream genes, including long- non-coding RNAs: TP53TG1 [87] and LOC102724938. The role of BORIS in the expression of small noncoding RNAs has never been studied. Therefore, we conducted a series of four additional RNA-seq experiments aimed at the identification of short ncRNA that would be differentially expressed upon BORIS KD, and focused on the ones that are relevant to gene expression regulation, namely miRNA and piRNA, for downstream analyses. While several surveys were made of the miRNA repertoire in CML [88-90], the miRNA molecular makeup of these cancers is far from completely understood. Thus, the goal of this analysis was to identify miRNAs, which change their expression and are known to be associated with the inhibition of the proliferation of cancer cells, and CML in particular. In our miRNA-seq data, 83 were up-regulated versus 111 down-regulated (Figure 3A, 3B), which included a number of uncharacterized/novel miRNA (Supplementary Table S4).
In order to get insight into the putative mechanism of BORIS control of miRNA expression we assessed the relative enrichment for BORIS binding in the vicinity of miRNA genes. The Figure 3C shows that the highest density of BORIS biding (ChIP) is not associated with the genes displaying the most extreme DE. This indicates that in K562 cells, similarly to protein-coding genes, BORIS is also acting in trans, via enhancers and/or 3D chromatin domain folding. For example, let-7i-5p is a part of a CpG island encompassing the TSS of the LINC01465 gene with a very strong BORIS and CTCF binding (Figure 3D). let-7a-3p, on the other hand, is expressed in an intergenic region harboring several ncRNAs and flanked by strong BORIS and CTCF binding sites, indicating a potential regional control of its expression by BORIS, while miR-142-5p is located in a 15-kb region expressing several noncoding RNA, without obvious binding of BORIS in the vicinity. Finally, the miR-1296-3p MiR is intragenic to the JMJD1C gene that has a strong BORIS binding site in its promoter, with a similar situation observed for miR-340-5p (the RNF130 gene), and for miR-224-3p (GABRE gene). Mature sequence miR-7-5p is encoded at three different loci in human genome, none of which appear to be associated with BORIS binding in cis.
Among miRNA downregulated upon BORIS KD, miR-7-2-3p and let-7a-5p stand out with highest fold (Supplementary Table S4). miR-7-2-3p information is presently limited to biomarker surveys [91, 92]. The let-7a-5p expression is normally maintained at a relatively stable level [93], and the three loci that encode it have no direct association with BORIS binding in K562. Nevertheless, the MIRLET7A2 locus is located within a nearly megabase region expressing several ncRNAs, which is flanked by strong BORIS and CTCF binding sites.
Multiple downstream pathways are regulated by miRNA under BORIS control
The chief value of miRNA analysis in our case was to have a measure of the regulatory reach by BORIS, mediated by miRNA expression. We used several tools (see Methods) to identify the targets of differentially expressed miRNAs after filtering for significance level. As all these methods are known to be prone for false-positive identification, we only focused on 4275 predicted by all three methods (Figure 3E). This still indicates that BORIS has a potential to control a sizeable genetic network in these BORIS-positive CML cells. As the network of DE miRNA resulting from BORIS KD is highly interconnected, with multiple pathways potentially affected (Supplementary Figure S2), it is a challenging task to extract the actionable components. One measure of the affected nodes of regulation that are relevant to the eventual K562 differentiation is the links between the DE mRNA and the DE ncRNAs, which are known/predicted to affect the corresponding mRNAs. Indeed, for most DE mRNAs there were from a dozen to a couple of hundred potential miRNA that were expressed in K562 (Figure 3F, 3G). While the compound effect of multiple miRNAs regulating the same gene but falling under a threshold of significance is not easy to evaluate, focusing on the pairs of significant DE MiRs and significant DE genes that were reciprocally changed (miRNA up with mRNA down, and vice versa) enables one to generate a snapshot of the putative regulatory network involved (Figure 4). This could also provide a plausible explanation for some genes with a changed expression, for which we don’t detect BORIS binding in the promoter vicinity or at the base of enclosing chromatin loop.
A number of piRNAs are regulated by BORIS in K562
Piwi-interacting RNA (piRNAs) are small non-coding regulatory RNAs that are abundantly present in testis [94, 95]. piRNAs are also involved in the control of transposable element (TE) expression [96]. We have previously showed that BORIS acts as a co-repressor for at least one TE family, SVA [36], however BORIS has not been implicated in regulating piRNA expression. Therefore, it was intriguing to see whether BORIS positively regulates piRNA expression in cancer cells, presumably by analogy with the spermatogenic germline. Upon analysis of the small ncRNA-seq data we identified 16 piRNA that were upregulated and 15 - downregulated (Figure 5A), which corresponded to 36 genomic locations. Only one known piRNA that showed a robust downregulation upon BORIS KD, piR-19521, was expressed in a large cluster of ncRNAs with no obvious genomic landmarks potentially linking its expression to BORIS (Figure 5B). However, the 3D structure of the corresponding locus in K562 is different from the BORIS-negative MCF7 cells, both for CTCF and RNAP II - mediated organization (Figure 5B). This may be due to the presence of BORIS in the K562 cells, but additional dissection of the pathway could be required to establish a specific link. In general, for all the DE piRNAs, there was little coincidence with BORIS binding in the immediate 2-kb window (Figure 5C). Following are known piRNAs that stand out with respect to their higher expression and fold of upregulation: piR-586 expressed from the same strand as several long noncoding RNAs (DGCR9 and DGCR5), which have strong BORIS binding peaks; piR-18780 overlapping with a MIR3 repeat; piR-1101 with several genomic locations corresponding to LINE1; and the piR-018569, a part of the last exon of the GLG1 gene, which has BORIS binding in the promoter. The information on novel piRNAs is included in Supplementary Table S4, with most of them originating from TE sequences.
Thus, it appears that piRNAs follow the same pattern of regulation by BORIS that was observed for mRNAs and miRNAs, i.e. while BORIS binding in cis is likely playing a role, the regulatory reach of BORIS via long-range interactions in the chromatin seems to be more dominant.
DISCUSSION
As a result of this work, we demonstrated that BORIS/CTCFL is controlling a sizeable genetic network in cancer cells. The K562 is the first cancer cell line that was shown to be addicted to a CT gene. Namely, the KO of the, normally testis-specific, BORIS gene resulted in a terminal differentiation of K562 cells [33]. At the same time, the transcriptome of the K562 differentiated after BORIS KO [33] did not enable the elucidation of the actual role played by BORIS in the cancer cell line itself, because cells differentiated and hence lost their potential to proliferate and self-renew. While BORIS is the CT gene that is most commonly activated in cancers among other CT genes [7], most cancers, unlike K562 cells, have low level of BORIS expression. Therefore, it was especially important to get inroads into understanding of the mechanism of K562 cells addiction to BORIS. The present detailed analysis of fast BORIS downregulation is helpful in better understanding the scope of CT-mediated regulation in cancer cells, as well as it insightful with respect to the differentiation therapy of CML.
In the present work, a nearly 5-fold rapid downregulation of main BORIS transcript by shRNA, which resulted in over tenfold reduction in BORIS protein levels, was shown to affect the expression of numerous transcripts, including both mRNAs and ncRNAs. That enabled us to survey the early response to BORIS depletion, and to pinpoint prospective pathways that could become dependent on BORIS in cancer. The normal development, differentiation, and maturation of precursors for blood cells are, essentially, an interplay between two reciprocal processes: the eventual loss of their self-renewal potential, and a sequential gain of gene expression patterns characteristic for specific lineages. While K562 cells are tumor cells, they, in a typical fashion for CML, retain the capacity to differentiate and mature (Figure 1A). Thus, one is to expect a similar interplay of genetic networks upon commitment to differentiation. One surprising result of the significant downregulation of BORIS in this work is, undoubtedly, the fact that relatively few genes were affected. It is especially striking, if one considers that the number of BORIS binding peaks in K562 runs in tens of thousands [33]. This inevitably leads to a conclusion that, while BORIS activation establishes epigenome-wide pattern of binding in a germline-like fashion [33, 35], the actual addiction of K562 to BORIS is restricted to a very limited number of pathways.
Among the upregulated mRNAs, a clear connection was evident to the genes that could potentially inhibit proliferation. Namely, SOX6, LIN28A, HDAC9 and UBA7 all directly interface with the proliferation control. SOX6 can suppress cell proliferation and induce differentiation upon overexpression in several model systems [50-53] including erythropoiesis [54]. The link of SOX6 dosage to differentiation potential is also bidirectional, as insufficient SOX6 can prevent differentiation [97]. Thus, it is not surprising that SOX6 transcription is tightly regulated by a negative auto-regulation loop [98]. Furthermore, it was already directly shown that ectopic SOX6 overexpression induced erythroid differentiation within 9 days in both K562 and primary erythroid cultures from human blood [99]. Thus, SOX6 is a suitable candidate for a strong downstream effector of BORIS KD that might be a part of the response to BORIS downregulation leading to cell differentiation.
LIN28A blocks the maturation of let-7 miRNA [100, 101] and so regulates a number of downstream cell cycle proteins including cyclin D1 (CCND1) and CDC25A. Furthermore, LIN28A also directly regulates several mRNAs leading to reprogramming of cellular metabolism [102] and may also control gene expression directly via epigenetic remodeling of promoters [103]. Thus, LIN28A could be one of the key factors enabling reprogramming in K562 upon BORIS KD. The UBA7/UBE1L gene was shown to reduce cyclin D1 protein levels, and it possibly affects other pathways via increasing ISGylation, thus having a pronounced anti-proliferation effect in cancer cell lines [104, 105]. HDAC9 is a histone deacetylase-like protein, which could be a significant factor in determining some of the downstream epigenetic effects of BORIS suppression in K562 acting through modulating transcription as other class IIa HDACs [106]. There is ample evidence directly linking HDAC9 to oncogenesis [107, 108, 109], including lymphomas [110]. Most recent data also indicate that overexpression of HDAC9 could directly interfere with cell cycle by inhibiting cyclin D1 expression via cooperation with PC3/Tis21 [111].
All four genes directly feed into the inhibition of cyclin D1 protein or the encoding gene, which is a common proto-oncogene. The corresponding CCND1 gene is known to cause malignant cell transformation in both CML and AML upon IGH fusion [112] and upon overexpression/dysregulation in other cancers, even in the absence of other major oncogenic hallmarks, such as p53, MYC and RAS [113]. However, the CCND1 mRNA level ends up not significantly changed upon BORIS KD, indicating the complexity of the regulation involved. One of the possible counterbalances could be a very high expression of CDC25A, regardless of the BORIS KD. Nevertheless, SOX6 is one of the most suitable candidate downstream BORIS, for inhibiting K562 proliferation upon BORIS KD. This gene is known to trigger differentiation in a number of systems [52, 53, 98]. The fact that BORIS regulates this gene in K562 cells is likely attributed not to the direct control of its promoter, but rather via invasion of the chromatin loop enclosing only the SOX6 locus and normally maintained by CTCF in somatic cells. This could be a reflection of the specific regulatory circuit potentially controlled by BORIS not just in myeloid progenitors, but also in other cancer cells that activate BORIS.
We also observed clear signs of changed regulation of a certain number of protein-encoding genes relevant to erythrocyte and megakaryocyte differentiation pathways, as well as a number of testis-specific genes, in response to BORIS KD. These finding shows that the involvement of BORIS in the maintenance of self-renewal and stemness of K562 is multifaceted, but still rather limited in scope, considering the abundance of BORIS binding to chromosomes.
Among the genes that were downregulated upon BORIS depletion we see a coordinated downregulation of some progenitor-specific genes as well as decreased expression of some proliferation factors, e.g. NUPR1, as well as genes that are normally germline specific (such as CCNYL1), but are activated in tumor cells owing, at least in part, to BORIS expression. CCNYL1 was recently identified as a germline-specific cyclin for CDK16 [78], and thus appears to be a bona fide testis target activated by BORIS, possibly representing an additional link to cell cycle controls that are facilitated by BORIS in K562 cells.
The other genes involved in the control of transcription or chromatin remodeling are FEV, CECR2, ZNF528, HEY2, and FOXO6. Of those, HEY2 acts upstream of Notch in blood lineages differentiation [114] and its KO in mice results in severe and early development defects [115]. Another interesting gene here is CECR2 encoding a bromodomain protein, which was implicated in chromatin remodeling and has a strong morphogenetic function in spermatogenesis [116].
In conclusion, as a result of surveying the mRNAs differentially regulated upon BORIS KD, it is evident that there are signs of both inhibition of proliferation and the commitment to differentiation. At present, it is unclear, what is the threshold of BORIS expression that becomes physiologically meaningful in cancer cells. As was demonstrated here, the nearly complete depletion of BORIS protein, still did not trigger K562 differentiation, despite showing the signs of cell commitment. This agrees well with low, even if pervasive, BORIS expression in tumors [117, 118], and indicates that the dependence of K562 on BORIS is not embodied by numerous pathways.
One possible mechanism that prevents BORIS from changing expression of thousands genes in K562 cells instead of hundreds observed, is some additional level of regulation that might work as feedback regulation. Non-coding RNAs in general, and micro-RNAs in particular, could provide such a mechanism. In this work, for the first time, we evaluated an involvement of BORIS in the control of the expression of small noncoding RNA. While it was hinted by the DE of some long noncoding RNAs (lncRNAs) upon mRNA-seq analysis, the revealed scope of BORIS involvement in short RNA expression is rather far-reaching, with thousands of downstream genes potentially affected. It is interesting, however, that the most well-characterized miRNA involved in hematopoiesis and blood cancers were not represented [119] in the significant DE set. This supports the idea that the massive control of miRNA expression by BORIS could be related to limiting the impact of BORIS activation.
The most dramatic effect among highly expressed known miRNA was observed for let-7a-3p, as well as: let-7i-5p, miR-1296-3p, miR-340-5p, miR-3184-3p, miR-7-5p, miR-142-5p, and miR-224-3p (Supplementary Table S4). The let-7i-5p has been placed in the molecular makeup of CML as one of several MiRs targeting NFKB1 [89]. The miR-1296-3p is known to play an tumor suppressing and inhibitory role in cancer [120, 121], as well as repress the expression of chromosome maintenance genes that are upregulated in tumors [122]. The higher levels of miR-340-5p are known to induce apoptosis [123]. miR-3184-3p is on the opposite strand relatively to the intron in the enclosing protein coding gene, and its antisense, miR-423-3p, is notably downregulated (Supplementary Table S4), indicating that BORIS mediated control is exerted differentially dependent on the direction of the gene. Incidentally, the miR-423-3p upregulation is known to promote proliferation of tumor cells [124, 125], while it’s inhibition has an opposite effect [126]. miR-7-5p is encoded at three different loci in human genome, none of which appear to be associated with BORIS binding in cis. Nevertheless, it is known to be p53-dependent [127], and its expression was shown to inhibit cancer cells [128, 129]. miR-142-5p has been shown to induce apoptosis in non-blood cancer cells [130] and inhibit cell cycle via beta-catenin/WNT signaling [131]. It has been also shown to be a component of macrophage-specific signaling program [132].
Thus, in general, it is evident that BORIS controls multiple species of miRNA, with a substantial spectrum of functions. This control is exerted through a number of mechanisms, from the whole locus-control functions to individual promoter occupancy of the host genes. This phenomenon may act as a buffer for the ability of BORIS to act as a repressor and activator of multiple genes, thus limiting the scope of BORIS activity, despite abundant expression.
In addition to miRNA, it was also revealed that piRNAs, which, when expressed in cancers, essentially qualify as non-protein-coding CT genes, are also regulated by BORIS. The degree, to which this control is direct, remains to be investigated further, as the knowledge of individual piRNAs’ regulation is extremely limited. Incidentally, while some piRNAs (such as piR-17033), and including most of the novel species, have originated from the endogenous retrovirus (ERV) sequences, none were mapped within or in the vicinity of SVA elements, reinforcing our original conclusion that BORIS directly regulates SVA by binding at SVA VNTRs [36].
