
INTRODUCTION
Lung cancer is the deadliest form of cancer for men and women of all races in the United States, causing more deaths than colorectal, breast, and prostate cancer combined [1, 2]. Lung cancer is usually aggressive and characterized by early progression and metastases, especially in patients diagnosed with non-small cell lung cancer (NSCLC). With chemotherapy providing only modest efficacy and associated with severe dose-limiting toxicities, new molecular targets are urgently needed to identify drug development candidates with potential for greater therapeutic efficacy and reduced toxicity [3-8].
Previous studies have shown that cyclic guanosine monophosphate (cGMP) signaling plays an important role in regulating tumor cell proliferation and survival [9-18]. Both particulate guanylyl cyclases (pGCs) and soluble guanylyl cyclases (sGCs) catalyze the biosynthesis of cGMP from GTP, while cGMP phosphodiesterases (PDEs) function to hydrolyze cGMP and terminate cGMP signaling [19, 20]. Activators of GCs or inhibitors of cGMP PDEs can increase intracellular cGMP levels to enhance the magnitude and/or duration of cGMP signaling to inhibit proliferation and induce apoptosis. Intracellular levels of cGMP are known to be aberrantly low in cancer cells resulting from the overexpression of one or more PDE isozymes, which may play an important role in tumorigenesis [9, 12, 17, 18]. The elevation of intracellular cGMP levels by cGMP PDE inhibitors can activate cGMP-dependent protein kinases (PKG), although the downstream pathways responsible for suppressing proliferation and inducing apoptosis of tumor cells are not well understood. Physiologically, cGMP signaling can be activated in the lung by natriuretic peptides binding to membrane receptors that can stimulate particulate GC activity, while nitric oxide activates soluble GC activity [19-22]. Although several cGMP degrading PDE isozymes have been reported in normal lung and lung tumors, including the cGMP-specific PDE isozyme, PDE5 [10, 18, 22, 23], the critical cGMP PDE isozymes that are induced during lung cancer progression and which are essential for tumor cell proliferation and survival have not been well studied, but may provide useful targets for the development of new anticancer drugs.
The human genome contains 21 PDE gene products grouped into 11 isozyme families having different substrate specificity, regulatory properties, tissue localization, and sensitivity to inhibitors [19, 20]. Compared with other PDE isozymes, the tissue distribution of PDE10 is limited with high expression in the brain striatum but low expression in most other normal tissues [20, 24-26]. Because PDE10 has been shown to play an important physiological role in the brain to affect motor function and cognition, PDE10 inhibitors have been developed and tested in clinical trials for the treatment of Huntington’s diseases and schizophrenia [27-29]. While there is no known physiological role of PDE10 in tissues outside the central nervous system, PDE10 may play an important role in lung disease as suggested by previous reports showing elevated PDE10 levels during pulmonary arterial hypertension [30].
We recently reported that PDE10 is elevated in colon tumor cell lines compared with primary cultures of cells derived from normal colonic mucosa [31]. PDE10 levels were also high in intestinal adenomas from ApcMin/+ mice, as well as human colon adenocarcinomas and metastatic lesions relative to normal intestinal mucosa [31, 32]. Consistent with the differential expression of PDE10 in normal and cancer cells, PDE10 inhibitors were found to selectively inhibit proliferation and induce apoptosis of colon tumor cells, while not affecting growth of normal colonocytes. PDE10 inhibition can increase intracellular levels of cGMP and cAMP to activate PKG and PKA signaling, respectively, although the mechanism responsible for its tumor cell growth inhibitory activity may be exclusively associated with cGMP/PKG signaling, given that cAMP PDE specific inhibitors (e.g. rolipram) or PKA activators (e.g. forskolin) do not inhibit colon tumor cell growth [12, 31-33]. Moreover, other studies reported that PKG inhibitors, but not PKA inhibitors, can attenuate the growth inhibitory activity of PDE10 inhibitors [32].
Here we show that PDE10 is overexpressed in NSCLC cell lines and human lung tumors compared with normal lung epithelial cells and tissues, respectively. Genetic silencing of PDE10 or inhibition by small molecules such as PQ10 selectively inhibits lung tumor cell growth and colony formation by activating cGMP/PKG signaling. We also show that PQ10 can phosphorylate β-catenin to induce degradation, leading to reduced levels of cyclin D1 and survivin and the induction of enzymes required for apoptosis. PQ10 can also suppress RAS-activated RAF/MAPK signaling within the same concentration range and kinetics as required to activate cGMP/PKG signaling.
RESULTS
PDE10 expression in human lung tumor and normal cells
The expression level of PDE10 and other cGMP degrading PDE isozymes was determined in a panel of human NSCLC cell lines, including H460, H1299, HOP62, A549 and H1975, and compared with primary cultures of normal human airway epithelial cells (NHAECs). Western blotting using PDE isozyme specific antibodies demonstrated that PDE10 was abundantly expressed in all five NSCLC cell lines, but undetectable in NHAECs (Figure 1A). Other cGMP degrading PDE isozymes, including PDE1A, 2A, 3A, 3B, 5A, 9A, and 11A were either not expressed in all of the NSCLC cell lines or did not show an appreciable difference in protein expression levels compared with NHAECs. To determine if higher PDE10 protein levels are attributed to increased transcriptional activity, levels of mRNA for the cGMP PDE isozymes expressed in HOP62 cells were measured by qRT-PCR using isozyme-specific primers and compared with mRNA levels of these same isozymes in NHAECs. As shown in Figure 1B, a large difference in PDE10 mRNA levels was detected between HOP62 lung tumor cells and NHAECs, while mRNA levels of other PDE isozymes, including PDE1, 2, 5, and 9 showed relatively minor differences.
PDE10 mRNA levels were also measured in cDNA arrays obtained from human lung tumor specimens by qRT-PCR and compared with levels in normal lung tissues. Consistent with results from cell lines, PDE10 mRNA levels were elevated in lung tumors (n = 19) compared with normal lung tissues (n = 4) as shown in Figure 1C. Increased PDE10 levels were confirmed by immunofluorescence microscopy in which labeling was readily apparent in human lung adenocarcinomas as shown in Figure 1D, while uninvolved lung tissue showed appreciably less labeling. Consistent with previous studies of the subcellular distribution of PDE10 in colon tumor cells [32], PDE10 was enriched primarily in the cytoplasm, although membrane labeling was also apparent.
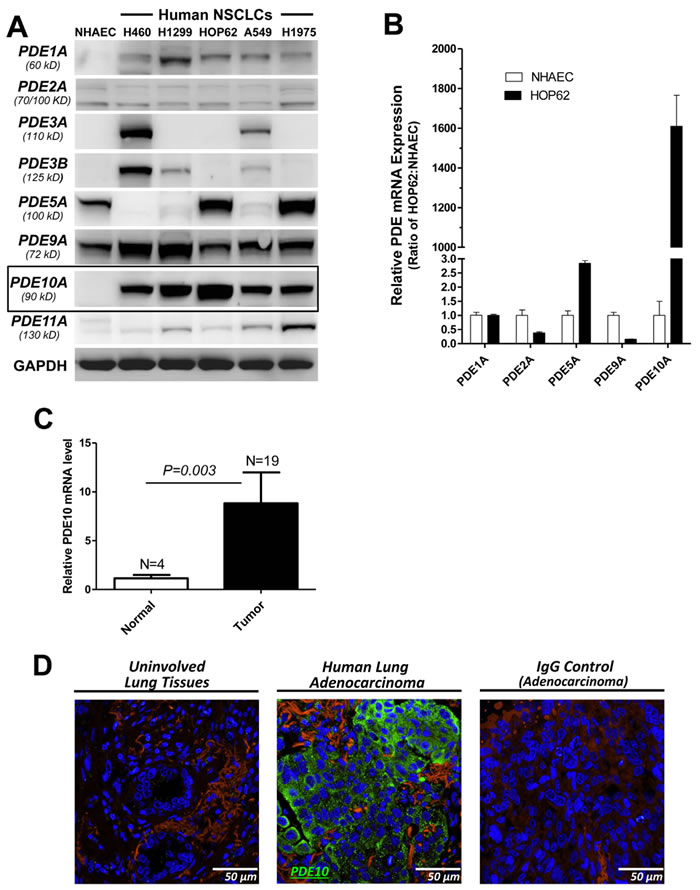
Figure 1: PDE10 overexpression in human lung tumor cells. A. Protein levels of eight major cGMP-degrading PDE isoforms, 1A, 2A, 3A, 3B, 5A, 9A, 10A and 11A, were measured in five NSCLC cell lines (H460, H1299, HOP62, A549 and H1975) and NHAECs by Western blotting. The frame highlights a protein band at the expected 90 kD molecular weight for PDE10 and increased levels in NSCLCs as compared with NHAECs. GAPDH was used as the loading and blotting control for the comparison of different PDE isoforms among these cell lines. Each isoform of PDE was detected by isoform-specific antibodies and the molecular weight of protein was indicated. B. The ratio of mRNA levels of PDE10 and other cGMP PDEs are compared between NHAEC and NSCLC (HOP62) cells by quantitative real time RT-PCR (qPCR). The relative amount of PDE mRNAs were normalized by PDE1A for the comparison of different PDE isoforms after measured using TATA-box binding protein (TBP) as the qPCR control between two cell lines. C. qPCR of patient samples show increased PDE10 mRNA levels in lung cancers (n = 19) vs. normal lung tissue (n = 4). Data are represented as mean ± SEM, P = 0.003 by F test. D. Specimens of primary human lung adenocarcinoma (middle) shows positive staining of PDE10 (green, Alexa 488) but negative staining in uninvolved normal lung tissues (left) with the autofluorescence (orange) and nuclear staining (DAPI).
