
INTRODUCTION
Diffuse large B-cell lymphoma (DLBCL), the most common type of non-Hodgkin lymphoma, is characterized by clinical, genetic, and biological heterogeneity [1]. The current frontline chemotherapy regimen for DLBCL is R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone). Using this regimen, about 60% of patients are cured with the remaining patients experiencing either refractory (~10%) or relapse (~30%) disease within 2–3 years. The salvage therapy options for patients with relapsed disease are poor (response rates < 20%), and survival is usually short. Notably, 2–3% of DLBCL cases with rearrangements of MYC and either the BCL2 or BCL6 gene, so-called double-hit lymphoma (DHL), are associated with the germinal center B-cell (GCB) phenotype, frequent extranodal and central nervous system involvement, higher International Prognostic Index scores, poor response to R-CHOP therapy, and overall dismal outcome [2–6]. Investigation of novel therapeutic approaches for relapsed/refractory DLBCL as well as DHL is underway, but lack of relevant human experimental models for understanding the biological basis of these cancers has hampered the identification of valid therapeutic regimens.
The ubiquitin-proteasome signaling pathway plays an important role in the proteolysis of key regulatory proteins [7, 8]. Importantly, dysregulation of this pathway is linked to the development of various diseases, including cancer, and targeting components of the pathway may offer therapeutic opportunities [8]. The development of the first-in-class proteasome inhibitor bortezomib is one of the major milestones of this approach; bortezomib is effective in the treatment of patients with new or relapsed/refractory multiple myeloma [9]. Bortezomib also inhibits cell growth and induces apoptosis in mantle cell lymphoma cells in vitro and has clinical efficacy in relapsed/refractory cases of this disease [10, 11]. However, the duration of response is limited, and peripheral neuropathy is a dose-limiting side effect [12, 13]. The good clinical outcome of bortezomib treatment gave impetus for the development of second-generation proteasome inhibitors, with the goals of enhancing antitumor activity and decreasing toxicity, as well as providing more flexible dosing schedules and greater patient convenience.
MLN9708 is a novel oral proteasome inhibitor that has shown promising preclinical and clinical activity in several types of cancers. