
INTRODUCTION
Pulmonary arterial hypertension (PAH) is a rare but fatal disease, with an estimated mean survival period in untreated patients of approximately 3 years [1]. Pulmonary vascular remodeling, occurring mostly in the small to mid-sized pulmonary arterioles (≤ 500μm), is a hallmark of PAH. This process is ascribed to the increased proliferation, migration and survival of pulmonary vascular cells within the pulmonary artery wall, i.e. pulmonary arterial smooth muscle cells (PA-SMCs), endothelial cells, myofibroblasts and pericytes [2]. However, the etiology and pathogenesis of PAH remain obscure.
Although many familial cases of heritable PAH exhibit an autosomal dominant mode of inheritance, with the majority having mutations in the Bone Morphogenetic Protein Receptor, Type 2 gene (BMPR2), [3] the penetrance of BMPR2 pathogenic variants is low and estimated to be 14% for males and 42% for females [4]. This low PAH penetrance in BMPR2 mutation carriers likely results from a combination of currently unknown genetic, environmental, and lifestyle factors [5].
Germain and colleagues have recently published the results of a genome-wide association study (GWAS) performed in patients with idiopathic PAH. They reported that the CBLN2 locus, which maps to 18q22.3, is a susceptibility locus for PAH, [6] however they excluded PAH patients carrying BMPR2 mutations from their study. Because additional common genetic variations may influence PAH development in patients either with or without a BMPR2 mutation, we decided to include idiopathic and heritable PAH (I/HPAH) patients carrying BMPR2 mutations in our GWAS.
Our objective was to identify new genetic variants that confer a predisposition to I/HPAH using a GWAS, and to validate this association in a second independent internal cohort of I/HPAH patients.
RESULTS
Discovery of novel PAH candidate genes
For discovery study, we genotyped 23 PAH patients (genotyped on the Illumina Human Omni2.5-8 Beadchip ver 1.1) and compared the results to genotypes from 2,002 healthy controls (JPDSC-Phase 2). Table 1 shows the clinical characteristics of the I/HPAH patients in the discovery study. Of the original 2,006 healthy controls, four subjects were excluded; two subjects because of first-degree relatives, and two subjects because they showed high-degree heterozygosities. We had planned to exclude samples that were judged to be outliers by PCA analysis using Eigenstrat software, [7] as well as samples with >1% missing genotypes, however such samples were not observed.
Table 1: Patients’ genetic and hemodynamic characteristics in the discovery cohort
ID |
Age |
Sex |
Ethnics |
BMPR2 mutation |
mPAP [mmHg] |
CO [l/min] |
PVR [dynes ▪ sec ▪ cm-5] |
|---|---|---|---|---|---|---|---|
1 |
63 |
Male |
Japanese |
+ |
42 |
2.72 |
1088 |
2 |
56 |
Female |
Japanese |
+ |
41 |
2.70 |
1064 |
3 |
66 |
Male |
Japanese |
+ |
54 |
1.96 |
2208 |
4 |
59 |
Female |
Japanese |
- |
54 |
4.40 |
856 |
5 |
30 |
Female |
Japanese |
- |
58 |
2.71 |
1563 |
6 |
31 |
Male |
Japanese |
- |
61 |
NA |
NA |
7 |
15 |
Female |
Japanese |
- |
75 |
2.38 |
2320 |
8 |
59 |
Female |
Japanese |
+ |
56 |
3.90 |
1008 |
9 |
48 |
Female |
Japanese |
- |
36 |
4.90 |
480 |
10 |
23 |
Female |
Japanese |
- |
70 |
2.90 |
1656 |
11 |
27 |
Female |
Japanese |
- |
74 |
1.86 |
2840 |
12 |
48 |
Female |
Japanese |
- |
45 |
3.27 |
1101 |
13 |
48 |
Female |
Japanese |
- |
60 |
NA |
2408 |
14 |
40 |
Female |
Japanese |
+ |
57 |
1.93 |
2152 |
15 |
53 |
Male |
Japanese |
+ |
66 |
3.20 |
NA |
16 |
48 |
Female |
Japanese |
- |
50 |
2.03 |
1728 |
17 |
62 |
Female |
Japanese |
- |
49 |
2.90 |
1200 |
18 |
35 |
Female |
Japanese |
- |
35 |
4.50 |
488 |
19 |
31 |
Female |
Japanese |
- |
42 |
4.32 |
593 |
20 |
22 |
Male |
Japanese |
+ |
85 |
1.85 |
3072 |
21 |
57 |
Female |
Japanese |
- |
86 |
1.63 |
3840 |
22 |
46 |
Female |
Japanese |
- |
49 |
6.39 |
330 |
23 |
17 |
Male |
Japanese |
+ |
83 |
5.90 |
1032 |
The hemodynamic data were evaluated in their initial right heart catheterization.
mPAP: mean pulmonary arterial pressure; CO: cardiac output; PVR: pulmonary vascular resistance; NA: not available.
To perform GWAS analyses, 1,347,690 SNPs were selected because they showed minor allele frequencies (MAF) of > 0.01 in controls, a Hardy-Weinberg equilibrium (HWE) P-value of > 0.001 in controls, and a SNP call rate of > 0.95. Furthermore, these SNPs were included in both platforms (ver 1.0 and 1.1). To exclude potential stratification of the study population, we performed QQ-plot analysis and calculated the genomic inflation factor (λ) [8]. Neither the QQ-plot of log P-values shown in Supplementary Figure 1, nor the genomic inflation factor (λ) of 0.927 indicate the presence of any biases related to population stratification. Under an additive model for the effect of the minor allele at each SNP, we identified seven loci, each of which had more than one SNP with P values less than 10-5 (Figure 1, Table 2). We did not select loci that had a single SNP with a low P-value, because they often result from errors. The seven loci selected were CDC73|KCNT2 on chr 1, PDE1A|DNAJC10 on chr 2, FAM184B on chr 4, MTCHI|FGD2 on chr 6, TLE4|TLE1 on chr 9, USP15 on chr 12 and AQP9|LIPC on chr 15. In Table 1, the SNP with the lowest P value for each locus is shown, although two SNPs are shown for the PDE1A|DNAJC10 locus, which is considered especially important. As will be discussed later, PDE5 inhibitors are known to be effective for IPAH, and PDE1A codes for a phosphodiesterase (PDE) that is within the same PDE superfamily as PDE5 [14].
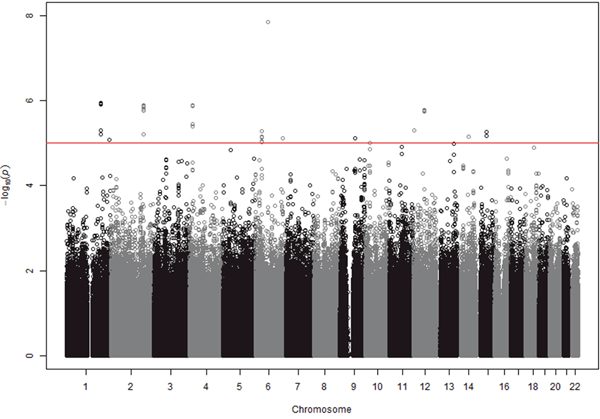
Figure 1: GWAS results at 1,347,690 SNPs with 23 cases and 2,002 controls under an additive model. SNPs above red line showed the P values < 10-5. Eight regions contained more than one SNP with P values < 10-5. The figure was drawn using the R environment (R Version 2.15.3).
Table 2: 9 SNPs at 8 loci, each of which had more than one SNP with low (< 10-5) P values, in the discovery GWAS, and the results of the validation study
Stagea |
rsID |
CHR |
POS |
Gene |
Minor allele |
Major allele |
MAFb |
HWEc |
Odds ratio |
P-value |
|---|---|---|---|---|---|---|---|---|---|---|
Discovery |
rs12402555 |
1 |
194053792 |
CDC73|KCNT2 |
A |
G |
0.044 |
0.038 |
5.864 |
1.120E-06 |
Validation |
rs12402555 |
1 |
194053792 |
|
T |
C |
0.057 |
0.766 |
0.883 |
8.714E-01 |
Meta analysis |
|
|
|
|
|
|
|
|
4.142 |
1.492E-05 |
Discovery |
rs71427857 |
2 |
183497840 |
PDE1A|DNAJC10 |
A |
C |
0.042 |
0.108 |
6.078 |
1.289E-06 |
Validation |
rs71427857 |
2 |
183497840 |
|
A |
C |
0.046 |
1.000 |
5.181 |
1.636E-03 |
Meta analysis |
|
|
|
|
|
|
|
|
5.759 |
7.927E-09 |
Discovery |
rs13023449 |
2 |
183499313 |
PDE1A|DNAJC10 |
G |
A |
0.042 |
0.111 |
6.071 |
1.320E-06 |
Validation |
rs13023449 |
2 |
183499313 |
|
G |
A |
0.046 |
1.000 |
5.163 |
1.674E-03 |
Meta analysis |
|
|
|
|
|
|
|
|
5.748 |
8.301E-09 |
Discovery |
rs11734719 |
4 |
17713542 |
FAM184B |
G |
A |
0.168 |
0.814 |
4.657 |
1.264E-06 |
Validation |