Furthermore, while we could hypothesize that BORIS is positively regulating piRNA expression in testis, that mode of regulation appears to be distorted in cancer cells, as multiple piRNA are upregulated upon BORIS KD. The involvement of BORIS in piRNA expression, at least in cancer cells, is a significant finding pertaining to BORIS biology in cancers. It could be directly related to the genomic instability in cancers expressing BORIS, due to the uncoupling of piRNA expression control from the normal somatic mechanisms.
In conclusion, it is evident that, while BORIS strongly binds promoter areas and TSS of thousands of genes in K562 cells, the resulting set of DE expressed genes upon the substantial depletion of BORIS is rather limited. It could be a reflection of the corresponding “buffering” role of multiple DE miRNAs that are concurrently regulated by BORIS. Nevertheless, this is a strong indication that the key to BORIS functions in cancer, and particularly to K562 addiction to BORIS, could be narrowed down to a tangible set of factors, rather than attributed to the whole-epigenome remodeling in a germline-like fashion.
materials and METHODS
Cell culture and generation of stable inducible knockdown cell lines
K562 cells and stably transfected derivatives were grown in IMDM (Hyclone) with 10% or 20% Tet-On certified FBS. BORIS knock-down vectors were constructed to express either of the two shRNA templates, GGAAATACCACGATGCAAATT (Site 1) or GGTGTGAAATGCTCCTCAACA (Site 2). To construct the vectors, the synthetic double-strand oligonucleotides were inserted into AgeI and EcoRI restriction sites of the pLKO-Tet-On-neo vector. To package lentivirus in HEK293T/17 cells they were co-transfected with the Tet-pLKO-Neo [133] (Addgene) vector (vector-only control) or with the same vector expressing either of two alternative anti-BORIS shRNA together with two packaging plasmids psPAX2 and Pmd2.G. The stocks of lentivirus were harvested 72 hr after the transfection. K562 cells grown to 40%-50% confluence were infected with 500µl of lentivirus stock using 8µg/ml polybrene (Sigma). Culture media supplemented with 600µg/ml G418 was replaced 12 hr after infection, and cells were selected for 4 weeks or more. Over ten independent stable cell lines expressing anti-BORIS shRNA were generated. For each, cells resistant to G418 were plated into in 96-well plates, and individual wells were analyzed by RT-qPCR and anti-BORIS immunoblotting. The wells that showed robust BORIS downregulation were pooled from one plate and frozen. For BORIS knockdown, stably infected cells were induced with 200ng/ml doxycycline resulting in the activation of the Tet-On promoter driving the expression of shRNAs.
Quantitative RT-PCR
Total RNA from cultured cells was extracted using Trizol (Invitrogen). cDNA was synthesized with the PrimescriptTM RT Reagent Kit with genomic DNA Eraser (Perfect Real Time) from TaKaRa. Quantitative PCR (qPCR) was done with the SYBR Premix Ex Taq (TaKaRa) on the Mx30005P qPCR workstation (Agilent).
Immunoblotting
For whole-cell protein extracts, cells were washed with PBS with 1X protease inhibitor cocktail (Roche Applied Science) and then lysed with SDS-PAGE sample buffer. Protein extracts were separated by SDS-PAGE and transferred to a PVDF membrane, followed by the incubation with the primary and then secondary antibodies fused to fluorochromes for LI-COR Odyssey (LI-COR Biosciences) detection and quantification.
The anti-BORIS antibodies were as described in [33]. Anti alpha-tubulin Abs were from Merck (Sigma Aldrich). Anti-CA2, anti-AKR1B1, and anti-SOX6 Abs were from Abcam.
RNA-seq analysis
For the RNA-seq, the inducible BORIS KD and empty vector control K562 transgenic cell lines were used. To induce BORIS KD, doxycycline was applied for 96 hr resulting in a reduction of BORIS mRNA level 2.5-4.5 fold, with the corresponding decrease of BORIS protein levels. At least 4 biological replicates were analyzed for each set of experimental conditions. For all NGS mRNA analyses, the inducible stable shRNA K562 cells were plated at ~40% confluence in DMEM media and induced in the presence of doxycycline (200 ng/ml) for 96 hr; the cells were then harvested and frozen. mRNA was extracted, libraries were prepared and sequenced at RiboBio (Guangzhou) using Illumina HiSeq3000. The initial assessment of quality of RNA was at the Agilent Genomics 2200 TapeStation. For the library quality testing, Agilent 2100 Bioanalyzer and ABI StepOnePlus Real-TimePCR System were used.
On average, 20 million reads were obtained from each experiment. The results of RNA-seq experiments were analyzed using a conventional pipeline. They were first analyzed for consistency and reproducibility, aligned to the human reference genome (hg38) using TopHat2 with the parameters —segment-length 20 -G GRCh38, then processed using Cufflinks 2.0.0 [134] (parameter settings: -g GRCh38), then Cuffmerge (-p 8 -o -g GRCh38 -s) and Cuffdiff (-p 8 -L -b -M, loci over 500k filtered out). The set of significantly changed DE genes set was then additionally filtered to remove low expression genes with expression < 0.5 FPKM. Expressed loci with no assigned gene symbol were also excluded from further analysis.
For ncRNA NGS analyses, the RNA preparations were done by BGI (Shenzhen) and sequenced using Illumina HiSeq4000. Different size RNA segments were separated by PAGE gel with 18-30nt stripe selected. Then, after the adapter ligation and RT-PCR procedures were recovered by PAGE gel and recycled products dissolved in the EB solution.
The resulting 49nt tags were cleaned from low quality tags and adaptor contaminants, afterwards they were categorized with all the small tags failed to fall into known category to be grouped as potentially novel miRNA with the following priority assumption: miRNA > piRNA > snoRNA > Rfam > other sRNA (Thus, each RNA falls into a single unique category). Additionally, for potential novel miRNAs their ability to form characteristic hairpin structure together with the Dicer cleavage site and the minimum free energy were analyzed via miRDeep2 [135]. Target prediction analysis with GO enrichment and KEGG [136] pathway analysis were made.
For piRNA prediction, the Piano [137] tool was used. To discover potential piRNAs [138], their structural features (namely, the fact, that each strand of this 22-24nt long dual-strand RNA is 2nt longer on the 3’ end than the paired one) were tested by aligning tags to each other.
To map the reads, either Bowtie2 [139] or cmsearch [140] (mapping to Rfam) were used with the following parameters: -q -L 16 —phred64 -p 6 (Bowtie2) and —cpu 6 —noali (cmsearch). The expression levels were calculated as TPM (transcripts per kb, million) [141]. For possible target prediction, RNAhybrid [142], miRanda [143] and TargetScan [144] software tools were used with the following parameters: -en -20 -strict (miRanda); -b 100 -c -f 2,8 -m 100000 -v 3 -u 3 -e -20 -p 1 -s 3utr_human (RNAhybrid). DESs screening and mapping were performed by using DEGseq [145] and pheatmap software, respectively.
The mRNA datasets were deposited at NCBI GEO with the accession number GSE99825, and the sRNA datasets and the corresponding metadata were deposited at NCBI GEO with the accession number GSE99899. Both datasets are accessible together through the series’ accession number GSE99900.
Abbreviations
Differential expression / differentially expressed (DE), cancer-testis (CT), zinc fingers (ZF), chronic myeloid leukemia (CML), knockdown (KD), knockout (KO), empty lentivirus vector (EV), breakpoint cluster region (BCR), the cancer genome atlas (TCGA), chromatin immuno-precipitation (ChIP), chromatin interaction analysis by paired-end tag/sequencing (ChIA-PET), transcription start site(s) (TSS), micro RNA (miRNA, MiR), Piwi-interacting RNA (piRNAs), transposable element(s) (TE), long noncoding RNAs (lncRNAs), endogenous retrovirus (ERV), SINE VNTR ALU element (SVA), variable number tandem repeat(s) (VNTR), transcript reads per kilobase per million (TPM), fragments per kilobase of transcript per million fragments mapped (FPKM).
Author contributions
ET, QFW, JL, EMP, AB - conducted experiments and analyzed data; DL and VL - analyzed data; AS - conceived and designed experiments, and wrote the manuscript.
ACKNOWLEDGMENTS
We thank Andrew P. Hutchins (SUSTC) and Gabriel Zentner (Indiana U.) for critical comments on the manuscript.
