
Introduction
Hepatocellular carcinoma (HCC) has been categorized as one of the five most common cancers in the world in recent years. The incidence of HCC has dramatically increased in Western countries and has become more prevalent in Asian countries in the last decades due to chronic liver disease, chronic hepatitis or cirrhosis induced by hepatotropic viruses, aflatoxin and alcohol consumption [1]. Even with aggressive therapy, the 5-year survival rate of patients with advanced HCC remains less than 10%[2]. This poor prognosis is due to high recurrent and metastatic rates even after the use of current treatment modalities such as surgery, trans-hepatic artery chemoembolzation, radiofrequency ablation, radiotherapy, and multitarget tyrosine kinase inhibitors[2-4]. Clearly, the development of novel therapeutics against HCC is an urgent task.
Lapatinib is a reversible tyrosine kinase inhibitor (TKI) developed for targeted therapy by GlaxoSmithKline [5-6]. Its known targets are tyrosine kinase in the cytoplastic domain of epidermal growth factor receptor (EGFR or ErbB1) and epidermal growth factor receptor-2 (ErbB2 or Her2). Due to the high incidence of EGFR/Her2 signaling amplification associated with a poor prognosis in some types of human cancers such as breast cancer, lapatinib in combination with capecitabine has been approved by the Food and Drug Administration as a therapy for patients with Her2-overexpressing or metastatic breast cancer that failed to respond to anthracycline, taxane, or the previous targeting agent, anti-ErbB2 monoclonal antibody transtuzumab (Herceptin) [5-6]. The advantages of lapatinib treatment include the specificity of the EGFR family members, better applicability through oral administration, and tolerability of adverse effects with high compliance. Lapatinib is well accepted in ongoing preclinical or clinical applications for treatment of various solid tumors, including those of the breast, lung, HCC, head and neck, vulva, colon, prostate, ovarian and gastric cancers [5, 7-9]. Compared to trastuzumab, lapatinib treatment also shows higher activity in Her2-positive breast cancer patients with PTEN mutations [5, 10]. Although the phase II trials for lapatinib and HCC show a marginal benefit to a subgroup of patients without predictive markers yet characterized [7-8], the role of lapatinib in the treatment of HCC remains to be elucidated.
Many anticancer drugs work by induction of programmed cell death in tumor cells. Apoptosis is the first well-characterized programmed cell death. Its unique point is cells with condensed and fragmented chromatin. Besides apoptosis, many studies have recently focused on anticancer-drug-induced non-apoptotic cell death, such as autophagic cell death and necroptosis [11-12]. In cells, autophagy was originally found to be a strategy to accelerate material and energy recycling by the digestion of aged organelles with autophagy-specific lysosomes, also called autophagosomes. Some cancer cells use this kind of autophagy to increase proliferation under stress or in some nutrient-insufficient microenvironments [11, 13]. However, in recent years, studies found autophagosome-associated autophagic cell death as the second type of well-studied programmed cell death. Compared to apoptosis, the different features of autophagic cell death include massive autophagic vacuolization (double-membraned vacuoles) inside the cytoplasm and the absence of chromosome condensation and nuclear fragmentation. Except for autophagic vacuoles, the specific features of autophagic cell death also include beclin-1 (ATG6), ATG5, ATG12, or ATG7 involvement and LC-I to LC-II conversion. More papers discuss breaking the apoptosis rules we knew a decade ago. For example, apoptotic cascade molecules, such as FADD and caspase-8, are also involved in autophagy, as mentioned above, since recruitment of TNFR molecule caspase-8 and FADD with autophagy-related proteins ATG5, ATG12, and ATG16L were observed in the development (generation) of early autophagosomes in certain autophagy signal pathways [14-15].
Potential targets for anticancer therapy in HCC include the EGFR and Her2 overexpressed in HCC and directly implicated in hepatocarcinogenesis. Previous investigations indicate that EGFR is actively expressed in human HCC cells and EGF is required for the growth of HCC cells [4, 16-17]. In addition, Her2 is expressed in a significant number of HCCs, and may be an independent prognostic factor [18]. In this study, we chose human HCC cells as a model system for dissecting the mechanisms of lapatinib-induced HCC cell death to extend the application of lapatinib in HCC, such as that for the administration of single therapeutic as reported in phase II clinical trials [7-8], as well as combinatory treatment with other drugs or radiotherapy. We found the mechanism of lapatinib-induced cytotoxicity of HCC is autophagic cell death.
Results
Cytotoxicity of lapatinib in HCC cells
First, we tested lapatinib-induced cytotoxicity in several kinds of HCC cell lines, including well differentiated Huh7, HepG2, and poorly differentiated HA22T-VGH (HA22T) cells. Relatively viable cells were tested by a MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt] tetrazolium compound assay. As shown in Fig. 1, after 3 days of lapatinib exposure, the viability of these cells was dramatically reduced in a dose dependent manner when compared with the relative viability of cells treated with the same volume of vehicle dimethyl sulfoxide (DMSO). According to this assay, the EC50 for growth inhibition in lapatinib-treated Huh7, HepG2, and HA22T was about 2.11, 3.42 and 4.85 μM, respectively. In addition, we also detected the targets of lapatinib, EGFR and ErbB2, in these HCC cells (Fig. S1).
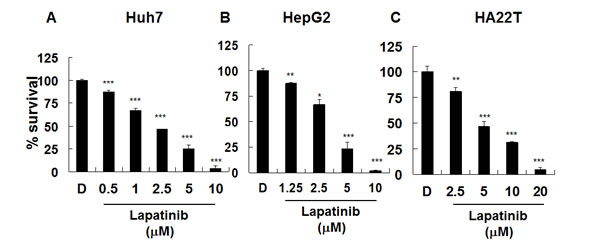
Figure 1: Inhibition of hepatoma cell proliferation by lapatinib. Huh7 (A), HepG2 (B), or HA22T (C) HCC cells were left untreated or treated with 0.1% DMSO (vehicle, D) or with DMSO containing various concentrations of lapatinib (1.25, 2.5, 5, or 10 μM) for 3 days. Relative amounts of viable cells were detected by MTS assay. O.D. values from DMSO-treated control cells were designated 100%.
Induction of non-apoptotic cell death by lapatinib in HCC cell lines
As shown in Fig. 2, lapatinib-induced more than 90% or 80% of dead (PI-positive) Huh7 or HA22T cells (left panel), respectively; however, around or less than 10% sub G1 (hypoploid) cells (or DNA laddering, right panel) were detected [19-21]. We found the similar pattern for lapatinib-treated HepG2 cells (data not shown). We also used the Annexin V assay to detect externalized phosphotidyl serine to confirm the apoptosis assay (data not shown). To further elucidate lapatinib-induced cell death, we found the involvement of mitochondria in the lapatinib-induced death pathway due to the loss of mitochondrial integrity in HCC cells (Fig. S2). These data suggest mitochondria-involved non-apoptotic cell death induction (like autophagy or necrosis) by lapatinib in HCC cells, similar to the effect of lapatinib in K562 cells in previous investigations.
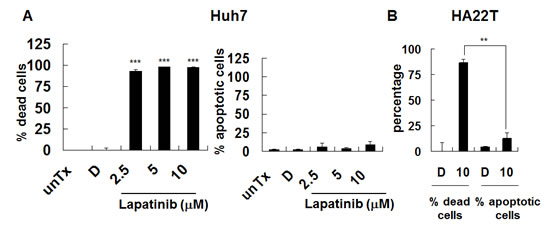
Figure 2: Induction of non-apoptotic cell death by lapatinib in HCC cells. Cells were left untreated (unTx) treated, with 0.1% DMSO (vehicle, D) or 2.5–10 μM lapatinib for 3 days. After collection, cells were split into 2 tubes and resuspended in either PI containing PBS buffer (A or left panel of C, for detection of percent of total dead cells) or PI-containing hypotonic buffer (B or right panel of C, for detection of percent of hypodiploid or apoptotic cells) by flow cytometry.
Lapatinib-induced autophagic cell death in HCC cells
Since we found lapatinib effective in killing HCC cells (near 100%, Fig. 2) with less than 10% attributed to apoptosis, we wondered if these cells could undergo autophagy or necrosis after lapatinib treatment. We tested whether lapatinib induced autophagic cell death in HCC cells by the detection of autophagic vacuoles-containing cells. As shown in Fig. 3, autolysosome formation (acridine orange positive cells) was detected after Huh7, HepG2 or HA22T cells were treated with lapatinib. In addition, punctuate LC3 aggregates (the evidence of LC3 conversion) were observed after 3 kinds of HCC cells were treated with lapatinib (Fig. 4)[22].