PDE10 knockdown suppresses lung tumor cell growth and colony formation
To study a potential functional role of PDE10 in lung tumor cell proliferation or survival, PDE10 protein levels were suppressed by transient transfection of HOP62 lung tumor cells with PDE10-specific siRNA. A reduction of PDE10 protein levels as determined by Western blotting coincided with an approximate 50% reduction of viable cell number following three days of transfection in comparison to parental HOP62 cells or HOP62 cells transfected with scrambled siRNA (Figure 2A and inset). Stable knockdown of PDE10 using a specific shRNA resulted in a 70% reduction of viable cell number relative to control cells (Figure 2B). Western blotting showed a greater reduction of PDE10 protein levels in the stable knockdown cells by shRNA (inset, Figure 2B) as compared with transient transfection by siRNA. Stable knockdown by PDE10 shRNA also significantly reduced colony formation of HOP62 cells in which a greater than 60% reduction of colony numbers was observed in PDE10 knockdown HOP62 cells compared with parental and shRNA vector control HOP62 cells (Figure 2C).
PDE10 inhibitors suppress lung tumor cell growth and colony formation
To further study the role of PDE10 in lung cancer cells, we synthesized a previously reported PDE10 inhibitor referred to as PQ10 [29, 34] with a structure as shown in Figure 2D and evaluated its ability to inhibit lung tumor cell growth. Initial studies were performed to determine the PDE10 isozyme selectivity of PQ10 using recombinant PDE isozymes. PQ10 inhibited PDE10 with IC50 values for cGMP and cAMP of 0.005 and 0.013 µmol/L, respectively, as shown in Figure 2D. Appreciably higher concentrations in the micromolar range were required to inhibit other PDE isozymes, demonstrating a high degree of selectivity for PDE10 (Table 1). PQ10 inhibited the growth of human lung tumor cells with IC50 values ranging from 0.32 -1.69 µmol/L, resulting in a maximum reduction of viable cell number greater than 75% following 3 days of treatment (Figure 2E). By comparison, NHAECs having reduced levels of PDE10 were as much as 50 fold less sensitive to PQ10 treatment, resulting in an IC50 value of 17.3 µmol/L in growth assays conducted under identical conditions as lung tumor cell lines (Figure 2E). PQ10 also caused a concentration-dependent inhibition of colony formation in HOP62 cells with an IC50 value of 0.098 µmol/L following 14 days of treatment (Figure 2F).
Table 1: PDE isozyme selectivity of PQ10
PDE Family |
Substrate |
IC50 of PQ10 |
|
cGMP (µmol/L) |
cAMP (µmol/L) |
||
1A |
cGMP/cAMP |
8.8 |
29.2 |
2A |
cGMP/cAMP |
3.7 |
7.0 |
3A |
cGMP/cAMP |
0.32 |
0.47 |
4B |
cAMP |
ND |
17.8 |
5A |
cGMP |
1.8 |
ND |
6A/B |
cGMP |
Inactive |
ND |
7A |
cAMP |
ND |
Inactive |
8A |
cAMP |
ND |
Inactive |
9A |
cGMP |
Inactive |
ND |
10A |
cGMP/cAMP |
0.005 |
0.013 |
11A |
cGMP/cAMP |
13.2 |
30.1 |
ND: not determined; Inactive: >100 µmol/L shows no inhibition.
To confirm that PDE10 inhibition is responsible for the tumor cell growth inhibitory activity of PQ10, HOP62 cells stably transfected with PDE10-shRNA were used to measure their sensitivity to PQ10 and compared with HOP62 cells transfected with vector alone. As shown in Figure 2B, PQ10 effectively inhibited the growth of vector control cells, while PDE10 knockdown cells were appreciably less sensitive to treatment. Similar differences were found in PQ10 treated HOP62 cells with PDE10-shRNA in colony formation assays (Figure 2F). For example, PQ10 treatment on HOP62 vector control cells at a concentration of 0.1 µmol/L reduced colony formation by 54.4% compared with control cells, while treatment of PDE10 knockdown cells with PQ10 resulted in only a 12.3% reduction of colony formation relative to control cells.
Similar growth inhibitory activity was measured with two other known PDE10 inhibitors, Pf2545920 and papaverine, as shown in Figure 2G and Figure 2H, respectively. Pf2545920 effectively inhibited the growth of a panel of nine human lung tumor cell lines derived from the NCI-60 panel. Papaverine also inhibited growth of NSCLC cells with IC50 values comparable with IC50 values for inhibition of total cGMP PDE enzymatic activity in whole cell lysates.
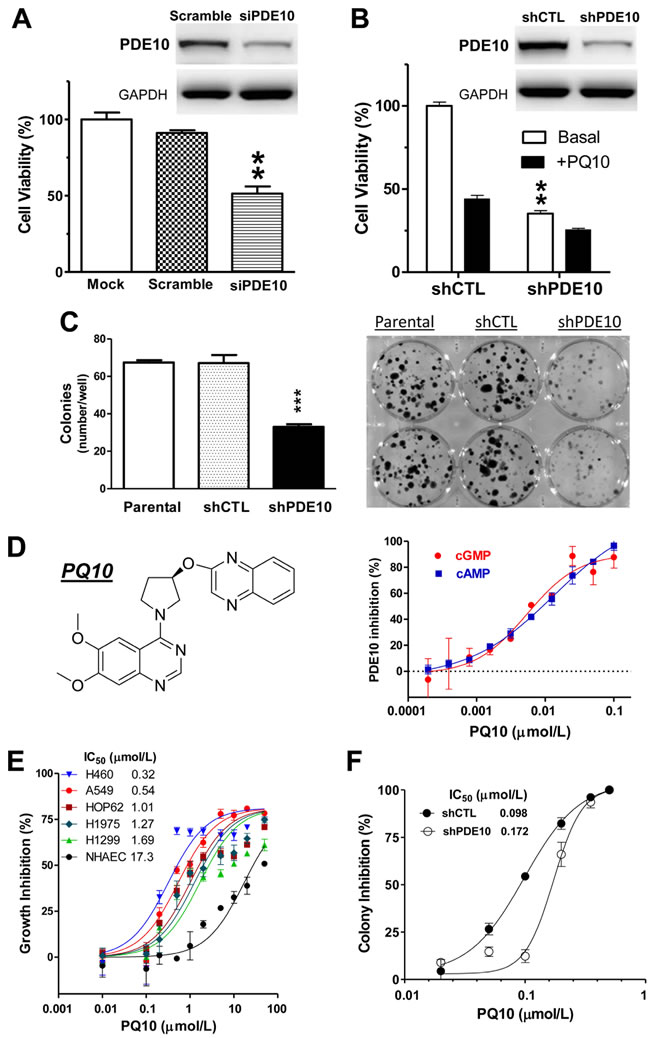
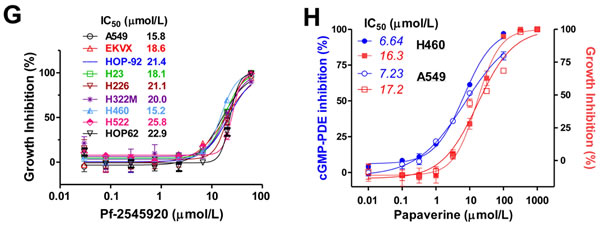
Figure 2: PDE10 inhibition suppresses lung tumor cell growth and colony formation. A. Genetic silencing PDE10 expression by transient knockdown with PDE10-specific siRNA (siPDE10) selectively inhibited NSCLC HOP62 cell growth as compared with parental cells (mock transfection) or HOP62 cells transfected with control siRNA (scramble). B. Stable knockdown of PDE10 by shRNA (shPDE10) in HOP62 cells also inhibited growth and attenuated the response to the PDE10 selective inhibitor, PQ10 (2 µmol/L), as compared with shRNA vector controls (shCTL). Insets of A/B, Western blotting confirmed that PDE10 protein levels in HOP62 cells were reduced after siRNA or shRNA treatment as compared with scrambled or shCTL control. C. Knockdown PDE10 by shRNA inhibited colony formation in HOP62 NSCLC cells. Stable PDE10 knockdown and parental/shCTL controls of HOP62 cells were grown in culture medium for 14 days followed by staining with crystal violet. Data analysis of colony numbers is shown in the left panel, while representative images are shown in the right panels. D. Chemical structure of PQ10 and inhibition of PDE10 by PQ10. E. Concentration-dependent growth inhibitory activities and IC50 values of PQ10 in NSCLCs vs. NHAECs. F. PQ10 inhibited colony formation in HOP62 cells, while PDE10 knockdown cells displayed reduced sensitivity. Results of A~F represent one of three repeated experiments. Data are represented as mean ± SEM, ** P<0.01, *** P<0.001 vs. parental, scramble siRNA or shCTL controls. G. NSCLC lung cancer cell panels showed growth inhibition and IC50 values after a treatment with another PDE10 selective inhibitor, Pf-2545920. H. Papaverine inhibited cell growth and cGMP-PDE activities in cell lysates from H460 and A549 NSCLC cells.