rs11734719 |
4 |
17713542 |
|
C |
T |
0.187 |
1.000 |
0.669 |
4.024E-01 |
Meta analysis |
|
|
|
|
|
|
|
|
2.580 |
3.444E-04 |
Discovery |
rs16868761 |
6 |
36955643 |
MTCH1|FGD2 |
A |
G |
0.010 |
1.000 |
12.310 |
5.134E-06 |
Validation |
rs16868761 |
6 |
36955643 |
|
T |
C |
0.011 |
1.000 |
0.000 |
9.971E-01 |
Meta analysis |
|
|
|
|
|
|
|
|
NA |
NA |
Discovery |
rs11138921 |
9 |
83547081 |
TLE4|TLE1 |
G |
A |
0.243 |
0.720 |
4.179 |
7.802E-06 |
Validation |
rs11138921 |
9 |
83547081 |
|
G |
A |
0.238 |
1.000 |
1.385 |
3.622E-01 |
Meta analysis |
|
|
|
|
|
|
|
|
2.559 |
8.152E-05 |
Discovery |
rs7316131 |
12 |
62669425 |
USP15 |
A |
G |
0.015 |
0.417 |
10.810 |
1.395E-06 |
Validation |
rs7316131 |
12 |
62669425 |
|
A |
G |
0.016 |
1.000 |
0.000 |
9.965E-01 |
Meta analysis |
|
|
|
|
|
|
|
|
NA |
NA |
Discovery |
rs74886773 |
15 |
58638467 |
AQP9|LIPC |
A |
G |
0.028 |
0.414 |
8.874 |
5.524E-06 |
Validation |
rs74886773 |
15 |
58638467 |
|
A |
G |
0.034 |
0.312 |
0.000 |
9.947E-01 |
Meta analysis |
|
|
|
|
|
|
|
|
NA |
NA |
Nine SNPs at seven loci identified by a discovery GWAS were validated by an independent study, followed by the calculation of P-value as a meta-analysis. MAF: minor allele frequency in the control group in the discovery phase (Phase 2). HME: P-value for the test of Hardy-Weinberg’s equilibirum (HWE) by the exact test.
A previous study reported that the CBLN2 locus at ch 18q22.3, was associated with PAH [6]. We attempted to replicate these results with our GWAS. Although SNP rs2217560 (Position 70150939 on chr 18 in build 37) which was reported to be associated with PAH by Germain and colleagues, was not included in our platform, proximal SNPs were examined. However, we did not find any association between the 34 SNPs that were located close to rs2217560 (from Position 70121268 to 70181319) with PAH in our study (P > 0.15). Thus, our data did not successfully replicate the previously described association.
Replication and validation of our GWAS study using another independent cohort
We next validated the associations observed in our discovery phase using another independent cohort of 21 patients and 991 controls (JPDSC-Phase 1). Table 3 shows the clinical characteristics of I/HPAH patients in the validation study. In this validation study, we examined the association of eight SNPs at seven loci (Table 2) with PAH using 21 cases and 991 controls (Table 1). The statistical method used to evaluate the association was identical to the one used for our first genome-wide study. Only rs71427857 and rs13023449, both at the PDE1A|DNAJC10 locus, showed significant associations in the validation study (P=1.64 × 10-3 and P=1.67 × 10-3, respectively) (Table 2). The P-value threshold for each of the eight SNPs in the validation study was 0.0056 (0.05/9), in accordance with Bonferroni’s correction method for multiple comparisons.