CONFLICTs OF INTEREST
Authors declare no conflicts of interest.
FUNDING
This work was supported by the PRC government’s “1000 Talents Program” grant to A.S., the Guangdong provincial government’s “Guangdong High Talent” award to A.S., the Guangzhou sciences and technology institutional grant 201508020131 to the Drug Discovery Center of the Guangzhou Institutes of Biomedicine and Health., and by the Intramural Program of the National Institute of Allergy and Infectious Diseases for V.L. E.T. was sponsored by CAS-TWAS President’s Fellowship for International Doctoral Students.
REFERENCES
1. Caballero OL, Chen YT. Cancer/testis (CT) antigens: potential targets for immunotherapy. Cancer Sci. 2009; 100: 2014-21.
2. Simpson AJ, Caballero OL, Jungbluth A, Chen YT, Old LJ. Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer. 2005; 5: 615-25.
3. Chen YT, Scanlan MJ, Venditti CA, Chua R, Theiler G, Stevenson BJ, Iseli C, Gure AO, Vasicek T, Strausberg RL, Jongeneel CV, Old LJ, Simpson AJ. Identification of cancer/testis-antigen genes by massively parallel signature sequencing. Proc Natl Acad Sci U S A. 2005; 102: 7940-5.
4. Gjerstorff MF, Andersen MH, Ditzel HJ. Oncogenic cancer/testis antigens: prime candidates for immunotherapy. Oncotarget. 2015; 6: 15772-87. doi: 10.18632/oncotarget.4694.
5. Hofmann O, Caballero OL, Stevenson BJ, Chen YT, Cohen T, Chua R, Maher CA, Panji S, Schaefer U, Kruger A, Lehvaslaiho M, Carninci P, Hayashizaki Y, et al. Genome-wide analysis of cancer/testis gene expression. Proc Natl Acad Sci U S A. 2008; 105: 20422-7.
6. Loukinov DI, Pugacheva E, Vatolin S, Pack SD, Moon H, Chernukhin I, Mannan P, Larsson E, Kanduri C, Vostrov AA, Cui H, Niemitz EL, Rasko JE, et al. BORIS, a novel male germ-line-specific protein associated with epigenetic reprogramming events, shares the same 11-zinc-finger domain with CTCF, the insulator protein involved in reading imprinting marks in the soma. Proc Natl Acad Sci U S A. 2002; 99: 6806-11.
7. Wang C, Gu Y, Zhang K, Xie K, Zhu M, Dai N, Jiang Y, Guo X, Liu M, Dai J, Wu L, Jin G, Ma H, et al. Systematic identification of genes with a cancer-testis expression pattern in 19 cancer types. Nat Commun. 2016; 7: 10499.
8. Lobanenkov VV, Nicolas RH, Adler VV, Paterson H, Klenova EM, Polotskaja AV, Goodwin GH. A novel sequence-specific DNA binding protein which interacts with three regularly spaced direct repeats of the CCCTC-motif in the 5’-flanking sequence of the chicken c-myc gene. Oncogene. 1990; 5: 1743-53.
9. Phillips JE, Corces VG. CTCF: master weaver of the genome. Cell. 2009; 137: 1194-211.
10. Phillips-Cremins JE, Corces VG. Chromatin insulators: linking genome organization to cellular function. Mol Cell. 2013; 50: 461-74.
11. Baniahmad A, Steiner C, Kohne AC, Renkawitz R. Modular structure of a chicken lysozyme silencer: involvement of an unusual thyroid hormone receptor binding site. Cell. 1990; 61: 505-14.
12. Bell AC, West AG, Felsenfeld G. The protein CTCF is required for the enhancer blocking activity of vertebrate insulators. Cell. 1999; 98: 387-96.
13. Kanduri C, Pant V, Loukinov D, Pugacheva E, Qi CF, Wolffe A, Ohlsson R, Lobanenkov VV. Functional association of CTCF with the insulator upstream of the H19 gene is parent of origin-specific and methylation-sensitive. Curr Biol. 2000; 10: 853-6.
14. Filippova GN, Thienes CP, Penn BH, Cho DH, Hu YJ, Moore JM, Klesert TR, Lobanenkov VV, Tapscott SJ. CTCF-binding sites flank CTG/CAG repeats and form a methylation-sensitive insulator at the DM1 locus. Nature genetics. 2001; 28: 335-43.
15. Rasko JE, Klenova EM, Leon J, Filippova GN, Loukinov DI, Vatolin S, Robinson AF, Hu YJ, Ulmer J, Ward MD, Pugacheva EM, Neiman PE, Morse HC 3rd, et al. Cell growth inhibition by the multifunctional multivalent zinc-finger factor CTCF. Cancer research. 2001; 61: 6002-7.
16. Ohlsson R, Renkawitz R, Lobanenkov V. CTCF is a uniquely versatile transcription regulator linked to epigenetics and disease. Trends Genet. 2001; 17: 520-7.
17. Chao W, Huynh KD, Spencer RJ, Davidow LS, Lee JT. CTCF, a candidate trans-acting factor for X-inactivation choice. Science. 2002; 295: 345-7.
18. Cuddapah S, Jothi R, Schones DE, Roh TY, Cui K, Zhao K. Global analysis of the insulator binding protein CTCF in chromatin barrier regions reveals demarcation of active and repressive domains. Genome research. 2009; 19: 24-32.
19. Taft RJ, Hawkins PG, Mattick JS, Morris KV. The relationship between transcription initiation RNAs and CCCTC-binding factor (CTCF) localization. Epigenetics & chromatin. 2011; 4: 13.
20. Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012; 485: 376-80.
21. Sanyal A, Lajoie BR, Jain G, Dekker J. The long-range interaction landscape of gene promoters. Nature. 2012; 489: 109-13.
22. Phillips-Cremins JE, Sauria ME, Sanyal A, Gerasimova TI, Lajoie BR, Bell JS, Ong CT, Hookway TA, Guo C, Sun Y, Bland MJ, Wagstaff W, Dalton S, et al. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell. 2013; 153: 1281-95.
23. Handoko L, Xu H, Li G, Ngan CY, Chew E, Schnapp M, Lee CW, Ye C, Ping JL, Mulawadi F, Wong E, Sheng J, Zhang Y, et al. CTCF-mediated functional chromatin interactome in pluripotent cells. Nature genetics. 2011; 43: 630-8.
24. Narendra V, Bulajic M, Dekker J, Mazzoni EO, Reinberg D. CTCF-mediated topological boundaries during development foster appropriate gene regulation. Genes Dev. 2016; 30: 2657-62.
25. Giorgetti L, Lajoie BR, Carter AC, Attia M, Zhan Y, Xu J, Chen CJ, Kaplan N, Chang HY, Heard E, Dekker J. Structural organization of the inactive X chromosome in the mouse. Nature. 2016; 535: 575-9.
26. Merkenschlager M, Nora EP. CTCF and Cohesin in Genome Folding and Transcriptional Gene Regulation. Annu Rev Genomics Hum Genet. 2016; 17: 17-43.
27. Lupianez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, Horn D, Kayserili H, Opitz JM, Laxova R, Santos-Simarro F, Gilbert-Dussardier B, Wittler L, et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. 2015; 161: 1012-25.
28. Suzuki T, Kosaka-Suzuki N, Pack S, Shin DM, Yoon J, Abdullaev Z, Pugacheva E, Morse HC 3rd, Loukinov D, Lobanenkov V. Expression of a testis-specific form of Gal3st1 (CST), a gene essential for spermatogenesis, is regulated by the CTCF paralogous gene BORIS. Mol Cell Biol. 2010; 30: 2473-84.
29. Pugacheva EM, Suzuki T, Pack SD, Kosaka-Suzuki N, Yoon J, Vostrov AA, Barsov E, Strunnikov AV, Morse HC 3rd, Loukinov D, Lobanenkov V. The structural complexity of the human BORIS gene in gametogenesis and cancer. PLoS One. 2010; 5: e13872.
30. Kosaka-Suzuki N, Suzuki T, Pugacheva EM, Vostrov AA, Morse HC 3rd, Loukinov D, Lobanenkov V. Transcription factor BORIS (Brother of the Regulator of Imprinted Sites) directly induces expression of a cancer-testis antigen, TSP50, through regulated binding of BORIS to the promoter. J Biol Chem. 2011; 286: 27378-88.