PQ10 increases intracellular cGMP levels at concentrations that inhibit tumor cell growth
To study the effects of PQ10 on cGMP signaling, intracellular cGMP levels were measured in lung tumor cells using a luciferase-based biosensor assay that allowed for real-time measurements of cGMP levels in live cells following treatment [35]. The luciferase construct was transfected into human H1299 lung tumor cells treated with PQ10. As shown in Figure 3A-3D, PQ10 caused a rapid increase in cGMP levels within 1 hour of treatment that was sustained for the duration of the experiment at 2 hours. The EC50 values of PQ10 to induce cGMP elevation were 1.31 µmol/L and 1.72 µmol/L for CNP and SNP (Figure 3B and Figure 3D), respectively, which coincided with the IC50 value of 1.69 µmol/L for PQ10 to inhibit the growth of H1299 lung tumor cells (refer to Figure 2E). PQ10 also increased cGMP levels in HEK293 cells transfected with the cGMP biosensor in combination with SNP (Figure 3E and Figure 3F).
The PDE5 specific inhibitor, sildenafil, was tested under the same conditions in the biosensor assay and provided a useful negative control having no effect on the growth of H1299 cells (not shown) that do not express PDE5 (refer to Figure 1A). Sildenafil at a concentration of 300-fold higher than its IC50 for PDE5 inhibition did not affect luminescence values as determined by the biosensor assay (Figure 3A). In addition, the non-selective PDE inhibitor, IBMX, increased luminescence levels in H1299 cells (Figure 3A), which provided additional evidence that the activity of PQ10 as measured in the biosensor assay was associated with a rise in intracellular cGMP levels.
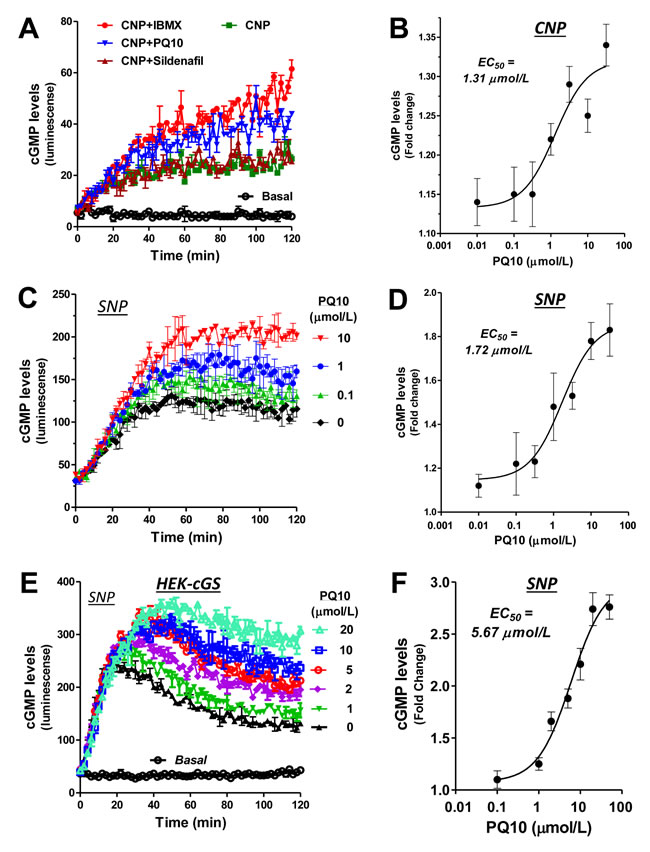
Figure 3: PDE10 inhibition elevates intracellular cGMP levels in NSCLCs as determined by a biosensor assay. A. Measurement of intracellular cGMP levels in live H1299 NSCLC cells with a stable transfection of cGMP biosensor (cGS) under a stimulation of particulate GC activator, CNP (100 nmol/L), in combined with PQ10 (10 µmol/L) and sildenafil (1 µmol/L, PDE5 inhibitor). PDE isozymes non-selective inhibitor IBMX (100 µmol/L) was used as positive control. B. EC50 calculation of PQ10 after CNP stimulation. C. and D. A similar dose-dependent manner and EC50 value of PQ10 was present in H1299 biosensor cells under a stimulation of soluble GC activator, sodium nitroprusside (SNP, 100 µmol/L). E. and F. PQ10 induced a similar manner of increases in intracellular cGMP levels in HEK293 biosensor cells under SNP (100 µmol/L) but the EC50 value was about 3.3-fold higher than H1299 cells transfected with the cGMP biosensor. Cells were cultured in 96 well plates to 80-90% confluence and were used for the biosensor assay which involved continuous measurements of luminescence values every two minutes for 2 hours. All cells used for these studies were cultured under similar conditions to obtain IC50 and EC50 values of PDE activities measurements in cell lysates, growth inhibition assay and biosensor analysis.
PQ10 activates PKG at concentrations that inhibit tumor cell growth
As previously reported, PKG activation occurs in response to an elevation of intracellular cGMP levels and appears to be the major signaling pathway responsible for the tumor cell growth inhibitory activity of cGMP PDE inhibitors [9, 14, 32, 33]. Experiments were therefore conducted to determine if PQ10 can activate PKG by Western blotting using a phospho-specific antibody against phosphorylated serine239 of vasodilator-stimulated phosphoprotein (pS239-VASP), a known cGMP-activated PKG specific site of phosphorylation and known biomarker of PKG activation [36]. As shown in Figure 4A, PQ10 treatment of H1299 lung tumor cells caused a rapid and concentration-dependent increase in pSer239-VASP levels without affecting total VASP protein levels. The effect occurred within 30 minutes of treatment, which paralleled increased levels of intracellular cGMP as measured by the biosensor assay (refer to Figure 3A-3D). The effective concentration range was also similar to EC50 and IC50 values required for increasing cGMP levels and inhibiting H1299 tumor cell growth, respectively. Given that PDE10 can also hydrolyze cAMP, we also determined if PQ10 can simultaneously activate PKA in H1299 cells using an antibody against pSer157-VASP, a known cAMP-activated PKA specific site of VASP phosphorylation. In addition, we used antibodies directed against phosphorylation sites on cAMP response element-binding protein (pSer133-CREB) and the microtubule-associated protein, Tau (pSer214-Tau) that are known to be phosphorylated by either PKA or PKG [37-39]. Consistent with the capacity of PDE10 to hydrolyze cAMP and cGMP, Western blotting showed that PQ10 increased levels of pSer157-VASP and pSer133-CREB in H1299 cells within 30 minutes of treatment, but longer treatment times of 1~4 hours were required to increase pSer214-Tau levels. These experiments show that PQ10 can activate both PKG and PKA signaling in lung tumor cells and are consistent with effects that are expected from a PDE10 inhibitor.