Table 3: Patients’ genetic and hemodynamic characteristics in the replication cohort
ID |
Age |
Sex |
Ethnics |
BMPR2 mutation |
mPAP [mmHg] |
CO [l/min] |
PVR [dynes ▪ sec ▪ cm-5] |
|---|---|---|---|---|---|---|---|
1 |
27 |
Female |
Japanese |
- |
69 |
4.69 |
1176 |
2 |
41 |
Female |
Japanese |
- |
74 |
3.40 |
1552 |
3 |
45 |
Female |
Japanese |
- |
72 |
2.00 |
2584 |
4 |
43 |
Female |
Japanese |
- |
82 |
2.20 |
2560 |
5 |
30 |
Female |
Japanese |
- |
59 |
3.81 |
1008 |
6 |
45 |
Female |
Japanese |
- |
42 |
3.50 |
824 |
7 |
59 |
Female |
Japanese |
- |
46 |
2.60 |
1200 |
8 |
36 |
Female |
Japanese |
+ |
66 |
1.95 |
2378 |
9 |
39 |
Female |
Japanese |
- |
49 |
3.10 |
1056 |
10 |
66 |
Female |
Japanese |
- |
32 |
2.84 |
734 |
11 |
47 |
Female |
Japanese |
- |
51 |
3.87 |
952 |
12 |
53 |
Female |
Japanese |
+ |
43 |
5.23 |
581 |
13 |
47 |
Female |
Japanese |
- |
51 |
2.52 |
1460 |
14 |
42 |
Female |
Japanese |
- |
39 |
4.50 |
622 |
15 |
53 |
Female |
Japanese |
- |
47 |
6.38 |
451 |
16 |
45 |
Female |
Japanese |
- |
54 |
3.62 |
1016 |
17 |
70 |
Female |
Japanese |
- |
45 |
4.17 |
614 |
18 |
60 |
Female |
Japanese |
- |
38 |
3.98 |
643 |
19 |
64 |
Female |
Japanese |
+ |
35 |
3.87 |
599 |
20 |
33 |
Female |
Japanese |
- |
54 |
4.93 |
698 |
21 |
49 |
Female |
Japanese |
- |
66 |
3.50 |
1412 |
The hemodynamic data were evaluated in their initial right heart catheterization.
mPAP: mean pulmonary arterial pressure; CO: cardiac output; PVR: pulmonary vascular resistance.
Meta-analysis and imputation study
Meta-analyses using data from the discovery and validation studies indicated that Pmeta for rs71427857 and rs13023449 were 7.93 × 10-9 and 8.30 × 10-9, respectively (Table 2). Both of these Pmeta values are lower than the required threshold of 5 x 10-8, and were considered to be significant as a result of the meta-analysis. Odds ratios obtained from the validation study were 5.18 (95%CI 1.86 – 14.42) and 5.16 (95%CI 1.86 – 14.37) for rs71427857 and rs13023449, respectively. Since odds ratios obtained in the discovery phase are often overestimated, we consider values from the validation study more reliable.
In our present study, regional plots near the PDE1A|DNAJC10 locus indicated that the associated SNPs are clustered in the 5’ region of PDE1A (Figure 2).
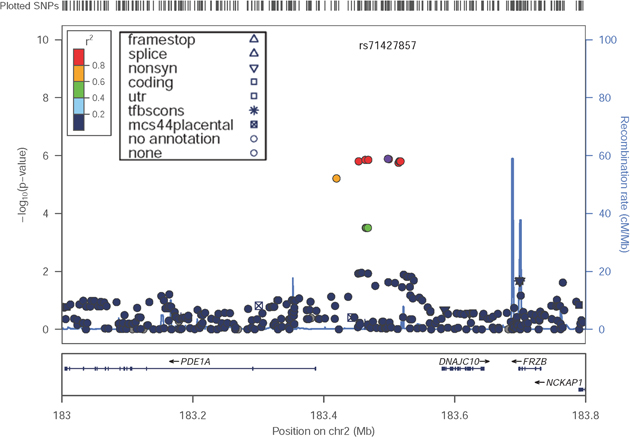
Figure 2: Locus-specific plots at the PDE1A|DNAJC10 locus in the discovery GWAS. The figure was drawn by LocusZoom Version 1.122.
We imputed genotypes for HapMap3 SNPs using IMPUTE version 2 (9) across the PDE1A|DNAJC10 locus (Supplementary Figure 2). In general, the imputation results were consistent with the genotyped SNP results, although some ungenotyped SNPs in the same region were added as associated SNPs. To exclude the possibility of genotyping mutations that were in fact false positives, which had arisen by calling errors from SNP chips, we performed direct genotyping by Sanger sequence. All cases were directly sequenceded at the rs71427857 and rs13023449 locus. As shown by the mutant sample in Supplementary Figure 3, all cases with minor alleles showed the same results, both at rs71427857 and rs13023449 by the direct sequence method.