31. Sleutels F, Soochit W, Bartkuhn M, Heath H, Dienstbach S, Bergmaier P, Franke V, Rosa-Garrido M, van de Nobelen S, Caesar L, van der Reijden M, Bryne JC, van Ijcken W, et al. The male germ cell gene regulator CTCFL is functionally different from CTCF and binds CTCF-like consensus sites in a nucleosome composition-dependent manner. Epigenetics Chromatin. 2012; 5: 8.
32. Moore JM, Rabaia NA, Smith LE, Fagerlie S, Gurley K, Loukinov D, Disteche CM, Collins SJ, Kemp CJ, Lobanenkov VV, Filippova GN. Loss of maternal CTCF is associated with peri-implantation lethality of Ctcf null embryos. PloS One. 2011; In Press.
33. Pugacheva EM, Rivero-Hinojosa S, Espinoza CA, Mendez-Catala CF, Kang S, Suzuki T, Kosaka-Suzuki N, Robinson S, Nagarajan V, Ye Z, Boukaba A, Rasko JE, Strunnikov AV, et al. Comparative analyses of CTCF and BORIS occupancies uncover two distinct classes of CTCF binding genomic regions. Genome Biol. 2015; 16: 161.
34. Renaud S, Pugacheva EM, Delgado MD, Braunschweig R, Abdullaev Z, Loukinov D, Benhattar J, Lobanenkov V. Expression of the CTCF-paralogous cancer-testis gene, brother of the regulator of imprinted sites (BORIS), is regulated by three alternative promoters modulated by CpG methylation and by CTCF and p53 transcription factors. Nucleic Acids Res. 2007; 35: 7372-88.
35. Rivero-Hinojosa S, Kang S, Lobanenkov VV, Zentner GE. Testis-specific transcriptional regulators selectively occupy BORIS-bound CTCF target regions in mouse male germ cells. Sci Rep. 2017; 7: 41279.
36. Pugacheva EM, Teplyakov E, Wu Q, Li J, Chen C, Meng C, Liu J, Robinson S, Loukinov D, Boukaba A, Hutchins AP, Lobanenkov V, Strunnikov A. The cancer-associated CTCFL/BORIS protein targets multiple classes of genomic repeats, with a distinct binding and functional preference for humanoid-specific SVA transposable elements. Epigenetics Chromatin. 2016; 9: 35.
37. Michael AK, Harvey SL, Sammons PJ, Anderson AP, Kopalle HM, Banham AH, Partch CL. Cancer/Testis Antigen PASD1 Silences the Circadian Clock. Mol Cell. 2015; 58: 743-54.
38. Liggins AP, Brown PJ, Asker K, Pulford K, Banham AH. A novel diffuse large B-cell lymphoma-associated cancer testis antigen encoding a PAS domain protein. Br J Cancer. 2004; 91: 141-9.
39. Yen PH, Allen E, Marsh B, Mohandas T, Wang N, Taggart RT, Shapiro LJ. Cloning and expression of steroid sulfatase cDNA and the frequent occurrence of deletions in STS deficiency: implications for X-Y interchange. Cell. 1987; 49: 443-54.
40. Heo I, Joo C, Cho J, Ha M, Han J, Kim VN. Lin28 mediates the terminal uridylation of let-7 precursor MicroRNA. Mol Cell. 2008; 32: 276-84.
41. Tsanov KM, Pearson DS, Wu Z, Han A, Triboulet R, Seligson MT, Powers JT, Osborne JK, Kane S, Gygi SP, Gregory RI, Daley GQ. LIN28 phosphorylation by MAPK/ERK couples signalling to the post-transcriptional control of pluripotency. Nat Cell Biol. 2017; 19: 60-7.
42. Zhang J, Ratanasirintrawoot S, Chandrasekaran S, Wu Z, Ficarro SB, Yu C, Ross CA, Cacchiarelli D, Xia Q, Seligson M, Shinoda G, Xie W, Cahan P, et al. LIN28 Regulates Stem Cell Metabolism and Conversion to Primed Pluripotency. Cell Stem Cell. 2016; 19: 66-80.
43. Balzeau J, Menezes MR, Cao S, Hagan JP. The LIN28/let-7 Pathway in Cancer. Front Genet. 2017; 8: 31.
44. Shyh-Chang N, Daley GQ. Lin28: primal regulator of growth and metabolism in stem cells. Cell Stem Cell. 2013; 12: 395-406.
45. Hayashi S, Tanaka J, Okada S, Isobe T, Yamamoto G, Yasuhara R, Irie T, Akiyama C, Kohno Y, Tachikawa T, Mishima K. Lin28a is a putative factor in regulating cancer stem cell-like properties in side population cells of oral squamous cell carcinoma. Exp Cell Res. 2013; 319: 1220-8.
46. Li G, Ruan X, Auerbach RK, Sandhu KS, Zheng M, Wang P, Poh HM, Goh Y, Lim J, Zhang J, Sim HS, Peh SQ, Mulawadi FH, et al. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell. 2012; 148: 84-98.
47. Al-Samir S, Papadopoulos S, Scheibe RJ, Meissner JD, Cartron JP, Sly WS, Alper SL, Gros G, Endeward V. Activity and distribution of intracellular carbonic anhydrase II and their effects on the transport activity of anion exchanger AE1/SLC4A1. J Physiol. 2013; 591: 4963-82.
48. Leppilampi M, Koistinen P, Savolainen ER, Hannuksela J, Parkkila AK, Niemela O, Pastorekova S, Pastorek J, Waheed A, Sly WS, Parkkila S, Rajaniemi H. The expression of carbonic anhydrase II in hematological malignancies. Clin Cancer Res. 2002; 8: 2240-5.
49. Cong X, Yan M, Yin X, Zhang DE. Hematopoietic cells from Ube1L-deficient mice exhibit an impaired proliferation defect under the stress of bone marrow transplantation. Blood Cells Mol Dis. 2010; 45: 103-11.
50. Wang J, Ding S, Duan Z, Xie Q, Zhang T, Zhang X, Wang Y, Chen X, Zhuang H, Lu F. Role of p14ARF-HDM2-p53 axis in SOX6-mediated tumor suppression. Oncogene. 2016; 35: 1692-702.
51. Li H, Zheng D, Zhang B, Liu L, Ou J, Chen W, Xiong S, Gu Y, Yang J. Mir-208 promotes cell proliferation by repressing SOX6 expression in human esophageal squamous cell carcinoma. J Transl Med. 2014; 12: 196.
52. Iguchi H, Urashima Y, Inagaki Y, Ikeda Y, Okamura M, Tanaka T, Uchida A, Yamamoto TT, Kodama T, Sakai J. SOX6 suppresses cyclin D1 promoter activity by interacting with beta-catenin and histone deacetylase 1, and its down-regulation induces pancreatic beta-cell proliferation. J Biol Chem. 2007; 282: 19052-61.
53. Hamada-Kanazawa M, Ishikawa K, Nomoto K, Uozumi T, Kawai Y, Narahara M, Miyake M. Sox6 overexpression causes cellular aggregation and the neuronal differentiation of P19 embryonic carcinoma cells in the absence of retinoic acid. FEBS Lett. 2004; 560: 192-8.
54. Yi Z, Cohen-Barak O, Hagiwara N, Kingsley PD, Fuchs DA, Erickson DT, Epner EM, Palis J, Brilliant MH. Sox6 directly silences epsilon globin expression in definitive erythropoiesis. PLoS Genet. 2006; 2: e14.
55. Cosset E, Hamdan G, Jeanpierre S, Voeltzel T, Sagorny K, Hayette S, Mahon FX, Dumontet C, Puisieux A, Nicolini FE, Maguer-Satta V. Deregulation of TWIST-1 in the CD34+ compartment represents a novel prognostic factor in chronic myeloid leukemia. Blood. 2011; 117: 1673-6.