PQ10 induces the phosphorylation and degradation of β-catenin
Previous studies have reported that PKG can phosphorylate β-catenin to suppress T-cell factor (TCF) transcriptional activity by inducing ubiquitin-mediated degradation [9, 31-33, 40, 41]. To determine if PQ10 can suppress oncogenic β-catenin signaling, we measured the phosphorylation of β-catenin in H1299 lung tumor cells in response to PQ10 treatment. As shown in Figure 4B, PQ10 increased the phosphorylation of β-catenin at pSer552 in a concentration-dependent manner at levels that were effective for activating PKG. However, there was no change in the phosphorylation level of β-catenin at pSer675, which is a known site of PKA-conserved phosphorylation [42]. Other experiments in which H1299 cells were pretreated with the PKG inhibitor, KT5823, or the PKA inhibitor, H89, showed partial inhibition of PQ10-induced phosphorylation β-catenin at pSer552 (Figure 4C), although the effective concentration of KT5823 was 10-fold lower than H89. Along with the inability of PQ10 to increase the phosphorylation of β-catenin at pSer675, these results suggest that PKG activation may be the predominant signaling pathway to phosphorylate β-catenin in response to PQ10 treatment and to inhibit tumor cell growth, which is supported by previous reports that cAMP elevation and PKA activation do not appear to be essential for the growth inhibitory activity of PDE10 inhibitors [31-33].
To determine if PDE10 inhibition can induce β-catenin degradation as previously reported for other cGMP PDE inhibitors [9, 13, 14, 33, 35], H1299 lung tumor cells were treated with PQ10 and total levels of β-catenin were measured by Western blotting as shown in Figure 4B. While β-catenin was rapidly phosphorylated within 30 minutes of treatment, total β-catenin levels began to decrease after 4 hours of treatment. Coinciding with the suppression of β-catenin levels, PQ10 treatment reduced the levels of cyclin D1 and survivin (Figure 4B), which are known regulated by β-catenin/Tcf transcriptional activity [31-33].
To determine if the suppression of cyclin D1 and survivin levels by PQ10 can induce apoptosis, cleaved PARP and caspase 3 levels were measured in H1299 cells treated with PQ10. As shown in Figure 4D, PQ10 treatment of H1299 cells increased the levels of cleaved PARP and caspase 3 from concentrations as low as 0.1 µmol/L following 24 hours of treatment. The effect of PQ10 reached a maximum at a 5 µmol/L concentration following 48 hours of treatment. Additional experiments showed that PKG inhibitor, KT5823, but not the PKA inhibitor, H89, attenuated the activation of PARP and caspase 3 by PQ10 following 24 hours of treatment (Figure 4E). These results suggest that PKG activation and PKG-mediated phosphorylation of β-catenin at pSer552 may mediate the growth inhibitory activity of PQ10, while PKA activation may not play a significant role.
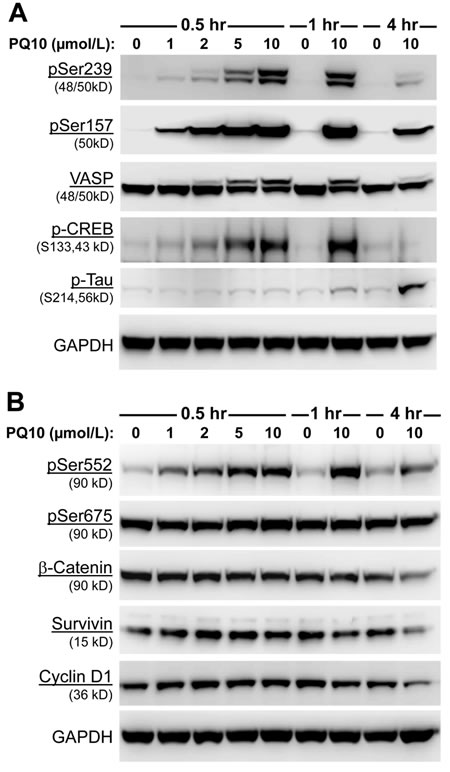
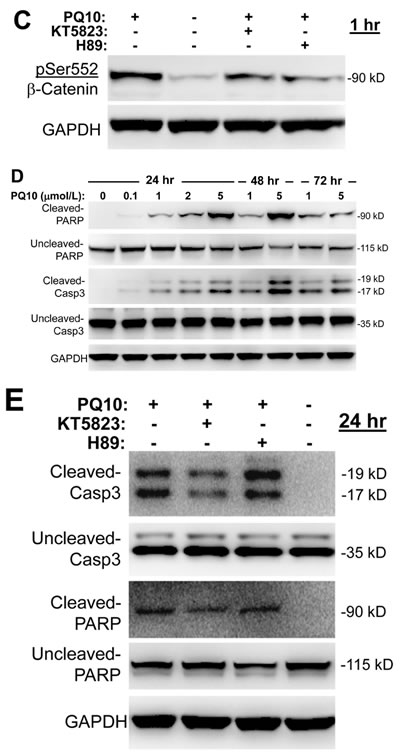
Figure 4: PQ10 activates PKG/PKA signaling, reduces β-catenin expression and induces apoptosis in NSCLC. A. Site-specific phosphorylation of vasodilator-stimulated phosphoprotein, VASP (Ser157 and Ser259), cAMP response element-binding protein, CREB (Ser133), and microtubule-associated protein, Tau (Ser214) in H1299 cells were measured for concentration-dependent and time-related phosphorylation manners after PQ10 treatments. B. PQ10 initiated a phosphorylation of β-catenin at Ser552 but did not show a change at Ser675 after 0.5~1 hour treatments. PQ10 also reduced total β-catenin, survivin and cyclin D1 expression after 4 hours of treatment. C. At 1 hour of treatment, PKG and PKA inhibitor, KT5823 (0.1 µmol/L) and H89 (1 µmol/L) showed partial inhibition of PQ10 (2 µmol/L) induced Ser552 phosphorylation of β-catenin. D. PQ10 induced apoptosis in H1299 NSCLC cells. The cleaved nuclear poly ADP ribose polymerase (PARP) and caspase 3 (Casp3) proteins were detected by Western blotting after 24~72 hours of treatment with different doses of PQ10. E. At 24 hours of treatment, PKG-inhibitor, KT5823, showed inhibition of PQ10 induced-cleaved Casp3 and PARP, but PKA-inhibitor H89 was ineffective.
PQ10 suppresses RAS-activated RAF/MAPK signaling
A high percentage of lung cancers harbor mutations in K-RAS that activate the RAF/MAPK pathway to stimulate tumor cell proliferation and survival. Previous studies showing that cGMP can suppress MAPK signaling by inhibiting RAF kinase activation [43] led us to determine the effect of PQ10 treatment on MAPK signaling in H1299 lung tumor cells. PQ10 treatment was found to cause a concentration- and time-dependent inhibition of MAPK signaling as evident by reduced levels of ERK1/2 (p42/44) phosphorylation at T202/Y204 (Figure 5A). The effect occurred rapidly within 30 minutes of PQ10 treatment that paralleled the kinetics of PKG activation. The level of phospho-p90RSK (Ser380), an essential downstream target of activated nuclear ERK1/2, was also reduced by PQ10 treatment with similar kinetics. The same effect was observed for phospho-MSK-1 at T581, another major downstream target of activated ERK1/2 [44]. PQ10 also inhibited the phosphorylation of upstream MEK1/2 (pSer217/Ser221). Consistent with a mechanism of growth suppression resulting from PDE10 inhibition that does not involve PKA signaling, PQ10 did not affect the phosphorylation of c-RAF at serine 338 and serine 259 as shown in Figure 5B. Given that these residues are known to be phosphorylated by PKA [45, 46], our results provide further evidence for a role of cGMP/PKG signaling in the growth inhibitory activity resulting from PDE10 inhibition.
