
Introduction
Lung cancer is a leading cause of cancer-related death worldwide [1, 2]. Metastasis is the primary cause of lung cancer treatment failure and mortality [1, 3]. Understanding the cellular mediators that contribute to the invasion and metastasis of lung cancer and the development of novel therapeutic agents targeting these mediators are urgently needed.
One of the potential mediators of metastasis is cyclooxygenase-2 (COX-2), the inducible isoform of COX. COX-2 is frequently found in early and advanced lung cancer tissues and is associated with poor prognosis [4-7]. Elevation of tumor COX-2 increases the level of its metabolite prostaglandin E2 (PGE2) that is a ligand of G protein-coupled receptors, such as EP1, EP2, EP3, and EP4. COX-2/PGE2 stimulates phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt) and extracellular signal-regulated kinase 1/2 (ERK1/2) signaling to induce tumor angiogenesis, cancer motility and invasiveness [8-11]. Recently COX-2/PGE2 has also been found to induce migration inducing gene-7 (MIG-7) protein that sustains the activation of PI3K/Akt/glycogen synthase kinase-3 β (GSK-3β) signaling via decreasing the activity of protein phosphatase 2A (PP2A) to increase lung cancer invasion/metastasis [12].
Recombinant DNA-derived VP1 (rVP1) of foot-and-mouth disease virus (FMDV) has previously been found to induce apoptosis of human cancer cell lines MCF-7, PC-3 and 22Rv1 via modulation of the integrin/Akt signaling pathway [13]. Recently, rVP1 has also been shown to suppress progression of hepatocellular carcinoma [14] and invasion of SKOV3 ovarian adenocarcinoma cells as well as SiHa and Caski cervical carcinoma cells [15, 16]. However, the molecular mechanisms underlying the inhibition of cancer invasion/metastasis by rVP1 remain largely unexamined.
In this study, we explored whether rVP1 has any effect on COX-2/PGE2 and MIG-7. Our results showed that rVP1 suppressed epithelial-mesenchymal transition (EMT), migration/invasion and metastasis of human lung cancer cells. Its mechanism of action involved modulation of PIP3 and ILK in the lipid rafts as well as suppression of IKK/NF-κB, COX-2/PGE2 and MIG-7. The role of rVP1-mediated decrease of COX-2/PGE2 and MIG-7 in inhibiting tumor growth and metastasis of lung adenocarcinoma was further substantiated by analyzing COX-2/PGE2 and MIG-7 in the lung cancer xenograft mice with or without rVP1 treatment.
Results
rVP1 suppresses EMT, MMP-2 and migration/invasion of human lung cancer cells
We previously showed that rVP1 suppresses growth and migration/invasion of ovarian and cervical cancer [15, 16]. To investigate whether rVP1 inhibits lung cancer cells in a similar manner, three human lung cancer cell lines, A549, H1299 and CL1-5 were treated with rVP1. Our results showed that rVP1 inhibited the cell viability of A549, H1299 and CL1-5 in a concentration-dependent manner (Figure 1A) with IC50 values of 0.93, 0.88 and 0.68 μM, respectively. Interestingly, even at concentrations (0.2 μM - 0.4 μM) that caused only minimal cell death (Figure 1A), rVP1 dramatically suppressed migration and invasion of A549, H1299 and CL1-5 cells (Figure 1B).
Since the downregulation of epithelial cell marker E-cadherin and a concomitant increase in the expression of the mesenchymal cell marker vimentin correlate with enhancement in EMT and migration/invasion of cancer cells [17], we determined the effect of rVP1 on the expression level of E-cadherin and vimentin in lung cancer cells. Immunoblotting analysis revealed that treatment with rVP1 (0.4 µM) for 24 h attenuated EMT by upregulating E-cadherin and downregulating vimentin expression levels in A549, H1299 and CL1-5 lung cancer cell lines (Figure 1C).
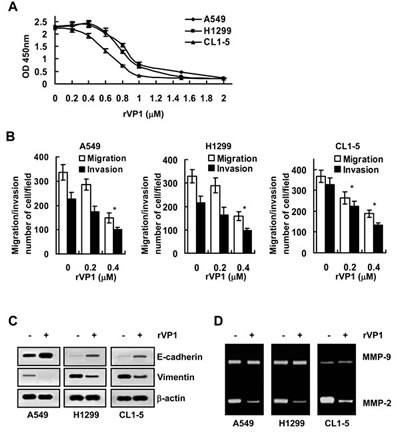
Figure 1: rVP1 suppresses the migration/invasion of lung cancer cells in vitro. Lung cancer cells (A549, H1299 and CL1-5; 2 × 105/mL) were treated with 0.4 µM rVP1 in 0.5% FBS medium for 24 h, unless specified otherwise. (A) Lung cancer cells were treated with serial concentrations of rVP1 (0–2 µM) as indicated. Cell proliferation was measured by WST-1 assay in vitro. Data represent means ± SD of three independent experiments. (B) The migration and invasion capability of cells with or without rVP1 treatment were measured by transwell migration assay. Data represent means ± SD of three independent experiments; *P < 0.05 by t-test. (C) The expression level of epithelial cell marker E-cadherin and mesenchymal cell marker vimentin were analyzed by immunoblotting. β-actin was used as a loading control. Blots are representative of three independent experiments. (D) The MMP-2 and MMP-9 enzyme activity in the cell-cultured medium was analyzed by a gelatin zymography assay. Data are representative of three independent experiments.