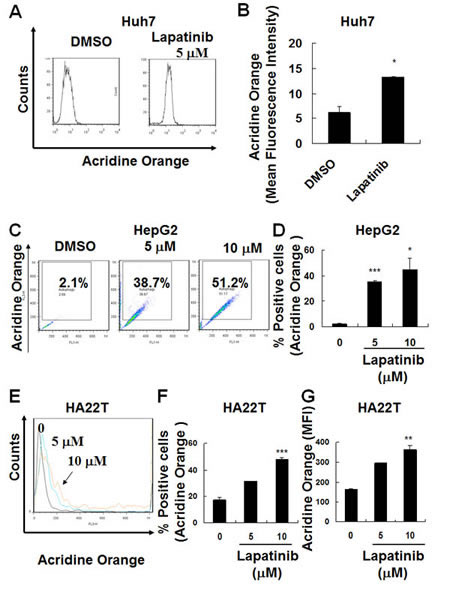
Figure 3: Induction of autophagy in lapatinib-treated HCC cells. After DMSO, 5 or 10 μM lapatinib treatment for 3 days, Huh7 (A-B), HepG2 (C-D) and HA22T (E-G) HCC cells were stained with acridine orange, and the autophagic cells was analyzed by flow cytometry. Data are expressed as the mean fluorescence intensity (B or G) or percentage of acridine orange positive cells (C, D or F).
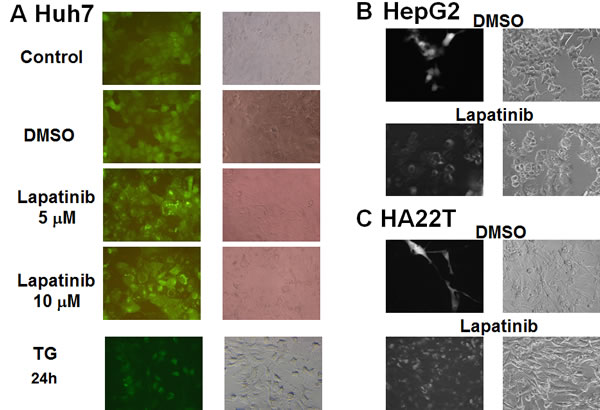
Figure 4: Induction of LC3 aggregations by lapatinib in HCC cells. The stable expression of GFP-LC3 in HCC cells was prepared by the transduction of a GFP-LC3-containing lentivirus. After DMSO, 5 or 10 μM lapatinib or 150 nM thapsigargin (TG, positive control) treatment for 3 days; Huh7 (A), HepG2 (B), or HA22T (C) cells were observed by fluorescence microscopy.
To further confirm lapatinib-caused autophagy in HCC cells, we checked the expression levels of some autophagy-related proteins in lapatinib-treated cells by immunoblotting [20]. As shown in Fig. 5, lapatinib induced LC3-II conversion, an increase of the autophagy-related proteins LC3 (LC3-II conversion), Beclin-1 (ATG6), ATG5, ATG7 and BNIP in HCC cells. Lapatinib also decreased the expression of p62 in Huh7 and HA22T cells. This is consistent with the degradation of p62 by autophagy[23]. We also checked the morphology of lapatinib-treated HCC cells by transmission electronic microscopy (TEM). As shown in Fig. 6, lapatinib induced a large amount of vacuoles in drug-treated HCC cells compared to the vehicle DMSO-treated cells. This phenomenon is specifically evident in both Huh7 and HepG2.
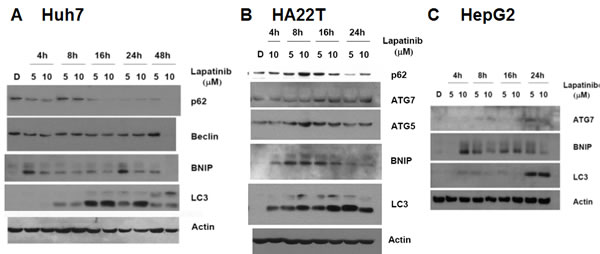
Figure 5: Induction of LC3 aggregations and autophagy-related genes by lapatinib in Huh7 (A), HA22T (B) and HepG2 (C) HCC cells. Cells were treated with DMSO or 5 or 10 μM lapatinib treatment for 4-48h as indicated. Cell lysates were then collected, subjected into 12% SDS-PAGE, and immunoblotted with antibodies against p62, Beclin, BNIP, ATG5, ATG7 LC3 and actin.
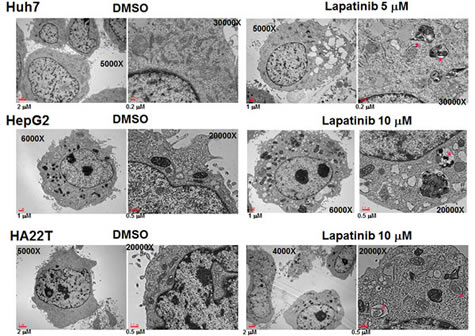
Figure 6: Microphotographs of lapatinib-treated HCC cells. (A) After DMSO or 5 or 10 μM lapatinib treatment for 2 days, Huh7 (A), HepG2 (B) or HA22T (C) HCC cells were fixed, stained and observed by transmission electron microscopy. Mitochondria with vacuolization were marked with red dots. The magnification information is as indicated in each figure.