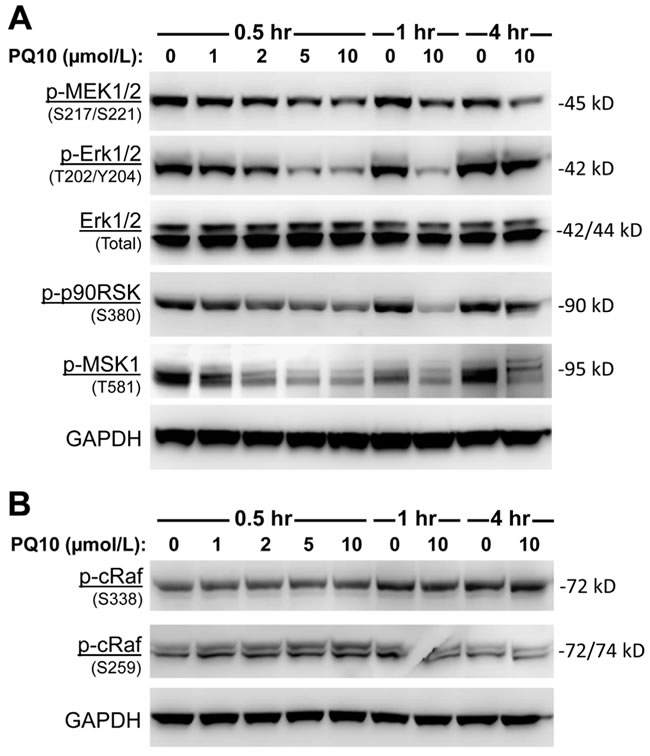
Figure 5: PQ10 suppresses RAF/MAPK signaling. A. PQ10 showed a dose-dependent and time-related inhibition of ERK1/2 (p42/44) phosphorylation at T202/Y204 in H1299 NSCLC cells without changing total protein levels. The level of downstream targets of activated nuclear phospho-p90RSK (S380) and phospho-MSK-1 (T581) were significantly reduced after PQ10 treatment that followed the kinetics of pERK1/2 reduction. PQ10 also reduced the phosphorylation level of MEK1/2 (S217/S221), the upstream kinases of ERK1/2. B. Phosphorylated c-RAF (S338 and S259) levels were also detected after the same treatment of PQ10.
DISCUSSION
We report here that PDE10 mRNA and protein levels are elevated in human lung cancer cell lines and lung tumors compared with primary cultures of normal airway epithelial cells and normal lung tissues, respectively. Genetic knockdown of PDE10 by either siRNA or shRNA strongly suppressed growth and colony formation of NSCLC cell lines. Three known inhibitors of PDE10, PQ10, Pf2545920, and papaverine, suppressed lung tumor cell growth by a mechanism that was confirmed to be mediated by PDE10 inhibition. PDE10 was not detected in normal airway epithelial cells, which displayed reduced sensitivity to PQ10 compared with NSCLC cells. PQ10 caused a rapid and sustained increase in intracellular cGMP levels as measured by a biosensor assay and activated PKG in lung tumor cells as measured by VASP phosphorylation. These effects of PQ10 occurred within the same concentration range as required for growth inhibition. PQ10 increased the phosphorylation of β-catenin with the same kinetics as VASP phosphorylation, while longer treatments reduced protein levels of β-catenin, as well as cyclin D1 and survivin levels, which preceded the activation of PARP and caspase 3 enzymes required for apoptosis. PDE10 inhibition by PQ10 also suppressed RAS-activated RAF/MAPK and downstream pathway components such as p90RSK/MSK-1 phosphorylation. Together, these results indicate that PDE10 expression is elevated in lung cancer cells relative to normal airway epithelial cells and essential for lung tumor cell proliferation and survival.
PDE10 mRNA and protein levels were essentially undetectable in NHAEC and normal lung epithelium, which is consistent with the limited expression of PDE10 as previously reported in normal tissues outside the central nervous system [24-26]. To our knowledge, lung tumors have never been previously studied for PDE10 expression. High PDE10 mRNA levels were measured in human lung tumors relative to normal lung tissues. PDE10 protein levels were confirmed by immunolabeling of lung adenocarcinomas using a PDE10 specific antibody and imaged by immunofluorescence microscopy, which showed cytoplasmic and membrane localization. Our results are consistent with recent observations of PDE10 overexpression in precancerous adenomas from the ApcMin/+ mouse model of colon cancer, as well as human colorectal adenocarcinomas [31]. These observations suggest that PDE10 is expressed during lung tumor development and essential for tumor growth.
Specific overexpression of the PDE10 isozyme in lung tumor cell lines was revealed by simultaneous detection of all known cGMP PDE isozymes (except PDE6) by Western blotting using isoform-specific antibodies. These experiments showed that while some isozymes could be detected in certain lung tumor cell lines, none were uniformly expressed in all NSCLC lines but not expressed in NHAEC, as the case of PDE10. Previous studies have shown that the cGMP specific PDE5 isozyme is expressed in various tumor cell lines, including those derived from lung, colon, and breast tumors [10-12]. We detected PDE5 in two of five NSCLC cell lines examined, but the levels were comparable to NHAEC. The PDE5 selective inhibitor, sildenafil has also been reported to inhibit tumorigenesis in an inflammation-driven model of colon cancer, which suggests a potentially important role of PDE5 in tumorigenesis [47]. However, the abundance of PDE10 in tumor cells may limit the sensitivity of tumor cells to PDE5 inhibitors, whereby combined treatment with PDE5 and PDE10 inhibitor or treatment with a dual PDE5/10 inhibitor may hold greater promise as a therapeutic strategy [33, 48]. In addition, we found that PDE9 was expressed in lung tumor cell lines, which is consistent with observations from other investigators reporting effects of PDE9 inhibitors on breast tumor cell growth [49-51]. However, since PDE9 is also expressed in NHAEC, it may provide a “house-keeping” function in the lung epithelium unrelated to tumorigenesis.
Experiments to determine if PDE10 represents a potential therapeutic target for lung cancer were performed using three known small molecule inhibitors of PDE10, including PQ10, Pf2545920 and papaverine. All three compounds effectively suppressed lung tumor cell growth with PQ10 showing the greatest potency. Because of a significance difference between IC50 values for PDE10 and growth inhibition, we performed additional experiments to confirm that PDE10 inhibition is indeed responsible for the growth inhibitory activity of PQ10. First, we profiled the inhibitory activity of PQ10 against a full panel of PDE isozymes, which confirmed that the compound is highly selective for inhibiting PDE10. We then showed that the tumor cell growth inhibitory activity of PQ10 occurred within the same concentration range that increased intracellular cGMP levels.
Relative intracellular cGMP levels were measured using a cGMP biosensor assay, which to our knowledge is the first report showing the utility of a luminescence-based assay for measuring cGMP levels in live cancer cells. PQ10 increased intracellular cGMP levels in H1299 cells with kinetics that paralleled the activation of PKG activation as measured by the phosphorylation of VASP at serine 239. Notably, the concentration ranges of both effects as measured by two distinct assay formats were comparable with IC50 values for growth inhibition. As compared with lung tumor H1299 cells, treatment of HEK293 cGMP biosensor cells with PQ10 showed an increase in cGMP levels at a higher EC50 value, coincident to a higher IC50 value for growth inhibition of cells derived from normal tissues (e.g. NHAECs). It is also noteworthy that with such specific cGMP biosensor analysis for PDE10 in H1299 cells, an undetectable change of intracellular cGMP levels was seen for sildenafil treatment at a concentration 300-fold higher than its IC50 to inhibit PDE5, in which the cells showed undetectable levels of PDE5 and no effect of sildenafil on growth.
Treatment of NSCLC cells with PQ10 to activate PKG was found to be associated with increased phosphorylation of β-catenin followed by reduction of protein levels. Under the same conditions, PQ10 reduced cyclin D1 and survivin levels, which occurred within a treatment period that preceded the activation of PARP and caspase 3 leading to apoptosis. These observations are consistent with previous reports that PDE10 inhibition can reduce the expression level, nuclear translocation, and TCF transcriptional activity of β-catenin in colon tumor cells [31-33]. Given that the activation of cGMP/PKG signaling has been reported to suppress oncogenic Wnt/β-catenin signaling in colon and breast cancer cells by inhibiting β-catenin expression at mRNA level and β-catenin/CTNNB1 promoter levels [14, 32], a similar mechanism may exist in lung tumor cells. We also report here for the first time that PDE10 inhibition by PQ10 can stimulate the phosphorylation of β-catenin at serine 552, which is considered to be a dual phosphorylation site for PKG and PKA. Interestingly, PQ10 did not affect the phosphorylation of the PKA-conserved site of β-catenin at serine 675. PQ10-induced phosphorylation of β-catenin at serine 552 was shown to be reduced by a PKG inhibitor at lower concentrations than a PKA inhibitor. Moreover, the time course paralleled the kinetics of cGMP/PKG-mediated VASP phosphorylation at serine 239, but occurred prior to the reduction of β-catenin expression. These results support the possibility of a PKG-mediated mechanism to suppress oncogenic Wnt/β-catenin signaling by ubiquitination and proteosomal degradation as previously reported by others [9, 40, 41]. It is unclear if this mechanism is distinct from the action of cAMP on the PKA-conserved phosphorylation at serine 675 of β-catenin as reported in non-tumor cells [42].