As MMP activity is positively associated with enhanced cellular invasion [18], we next evaluated the effect of rVP1 on MMP activity of lung cancer cells. Gelatin zymographic analysis showed that the MMP-2 activity in A549, H1299 and CL1-5 cells was reduced by rVP1 treatment (Figure 1D). These results thus demonstrated that rVP1 decreases EMT and MMP-2 activity to suppress the migration and invasion of lung cancer cells.
rVP1 downregulates integrin β1/Akt, COX-2/PGE2 and MIG-7 to suppress lung cancer cell migration/invasion
rVP1 suppresses migration/invasion of cervical cancer cells via downregulating the Akt signaling pathway through integrin β1 [15]. Since inhibition of integrin β1 signaling has been reported to attenuate COX-2 and MIG-7 levels [19, 20], we examined whether rVP1 suppresses expression of COX-2 and MIG-7 in human lung cancer cells through the integrin β1/Akt pathway. Our results showed that phospho-AktS473, COX-2, PGE2 and MIG-7 were decreased in lung cancer cells treated with rVP1 (0.4 µM) for 24 h (Figure 2A). The rVP-1-mediated reduction of AktS473 phosphorylation as well as COX-2, PGE2 and MIG-7 expression was reversed by anti-integrin β1 antibodies, but not by the control immunoglobulin G (IgG) (Figure 2A). Constitutive phosphorylation of Akt at serine 473 (AktS473) by transfecting lung cancer cells with dominant active Akt plasmid (pAkt-DA) increased the EMT, MMP2 activity and invasion ability of cancer cells and increased expression of COX-2 and MIG-7 (Figure 2B and 2C). These effects of constitutively active Akt were reversed by knockdown of COX-2 or MIG-7 with transfection of COX-2 siRNA (siCOX-2) or MIG-7 siRNA (siMIG-7) (Figure 2B and 2C). As anticipated, the constitutively active-Akt-mediated COX-2 and MIG-7 expression was not attenuated by rVP1 (0.4 µM) (Figure 2D). These results suggest that rVP1 inhibits EMT, MMP2 activity and the invasion of lung cancer cells via downregulating phosphorylated Akt as well as suppression of COX-2 and MIG-7 expression.
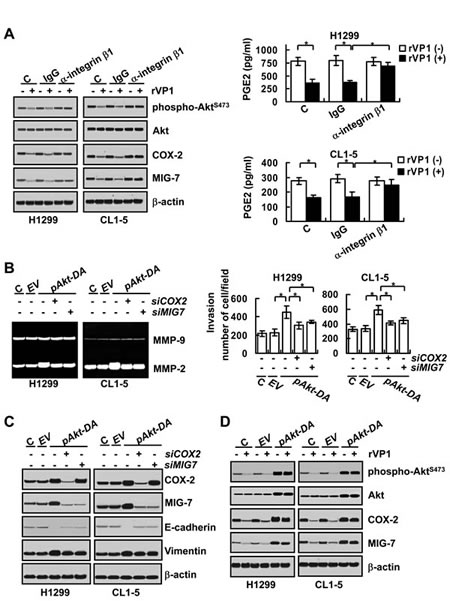
Figure 2: rVP1 binds to integrin to inhibit COX-2 and MIG-7 and decrease lung cancer cell invasion. Lung cancer cells (H1299 and CL1-5; 2 × 105/mL) were treated with control IgG or anti-integrin β1 antibodies (2 μg/ml) for 30 min followed by 0.4 µM rVP1 for 24 h in 0.5% FBS medium. Proteins were determined by immunoblotting. β-actin was used as a loading control. Blots are representative of three independent experiments. Cell-conditioned media were examined for PGE2 by PGE2 EIA kit (right panels). Data represent means ± SD of three independent experiments; *P < 0.05 by t-test. (B and C) Parental lung cancer cells (H1299 and CL1-5; C) were transfected with empty vector (EV), dominant-active Akt (pAkt-DA) plasmids, control scrambled siRNAs (-siCOX2 and -siMIG7), COX2 siRNAs (+siCOX2) or MIG7 siRNAs (+siMIG7) for 48 h. (D) Cells (H1299 and CL1-5; C) transfected with empty vector (EV) or dominant-active Akt (pAkt-DA) plasmids for 48 h were treated with 0.4 µM rVP1 for 24 h in 0.5% FBS medium. Immunoblotting was used for determining proteins, zymography assay for MMP activity and transwell assay for cell invasion as described in Materials and Methods. Blots are representative of three independent experiments. Data represent means ± SD of three independent experiments; *P < 0.05 by t-test.
PIP3 and ILK play essential roles in rVP1-mediated downregulation of IKK/NF-κB signaling and COX-2
Phosphatidylinositol 3-kinase (PI3K) phosphorylates phosphatidylinositol (4,5)-biphosphate (PIP2) to generate phosphatidylinositol (3,4,5)-triphosphate (PIP3) that can bind to the pleckstrin-homology domains of integrin-linked kinase (ILK) and Akt to increase the attachment of ILK and Akt to lipid rafts, leading to the activation of Akt signaling pathways [21-23]. To elucidate the mechanism by which rVP1 acts on the integrin β1/Akt signaling of lung cancer cells, we examined the effects of rVP1 on integrin downstream targets, PI3K, PIP3, ILK and Akt. Our results showed that rVP1 increased phosphorylation of phosphatase PTEN accompanied by a decrease in phosphorylation of focal adhesion kinase (FAK), a molecule upstream of PI3K, as well as PI3K-p85Y458 and AktS473 (Figure 3A). Further studies showed that rVP1 decreased PIP3 and prevented accumulation of ILK, Akt and phospho-AktS473 in the raft domain (Figure 3B). Addition of PIP3 reversed the inhibitory effect of rVP1 on the level of ILK, Akt and phospho-AktS473 in the lipid rafts (Figure 3B). These results demonstrated that rVP1 decreases phosphorylation of FAK, PI3K and PIP3 leading to a decline in ILK and phospho-AktS473 in the raft domains.