To further confirm lapatinib-induced autophagic cell death, we tested the effects of two inhibitors, 3-methyladenine (3-MA) and chloroquine, which block the early stages of autophagic processes with a PtdIns3K inhibitor (3-MA) or a lysosomotropic compound for elevating pH in lysosomes/autophagosomes (chloroquine) [20]. Lapatinib-induced growth inhibition was effectively blocked when HCC cells were incubated with 3-MA or chloroquine (Fig. 7). This data suggests lapatinib induced autophagic cell death in HCC cells.
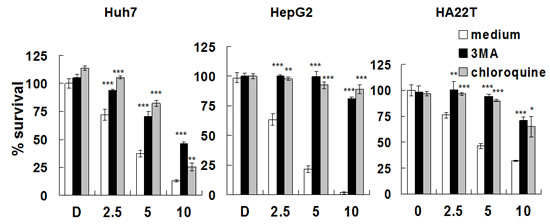
Figure 7: Rescue of Huh7, HepG2 or HA22T cells from lapatinib-induced cytotoxicity by the autophagy inhibitors. Huh7, HepG2 or HA22T cells were left untreated or were treated with various concentrations of lapatinib in the presence or absence of 1.25-mM 3-MA or 5 μM chloroquine for 48h. Relative amounts of viable cells were detected using the MTS assay, and the relative percentage of growth inhibition was calculated as described in Fig. 1.
To further test whether lapatinib induced autophagic cell death, we explored the role of autophagic proteins, such as ATG5, ATG7 and Beclin-1. In the vehicle- or lapatinib-treated cells, we knocked down expression of these autophagy-related proteins (Fig. S3) after transduction with a short hairpin RNA (shRNA) expression lentivirus system [21]. As shown in Fig. 8, the specific knockdown of ATG5, ATG7, and Beclin-1 mRNA, but not the non-targeting red fluorescent protein (RFP) mRNA, rescued the cells from lapatinib-mediated cell death. These data indicate, consistent with our previous figures, the involvement of these autophagic proteins with lapatinib-induced autophagic cell death in HCC cells.
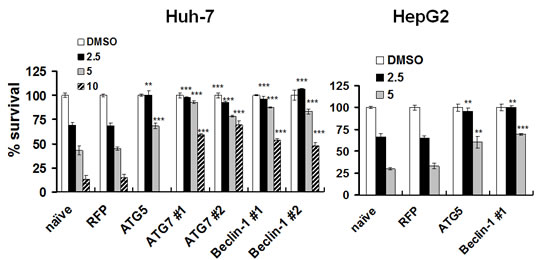
Figure 8: Protection of HCC cells from lapatinib-induced cytotoxicity by the knockdown of autophagy-related proteins. After transduction with shRNA expression lentivirus as indicated, Huh7 or HepG2 cells were selected and were treated with DMSO or 2.5 - 10 μM lapatinib for 48h, and then the relative percentages of growth inhibition were detected using the MTS assay and calculated as described in Fig. 1.
Lapatinib inhibits hepatoma tumor growth in vivo xenografts
Since we have demonstrated the cytotoxicity effect of lapatinib in hepatoma cells, we wondered whether lapatinib treatment reduces HCC tumor growth in vivo. We used a xenograft system to inoculate human HepG2 cells into nude mice. As shown in Fig. 9, after being orally administered 100 or 200 mg/kg lapatinib for 3 weeks, lapatinib effectively reduced the tumor volumes in a dose dependent manner compared to the vehicle administration while lapatinib did not dramatically affect body weight and numbers of white blood cells in these mice (data not shown). This demonstrates a reduction of tumorigenesis by lapatinib in vivo.
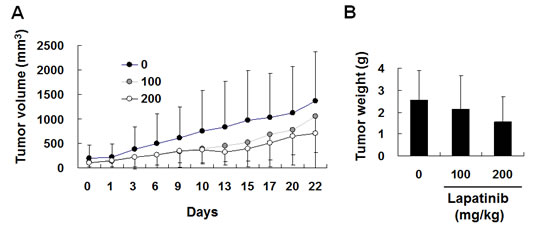
Figure 9: In vivo inhibition of HCC xenografts by lapatinib. After inoculation of HepG2 cells (luciferase stably expressed) for 3 weeks, 12 mice were randomly divided into 3 groups and were orally administrated DMSO, 100 or 200 mg/kg of lapatinib for another 22 days (days 0-22). Tumor volumes were monitored every 2 days by detection of luciferase-activity-containing cells in vivo (A) or weighted after sacrificing on day 42 (B).