Immunohistochemical studies and quantitative RT-PCR
To confirm the contribution of the PDE1A gene to the pathogenesis of PAH, confocal microscopic analyses and double labeling with PDE1A and SM22 alpha, a marker of adult smooth muscle, were used to investigate the expression of PDE1A protein in lung specimens from 5 patients with PAH, and control subjects. We found strong staining of PDE1A in paraffin embedded lung tissue from patients with I/HPAH when compared to control subjects (Figure 3A). Interestingly, more intense immunoreactivity was noted for PDE1A protein in distal remodeled pulmonary artery walls from IPAH patients versus controls (Figure 3B). Also, the mRNA expression levels of PDE1A in PA-SMCs from IPAH were significantly higher than those from controls (Figure 3C), suggesting that the increased expression of PDE1A in PA-SMCs from the patients with IPAH was associated with the transcriptional activation.
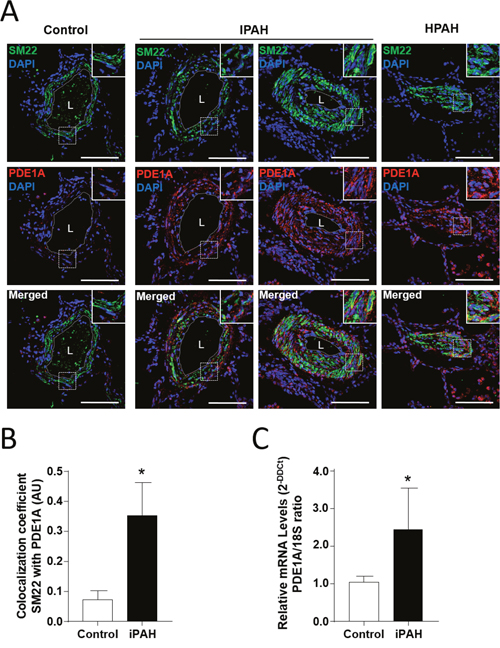
Figure 3: The augmentation of PDE1A expressions in pulmonary arterial smooth muscle cells (PA-SMCs) from patients with I/HPAH. (A) Representative photomicrograph of PDE1A immunostaining in distal pulmonary arteries in lung sections of controls and idiopathic and heritable pulmonary arterial hypertension (I/HPAH) patients. Scale bars= 50 μm in all sections. L=vessel lumens. (B) Ratio of colocalization cells of PDE1A and SM22 positive cells in distal pulmonary arteries in lung sections from controls and IPAH patients. (C) PDE1A mRNA levels detected by quantitative reverse transcription PCR (RT-qPCR) in cultured PA-SMCs from controls and IPAH patients. * p-value < 0.05.
DISCUSSION
In this study, we performed GWAS in a Japanese population comprising 44 individuals with I/HPAH, and 2,993 controls. Out of seven loci identified in the discovery study, a novel susceptibility locus, PDE1A|DNAJC10, was identified in the replication study. More intense immunochemical staining for PDE1A protein in distal remodeled pulmonary artery walls was observed in PAH patients than controls.
Cyclic nucleotide PDEs play important roles in signal transduction by regulating intracellular cyclic nucleotide concentrations through the hydrolysis of cAMP and/or cGMP to their respective nucleoside monophosphates [9]. There are at least 11 families of one or more genes that encode PDE superfamily members. Furthermore, different proteins are synthesized from a single gene by different modifications of the encoding mRNA [14]. Members of the PDE1 family, such as PDE1A, are Ca2+/calmodulin-dependent PDEs that are activated by calmodulin in the presence of Ca2+ [10, 11].
PDEs have attracted attention from researchers, as well as pharmaceutical companies, because cAMP and cGMP are important signaling molecules in many tissues and organs [9]. Inhibitors of PDEs are likely to have a variety of pharmacological effects, many of which may be of clinical use. Nevertheless, at present, only a few PDE inhibitors are in widespread clinical use. For example, the PDE3 inhibitors, milrinone and cilostazol, are used to treat cardiovascular disease, [12, 13] and the PDE5 inhibitors, sildenafil, vardenafil and tadalafil, are useful for treating male erectile dysfunction [9]. PDE5 inhibitors are also useful for treating patients with pulmonary arterial hypertension [14].
Increase in the expression of PDE1A, as well as PDE5, has been shown in pulmonary arterial smooth muscle cells from patients with PAH [15]. In addition, Selective inhibition of PDE1 is known to augment the vasodilatory effect of inhaled nitric oxide in a PH model. Therefore, it is suggested that PDE1A is related with the development of PAH [16].