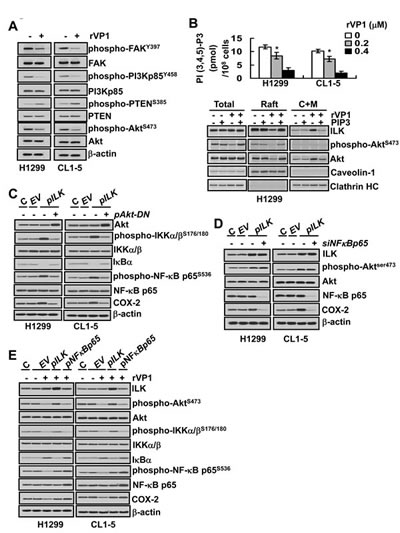
Figure 3: rVP1 decreases COX-2 via modulating PIP3 and IKK/NF-B signaling in lung cancer cells. (A) H1299 and CL1-5 cells were treated with or without 0.4 µM rVP1 for 24 h in 0.5% FBS medium. Proteins were determined by immunoblotting. β–actin was used as a loading control. (B) After treatment with 0.2 µM or 0.4 µM rVP1 for 24 h in 0.5% FBS medium, cellular phospholipids were extracted and PIP3 levels determined by ELISA (upper panel). Data represent means ± SD of three independent experiments; *P < 0.05 by t-test. H1299 and CL1-5 cells were treated with 0.4 µM rVP1, 5 µM PIP3 or rVP1 plus PIP3 as indicated for 24 h in 0.5% FBS medium. Proteins in the membrane raft or cytosol plus non-raft membrane (C+M) fractions were determined by immunoblotting. Caveolin-1 served as a membrane raft marker and clathrin-HC served as a membrane non-raft marker (lower panel). (C and D) Parental lung cancer cells (H1299 and CL1-5; C) were transfected with empty vector (EV and -pAkt-DN), pILK plasmids (pILK), dominant-negative Akt (+pAkt-DN) plasmids, control scrambled siRNAs (-siNFκBp65), or NFκBp65 siRNA (+siNFκBp65) for 48 h. (E) Parental cells (H1299 and CL1-5; C) transfected with empty vector (EV), pILK or pNF-κBp65 for 48 h were treated with 0.4 µM rVP1 for 24 h in 0.5% FBS medium. Proteins were examined by immunoblotting. Blots are representative of three independent experiments.
To further understand how rVP1 could reduce expression of COX-2, we next examined whether ILK or the IKK/NF-κB pathway plays any role in the inhibitory effect of rVP1 on COX-2 expression in lung cancer cells. We found that overexpression of ILK by transfection of ILK plasmid (pILK) in H1299 and CL1-5 lung cancer cells increased phosphorylation of AktS473, IKKα/βS176/180 and NF-κB p65S536 as well as degradation of IκB, which were positively correlated with elevation of COX-2 level (Figure 3C and 3D). Transduction of inactive Akt by transfection of Akt-DN plasmid (pAkt-DN) blocked the ILK-mediated induction of IKKα/β-IκB-NF-κB signaling and expression of COX-2 (Figure 3C). Even though knockdown of NF-κB p65 by transfection of NF-κB p65 siRNA (+siNFκBp65) did not affect phosphorylation of AktS473, it also suppressed the ILK-induced COX-2 expression (Figure 3D). Overexpression of ILK or NF-κB p65 by transfection with pILK or pNFκBp65 blocked the inhibition of COX-2 expression by rVP1 (Figure 3E). Taken together, these results indicate that rVP1 inhibits integrin/FAK/PI3K to decrease the level of PIP3 which in turn modulates ILK level in the lipid rafts to regulate IKK/NF-κB pathway signaling and COX-2 expression.
rVP-1 suppresses COX-2 and MIG-7 to mitigate EMT and migration/invasion of lung cancer cells
We recently showed that COX-2/PGE2 activates EP4 to enhance Akt and GSK-3β phosphorylation and β-catenin/LEF/TCF signaling leading to MIG-7 upregulation [12]. To investigate whether rVP1-mediated decrease of COX-2/PGE2 (and/or MIG-7) is functionally associated with rVP1/integrin β1/Akt-mediated reduction in EMT, MMP2 and invasion of lung cancer cells, we transfected lung cancer cell lines (H1299 and CL1-5) with COX-2 or MIG-7 plasmids (pCOX2 or pMIG7) to overexpress COX-2 or MIG-7 protein. We found that transfection with pCOX2 increased COX-2 together with MIG-7 protein, EMT, MMP-2 activity and invasion of lung cancer cells (Figure 4A and Supplementary Figure 1). PGE2 treatment also increased MIG-7 protein, EMT, MMP-2 activity and invasion of lung cancer cells (Supplementary Figure 1). MIG-7 knockdown (+siMIG7) attenuated the effects of COX-2 overexpression and PGE2 treatment on MIG-7, EMT, MMP-2 and the invasion of the cells, but did not affect expression level of COX-2 (Supplementary Figure 1). The rVP-1-mediated suppression of EMT as shown by induction of E-cadherin expression and reduction of vimentin could be reversed in cancer cells by transduction of pCOX-2 or pMIG-7 (Figure 4A and 4B). In addition, overexpression of COX-2 and MIG-7 reversed the inhibition of MMP2 enzyme activity and invasion ability of H1299 and CL1-5 cells by rVP1 (Figure 4A and 4B). Collectively, these results suggest that rVP1 modulates integrin β1/Akt signaling to downregulate COX-2/PGE2 that then attenuates MIG-7 protein level resulting in inhibition of the EMT, MMP-2 activity and invasion of lung cancer cells.
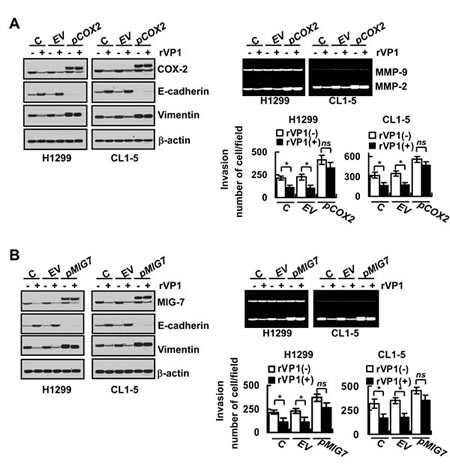
Figure 4: Overexpression of COX-2 and MIG-7 reverses rVP1-mediated inhibition on EMT, MMP-2 activity and lung cancer cell invasion. (A) Parental cells (H1299 and CL1-5; C) transfected with empty vector (EV) and pCOX2 plasmids (pCOX2) for 48 h were treated with 0.4 µM rVP1 as indicated for 24 h in 0.5% FBS medium. (B) Cells transfected with empty vector (EV) and pMIG7 plasmids (pMIG7) for 48 h were treated with 0.4 µM rVP1 as indicated for 24 h in 0.5% FBS medium. Cells were examined by immunoblotting for protein expression, zymography for MMP activity and transwell assay for cell invasion. Blots are representative of three independent experiments. Data represent means ± SD of three independent experiments; *P < 0.05 by t-test.
rVP-1 reduces COX-2 and MIG-7 and suppresses metastasis and lethal effects of lung cancer cells in vivo
To examine whether rVP1 inhibits lung cancer metastasis via downregulating COX-2 and MIG-7 in vivo, we generated CL1-5GL cells stably expressing green fluorescent protein and luciferase (GL). Some CL1-5GL cells were pretreated for 24 h with rVP1 at such a low concentration (0.3 µM) that inhibited cell invasion (Supplementary Figure 2A) but not cell proliferation (Supplementary Figure 2B). The CL1-5GL cells with or without rVP1 pretreatment, respectively, were then implanted into two groups of SCID mice via tail-vein injection. After implantation, a significant reduction of tumor nodules in the lung was observed in mice injected with rVP1-pretreated-CL1-5GL cells (Figure 5A and 5B) as compared to those injected with CL1-5GL cells that were not pretreated with rVP1. Analysis of lung tissue lysates showed that mice implanted with rVP1-pretreated CL1-5GL had lower EMT, phospho-AktS473, phospho-NF-κBS536, COX-2 and MIG-7 than those implanted with CL1-5GL without rVP1 pretreatment (Figure 5C). Histopathologic examination showed that there was more downregulation of Ki67 and upregulation of E-cadherin in in the rVP1-pretreated group than in the control group (Figure 5D). Moreover, all the mice implanted with CL1-5GL without rVP1 pretreatment died on or before day 50 post-inoculation; whereas all the mice implanted with rVP1-pretreated-CL1-5GL cells were disease free on day 250.
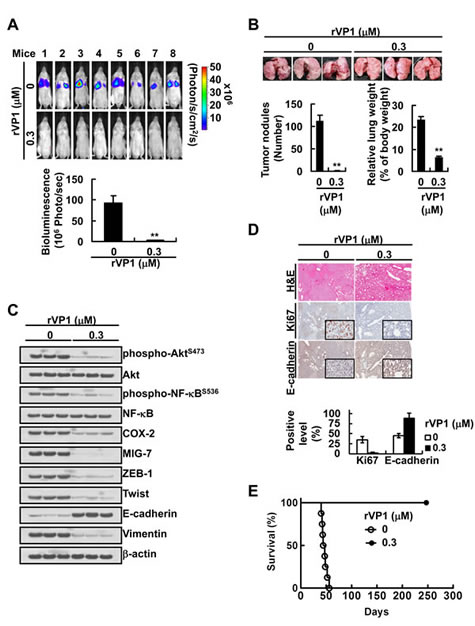
Figure 5: Treatment of lung cancer cells with low concentration of rVP1 decreases their tumor forming capability and lethality in vivo. CL1-5GL cells stably expressing green fluorescent protein and luciferase (GL) were generated as described in Materials and Methods. CL1-5GL cells were pretreated with or without 0.3 µM rVP1 for 24 h in 0.5% FBS medium and were implanted into SCID mice (1 × 106 cells/50 µL/mouse; n = 11) via tail-vein injection. (A and B) The whole bodies of mice underwent bioluminescent imaging 5 weeks after inoculation and the lungs of mice were dissected from the surrounding tissue to measure weight and count tumor nodules. Data represent means SD of at least 3 mice of each group; **P < 0.01 by t-test. (C and D) The murine lung sections were analyzed after H&E (at ×40 magnification) and immunohistochemistry staining (at ×40 magnification; insets ×400 magnification) and cell lysates were extracted and analyzed by immunoblotting. Blots of three tissue samples of each group are representative of three independent experiments. (E) Percentage of survival of mice inoculated with rVP1-pretreated CL1-5GL cells was compared with that of mice inoculated with cells without rVP1 pretreatment; P < 0.0001, n = 6 by t-test.
To further investigate the in vivo effect of rVP1, instead of pretreating CL1-5GL cells with rVP1, we implanted the cancer cells into SCID mice and one week later start administering rVP1 (5 or 15 mg/kg body weight) intravenously via the tail vein three times per week for 4 weeks. Although not as effective as pretreatment, rVP1 still significantly reduced tumor growth in the lungs of CL1-5GL-bearing mice as compared to those treated with control vehicle (Figure 6A and 6B). Analysis of tumor lysates obtained from the vehicle- and rVP1-treated mice showed that the levels of EMT, phospho-AktS473, phospho-NF-κBS536, COX-2 and MIG-7 in tumors from the rVP1-treated mice were lower than those in tumors from the vehicle-treated mice (Figure 6C). Histological analysis of lungs revealed that there was less dissemination of tumor cells in lung tissue sections from rVP1-treated mice whereas many more cancer cells were found in vehicle-treated mice (Figure 6D) as indicated by Ki67 level. Comparison of the survival curve of rVP1-treated mice with that of vehicle-treated mice showed that rVP1 treatment significantly prolonged the survival of tumor-bearing mice (Figure 6E). Taken together, these results suggest that rVP1-mediated downregulation of COX-2 and MIG-7 suppressed EMT and the metastatic capability of human lung cancer cells, and therefore prolonged the survival of lung cancer xenograft mice.
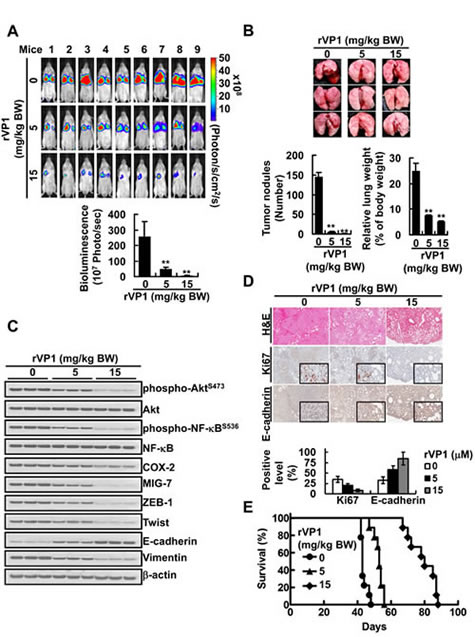
Figure 6: rVP1 decreases COX-2 and MIG-7 and suppresses lung cancer metastasis in xenograft mice. CL1-5GL stable cells were injected into SCID mice tail vein (n = 12). One week after implantation, rVP1 (5 or 15 mg/kg body weight) was administered via tail-vein injection three times per week for four weeks. (A and B) The whole bodies of mice underwent bioluminescent imaging and the lungs of mice were dissected from the surrounding tissue to measure weight and count tumor nodules. Data represent means ± SD of at least 3 mice from each group; **P < 0.01 by t-test. (C and D) The murine lung sections were analyzed after H&E (at ×40 magnification) and immunohistochemistry staining (at ×40 magnification; insets ×400 magnification) and cell lysates were extracted and analyzed by immunoblotting. Blots of three tissue samples of each group are representative of three independent experiments. (E) Percentage of survival of CL1-5GL-bearing mice treated with vehicle was compared with those treated with rVP1. Without rVP1 treatment t50 = 42 day, 5 and 15 mg/kg body weight of rVP1 treatment t50 = 55 and 78 day, respectively; n = 9; P < 0.001.
Discussion
A variety of scenarios, notably smoking and inflammation induce high levels of COX-2/PGE2 [24]. Although we previously reported that binding of rVP1 to integrins induces cancer apoptosis [13] and decreases cancer invasion/metastasis [16], the relationship between rVP-mediated effects and COX-2/PGE2 was unclear. The results of this study demonstrate for the first time that rVP1 is able to suppress COX-2/PGE2-mediated signaling transduction to attenuate cancer invasiveness (Figures 1, 2 and 3). In view of these findings, it might be worthwhile exploring whether the COX-2/PGE2-related adverse effects of smoking and inflammation can be modulated by rVP1.
rVP1 inhibits cancer cell migration/invasion by downregulating integrin/FAK/Akt/GSK-3β signaling and MMP-2 activity [16]. However, it was not clear how interaction of rVP1 with integrin could attenuate Akt/GSK-3β signaling. Here we showed that rVP1 inhibited lung cancer invasion by decreasing FAK/PI3K/PIP3 to downregulate ILK and phospho-AktS473 in the lipid rafts (Figure 2 and 3). ILK is known to regulate not only cell survival and proliferation but also migration by connecting the cytoplasmic domains of β-integrins to the actin cytoskeleton in diverse cell types [22]. Our results, however, demonstrated that rVP1-mediated inhibition of ILK resulted in the downregulation of IKK/NF-κB signaling and COX-2/PGE2 expression (Figure 3C and 3D). Overexpression of ILK or NF-κB p65 blocked the inhibitory effect of rVP1 on COX-2 expression (Figs. 3E) to induce MIG-7, EMT and MMP2 activity as well as invasiveness of lung cancer cells (Supplementary Figure 1). Moreover, overexpression of COX-2 or MIG-7 reversed the inhibitory effects of rVP1 on invasion/migration (Figure 4A and 4B). ILK may thus upregulate COX-2 and MIG-7 to enhance EMT and migration/invasion of cancer cells. Whether rVP1 can regulate the actin cytoskeleton to attenuate COX-2 and MIG-7 or regulate COX-2 and MIG-7 to attenuate the actin cytoskeleton remains to be elucidated.
EMT is enhanced by transcription repressors notably, ZEB1, Twist and Snail that interact with E-box elements located within the proximal region of the E-cadherin (CDH1) promoter to suppress E-cadherin [17, 25]. Phosphorylated prohibitin has been shown to decrease E-cadherin via increasing ZEB1 and Snail but not Twist [15]. In comparison, MIG-7 decreases E-cadherin by increasing ZEB1 and Twist but not Snail [12]; (Supplementary Figure 3). rVP1 suppresses cervical cancer cell metastasis by decreasing membrane bound PIP3 leading to modulation of phospho-PHBT258 in the lipid rafts of the cervical cancer cells and inactivation of Raf-1/ERK [15]. Since rVP1 also downregulated MIG-7 whereas overexpression of MIG-7 reversed the inhibitory effects of rVP1 on EMT and invasion (Figure 4A and 4B) it is likely that MIG-7 and phosphorylated prohibitin might have a synergistic or additive effect on E-cadherin, EMT and cell migration/invasion. Whether there is a functional relationship between phospho-PHBT258 and MIG-7 and whether rVP1 attenuates either one to affect the other or both via different pathways are currently under investigation.
In summary, our study demonstrates that rVP1 downregulates integrin/FAK/PI3K and PIP3 as well as ILK and phospho-Akt in the raft domains to attenuate IKK/NF-κB signaling and reduce COX-2 and MIG-7 level leading to suppression of EMT and the invasion/metastasis of lung cancer cells in vitro and in vivo. These results suggest that agents such as rVP1 that selectively modulate COX-2/PGE2 and MIG-7 level may have great potential for development as novel therapeutic agents for metastatic cancer.