Discussion
In this study, we clarified the mode of cell death and the mechanism of action of lapatinib against human HCC cells[24]. It could be regarded as translational information for the design of combinatory treatments with other regimens or radiotherapy for HCC.
The evidence of lapatinib-induced autophagic cell death includes a massive amount of dead cells with a limited amount of apoptotic cells (Fig. 2) [19-21]; the loss of mitochondrial integrity (Fig. S2)[25]; the detection of punctuate LC3 aggregates (Fig. 4) and acridine orange positive cells (Fig. 3)[22, 26]; the decreased expression of p62 and the elevated expression of autophagy-related proteins such as LC3, ATG5, ATG7, Beclin-1, and BNIP (Fig. 5); and the observation of massive vacuoles by TEM (Fig. 6) [11, 20, 27]. We also confirmed autophagic cell death by effectively reducing growth inhibition with both autophagy inhibitor 3-MA and lysosomal inhibitor chloroquine (Fig. 7) and by the expression of shRNA against autophagy-related proteins ATG5, ATG7 and Beclin-1 (Fig. 8)[11, 20-21, 27]. Most of all, we found a significant reduction of tumor growth in a dose-dependent manner in vivo after oral administration of lapatinib in a human HCC xenograft system (Fig. 9). We also found elevated E-cadherin expression in lapatinib-treated Huh7 HCC cells (Fig. S4), implicating the potential of lapatinib against the metastasis of HCC. This point will be further studied in the future.
According to our previous study, tumor cells are relatively sensitive to lapatinib as compared to normal counterparts like human CD14+ monocytes or mouse bone-marrow cells [21]. For attached cells, we also found lapatinib easily kills E1A/Ras transformated[28-29], but not its normal diploid counterpart, wild-type mouse embryonic fibroblasts with the same genetic background (data not shown). This correlates with the safety advantage for the administration of lapatinib to patients.
Although there are some reports about apoptotic cell death in lapatinib-treated cancer cells, autophagic cell death has also been induced in lapatinib-treated leukemia, HCT116 colon and bladder cancer cells[21, 30-31] and breast cancer cells co-treated with lapatinib and obatoclaxs[10], similar to our present study. Autophagic cell death is also induced by many other stimuli, such as platonin-treated leukemia cells[26], TNFα-treated cells, and death receptor-mediated cell death in cells lacking FADD, caspase activation, or NF-κB activation[15, 32-33].
Although unlike the obvious overexpression of ErbB2 in about 30% of breast or lung cancer patients[5], there are still accumulated studies demonstrating mutations or aberrant expressions of EGFR superfamily members in HCC, such as ErbB2 overexpression or truncated ErbB2 in a certain population of HCC patients [16-18, 34]. According to our data, we detected similar expression levels of EGFR and ErbB2 in our HCC cells (Fig. S1) as compared to A549 lung adenocarcinoma cells, which belong to cells expressing low levels of these receptors [9]. Since those human tumor cell lines with EGFR/ErbB2-overexpressing are more sensitive to lapatinib cytotoxicity, according to our study, lapatinib blocks growth signals mediated by either important EGFR/ErbB2 signaling through moderate amounts of EGFR and ErbB2 receptors or unveiled targets associated with HCC proliferation. Moreover, although HepG2 cells have lower EGFR and Her2 expression (Fig. S1), their lapatinib sensitivity is similar to Huh7 and HA22T (Fig. 1). Further work includes exploring the correlations between lapatinib and the mutations or amplifications of EGFR/ErbB2 and their downstream signaling in HCC cell proliferation [34]. This may further clarify the roles of EGFR/ErbB2 signaling in the development of HCC.
HCC cells were reported autophagy-defective with impaired autophagy responses[35]. Therefore, induction of autophagy or autophagic cell death might have a role in enhancing treatment responses for HCC.
Our study demonstrates lapatinib-induced cytotoxicity of HCC through the mechanisms of autophagic cell death. To extend the clinical application of lapatinib against HCC, our results could be a reference and translational information for the design of combinatory treatments with other regimens or radiotherapy. From point view of drug re-positioning by using lapatinib, the strategy about combination of lapatinib and other treatments or radiotherapy against hepatoma will be tested in the future.
Methods
Tissue culture and agent treatments
Human hepatoma Huh-7, HepG2 and HA22T cells were cultured in Dulbeco’s modified Eagles’ medium (Gibco) supplemented with 10% fetal calf serum (Hyclone), 100 IU/ml of penicillin (Gibco), 100 mg/ml of streptomycin (Gibco), and 1% nonessential amino acid (Gibco) . One thousand-fold stock solution of lapatinib was prepared by dissolving a ground lapatinib tablet (GlaxoSmithKline) in DMSO (Sigma). For lapatinib and 3-MA (Sigma) or chloroquine co-treatment experiments, 20-mM of 3-MA or 500-fold chloroquine stock solution was prepared by dissolving both drugs in a culture medium.
Growth inhibition analysis
After drug treatments, the viability of the cells was measured using the MTS assay (Promega), according to the manufacturer’s instructions. The optical density (OD) value of DMSO-vehicle treated cells was calculated as 100% viability.
Flow cytometry
For simultaneous detection of both dead cells and apoptotic cells by flow cytometry (FACSCalibur, Becton Dickenson), HCC cells with intact plasma membranes (live cells) or with DNA laddering (hypodiploid or sub G1 phase) were detected by cell-cycle detection assay as previously described[20-21]. After treatment, all floating and attaching HCC cells were collected, split into 2 tubes, and resuspended in 1 μg/ml propidium iodide (PI, Sigma) containing either phosphate buffered saline (PBS) or hypotonic buffer (0.1% sodium citrate, 0.1% Triton X-100, and 5 μg/ml PI). The former was used to detect the percentage of total dead cells (PI-positive cells) without intact plasma membranes, while the latter was used to detect apoptotic (hypodiploid) cells [20-21]. To detect the percent of acridine orange positive cells, cells were collected, stained with 10 μg/ml acridine orange, and then analyzed by flow cytometry. All data measured by flow cytometry were analyzed using FlowJo software (Tree Star).
Western blot analysis
After lapatinib treatment, HCC cells were collected and resuspended in a lysis buffer (Sigma) with protease inhibitors (Roche Biochemicals). Cell lysates with 30 or 50 μg of protein were subjected to 12% sodium dodecylsulfate-polyacrylamide gel (SDS-PAGE) electrophoresis. For immunoblotting, the following antibodies were used: actin (Sigma), ATG5 (Cell Signaling), ATG7 (Santa Cruz Biochemicals), Beclin-1 (or ATG6, Cell Signaling), LC3 (or ATG8, Abgent), and nucleoporin p62 (Genetax).
Transmission electronic microscopy (TEM)
After treatment, HCC cells were collected from culture dishes using a PBS washing buffer and a Trypsin-EDTA detaching buffer. According to the manufacturer’s instructions (Electron Microscopy Science), cells were fixed by 2.5% glutaraldehyde-containing cacodylate buffer (pH 7.4), stained with 1% osmium tetroxide, and then embedded in Spurr’s resin after dehydration. The morphology of cells was then observed by TEM.
Gene knockdown by shRNA expression system
As described previously [21], a lentivirus-based shRNA expression system from the RNAi Consortium (TRC) was used to knockdown the expression of the following genes: ATG5, ATG7, Beclin-1 and RFP (as non-targeting shRNA control). These pLKO.1-shRNA constructions were from the National RNAi Core Facility, Academia Sinica, Taiwan[21]. We used 2 different shRNA constructs (#1 and #2, as indicated in Figs. 8 and S3) for ATG7 and Beclin-1 (TRCN0000007584, TRCN0000007587, TRCN0000033549 and TRCN0000033552, respectively)[21]. Lentiviral production, viral infection and puromycin selection were performed as per TRC’s instructions. Finally, cells were treated with lapatinib, and viability was tested by the MTS assay as described above.
In vivo human tumor xenograft system
Nude mice experiments were done in the animal colonies of Mackay Memorial Hospital after approval by the Institutional Animal Experimentation Committee of the hospital. Human HepG2 cells that stably expressed luciferase were first enriched after transfection and puromycin selection. As described previously[36], after inoculation of HepG2 luciferase stably expressed cells (106 in 0.1 ml PBS) into the right hind limbs of Balc/c nude mice for 3 weeks, 12 mice with similar tumor volumes were selected and randomly divided into 3 groups. They were then orally administered a vehicle (0.5% hydroxypropyl-methylcellulose with 0.1% Tween-80), or 100 or 200 mg/kg lapatinib in vehicle every day for the next 3 weeks (days 0-22). Tumor volumes were monitored every 2 days in vivo by detection of the luciferase activity of cells with Xenogeny IVIS 200 Imaging System (PerkinElmer). The mice were left untreated for days 23-42. After sacrificing at day 42, tumor volumes inside the mice were double-checked with weight and caliper measurements.
Statistical analysis
All data gathered from triplicate (or more) experiments were expressed as means ± S.D. P values (*P < 0.05, **P < 0.01, ***P < 0.001) and were analysed by Student’s t-test between lapatinib-treated and DMSO control cells or as indicated in each figure. Values of EC50 were calculated from MTS assay data at day 3.