56. Wang N, Guo D, Zhao YY, Dong CY, Liu XY, Yang BX, Wang SW, Wang L, Liu QG, Ren Q, Lin YM, Ma XT. TWIST-1 promotes cell growth, drug resistance and progenitor clonogenic capacities in myeloid leukemia and is a novel poor prognostic factor in acute myeloid leukemia. Oncotarget. 2015; 6: 20977-92. doi: 10.18632/oncotarget.4007.
57. Xu J, Zhang W, Yan XJ, Lin XQ, Li W, Mi JQ, Li JM, Zhu J, Chen Z, Chen SJ. DNMT3A mutation leads to leukemic extramedullary infiltration mediated by TWIST1. J Hematol Oncol. 2016; 9: 106.
58. Zhou X, Marks PA, Rifkind RA, Richon VM. Cloning and characterization of a histone deacetylase, HDAC9. Proc Natl Acad Sci U S A. 2001; 98: 10572-7.
59. Siekmann TE, Gerber MM, Toland AE. Variants in an Hdac9 intronic enhancer plasmid impact Twist1 expression in vitro. Mammalian genome : official journal of the International Mammalian Genome Society. 2016; 27: 99-110.
60. Junier I, Dale RK, Hou C, Kepes F, Dean A. CTCF-mediated transcriptional regulation through cell type-specific chromosome organization in the beta-globin locus. Nucleic Acids Res. 2012; 40: 7718-27.
61. Valdes-Quezada C, Arriaga-Canon C, Fonseca-Guzman Y, Guerrero G, Recillas-Targa F. CTCF demarcates chicken embryonic alpha-globin gene autonomous silencing and contributes to adult stage-specific gene expression. Epigenetics. 2013; 8: 827-38.
62. Guo Y, Xu Q, Canzio D, Shou J, Li J, Gorkin DU, Jung I, Wu H, Zhai Y, Tang Y, Lu Y, Wu Y, Jia Z, et al. CRISPR Inversion of CTCF Sites Alters Genome Topology and Enhancer/Promoter Function. Cell. 2015; 162: 900-10.
63. Yu M, Riva L, Xie H, Schindler Y, Moran TB, Cheng Y, Yu D, Hardison R, Weiss MJ, Orkin SH, Bernstein BE, Fraenkel E, Cantor AB. Insights into GATA-1-mediated gene activation versus repression via genome-wide chromatin occupancy analysis. Mol Cell. 2009; 36: 682-95.
64. Mak VC, Wong OG, Siu MK, Wong ES, Ng WY, Wong RW, Chan KK, Ngan HY, Cheung AN. FBI-1 Is Overexpressed in Gestational Trophoblastic Disease and Promotes Tumor Growth and Cell Aggressiveness of Choriocarcinoma via PI3K/Akt Signaling. Am J Pathol. 2015; 185: 2038-48.
65. Luc S, Huang J, McEldoon JL, Somuncular E, Li D, Rhodes C, Mamoor S, Hou S, Xu J, Orkin SH. Bcl11a Deficiency Leads to Hematopoietic Stem Cell Defects with an Aging-like Phenotype. Cell Rep. 2016; 16: 3181-94.
66. Basak A, Sankaran VG. Regulation of the fetal hemoglobin silencing factor BCL11A. Ann N Y Acad Sci. 2016; 1368: 25-30.
67. Tong WG, Wierda WG, Lin E, Kuang SQ, Bekele BN, Estrov Z, Wei Y, Yang H, Keating MJ, Garcia-Manero G. Genome-wide DNA methylation profiling of chronic lymphocytic leukemia allows identification of epigenetically repressed molecular pathways with clinical impact. Epigenetics. 2010; 5: 499-508.
68. Zhao Y, Londono P, Cao Y, Sharpe EJ, Proenza C, O’Rourke R, Jones KL, Jeong MY, Walker LA, Buttrick PM, McKinsey TA, Song K. High-efficiency reprogramming of fibroblasts into cardiomyocytes requires suppression of pro-fibrotic signalling. Nat Commun. 2015; 6: 8243.
69. Varadaraj A, Patel P, Serrao A, Bandyopadhay T, Lee NY, Jazaeri AA, Huang Z, Murphy SK, Mythreye K. Epigenetic Regulation of GDF2 Suppresses Anoikis in Ovarian and Breast Epithelia. Neoplasia. 2015; 17: 826-38.
70. Liu M, Li Y, Chen L, Chan TH, Song Y, Fu L, Zeng TT, Dai YD, Zhu YH, Li Y, Chen J, Yuan YF, Guan XY. Allele-specific imbalance of oxidative stress-induced growth inhibitor 1 associates with progression of hepatocellular carcinoma. Gastroenterology. 2014; 146: 1084-96.
71. Tan JX, Wang XY, Li HY, Su XL, Wang L, Ran L, Zheng K, Ren GS. HYAL1 overexpression is correlated with the malignant behavior of human breast cancer. Int J Cancer. 2011; 128: 1303-15.
72. Wang F, Grigorieva EV, Li J, Senchenko VN, Pavlova TV, Anedchenko EA, Kudryavtseva AV, Tsimanis A, Angeloni D, Lerman MI, Kashuba VI, Klein G, Zabarovsky ER. HYAL1 and HYAL2 inhibit tumour growth in vivo but not in vitro. PLoS One. 2008; 3: e3031.
73. Vega FM, Ridley AJ. The RhoB small GTPase in physiology and disease. Small GTPases. 2016: 1-10.
74. Arsic N, Ho-Pun-Cheung A, Evelyne C, Assenat E, Jarlier M, Anguille C, Colard M, Pezet M, Roux P, Gadea G. The p53 isoform delta133p53ss regulates cancer cell apoptosis in a RhoB-dependent manner. PLoS One. 2017; 12: e0172125.
75. Li L, Dong X, Leong MC, Zhou W, Yang Z, Chen F, Bao Y, Jia W, Hu R. Identification of the novel protein FAM172A, and its up-regulation by high glucose in human aortic smooth muscle cells. Int J Mol Med. 2010; 26: 483-90.
76. Cui C, Ye L, Huang Z, Huang S, Liu H, Yu J. FAM172A is a tumor suppressor in colorectal carcinoma. Tumour Biol. 2016; 37: 6501-10.
77. Qian K, Zhang J, Lu J, Liu W, Yao X, Chen Q, Lu S, Xiang G, Liu H. FAM172A modulates apoptosis and proliferation of colon cancer cells via STAT1 binding to its promoter. Oncol Rep. 2016; 35: 1273-80.
78. Zi Z, Zhang Z, Li Q, An W, Zeng L, Gao D, Yang Y, Zhu X, Zeng R, Shum WW, Wu J. CCNYL1, but Not CCNY, Cooperates with CDK16 to Regulate Spermatogenesis in Mouse. PLoS Genet. 2015; 11: e1005485.
79. Emma MR, Iovanna JL, Bachvarov D, Puleio R, Loria GR, Augello G, Candido S, Libra M, Gulino A, Cancila V, McCubrey JA, Montalto G, Cervello M. NUPR1, a new target in liver cancer: implication in controlling cell growth, migration, invasion and sorafenib resistance. Cell Death Dis. 2016; 7: e2269.
80. Li J, Ren S, Liu Y, Lian Z, Dong B, Yao Y, Xu Y. Knockdown of NUPR1 inhibits the proliferation of glioblastoma cells via ERK1/2, p38 MAPK and caspase-3. J Neurooncol. 2017; 132: 15-26.
81. Cano CE, Hamidi T, Sandi MJ, Iovanna JL. Nupr1: the Swiss-knife of cancer. J Cell Physiol. 2011; 226: 1439-43.
82. Grasso D, Bintz J, Lomberk G, Molejon MI, Loncle C, Garcia MN, Lopez MB, Urrutia R, Iovanna JL. Pivotal Role of the Chromatin Protein Nupr1 in Kras-Induced Senescence and Transformation. Sci Rep. 2015; 5: 17549.
83. Aguado-Llera D, Hamidi T, Domenech R, Pantoja-Uceda D, Gironella M, Santoro J, Velazquez-Campoy A, Neira JL, Iovanna JL. Deciphering the binding between Nupr1 and MSL1 and their DNA-repairing activity. PLoS One. 2013; 8: e78101.
84. Fujii S, Luo RZ, Yuan J, Kadota M, Oshimura M, Dent SR, Kondo Y, Issa JP, Bast RC Jr, Yu Y. Reactivation of the silenced and imprinted alleles of ARHI is associated with increased histone H3 acetylation and decreased histone H3 lysine 9 methylation. Hum Mol Genet. 2003; 12: 1791-800.
85. Tesfaigzi J, Carlson DM. Expression, regulation, and function of the SPR family of proteins. A review. Cell Biochem Biophys. 1999; 30: 243-65.
86. Han K, Chen H, Gennarino VA, Richman R, Lu HC, Zoghbi HY. Fragile X-like behaviors and abnormal cortical dendritic spines in cytoplasmic FMR1-interacting protein 2-mutant mice. Hum Mol Genet. 2015; 24: 1813-23.
87. Diaz-Lagares A, Crujeiras AB, Lopez-Serra P, Soler M, Setien F, Goyal A, Sandoval J, Hashimoto Y, Martinez-Cardus A, Gomez A, Heyn H, Moutinho C, Espada J, et al. Epigenetic inactivation of the p53-induced long noncoding RNA TP53 target 1 in human cancer. Proc Natl Acad Sci U S A. 2016; 113: E7535-E44.
88. Litwinska Z, Machalinski B. miRNAs in chronic myeloid leukemia: small molecules, essential function. Leuk Lymphoma. 2017; 58: 1297-305.
89. Wang K, Xu Z, Wang N, Tian Y, Sun X, Ma Y. Analysis of microRNA and gene networks in human chronic myelogenous leukemia. Mol Med Rep. 2016; 13: 453-60.
90. Agatheeswaran S, Pattnayak NC, Chakraborty S. Identification and functional characterization of the miRNA-gene regulatory network in chronic myeloid leukemia lineage negative cells. Sci Rep. 2016; 6: 32493.
91. Matsui D, Zaidi AH, Martin SA, Omstead AN, Kosovec JE, Huleihel L, Saldin LT, DiCarlo C, Silverman JF, Hoppo T, Finley GG, Badylak SF, Kelly RJ, et al. Primary tumor microRNA signature predicts recurrence and survival in patients with locally advanced esophageal adenocarcinoma. Oncotarget. 2016; 7: 81281-91. doi: 10.18632/oncotarget.12832.
92. Saiselet M, Gacquer D, Spinette A, Craciun L, Decaussin-Petrucci M, Andry G, Detours V, Maenhaut C. New global analysis of the microRNA transcriptome of primary tumors and lymph node metastases of papillary thyroid cancer. BMC Genomics. 2015; 16: 828.
93. Babapoor S, Wu R, Kozubek J, Auidi D, Grant-Kels JM, Dadras SS. Identification of microRNAs associated with invasive and aggressive phenotype in cutaneous melanoma by next-generation sequencing. Lab Invest. 2017.
94. Girard A, Sachidanandam R, Hannon GJ, Carmell MA. A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature. 2006; 442: 199-202.
95. Aravin A, Gaidatzis D, Pfeffer S, Lagos-Quintana M, Landgraf P, Iovino N, Morris P, Brownstein MJ, Kuramochi-Miyagawa S, Nakano T, Chien M, Russo JJ, Ju J, et al. A novel class of small RNAs bind to MILI protein in mouse testes. Nature. 2006; 442: 203-7.
96. Aravin AA, Sachidanandam R, Girard A, Fejes-Toth K, Hannon GJ. Developmentally regulated piRNA clusters implicate MILI in transposon control. Science. 2007; 316: 744-7.
97. Hamada-Kanazawa M, Ishikawa K, Ogawa D, Kanai M, Kawai Y, Narahara M, Miyake M. Suppression of Sox6 in P19 cells leads to failure of neuronal differentiation by retinoic acid and induces retinoic acid-dependent apoptosis. FEBS Lett. 2004; 577: 60-6.
98. Cantu C, Grande V, Alborelli I, Cassinelli L, Cantu I, Colzani MT, Ierardi R, Ronzoni L, Cappellini MD, Ferrari G, Ottolenghi S, Ronchi A. A highly conserved SOX6 double binding site mediates SOX6 gene downregulation in erythroid cells. Nucleic Acids Res. 2011; 39: 486-501.
99. Cantu C, Ierardi R, Alborelli I, Fugazza C, Cassinelli L, Piconese S, Bose F, Ottolenghi S, Ferrari G, Ronchi A. Sox6 enhances erythroid differentiation in human erythroid progenitors. Blood. 2011; 117: 3669-79.
100. Li N, Zhong X, Lin X, Guo J, Zou L, Tanyi JL, Shao Z, Liang S, Wang LP, Hwang WT, Katsaros D, Montone K, Zhao X, et al. Lin-28 homologue A (LIN28A) promotes cell cycle progression via regulation of cyclin-dependent kinase 2 (CDK2), cyclin D1 (CCND1), and cell division cycle 25 homolog A (CDC25A) expression in cancer. J Biol Chem. 2012; 287: 17386-97.
101. Piskounova E, Polytarchou C, Thornton JE, LaPierre RJ, Pothoulakis C, Hagan JP, Iliopoulos D, Gregory RI. Lin28A and Lin28B inhibit let-7 microRNA biogenesis by distinct mechanisms. Cell. 2011; 147: 1066-79.
102. Shyh-Chang N, Zhu H, Yvanka de Soysa T, Shinoda G, Seligson MT, Tsanov KM, Nguyen L, Asara JM, Cantley LC, Daley GQ. Lin28 enhances tissue repair by reprogramming cellular metabolism. Cell. 2013; 155: 778-92.
103. Zeng Y, Yao B, Shin J, Lin L, Kim N, Song Q, Liu S, Su Y, Guo JU, Huang L, Wan J, Wu H, Qian J, et al. Lin28A Binds Active Promoters and Recruits Tet1 to Regulate Gene Expression. Mol Cell. 2016; 61: 153-60.
104. Feng Q, Sekula D, Guo Y, Liu X, Black CC, Galimberti F, Shah SJ, Sempere LF, Memoli V, Andersen JB, Hassel BA, Dragnev K, Dmitrovsky E. UBE1L causes lung cancer growth suppression by targeting cyclin D1. Mol Cancer Ther. 2008; 7: 3780-8.
105. Zhou MJ, Chen FZ, Chen HC, Wan XX, Zhou X, Fang Q, Zhang DZ. ISG15 inhibits cancer cell growth and promotes apoptosis. Int J Mol Med. 2017; 39: 446-52.
106. Parra M. Class IIa HDACs - new insights into their functions in physiology and pathology. FEBS J. 2015; 282: 1736-44.
107. Gu Z, Churchman M, Roberts K, Li Y, Liu Y, Harvey RC, McCastlain K, Reshmi SC, Payne-Turner D, Iacobucci I, Shao Y, Chen IM, Valentine M, et al. Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat Commun. 2016; 7: 13331.
108. Rastogi B, Raut SK, Panda NK, Rattan V, Radotra BD, Khullar M. Overexpression of HDAC9 promotes oral squamous cell carcinoma growth, regulates cell cycle progression, and inhibits apoptosis. Mol Cell Biochem. 2016; 415: 183-96.
109. Lapierre M, Linares A, Dalvai M, Duraffourd C, Bonnet S, Boulahtouf A, Rodriguez C, Jalaguier S, Assou S, Orsetti B, Balaguer P, Maudelonde T, Blache P, et al. Histone deacetylase 9 regulates breast cancer cell proliferation and the response to histone deacetylase inhibitors. Oncotarget. 2016; 7: 19693-708. doi: 10.18632/oncotarget.7564.
110. Gil VS, Bhagat G, Howell L, Zhang J, Kim CH, Stengel S, Vega F, Zelent A, Petrie K. Deregulated expression of HDAC9 in B cells promotes development of lymphoproliferative disease and lymphoma in mice. Dis Model Mech. 2016; 9: 1483-95.
111. Micheli L, D’Andrea G, Leonardi L, Tirone F. HDAC1, HDAC4, and HDAC9 Bind to PC3/Tis21/Btg2 and Are Required for Its Inhibition of Cell Cycle Progression and Cyclin D1 Expression. J Cell Physiol. 2017; 232: 1696-707.