Materials and methods
Materials
Mouse anti-vimentin (V9) antibodies and recombinant PGE2 were purchased from Sigma-Aldrich (St Louis, MO). Rabbit anti-MIG-7 antibodies were obtained from Abcam (Cambridge, UK). Mouse anti-β-actin, goat anti-COX-2 (M-19), rabbit anti-E-cadherin (H-108), rabbit anti-ZEB1 (H-102), rabbit anti-Snail (H-130) antibodies as well as horseradish peroxidase-conjugated anti-mouse IgG, horseradish peroxidase-conjugated anti-goat IgG and horseradish peroxidase-conjugated anti-rabbit IgG antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit anti-FAK and mouse anti-phospho-FAKY397 antibodies were obtained from BD Biosciences (Bedford, MA). Rabbit-anti-ILK, rabbit anti-phospho-PI3Kp85, rabbit anti-phospho-PI3Kp85Y458, rabbit anti-PTEN, rabbit anti-phospho-PTENS385, rabbit anti-Akt, rabbit anti-phospho-Akt Ser473, rabbit anti-phospho-IKKα/βSer176/180, rabbit anti-phospho-NF-κB p65Ser536 and rabbit anti-clathrin HC antibodies were obtained from Cell Signaling Technology (Beverly, MA). Mouse anti-integrin-β1 (MAB1965), mouse anti-IKK/, rabbit anti-IκBα and rabbit anti-NF-κB p65 antibodies were obtained from Millipore (Bedford, MA). Rabbit anti-caveolin-1 antibodies were obtained from Upstate Biotechnology (Charlottesville, VA). Phosphatidylinositol (3,4,5)-trisphosphate diC16 (PI(3,4,5)P3 diC16) was purchased from Echelon Biosciences (Salt Lake City, UT). Cell proliferation was determined routinely by WST-1 reagent (Roche Diagnostics, Mannheim, Germany) as described previously [13, 26].
Human lung cancer cell lines and mice
A549 cell line (ATCC: CCL-185) was maintained in DMEM medium (GibcoBRL Life Technologies, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS; GibcoBRL Life Technologies) and 1% penicillin-streptomycin-neomycin (GibcoBRL Life Technologies). H1299 (ATCC: CRL-5803) and CL1-5 [27] were maintained in RPMI medium (GibcoBRL Life Technologies) supplemented with 10% FBS and 1% penicillin-streptomycin-neomycin. All cells were cultured in a humidified incubator containing 5% CO2 at 37°C. Male SCID mice (C.B17/lcr-Prkdc scid/CrlNarl) were purchased from the National Laboratory Animal Center (Taipei, Taiwan). All animal care and in vivo experiments were performed in compliance with the guidelines of the Academia Sinica Institutional Animal Care and Utilization Committee. Mice were provided a standard laboratory diet and distilled water and kept on a 12-h light/dark cycle at 25 ± 2°C and 55 ± 5% relative humidity.
Purification of recombinant VP1 protein
Purification of recombinant VP1 protein was carried out as described previously [28]. In brief, the VP1 gene in the expression vector pET24a(+) (Novagen, Madison, WI) was expressed in Escherichia coli. After breaking up the bacteria with a microfluidizer in TEN buffer (50 mM Tris–HCl, pH 8.0, 1 mM EDTA, 0.1 M NaCl), the pellet was washed with 0.5% deoxycholate in TEN buffer, followed by rinsing with TEN buffer and resuspended in binding buffer (20 mM Tris–HCl, pH 8.0, 0.5 M NaCl, 8 M urea). The solution was then applied to a metal-chelating affinity column and eluted with a gradient of 0–0.2 M imidazole. SDS was then added to the protein solution to a final concentration of 1%. The protein solution was subsequently applied to a Superdex 200 column (GE Healthcare, Piscataway, NJ) and eluted with a buffer solution containing 25 mM Tris–HCl, pH 8.0, 1 mM EDTA, 0.1 M NaCl and 0.05% SDS. The fractions containing rVP1 protein were pooled, concentrated and dialyzed against PBS before use.
Knockdown and overexpression of proteins
Full-length human COX-2 cDNA (NM 000963.2) and human MIG-7 (DQ080207.2) cDNA derived from A549 cells were amplified by using specific primers (Supplementary Table 1) (Sigma-Proligo, St Louis, MO) and subcloned into pcDNATM6/BioEaseTM-DEST by Gateway cloning technology (Invitrogen, Carlsbad, CA) to generate pCOX2 and pMIG7 plasmids. The insert sequences in the plasmids were confirmed by automated DNA sequencing. pUSEamp, pUSEamp-myr-Akt1 (dominant active) and pUSEamp-Akt1 K179M (dominant negative) were purchased from Upstate Biotechnology. Full-length ILK (BC001554) cDNA and NF-κBp65 (BC110830) cDNA cloned in pCMV-SPORT6 vectors (pILK and pNF-κBp65) were purchased from Open Biosystems (Huntsville, AL). Control siRNAs, COX2-siRNA and NF-κBp65-siRNA were obtained from Santa Cruz Biotechnology. The plasmids or siRNAs were transfected into cell lines by PolyJet In Vitro DNA Transfection Reagent or GeneMute siRNA and DNA Transfection Reagent (SignaGen laboratories, Ijamsville, MD). MIG-7 siRNA and scrambled control siRNA were purchased from Dharmacon (Thermo Fisher Scientific, Lafayette, CO). The sequence of the MIG-7 siRNA is: GUCGAAGAAAUGAAACUUUUU. MIG-7 siRNA transfection was undertaken using Dharmacon Accell SMARTpool siRNA reagent (Thermo Fisher Scientific) according to the protocol recommended by Dharmacon (Heidelberg, Germany). The transfected cells were used for a variety of experiments as indicated and expression of target proteins in the transfected cells was determined 48 h after transfection, unless specified otherwise.
Extraction of cellular proteins
Sixty milligrams of frozen tissue was immersed in 2 ml RIPA buffer (Santa Cruz Biotechnology) containing proteinase and phosphatase inhibitor cocktail (Roche, Mannheim, Germany) and then lysed with homogenizer at 4°C. After centrifugation at 14000 × g for 60 min at 4°C, the pellets were discarded and the supernatant containing proteins was stored -80°C until needed. Equal amounts of protein (60 μg) were loaded onto 10% SDS-PAGE and transferred to PVDF membrane (Millipore, Bedford, MA) for western blot (immunoblot) analysis.