Finally, we determined the effect of PDE10 inhibition on the phosphorylation of MEK1/2 and ERK1/2 because of previous reports showing that cGMP/PKG signaling can inhibit RAS-activated RAF/MAPK-kinases [43, 52, 53]. PQ10 was found to inhibit the phosphorylation of MEK1/2 and ERK1/2 in lung tumor cells within the same concentration range that was effective for suppressing β-catenin transcriptional activity. PQ10 also strongly inhibited the phosphorylation of downstream p90RSK and MSK-1, which are important for receptor tyrosine kinase stimulated nuclear transcription and ribosomal translation [44]. In addition, the timeline of PQ10 suppression of RAF/MAPK signaling followed the time required to increase intracellular cGMP levels and activate PKG, which paralleled the time required for β-catenin phosphorylation (Ser552) and the suppression of β-catenin, survivin and cyclin D1 levels, leading to the induction of apoptosis.
In conclusion, PDE10 is overexpressed in lung tumor cells and appears to be essential for lung tumor cell proliferation and survival. The growth inhibitory activity resulting from PDE10 inhibition in lung tumor cells was associated with the elevation of intracellular cGMP levels and activation of PKG to inhibit two important oncogenic pathways involving β-catenin and MAPK signaling. The ability of PDE10 inhibitors to simultaneously suppress both oncogenic pathways may result in greater antitumor efficacy and reduced potential for resistance compared with other targeted drugs that are selective for one or the other pathway. We conclude that PDE10 represents a novel molecular target for the treatment of lung cancer.
MATERIALS AND METHODS
Reagents and drugs
The isozyme specific antibodies for PDE1, 2, 3, 9, 10, and 11 were purchased from GeneTex. Other antibodies, including a PDE5 specific antibody and phospho-specific antibodies for protein phosphorylation and their non-phosphorylated forms were purchased from Cell Signaling Technologies. Small interfering RNA (siRNA) targeting human PDE10 (siPDE10) and scramble control siRNA were purchased from Dharmacon and Qiagen as previously described [31]. Papaverine was purchased from Sigma-Aldrich. Pf-2545920 was purchased from Selleck Chemicals, while PQ10 was synthesized at our institution as described previously [34]. DMSO was used as vehicle for all compounds unless otherwise noted. All other reagents were purchased from Sigma-Aldrich or as indicated.
Cells and cell culture
Human non-small cell lung cancer (NSCLC) cell lines were purchased from the American Type Culture Collection (ATCC) and grown under standard cell culture conditions in RPMI1640 medium containing 5 % fetal bovine serum at 37°C in a humidified atmosphere with 5% CO2. All NSCLC cell lines were expanded upon delivery, and numerous vials of low-passage cells were preserved in liquid nitrogen. Cells were passaged in culture no longer than 2 months. Cell line characterization is conducted by ATCC through short tandem repeat profiling. No additional authentication of NSCLC cell lines was conducted except for experimental reasons (e.g. confirmation of PDE10 expression levels and sensitivity to PDE10 inhibitors, etc.). Normal human airway epithelial cells (NHAECs) were obtained from an adult donor and established as primary cultures (P0) from the University of Alabama at Birmingham. NHAECs were grown in serum-free Bronchial Epithelial Cell Basal medium (Lonza) supplemented with growth factors and other nutrients (SingleQuots) for additional passaging (P1~P3). All cell passages used for experiments were maintained below passage 4.
Knockdown of PDE10 expressions by siRNA and shRNA
The transient knockdown of PDE10 in NSCLC cells by PDE10-specific siRNA and scramble siRNA control was performed using RNAiMAX reagent (Invitrogen) as previously described [31]. Effects of siRNA on NSCLC cell growth and PDE10 expression levels were measured after 72 hours of treatment. For a stable knockdown of PDE10, GIPZ lentiviral short hairpin RNA (shRNA) vectors targeting human PDE10, nonsilencing GPIZ lentiviral control vector, and transduction kit were purchased from Open Biosystems [31]. Lentiviral particles of shRNA were produced in HEK293T cells (ATCC) according to the manufacturer’s instructions. HOP62 NSCLC cells were transfected by the lentivirus particles followed by puromycin (5 μg/mL) selection. Stable expression cell lines with shRNA of PDE10 (shPDE10) or lentiviral vector control (shCTL) were obtained by limited dilution for single cell clone and confirmed by Western blotting.
TissueScan lung cancer cDNA array and real-time PCR
TissueScan lung cancer cDNA arrays were purchased from Origene containing 19 lung tumors (with 8 adenocarcinomas as the major subtype) and 4 normal lung tissues that were used to evaluate PDE10 mRNA overexpression in human lung cancer. High quality total RNAs were extracted from pathologist-verified tissues and used for the synthesis of cDNAs for real-time PCR. Real-time PCR was performed using Bio-Rad Chromo 4 system and quantified using the 2-ΔΔCt method as we previously described [31]. PDE10 levels were normalized using β-actin. Human PDE10 primers and RT2 SYBR Green qPCR Mastermix were purchased from Qiagen. The mRNA levels of PDE10 and other PDE isozymes in cultured HOP62 and NHAECs were evaluated by the same method. Primers used for testing the expression of various PDE isoforms in cultured cell studies were self-designed and synthesized. Forward primers are: GCATACAGGGACAACAAACAAC (PDE1A), CACTTCTGCTACCTGCTCTAC (PDE2A), CAGGAAACGGTGGGACATTTA (PDE5A), CACTTGGCTGTCCTAGAGAAAC (PDE9A), GGACAGAGACAAGAAGGATGAA (PDE10A); reverse primers are: GTGGTGATTCTCAA-GGACAGAG (PDE1A), GTCATGACACATGCAGGAAATAA (PDE2A), GAGCTGAGCAT-TATGAAGAACAATAC (PDE5A), GCTCCTCCCTCATCTTCTTAATG (PDE9A), GGATC-TGGGTAAGGGTTGTATAG (PDE10A). The primers for control TATA-binding protein (TBP) are: CTTCGGAGAGTTCTGGGATTG (F) and CACGAAGTGCAATGGTCTTTAG (R).
Growth assays
Cells were plated in half area 96-well microtiter plates (Corning) at a density of 3, 000 cells per well. For drug treatment, cells were treated with compound or DMSO as vehicle (final concentration at 0.2%). For siRNA assays, cells were transfected with siRNAs under the same condition as described above. The effect of treatment on cell growth was measured using the Cell Titer Glo (CTG) assay (Promega) as specified by the measurement of viable cells based on ATP content after 72 hours standard culture [31]. All growth assay involving NSCLCs and NHAECs were performed in the presence of 5% serum.
Colony formation assays
For colony formation assays, 200 viable cells were plated in 6-well tissue culture plates and were cultured in RPMI medium supplemented with 5% serum. DMSO (0.2%) as the vehicle and different concentrations of PQ10 were added to the cell culture medium for 14 days of treatment. Colonies were counted after staining with crystal violet as previously described [31].
cGMP biosensor assay
Stable cell lines with an expression of luciferase-based cGMP biosensor were established from H1299 NSCLC cells and HEK293 cells using a genetically encoded GAF-Luc construct (Promega), containing a cGMP-specific binding domain from human PDE5A GAF-A domain fused to modified firefly luciferase (35). Cells were pre-incubated at room temperature in CO2 independent media supplemented with 10% FBS and 5 mmol/L luciferin. For the measurement of intracellular cGMP levels, sodium nitroprusside (SNP) or C-type natriuretic peptide (CNP) in combination with PDE inhibitors were added to initiate the experiment and the increased luminescence values in live cells were detected constantly over a period time of 2 hours.
PDE assay
The enzymatic activity of recombinant PDE or PDE activities in cell lysates were measured using the IMAP fluorescence polarization (FP) PDE assay (Molecular Devices) as previously described [12, 13]. PDE10 inhibitors and DMSO as the vehicle were added to assay buffer 10 minutes before the incubation with the enzyme reaction. Fluorescence polarization values were measured using Synergy H4 Hybrid Reader (BioTek).
Western blotting
Whole cell extraction was performed according to a previously described method [12, 23, 35] using cell lysing buffer (Cell Signaling) supplemented with 10 mmol/L NaF for protein phosphorylation studies. Cell lysates were separated by 4-12% gradient, 7% or 10% pre-casting PAGE (Novex/Invitrogen) followed by electrophoretic transfer to a nitrocellulose membrane [13, 23]. Western blotting procedures were performed using Chemiluminescence Reagent (Pierce) and visualized using Chemi Doc Imaging System (BioRad).