Acknowledgements
We thank National RNAi Core Facility, Academia Sinica, Taiwan for the pLKO.1-shRNA constructions from TRC. We also acknowledge Dr. King-Song Jeng for experimental assistance. This study was supported by grants MMH-E-102-13 and MMH-E-103-13 from Mackay Memorial Hospital, and grants NSC 97-2314-B-309-003-, NSC 100-2314-B-309-004- and NSC 100-2314-B-195-007-MY3 from the National Science Council of Taiwan, ROC, MOST 103-2313-B-309-001.
References
1. Herath NI, Leggett BA and MacDonald GA. Review of genetic and epigenetic alterations in hepatocarcinogenesis. J Gastroenterol Hepatol. 2006; 21(1 Pt 1):15-21.
2. Huitzil-Melendez FD, Capanu M, O’Reilly EM, Duffy A, Gansukh B, Saltz LL and Abou-Alfa GK. Advanced hepatocellular carcinoma: which staging systems best predict prognosis? J Clin Oncol. 2010; 28(17):2889-2895.
3. Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS, Luo R, Feng J, Ye S, Yang TS, Xu J, Sun Y, Liang H, Liu J, Wang J, Tak WY, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009; 10(1):25-34.
4. Llovet JM and Bruix J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology. 2008; 48(4):1312-1327.
5. Kopper L. Lapatinib: a sword with two edges. Pathol Oncol Res. 2008; 14(1):1-8.
6. MacFarlane RJ and Gelmon KA. Lapatinib for breast cancer: a review of the current literature. Expert Opin Drug Saf. 2011; 10(1):109-121.
7. Ramanathan RK, Belani CP, Singh DA, Tanaka M, Lenz HJ, Yen Y, Kindler HL, Iqbal S, Longmate J, Mack PC, Lurje G, Gandour-Edwards R, Dancey J and Gandara DR. A phase II study of lapatinib in patients with advanced biliary tree and hepatocellular cancer. Cancer Chemother Pharmacol. 2009; 64(4):777-783.
8. Bekaii-Saab T, Markowitz J, Prescott N, Sadee W, Heerema N, Wei L, Dai Z, Papp A, Campbell A, Culler K, Balint C, O’Neil B, Lee RM, Zalupski M, Dancey J, Chen H, et al. A multi-institutional phase II study of the efficacy and tolerability of lapatinib in patients with advanced hepatocellular carcinomas. Clin Cancer Res. 2009; 15(18):5895-5901.
9. Rusnak DW, Alligood KJ, Mullin RJ, Spehar GM, Arenas-Elliott C, Martin AM, Degenhardt Y, Rudolph SK, Haws TF, Jr., Hudson-Curtis BL and Gilmer TM. Assessment of epidermal growth factor receptor (EGFR, ErbB1) and HER2 (ErbB2) protein expression levels and response to lapatinib (Tykerb, GW572016) in an expanded panel of human normal and tumour cell lines. Cell Prolif. 2007; 40(4):580-594.
10. Cruickshanks N, Tang Y, Booth L, Hamed H, Grant S and Dent P. Lapatinib and Obatoclax Kill Breast Cancer Cells through Reactive Oxygen Species-Dependent Endoplasmic Reticulum Stress. Mol Pharmacol. 2012; 82(6):1217-1229.
11. Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, Ahn HJ, Ait-Mohamed O, Ait-Si-Ali S, Akematsu T, Akira S, Al-Younes HM, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012; 8(4):445-544.
12. Declercq W, Vanden Berghe T and Vandenabeele P. RIP kinases at the crossroads of cell death and survival. Cell. 2009; 138(2):229-232.
13. Chen S, Li X, Feng J, Chang Y, Wang Z and Wen A. Autophagy facilitates the Lapatinib resistance of HER2 positive breast cancer cells. Med Hypotheses. 2011; 77(2):206-208.
14. Bell BD and Walsh CM. Coordinate regulation of autophagy and apoptosis in T cells by death effectors: FADD or foundation. Autophagy. 2009; 5(2):238-240.
15. Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH and Lenardo MJ. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. 2004; 304(5676):1500-1502.
16. Altimari A, Fiorentino M, Gabusi E, Gruppioni E, Corti B, D’Errico A and Grigioni WF. Investigation of ErbB1 and ErbB2 expression for therapeutic targeting in primary liver tumours. Dig Liver Dis. 2003; 35(5):332-338.
17. Matsuo M, Sakurai H and Saiki I. ZD1839, a selective epidermal growth factor receptor tyrosine kinase inhibitor, shows antimetastatic activity using a hepatocellular carcinoma model. Mol Cancer Ther. 2003; 2(6):557-561.
18. Bekaii-Saab T, Williams N, Plass C, Calero MV and Eng C. A novel mutation in the tyrosine kinase domain of ERBB2 in hepatocellular carcinoma. BMC Cancer. 2006; 6:278.
19. Nicoletti I, Migliorati G, Pagliacci MC, Grignani F and Riccardi C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods. 1991; 139(2):271-279.
20. Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS, Golstein P, Green DR, Hengartner M, Knight RA, Kumar S, Lipton SA, Malorni W, Nunez G, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009; 16(1):3-11.
21. Huang HL, Chen YC, Huang YC, Yang KC, Pan HY, Shih SP and Chen YJ. Lapatinib induces autophagy, apoptosis and megakaryocytic differentiation in chronic myelogenous leukemia k562 cells. PLoS One. 2011; 6(12):e29014.
22. Su WC, Chao TC, Huang YL, Weng SC, Jeng KS and Lai MM. Rab5 and class III phosphoinositide 3-kinase Vps34 are involved in hepatitis C virus NS4B-induced autophagy. J Virol. 2011; 85(20):10561-10571.
23. Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H and Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005; 171(4):603-614.
24. Lo SJ, Fan LC, Tsai YF, Lin KY, Huang HL, Wang TH, Liu H, Chen TC, Huang SF, Chang CJ, Lin YJ, Yung BY and Hsieh SY. A novel interaction of nucleophosmin with BCL2-associated X protein regulating death evasion and drug sensitivity in human hepatoma cells. Hepatology. 2013; 57(5):1893-1905.
25. Li ZY, Yang Y, Ming M and Liu B. Mitochondrial ROS generation for regulation of autophagic pathways in cancer. Biochem Biophys Res Commun. 2011; 414(1):5-8.
26. Chen YJ, Huang WP, Yang YC, Lin CP, Chen SH, Hsu ML, Tseng YJ, Shieh HR, Chen YY and Lee JJ. Platonin induces autophagy-associated cell death in human leukemia cells. Autophagy. 2009; 5(2):173-183.
27. Colosetti P, Puissant A, Robert G, Luciano F, Jacquel A, Gounon P, Cassuto JP and Auberger P. Autophagy is an important event for megakaryocytic differentiation of the chronic myelogenous leukemia K562 cell line. Autophagy. 2009; 5(8):1092-1098.
28. Huang HL, Yeh WC, Lai MZ, Mirtsos C, Chau H, Chou CH and Benchimol S. Impaired TNFalpha-induced A20 expression in E1A/Ras-transformed cells. Br J Cancer. 2009; 101(9):1555-1564.
29. Chau H, Mirtsos C and Huang HL. Regulation of death complexes formation in tumor necrosis factor receptor signaling. Exp Cell Res. 2011; 317(13):1841-1850.
30. Martin AP, Mitchell C, Rahmani M, Nephew KP, Grant S and Dent P. Inhibition of MCL-1 enhances lapatinib toxicity and overcomes lapatinib resistance via BAK-dependent autophagy. Cancer Biol Ther. 2009; 8(21):2084-2096.
31. Becker MN, Wu KJ, Marlow LA, Kreinest PA, Vonroemeling CA, Copland JA and Williams CR. The combination of an mTORc1/TORc2 inhibitor with lapatinib is synergistic in bladder cancer in vitro. Urol Oncol. 2014; 32(3):317-326.
32. Djavaheri-Mergny M, Amelotti M, Mathieu J, Besancon F, Bauvy C, Souquere S, Pierron G and Codogno P. NF-kappaB activation represses tumor necrosis factor-alpha-induced autophagy. J Biol Chem. 2006; 281(41):30373-30382.
33. Pyo JO, Jang MH, Kwon YK, Lee HJ, Jun JI, Woo HN, Cho DH, Choi B, Lee H, Kim JH, Mizushima N, Oshumi Y and Jung YK. Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J Biol Chem. 2005; 280(21):20722-20729.
34. Berasain C and Avila MA. The EGFR signalling system in the liver: from hepatoprotection to hepatocarcinogenesis. J Gastroenterol. 2014; 49(1):9-23.
35. Bao L, Chandra PK, Moroz K, Zhang X, Thung SN, Wu T and Dash S. Impaired autophagy response in human hepatocellular carcinoma. Exp Mol Pathol. 2014; 96(2):149-154.
36. Chi CW, Chen CC and Chen YJ. Therapeutic and radiosensitizing effects of armillaridin on human esophageal cancer cells. Evid Based Complement Alternat Med. 2013; 2013:459271.