Extraction of membrane raft proteins
Membrane raft proteins were extracted as described previously [29]. Briefly, cells (2 × 106) were washed in ice-cold PBS and lysed by incubation for 30 min in ice-cold lysis buffer (0.5% Triton X-100, 150 mM NaCl, 20 mM Tris–HCl, pH 7.5) containing proteinase and phosphatase inhibitor cocktail (Roche). After centrifugation at 14,000 rpm for 30 min at 4°C, the supernatants (containing the Triton X-100 soluble fractions) were collected and referred to as the cytosolic plus non-raft membrane (C+M) fraction. The insoluble pellets were resuspended in the same lysis buffer supplemented with 0.5% SDS and 2 mM DTT and sonicated for 10 min at 4°C. After centrifugation at 14,000 rpm for 30 min at 4°C, the supernatants consisting of membrane raft proteins were collected and analyzed by immunoblotting.
Immunoblotting and gelatin zymographic analysis
Immunoblotting and gelatinolytic activities of matrix metalloproteinase-2 (MMP-2) and MMP-9 were performed as described previously [12, 30].
Migration and invasion assays
In vitro migration and invasion assays were performed as described previously [12, 30].
ELISA for PGE2
The levels of intracellular and secreted PGE2 in the cell lysate and cell-conditioned medium were measured using a commercially available PGE2 EIA assay kit (Amersham, Arlington Heights, IL) according to the manufacturer’s instructions.
ELISA for PIP3
H1299 and CL1-5 cells (1 108) were treated with or without rVP1 for 24 h in 0.5% FBS medium. The cells were washed twice with ice-cold PBS, and phospholipids were extracted and PIP3 levels were measured by ELISA assay using PIP3 Mass ELISA Kit (Echelon Biotechnology) according to the manufacturer’s instructions.
Preparation of pCMV-GFP/luciferase-lentivirus and establishment of stable CL1-5GL cell lines
To generate CL1-5GL cells expressing both green fluorescent protein (GFP) and luciferase in CL1-5 cells, pCMV-GFP/luciferase-lentivirus was prepared as described previously [16]. CL1-5GL cells were then produced by infecting CL1-5 cells with pCMV-GFP/luciferase-lentivirus. Flow cytometric analysis revealed that more than 99% of cells expressed GFP. Cell lysates were also harvested for validation of luciferase activity using a Minilumat LB 9506 luminometer (Berthold, Wildbach, Germany). Luciferase activity was confirmed by measuring the photon counts of serial cell dilutions (1~105) by the IVIS Imaging System (Xenogen, Alameda, CA).
In vivo experimental metastasis assay for rVP1-pretreated cancer cells
Human lung cancer cells (CL1-5GL cells) were pretreated with or without 0.3µM rVP1 in 0.5% FBS-medium at 37°C for 24 h. The cells (1 × 106 cells/50 µl PBS/mouse) were injected into male SCID mice per group via the tail vein. Metastatic progression was monitored weekly and quantified using a noninvasive bioluminescence IVIS Imaging System (Xenogen, Alameda, CA) as described previously [16]. After 5 weeks, three mice were killed for necropsy and the other 8 mice were kept for survival studies. The lungs of the sacrificed mice were isolated, collected and processed for H&E/immunohistochemistry staining. The number of tumor colonies in the lungs was counted, and relative lung weight (lung weight/body weight) was recorded.
Experimental xenograft murine metastasis assay to test in vivo effects of rVP1
Male SCID mice (n = 11/group) were implanted with 50 µl RPMI medium (vehicle) or 1 × 106/50 µl tumor cells (CL1-5GL) on day 0 by lateral tail vein injection. Metastatic progression was monitored weekly and quantified using a noninvasive bioluminescence IVIS Imaging System (Xenogen, Alameda, CA) as described previously [16]. One week after lateral tail vein injection, one group of mice was treated with vehicle and another group was treated with rVP1 (5 or 15 mg/kg body weight in 100 µl PBS) via tail-vein injection three times per week for 4 weeks. Three mice in each group were killed for necropsy and the other 9 mice were kept for survival studies. The lungs of the sacrificed mice were isolated, collected and processed for H&E/immunohistochemistry staining. The number of tumor colonies in the lungs was counted, and relative lung weight (lung weight/body weight) recorded.
Immunohistochemistry and histopathology examination
The mouse tissue samples were embedded in paraffin and cut to 4 μm thicknesses. Samples were stained with hematoxylin and eosin (H&E) and immunostained with different antibodies, and examined in the same manner as described previously [16, 30]. The images were scanned into a digital format by Scanscope XT system (Aperio Technologies, Vista, CA) and analyzed using Aperio ImageScope 9.1 software (Aperio Technologies).
Statistics
All statistical comparisons were made with two-tailed tests. The survival time was assessed using Kaplan-Meier curves and tested for significance by the log-rank test. Statistical evaluation was performed using GraphPad Prism version 5.0 for Microsoft Windows (GraphPad Software, La Jolla, CA). Differences between groups were considered statistically significant at *P < 0.05 or **P < 0.01.
Conflict of interest statement
The authors have no conflicts of interest to declare.
Acknowledgments
This work was supported by grants from Academia Sinica (to S.-M. L. and C.-M. L.). The authors thank Dr. Michael Hsiao for offering the pCMV-GFP/luciferase-lentivirus; Ms. Chiao-Li Chu and Mr. Chih-Yuan Chen for preparing the rVP1; Ms.Tzu-Wen Tai of the Scientific Instrument Center, Academia Sinica for her assistance with flow cytometry analysis; and Ms. Miranda Loney of the English Editors’ Office of the Agricultural Biotechnology Research Center, Academia Sinica for English editorial assistance.
References
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E and Forman D. Global cancer statistics. CA: a cancer journal for clinicians. 2011; 61(2):69-90.
2. Sun S, Schiller JH, Spinola M and Minna JD. New molecularly targeted therapies for lung cancer. The Journal of clinical investigation. 2007; 117(10):2740-2750.
3. Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nature reviews Cancer. 2003; 3(6):453-458.
4. Brown JR and DuBois RN. Cyclooxygenase as a target in lung cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2004; 10(12 Pt 2):4266s-4269s.
5. Castelao JE, Bart RD, 3rd, DiPerna CA, Sievers EM and Bremner RM. Lung cancer and cyclooxygenase-2. The Annals of thoracic surgery. 2003; 76(4):1327-1335.