Genetic studies in human diseases may lead to an understanding of a protein’s function in the human body. For example, mutations in PDE6A and PDE6B are associated with autosomal recessive retinitis pigmentosa [17, 18]. Similarly, human PDE4D haplotypes and single-nucleotide polymorphisms (SNPs) correlate with ischemic stroke, and PDE4B SNPs are associated with schizophrenia [19, 20].
This study had several limitations. First, the number of I/HPAH patients included in this study was relatively small. One of the reasons why we could identify a novel susceptibility locus is that we used data of a large number of healthy controls. And since the ethnics of the patients were all Japanese, they were ethnically matched with the controls. In a previous GWAS study of Japanese population which identified genes associated with allopurinol-related Stevens-Johnson syndrome and toxic epidermal necrolysis, the identification was enabled by comparing only 14 patients with 991 healthy controls [21]. Second, we included not only young patients but also older populations both in discovery cohort and replication cohort suggesting that there is a possibility to include atypical I/HPAH patients among the cohort. However, all of the patients included in this study were diagnosed in experienced pulmonary hypertension centers, and the fact certifies all of the cases obtained typical characteristics of I/HPAH independent from the effect from other diseases such as left ventricular dysfunctions or pulmonary diseases regardless of the older ages. Third, we screened all I/HPAH cases for BMPR2 mutations with Sanger sequencing. However, Sanger sequencing is not enough to identify patients with large deletions or duplications, and combination use of multiplex ligation-dependent prove amplication with Sanger sequencing might be more appropriate. Fourth, our data did not successfully replicate the CBLN2 finding in the previous study [6]. The different result might come from the difference of races included in each study; the genetic factors which modify PAH disease expression could be different between races. Further analysis is required to confirm the association of the locus and development of I/HPAH in it other cohorts with different races.
In conclusion, this GWAS in I/HPAH patients, irrespective of the presence of a BMPR2 mutation, provided additional information about the association between a genetic locus, PDE1A|DNAJC10, and the disease, suggesting that PDE1A could be a novel therapeutic target of PAH.
MATERIALS AND METHODS
Study populations
We included patients with I/HPAH in this study. Diagnosis of PAH was hemodynamically confirmed for all cases included in the study by right-heart catheterization (discovery and validation stages). Cases were identified at three major pulmonary hypertension centers: Keio University, Chiba University, and Kyorin University in Japan. For all cases, PAH was defined as a mean pulmonary arterial pressure not lower than 25 mm Hg, and pulmonary capillary wedge pressure not higher than 15 mm Hg at baseline, according to protocols previously described [22]. A diagnosis of I/HPAH was declared only after clinical and biological investigations had eliminated all known causes of PAH. We excluded cases with complications such as lung or liver disease, even if not severe. After a confirmed diagnosis of I/HPAH, all cases were screened for BMPR2 mutations by Sanger sequencing. Eight of 23 patients included in the discovery GWAS had mutations in the BMPR2 gene, while 3 of the 21 patients included in the validation study had BMPR2 mutations. We included only a single case from a family for the present study.
Controls consisted of 2,002 (Phase 2) and 991 (Phase 1) healthy Japanese individuals. Control samples were collected at two different times by the Japan PGx Data Science Consortium (JPDSC) [23]. Written informed consent was provided for enrollment in the study, and the GWAS study was approved by IRB of Keio University.
GWAS and validation genotyping
Genomic DNA was isolated from whole blood and applied to the Illumina Human Omni2.5-8 Beadchip ver 1.1 SNP chips (cases) or Illumina Human Omni2.5-8 Beadchip ver 1.0 (controls Phase 2) SNP arrays. Genotyping conditions were as described in the manufacturer’s manual.
Validation genotyping was carried out at Keio University by Sanger sequencing of PCR products generated with forward primer 5’- GCAATGCTGCTTTGTTTCTCTG -3’ and reverse primer 5’ - TGACTGAGACTAGTGGGGAGTC-3’. The appropriate temperature for successful resolution were determined by the dHPLC melting algorithm. Samples with an altered dHPLC profile were sequenced using the BigDye Terminator cycle sequencing kit (Applied Biosystems, Foster city, CA) on an ABI 3730xl DNA sequencer (Applied Biosystems). The resulting sequences were compared with the reference sequence of the SNPs sites with the ABI SeqScape software (Applied Biosystems).