112. Manda-Mapalo MT, Khalili P, Quintana D, Rabinowitz I, Zhang QY. Chronic myelogenous leukemia with acquired t(11;14)(q13;q32) CCND1-IGH: A case report and literature review. Cancer Genet. 2016; 209: 481-5.
113. Junk DJ, Cipriano R, Stampfer M, Jackson MW. Constitutive CCND1/CDK2 activity substitutes for p53 loss, or MYC or oncogenic RAS expression in the transformation of human mammary epithelial cells. PLoS One. 2013; 8: e53776.
114. Rowlinson JM, Gering M. Hey2 acts upstream of Notch in hematopoietic stem cell specification in zebrafish embryos. Blood. 2010; 116: 2046-56.
115. Gessler M, Knobeloch KP, Helisch A, Amann K, Schumacher N, Rohde E, Fischer A, Leimeister C. Mouse gridlock: no aortic coarctation or deficiency, but fatal cardiac defects in Hey2 -/- mice. Curr Biol. 2002; 12: 1601-4.
116. Thompson PJ, Norton KA, Niri FH, Dawe CE, McDermid HE. CECR2 is involved in spermatogenesis and forms a complex with SNF2H in the testis. J Mol Biol. 2012; 415: 793-806.
117. Alberti L, Losi L, Leyvraz S, Benhattar J. Different Effects of BORIS/CTCFL on Stemness Gene Expression, Sphere Formation and Cell Survival in Epithelial Cancer Stem Cells. PLoS One. 2015; 10: e0132977.
118. Alberti L, Renaud S, Losi L, Leyvraz S, Benhattar J. High expression of hTERT and stemness genes in BORIS/CTCFL positive cells isolated from embryonic cancer cells. PLoS One. 2014; 9: e109921.
119. Rasko JE, Wong JJ. Nuclear microRNAs in normal hemopoiesis and cancer. J Hematol Oncol. 2017; 10: 8.
120. Phan B, Majid S, Ursu S, de Semir D, Nosrati M, Bezrookove V, Kashani-Sabet M, Dar AA. Tumor suppressor role of microRNA-1296 in triple-negative breast cancer. Oncotarget. 2016; 7: 19519-30. doi: 10.18632/oncotarget.6961.
121. Shan X, Wen W, Zhu D, Yan T, Cheng W, Huang Z, Zhang L, Zhang H, Wang T, Zhu W, Zhu Y, Zhu J. miR 1296-5p Inhibits the Migration and Invasion of Gastric Cancer Cells by Repressing ERBB2 Expression. PLoS One. 2017; 12: e0170298.
122. Majid S, Dar AA, Saini S, Chen Y, Shahryari V, Liu J, Zaman MS, Hirata H, Yamamura S, Ueno K, Tanaka Y, Dahiya R. Regulation of minichromosome maintenance gene family by microRNA-1296 and genistein in prostate cancer. Cancer Res. 2010; 70: 2809-18.
123. Wang W, Li R, Meng M, Wei C, Xie Y, Zhang Y, Jiang L, Dong R, Wang C, Zhong Y, Yang F, Tang W, Jin X, et al. MicroRNA profiling of CD3+ CD56+ cytokine-induced killer cells. Sci Rep. 2015; 5: 9571.
124. Murria R, Palanca S, de Juan I, Alenda C, Egoavil C, Segui FJ, Garcia-Casado Z, Juan MJ, Sanchez AB, Segura A, Santaballa A, Chirivella I, Llop M, et al. Immunohistochemical, genetic and epigenetic profiles of hereditary and triple negative breast cancers. Relevance in personalized medicine. Am J Cancer Res. 2015; 5: 2330-43.
125. Zhao H, Gao A, Zhang Z, Tian R, Luo A, Li M, Zhao D, Fu L, Fu L, Dong JT, Zhu Z. Genetic analysis and preliminary function study of miR-423 in breast cancer. Tumour Biol. 2015; 36: 4763-71.
126. Li HT, Zhang H, Chen Y, Liu XF, Qian J. MiR-423-3p enhances cell growth through inhibition of p21Cip1/Waf1 in colorectal cancer. Cell Physiol Biochem. 2015; 37: 1044-54.
127. Blume CJ, Hotz-Wagenblatt A, Hullein J, Sellner L, Jethwa A, Stolz T, Slabicki M, Lee K, Sharathchandra A, Benner A, Dietrich S, Oakes CC, Dreger P, et al. p53-dependent non-coding RNA networks in chronic lymphocytic leukemia. Leukemia. 2015; 29: 2015-23.
128. Giles KM, Brown RA, Ganda C, Podgorny MJ, Candy PA, Wintle LC, Richardson KL, Kalinowski FC, Stuart LM, Epis MR, Haass NK, Herlyn M, Leedman PJ. microRNA-7-5p inhibits melanoma cell proliferation and metastasis by suppressing RelA/NF-kappaB. Oncotarget. 2016; 7: 31663-80. doi: 10.18632/oncotarget.9421.
129. Shi Y, Luo X, Li P, Tan J, Wang X, Xiang T, Ren G. miR-7-5p suppresses cell proliferation and induces apoptosis of breast cancer cells mainly by targeting REGgamma. Cancer Lett. 2015; 358: 27-36.
130. Lou K, Chen N, Li Z, Zhang B, Wang X, Chen Y, Xu H, Wang D, Wang H. MicroRNA-142-5p Overexpression Inhibits Cell Growth and Induces Apoptosis by Regulating FOXO in Hepatocellular Carcinoma Cells. Oncol Res. 2017; 25: 65-73.
131. Hu T, Phiwpan K, Guo J, Zhang W, Guo J, Zhang Z, Zou M, Zhang X, Zhang J, Zhou X. MicroRNA-142-3p Negatively Regulates Canonical Wnt Signaling Pathway. PLoS One. 2016; 11: e0158432.
132. Su S, Zhao Q, He C, Huang D, Liu J, Chen F, Chen J, Liao JY, Cui X, Zeng Y, Yao H, Su F, Liu Q, et al. miR-142-5p and miR-130a-3p are regulated by IL-4 and IL-13 and control profibrogenic macrophage program. Nat Commun. 2015; 6: 8523.
133. Wee S, Wiederschain D, Maira SM, Loo A, Miller C, deBeaumont R, Stegmeier F, Yao YM, Lengauer C. PTEN-deficient cancers depend on PIK3CB. Proc Natl Acad Sci U S A. 2008; 105: 13057-62.
134. Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012; 7: 562-78.
135. Friedlander MR, Chen W, Adamidi C, Maaskola J, Einspanier R, Knespel S, Rajewsky N. Discovering microRNAs from deep sequencing data using miRDeep. Nat Biotechnol. 2008; 26: 407-15.
136. Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T, Yamanishi Y. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008; 36: D480-4.
137. Wang K, Liang C, Liu J, Xiao H, Huang S, Xu J, Li F. Prediction of piRNAs using transposon interaction and a support vector machine. BMC Bioinformatics. 2014; 15: 419.
138. Jagla B, Aulner N, Kelly PD, Song D, Volchuk A, Zatorski A, Shum D, Mayer T, De Angelis DA, Ouerfelli O, Rutishauser U, Rothman JE. Sequence characteristics of functional siRNAs. RNA. 2005; 11: 864-72.
139. Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009; 10: R25.
140. Nawrocki EP, Eddy SR. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013; 29: 2933-5.
141. t Hoen PA, Ariyurek Y, Thygesen HH, Vreugdenhil E, Vossen RH, de Menezes RX, Boer JM, van Ommen GJ, den Dunnen JT. Deep sequencing-based expression analysis shows major advances in robustness, resolution and inter-lab portability over five microarray platforms. Nucleic Acids Res. 2008; 36: e141.
142. Kruger J, Rehmsmeier M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res. 2006; 34: W451-4.
143. John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human MicroRNA targets. PLoS Biol. 2004; 2: e363.
144. Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015; 4.
145. Wang L, Feng Z, Wang X, Wang X, Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010; 26: 136-8.
146. Delgado MD, Leon J. Myc roles in hematopoiesis and leukemia. Genes Cancer. 2010; 1: 605-16. doi: 10.1177/1947601910377495.