6. Hida T, Yatabe Y, Achiwa H, Muramatsu H, Kozaki K, Nakamura S, Ogawa M, Mitsudomi T, Sugiura T and Takahashi T. Increased expression of cyclooxygenase 2 occurs frequently in human lung cancers, specifically in adenocarcinomas. Cancer research. 1998; 58(17):3761-3764.
7. Wolff H, Saukkonen K, Anttila S, Karjalainen A, Vainio H and Ristimaki A. Expression of cyclooxygenase-2 in human lung carcinoma. Cancer research. 1998; 58(22):4997-5001.
8. Chell S, Kaidi A, Williams AC and Paraskeva C. Mediators of PGE2 synthesis and signalling downstream of COX-2 represent potential targets for the prevention/treatment of colorectal cancer. Biochimica et biophysica acta. 2006; 1766(1):104-119.
9. Fresno Vara JA, Casado E, de Castro J, Cejas P, Belda-Iniesta C and Gonzalez-Baron M. PI3K/Akt signalling pathway and cancer. Cancer treatment reviews. 2004; 30(2):193-204.
10. Hanada M, Feng J and Hemmings BA. Structure, regulation and function of PKB/AKT--a major therapeutic target. Biochimica et biophysica acta. 2004; 1697(1-2):3-16.
11. Wang D and Dubois RN. Eicosanoids and cancer. Nature reviews Cancer. 2010; 10(3):181-193.
12. Ho MY, Liang SM, Hung SW and Liang CM. MIG-7 controls COX-2/PGE2-mediated lung cancer metastasis. Cancer research. 2013; 73(1):439-449.
13. Peng JM, Liang SM and Liang CM. VP1 of foot-and-mouth disease virus induces apoptosis via the Akt signaling pathway. The Journal of biological chemistry. 2004; 279(50):52168-52174.
14. Chen TA, Wang JL, Hung SW, Chu CL, Cheng YC and Liang SM. Recombinant VP1, an Akt inhibitor, suppresses progression of hepatocellular carcinoma by inducing apoptosis and modulation of CCL2 production. PloS one. 2011; 6(8):e23317.
15. Chiu CF, Peng JM, Hung SW, Liang CM and Liang SM. Recombinant viral capsid protein VP1 suppresses migration and invasion of human cervical cancer by modulating phosphorylated prohibitin in lipid rafts. Cancer letters. 2012; 320(2):205-214.
16. Peng JM, Chen YH, Hung SW, Chiu CF, Ho MY, Lee YJ, Lai TC, Hsiao M, Liang CM and Liang SM. Recombinant viral protein promotes apoptosis and suppresses invasion of ovarian adenocarcinoma cells by targeting alpha5beta1 integrin to down-regulate Akt and MMP-2. British journal of pharmacology. 2012; 165(2):479-493.
17. Thiery JP and Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nature reviews Molecular cell biology. 2006; 7(2):131-142.
18. Bauvois B. New facets of matrix metalloproteinases MMP-2 and MMP-9 as cell surface transducers: outside-in signaling and relationship to tumor progression. Biochimica et biophysica acta. 2012; 1825(1):29-36.
19. Crouch S, Spidel CS and Lindsey JS. HGF and ligation of alphavbeta5 integrin induce a novel, cancer cell-specific gene expression required for cell scattering. Experimental cell research. 2004; 292(2):274-287.
20. Han S, Sidell N, Roser-Page S and Roman J. Fibronectin stimulates human lung carcinoma cell growth by inducing cyclooxygenase-2 (COX-2) expression. International journal of cancer Journal international du cancer. 2004; 111(3):322-331.
21. Delcommenne M, Tan C, Gray V, Rue L, Woodgett J and Dedhar S. Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proceedings of the National Academy of Sciences of the United States of America. 1998; 95(19):11211-11216.
22. Hannigan G, Troussard AA and Dedhar S. Integrin-linked kinase: a cancer therapeutic target unique among its ILK. Nature reviews Cancer. 2005; 5(1):51-63.
23. Persad S, Attwell S, Gray V, Mawji N, Deng JT, Leung D, Yan J, Sanghera J, Walsh MP and Dedhar S. Regulation of protein kinase B/Akt-serine 473 phosphorylation by integrin-linked kinase: critical roles for kinase activity and amino acids arginine 211 and serine 343. The Journal of biological chemistry. 2001; 276(29):27462-27469.
24. Lin CC, Lee IT, Yang YL, Lee CW, Kou YR and Yang CM. Induction of COX-2/PGE(2)/IL-6 is crucial for cigarette smoke extract-induced airway inflammation: Role of TLR4-dependent NADPH oxidase activation. Free radical biology & medicine. 2010; 48(2):240-254.
25. Peinado H, Olmeda D and Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nature reviews Cancer. 2007; 7(6):415-428.
26. Ho MY, Leu SJ, Sun GH, Tao MH, Tang SJ and Sun KH. IL-27 directly restrains lung tumorigenicity by suppressing cyclooxygenase-2-mediated activities. Journal of immunology. 2009; 183(10):6217-6226.
27. Chu YW, Yang PC, Yang SC, Shyu YC, Hendrix MJ, Wu R and Wu CW. Selection of invasive and metastatic subpopulations from a human lung adenocarcinoma cell line. American journal of respiratory cell and molecular biology. 1997; 17(3):353-360.
28. Wang JH, Liang CM, Peng JM, Shieh JJ, Jong MH, Lin YL, Sieber M and Liang SM. Induction of immunity in swine by purified recombinant VP1 of foot-and-mouth disease virus. Vaccine. 2003; 21(25-26):3721-3729.
29. Huang Q, Shen HM, Shui G, Wenk MR and Ong CN. Emodin inhibits tumor cell adhesion through disruption of the membrane lipid Raft-associated integrin signaling pathway. Cancer research. 2006; 66(11):5807-5815.
30. Chiu CF, Ho MY, Peng JM, Hung SW, Lee WH, Liang CM and Liang SM. Raf activation by Ras and promotion of cellular metastasis require phosphorylation of prohibitin in the raft domain of the plasma membrane. Oncogene. 2013; 32(6):777-787.