Histological immunofluorescence microscopy
Portions of the lung tissue with tumors were immersed in formalin, embedded in paraffin, and 5 μm sections were placed on glass slides for histological immunofluorescence. Slides were deparaffinized in xylene and rehydrated by decreasing concentrations of ethanol in water. All slides were blocked with background sniper (Biocare) for 30 minutes and incubated overnight in a 1:200 dilution of the primary antibody against the carboxyl terminus of human PDE10A (GeneTex) in diluent solution (Fisher). Slides were then rinsed 3 times with 1X tris-buffered saline (TBS) and incubated in a 1:500 dilution of the Alexa Fluor 488 conjugated secondary antibody (Invitrogen) for 1.5 hours. Slides were counterstained with DAPI (NucBlue Fixed Cell ReadyProbe Reagent; Fisher) and fluorescent mounting medium (Vectashield; Vector) was used to apply the coverslips. Spectral images of fluorescence emission were acquired at 10 nm increments, from 425-685 nm, using an A1R spectral confocal microscope. NIS Elements software (Nikon Instruments, Inc., Melville, NY, USA) was used to acquire all confocal images and hyperspectral analysis was used to identify fluorophores of interest in highly autofluorescent lung tissue as previously described [54].
Experimental design and data analysis
Drug effects on cell growth and IC50 values were determined as described previously [31]. All experiments were repeated a minimum of three times to determine the reproducibility of the results. All error bars represent standard error of the mean (SEM). Statistical analysis was performed using Student’s t test or other as indicated. A P value of <0.05 was considered statistically significant.
Abbreviations
cAMP, cyclic adenosine monophosphate; cGMP, cyclic guanosine monophosphate; cGS, cGMP biosensor; pGC, particulate guanylyl cyclase; sGC, soluble guanylyl cyclase; PDE, phosphodiesterase; PKA, cAMP dependent protein kinase; PKG, cGMP dependent protein kinase; shRNA, short hairpin RNA; siRNA, small interfering RNA; VASP, vasodilator-stimulated phosphoprotein.
Authors’ contributions
Conception design and Study supervision: B. Zhu, G.A. Piazza.
Development methodology and acquisition of data: B. Zhu, A. Lindsey, N. Li, K. Lee, J.C. Canzoneri, A. Fajardo, M. Thomas, J.T. Piazza, E. Gurpinar, D. Otali, W. Grizzle.
Analysis and interpretation of data: B. Zhu, A. Lindsey, N. Li, K. Lee, V. Ramirez-Alcantara, J.C. Canzoneri, L. Madeira da Silva, A.B. Keeton, G.A. Piazza.
Writing, review, and/or revision of the manuscript: B. Zhu, A. Lindsey, K. Lee, V. Ramirez-Alcantara, W. Grizzle, X. Chen, A.B. Keeton, G.A. Piazza.
Other (chemistry development and synthesis): L. Yet, B.T. Eberhardt, J. Valiyaveettil, X. Chen.
CONFLICTS OF INTEREST
G.A. Piazza, A.B. Keeton, and X. Chen are co-founders of ADT Pharmaceuticals Inc.; G.A. Piazza is a founder of PDEi Pharmaceuticals Inc.
GRANT SUPPORT
Research reported here was supported by the National Cancer Institute of the National Institutes of Health under Award Numbers 1R01CA131378, 1R01CA148817, 1R01CA197147 and 1R01CA155638 to G.A. Piazza; and 1R21CA182941 to G.A. Piazza and B. Zhu. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
REFERENCES
1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012; 62:10-29.
2. Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008; 359:1367-1380.
3. Imai K, Takaoka A. Comparing antibody and small-molecule therapies for cancer. Nat Rev Cancer. 2006; 6:714-727.
4. Gatzemeier U, Groth G, Butts C, Van Zandwijk N, Shepherd F, Ardizzoni A, Barton C, Ghahramani P, Hirsh V. Randomized phase II trial of gemcitabine-cisplatin with or without trastuzumab in HER2-positive non-small-cell lung cancer. Ann Oncol. 2004; 15:19-27.
5. Young A, Lyons J, Miller AL, Phan VT, Alarcón IR, McCormick F. Ras signaling and therapies. Adv Cancer Res. 2009; 102:1-17.
6. Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol. 2010; 28:1075-1083.
7. Khuder SA, Herial NA, Mutgi AB, Federman DJ. Nonsteroidal antiinflammatory drug use and lung cancer: a metaanalysis. Chest. 2005; 127:748-754.
8. Tennis M, Van Scoyk M, Winn RA. Role of the Wnt signaling pathway and lung cancer. J Thorac Oncol. 2007; 2:889-892.
9. Thompson WJ, Piazza GA, Li H, Liu L, Fetter J, Zhu B, Sperl G, Ahnen D, Pamukcu R. Exisulind induction of apoptosis involves guanosine 3’, 5’-cyclic monophosphate phosphodiesterase inhibition, protein kinase G activation, and attenuated beta-catenin. Cancer Res. 2000; 60:3338-3342.
10. Whitehead CM, Earle KA, Fetter J, Xu S, Hartman T, Chan DC, Zhao TL, Piazza G, Klein-Szanto AJ, Pamukcu R, Alila H, Bunn PA Jr, Thompson WJ. Exisulind-induced apoptosis in a non-small cell lung cancer orthotopic lung tumor model augments docetaxel treatment and contributes to increased survival. Mol Cancer Ther. 2003; 2:479-488.
11. Tinsley HN, Gary BD, Keeton AB, Zhang W, Abadi AH, Reynolds RC, Piazza GA. Sulindac sulfide selectively inhibits growth and induces apoptosis of human breast tumor cells by phosphodiesterase 5 inhibition, elevation of cyclic GMP, and activation of protein kinase G. Mol Cancer Ther. 2009; 8:3331-3340.
12. Tinsley HN, Gary BD, Thaiparambil J, Li N, Lu W, Li Y, Maxuitenko YY, Keeton AB, Piazza GA. Colon tumor cell growth-inhibitory activity of sulindac sulfide and other nonsteroidal anti-inflammatory drugs is associated with phosphodiesterase 5 inhibition. Cancer Prev Res. 2010; 3:1303-1313.
13. Li N, Xi Y, Tinsley HN, Gurpinar E, Gary BD, Zhu B, Li Y, Chen X, Keeton AB, Abadi AH, Moyer MP, Grizzle WE, Chang WC, et al. Sulindac selectively inhibits colon tumor cell growth by activating the cGMP/PKG pathway to suppress Wnt/beta-catenin signaling. Mol Cancer Ther. 2013; 12:1848-1859.
14. Tinsley HN, Gary BD, Keeton AB, Lu W, Li Y, Piazza GA. Inhibition of PDE5 by sulindac sulfide selectively induces apoptosis and attenuates oncogenic Wnt/β-catenin-mediated transcription in human breast tumor cells. Cancer Prev Res. 2011; 4:1275-1284.
15. Soh JW, Kazi JU, Li H, Thompson WJ, Weinstein IB. Celecoxib-induced growth inhibition in SW480 colon cancer cells is associated with activation of protein kinase G. Mol Carcinog. 2008; 47:519-525.
16. Kwon IK, Schoenlein PV, Delk J, Liu K, Thangaraju M, Dulin NO, Ganapathy V, Berger FG, Browning DD. Expression of cyclic guanosine monophosphate-dependent protein kinase in metastatic colon carcinoma cells blocks tumor angiogenesis. Cancer. 2008; 112:1462-1470.
17. Zhu B, Vemavarapu L, Thompson WJ, Strada SJ. Suppression of cyclic GMP-specific phosphodiesterase 5 promotes apoptosis and inhibits growth in HT29 cell. J Cell Biochem. 2005; 94:336-350.
18. Zhu B, Strada SJ. The novel functions of cGMP-specific phosphodiesterase 5 and its inhibitors in carcinoma and pulmonary/cardiovascular vessels. Curr Top Med Chem. 2007; 7:437-454.
19. Beavo JA. Cyclic nucleotide phosphodiesterases: functional implications of multiple isoforms. Physiol Rev. 1995; 75:725-748.
20. Omori K, Kotera J. Overview of PDEs and their regulation. Circ Res. 2007; 100:309-327.
21. Weinberger B, Heck DE, Laskin DL, Laskin JD. Nitric oxide in the lung: therapeutic and cellular mechanisms of action. Pharmacol Ther. 1999; 84:401-411.
22. Zhu B, Strada SJ, Stevens T. Cyclic GMP-specific phosphodiesterase 5 regulates cell growth and apoptosis of pulmonary endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2005; 289:L196-L206.
23. Zhu B, Zhang L, Alexeyev M, Alvarez D, Strada SJ, Stevens T. Type 5 phosphodiesterase is a critical determinant of the lung endothelial cell angiogenic phenotype. Am J Physiol Lung Cell Mol Physiol. 2009; 296:L220-L228.
24. Soderling SH, Bayuga SJ, Beavo JA. Isolation and characterization of a dual-substrate phosphodiesterase gene family: PDE10A. Proc Natl Acad Sci USA. 1999; 96:7071-7076.
25. Seeger TF, Bartlett B, Coskran TM, Culp JS, James LC, Krull DL, Lanfear J, Ryan AM, Schmidt CJ, Strick CA, Varghese AH, Williams RD, Wylie PG, et al. Immunohistochemical localization of PDE10A in the rat brain. Brain Res. 2003; 985:113-126.
26. Xie Z, Adamowicz WO, Eldred WD, Jakowski AB, Kleiman RJ, Morton DG, Stephenson DT, Strick CA, Williams RD, Menniti FS. Cellular and subcellular localization of PDE10A, a striatum-enriched phosphodiesterase. Neuroscience. 2006; 139:597-607.
27. Bollen E, Prickaerts J. Phosphodiesterases in neurodegenerative disorders. IUBMB Life. 2012; 64:965-970.
28. Siuciak JA. The role of phosphodiesterases in schizophrenia: therapeutic implications. CNS Drugs. 2008; 22:983-993.
29. Kehler J, Ritzen A, Langgård M, Petersen SL, Farah MM, Bundgaard C, Christoffersen CT, Nielsen J, Kilburn JP. Triazoloquinazolines as a novel class of phosphodiesterase 10A (PDE10A) inhibitors. Bioorg Med Chem Lett. 2011; 21:3738-3742.
30. Tian X, Vroom C, Ghofrani HA, Weissmann N, Bieniek E, Grimminger F, Seeger W, Schermuly RT, Pullamsetti SS. Phosphodiesterase 10A upregulation contributes to pulmonary vascular remodeling. PLoS One. 2011; 6:e18136.
31. Li N, Lee K, Xi Y, Zhu B, Gary BD, Ramirez-Alcantara V, Gurpinar E, Canzoneri JC, Fajardo A, Sigler S, Piazza JT, Chen X, Andrews J, et al. Phosphodiesterase 10A: a novel target for selective inhibition of colon tumor cell growth and beta-catenin-dependent TCF transcriptional activity. Oncogene. 2015; 34:1499-1509.
32. Lee K, Lindsey AS, Li N, Gary B, Andrews J, Keeton AB, Piazza GA. β-catenin nuclear translocation in colorectal cancer cells is suppressed by PDE10A inhibition, cGMP elevation, and activation of PKG. Oncotarget. 2016; 7:5353-5365. https://doi.org/10.18632/oncotarget.6705.
33. Li N, Chen X, Zhu B, Ramírez-Alcántara V, Canzoneri JC, Lee K, Sigler S, Gary B, Li Y, Zhang W, Moyer MP, Salter EA, Wierzbicki A, et al. Suppression of β-catenin/TCF transcriptional activity and colon tumor cell growth by dual inhibition of PDE5 and 10. Oncotarget. 2015; 6:27403-27415. https://doi.org/10.18632/oncotarget.4741.
34. Chappie TA, Humphrey JM, Allen MP, Estep KG, Fox CB, Lebel LA, Liras S, Marr ES, Menniti FS, Pandit J, Schmidt CJ, Tu M, William RD, et al. Discovery of a series of 6, 7-dimethoxy-4-pyrrolidylquinazoline PDE10A inhibitors. J Med Chem. 2007; 50:182-185.
35. Whitt JD, Li N, Tinsley HN, Chen X, Zhang W, Li Y, Gary BD, Keeton AB, Xi Y, Abadi AH, Grizzle WE, Piazza GA. A novel sulindac derivative that potently suppresses colon tumor cell growth by inhibiting cGMP phosphodiesterase and beta-catenin transcriptional activity. Cancer Prev Res. 2012; 5:822-833.
36. Oelze M, Mollnau H, Hoffmann N, Warnholtz A, Bodenschatz M, Smolenski A, Walter U, Skatchkov M, Meinertz T, Münzel T. Vasodilator-stimulated phosphoprotein serine 239 phosphorylation as a sensitive monitor of defective nitric oxide/cGMP signaling and endothelial dysfunction. Circ Res. 2000; 87:999-1005.
37. Gudi T, Casteel DE, Vinson C, Boss GR, Pilz RB. NO activation of fos promoter elements requires nuclear translocation of G-kinase I and CREB phosphorylation but is independent of MAP kinase activation. Oncogene. 2000; 19:6324-6333.
38. Zhu B, Zhang L, Creighton J, Alexeyev M, Strada SJ, Stevens T. Protein kinase A phosphorylation of Tau-serine 214 reorganizes microtubules and disrupts the endothelial cell barrier. Am J Physiol Lung Cell Mol Physiol. 2010; 299:L493-501.
39. Balczon R, Prasain N, Ochoa C, Prater J, Zhu B, Alexeyev M, Sayner S, Frank DW, Stevens T. Pseudomonas aeruginosa exotoxin Y-mediated tau hyperphosphorylation impairs microtubule assembly in pulmonary microvascular endothelial cells. PLoS One. 2013; 8:e74343.
40. Li H, Pamukcu R, Thompson WJ. β-Catenin signaling: therapeutic strategies in oncology. Cancer Biol Ther. 2002; 1:621-625.
41. Li H, Liu L, David ML, Whitehead CM, Chen M, Fetter JR, Sperl GJ, Pamukcu R, Thompson WJ. Pro-apoptotic actions of exisulind and CP461 in SW480 colon tumor cells involve β-catenin and cyclin D1 down-regulation. Biochem Pharmacol. 2002; 64:1325-1336.
42. Hino SI, Tanji C, Nakayama KI, Kikuchi A. Phosphorylation of β-catenin by cyclic AMP-dependent protein kinase stabilizes β-catenin through inhibition of its ubiquitination. Mol Cell Biol. 2005; 25:9063-9072.
43. Suhasini M, Li H, Lohmann SM, Boss GR, Pilz RB. Cyclic-GMP-dependent protein kinase inhibits the Ras/mitogen-activated protein kinase pathway. Mol Cell Biol. 1998; 18:6983-6994.
44. Anjum R, Blenis J. The RSK family of kinases: emerging roles in cellular signaling. Nat Rev Mol Cell Biol. 2008; 9:747-758.
45. Dumaz N, Marais R. Integrating signals between cAMP and the RAS/RAF/MEK/ERK signalling pathways. FASEB J. 2005; 272:3491-3504.
46. Dumaz N, Marais R. Protein kinase A blocks Raf-1 activity by stimulating 14-3-3 binding and blocking Raf-1 interaction with Ras. J Biol Chem. 2003; 278:29819-29823.
47. Islam BN, Sharman SK, Hou Y, Bridges AE, Singh N, Kim S, Kolhe R, Trillo-Tinoco J, Rodriguez PC, Berger FG, Sridhar S, Browning DD. Sildenafil suppresses inflammation-driven colorectal cancer in mice. Cancer Prev Res. 2017; 10:377-388.
48. Piazza GA. Validation of PDE5 as a chemoprevention target. Cancer Prev Res. 2017; 10:373-376.
49. Karami-Tehrani F, Moeinifard M, Aghaei M, Atri M. Evaluation of PDE5 and PDE9 expression in benign and malignant breast tumors. Arch Med Res. 2012; 43:470-475.
50. Saravani R, Karami-Tehrani F, Hashemi M, Aghaei M, Edalat R. Inhibition of phosphodiestrase 9 induces cGMP accumulation and apoptosis in human breast cancer cell lines, MCF-7 and MDA-MB-468. Cell Prolif. 2012; 45:199-206.
51. Hassaan EA, Sigler SC, Ibrahim TM, Lee KJ, Cichon LK, Gary BD, Canzoneri JC, Piazza GA, Abadi AH. Mining ZINC database to discover potential phosphodiesterase 9 inhibitors using structure-based drug design approach. Med Chem. 2016; 10:472-477.
52. Wu Y, Chen Y, Qu R, Lan T, Sang J. Type II cGMP-dependent protein kinase inhibits EGF-triggered signal transduction of the MAPK/ERK-mediated pathway in gastric cancer cells. Oncol Rep. 2012; 27:553-558.
53. Jiang L, Lan T, Chen Y, Sang J, Li Y, Wu M, Tao Y, Wang Y, Qian H, Gu L. PKG II inhibits EGF/EGFR-induced migration of gastric cancer cells. PLoS One. 2013; 8:e61674.
54. Leavesley SJ, Annamdevula N, Boni J, Stocker S, Grant K, Troyanovsky B, Rich TC, Alvarez DF. HyperSpectral imaging microscopy for identification and quantitative analysis of fluorescently-labeled cells in highly autofluorescent tissue. J Biophotonics. 2012; 5:67-84.