Immunostaining
Lung specimens were fixed in 4% paraformaldehyde and embedded in Paraffin. Sections (5-μm) were dewaxed and progressively rehydrated. Lung sections were then blocked in 5% bovine serum albumin for 30 minutes at room temperature and stained with anti-PDE1A (Novus Biologicals, Lilles, France) and anti-SM22 (Santa Cruz Biotechnology, Heidelberg, Germany) antibodies overnight at 4°C. Secondary antibodies, conjugated with a fluorescent label (Interchim, Montluçon, France), were applied for 1 hour at room temperature. Nuclei were labelled using DAPI (Life technologies). Sections were mounted using ProLong Gold antifade reagent (Life technologies). Images were taken using a LSM700 confocal microscope (Zeiss). Overlap between the PDE1A and SM22 proteins, as represented by the Manders coefficient, was determined using the ZEN software (Blue edition, version 2012, Zeiss) from a minimum of 8 distal pulmonary arteries for each group.
Quantitative RT-PCR
Total RNA was isolated from cultured PA-SMCs by RNeasy mini kit (Qiagen, Courtaboeuf, France). Total RNA (2 μg) was reverse-transcribed using High Capacity RNA-to-cDNA™ Kit (Invitrogen) per manufacturer’s instructions. Gene expression levels of PDE1A was quantified using pre-verified Assays-on-Demand TaqMan primer/probe sets (Applied Biosystems, St. Aubin, France) and normalized to 18S ribosomal RNA using the comparative Cycle threshold (Ct) method (2-ΔΔCt).
Statistical methods
Associations in GWAS were tested by multivariate logistic regression using PLINK software (v 1.0.7, PLINK) [24]. Age and gender were used as covariates. Manhattan plots and QQ-plots were drawn using the R environment (v 2.15.2). Regional plots were drawn by LocusZoom Version 1.110. Meta-analysis was performed by the inverse-variance method. Other statistical methods were the same as those in our previous papers [25, 26].
Author contributions
Mai Kimura drafted the manuscript and performed the genotyping for the GWAS; Yuichi Tamura designed the study and wrote the manuscript; Makoto Takei collected subjects and participated in the diagnostic evaluations; Kenjiro Kosaki performed the genotyping for the GWAS; Nobuhiro Tanabe collected subjects and participated in the diagnostic evaluations; Koichiro Tatsumi collected subjects and participated in the diagnostic evaluations; Tsutomu Saji collected subjects and participated in the diagnostic evaluations; Toru Satoh collected subjects and participated in the diagnostic evaluations; Masaharu Kataoka collected subjects and participated in the diagnostic evaluations; Shigeo Kamitsuji analyzed the GWAS data; Naoyuki Kamatani contributed to the overall GWAS study design; Christophe Guiganbert, Raphaël Thuillet, Ly Tu and Marc Humbert contributed to the demonstration of increased PDE1A protein expression in patients with PAH; Keiichi Fukuda contributed to the overall GWAS study design; and Motoaki Sano contributed to the overall GWAS study design.
ACKNOWLEDGMENTS
We acknowledge the medical staff of the Department of Cardiology, Keio University Hospital, Tokyo, Japan for their work on completion of medical records.
CONFLICTS OF INTEREST
The authors declare no competing financial interests.
FUNDING
This work was supported by Health and Labor Sciences Research Grants in Japan.
REFERENCES
1. D’Alonzo GE, Barst RJ, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, Fishman AP, Goldring RM, Groves BM, Kernis JT, Levy PS, Pietra GG, Reid LM, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med. 1991; 115:343-349.
2. Guignabert C, Tu L, Girerd B, Ricard N, Huertas A, Montani D, Humbert M. New molecular targets of pulmonary vascular remodeling in pulmonary arterial hypertension: importance of endothelial communication. Chest. 2015; 147:529-537.
3. Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, Kalachikov S, Cayanis E, Fischer SG, Barst RJ, Hodge SE, Knowles JA. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet. 2000; 67:737-744.
4. Larkin EK, Newman JH, Austin ED, Hemnes AR, Wheeler L, Robbins IM, West JD, Phillips JA 3rd, Hamid R, Loyd JE. Longitudinal analysis casts doubt on the presence of genetic anticipation in heritable pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012; 186:892-896.
5. Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA 3rd, Loyd JE, Nichols WC, Trembath RC. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat Genet. 2000; 26:81-84.
6. Germain M, Eyries M, Montani D, Poirier O, Girerd B, Dorfmuller P, Coulet F, Nadaud S, Maugenre S, Guignabert C, Carpentier W, Vonk-Noordegraaf A, Levy M, et al. Genome-wide association analysis identifies a susceptibility locus for pulmonary arterial hypertension. Nat Genet. 2013; 45:518-521.
7. Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006; 38:904-909.
8. Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999; 55:997-1004.
9. Maurice DH, Ke H, Ahmad F, Wang Y, Chung J, Manganiello VC. Advances in targeting cyclic nucleotide phosphodiesterases. Nat Rev Drug Discov. 2014; 13:290-314.
10. Michibata H, Yanaka N, Kanoh Y, Okumura K, Omori K. Human Ca2+/calmodulin-dependent phosphodiesterase PDE1A: novel splice variants, their specific expression, genomic organization, and chromosomal localization. Biochim Biophys Acta. 2001; 1517:278-287.
11. Fidock M, Miller M, Lanfear J. Isolation and differential tissue distribution of two human cDNAs encoding PDE1 splice variants. Cell Signal. 2002; 14:53-60.
12. Movsesian MA, Kukreja RC. Phosphodiesterase inhibition in heart failure. Handb Exp Pharmacol. 2011; 204:237-249.
13. Kanlop N, Chattipakorn S, Chattipakorn N. Effects of cilostazol in the heart. J Cardiovasc Med (Hagerstown). 2011; 12:88-95.
14. Archer SL, Michelakis ED. Phosphodiesterase type 5 inhibitors for pulmonary arterial hypertension. N Engl J Med. 2009; 361:1864-1871.
15. Murray F, Patel HH, Suda RY, Zhang S, Thistlethwaite PA, Yuan JX, Insel PA. Expression and activity of cAMP phosphodiesterase isoforms in pulmonary artery smooth muscle cells from patients with pulmonary hypertension: role for PDE1. Am J Physiol Lung Cell Mol Physiol. 2007; 292:L294-303.
16. Evgenov OV, Busch CJ, Evgenov NV, Liu R, Petersen B, Falkowski GE, Petho B, Vas A, Bloch KD, Zapol WM, Ichinose F. Inhibition of phosphodiesterase 1 augments the pulmonary vasodilator response to inhaled nitric oxide in awake lambs with acute pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2006; 290:L723-L729.
17. Cote RH. Cyclic Nucleotide Phosphodiesterases in Health and Disease. CRC Press, 2007, p.165-194.
18. Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006; 368:1795-1809.
19. Gretarsdottir S, Thorleifsson G, Reynisdottir ST, Manolescu A, Jonsdottir S, Jonsdottir T, Gudmundsdottir T, Bjarnadottir SM, Einarsson OB, Gudjonsdottir HM, Hawkins M, Gudmundsson G, Gudmundsdottir H, et al. The gene encoding phosphodiesterase 4D confers risk of ischemic stroke. Nat Genet. 2003; 35:131-138.
20. Fatemi SH, King DP, Reutiman TJ, Folsom TD, Laurence JA, Lee S, Fan YT, Paciga SA, Conti M, Menniti FS. PDE4B polymorphisms and decreased PDE4B expression are associated with schizophrenia. Schizophr Res. 2008; 101:36-49.
21. Tohkin M, Kaniwa N, Saito Y, Sugiyama E, Kurose K, Nishikawa J, Hasegawa R, Aihara M, Matsunaga K, Abe M, Furuya H, Takahashi Y, Ikeda H, et al. A whole-genome association study of major determinants for allopurinol-related Stevens-Johnson syndrome and toxic epidermal necrolysis in Japanese patients. Pharmacogenomics J. 2013; 13:60-69.
22. Tamura Y, Ono T, Kuwana M, Inoue K, Takei M, Yamamoto T, Kawakami T, Fujita J, Kataoka M, Kimura K, Sano M, Daida H, Satoh T, Fukuda K. Human pentraxin 3 (PTX3) as a novel biomarker for the diagnosis of pulmonary arterial hypertension. PLoS One. 2012; 7:e45834.
23. Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007; 81:559-575.
24. Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, Boehnke M, Abecasis GR, Willer CJ. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010; 26:2336-2337.
25. Sano M, Kamitsuji S, Kamatani N, Hong KW, Han BG, Kim Y, Kim JW, Aizawa Y, Fukuda K. Genome-wide association study of electrocardiographic parameters identifies a new association for PR interval and confirms previously reported associations. Hum Mol Genet. 2014; 23:6668-6676.
26. Kou I, Takahashi Y, Johnson TA, Takahashi A, Guo L, Dai J, Qiu X, Sharma S, Takimoto A, Ogura Y, Jiang H, Yan H, Kono K, et al. Genetic variants in GPR126 are associated with adolescent idiopathic scoliosis. Nat Genet. 2013; 45:676-679.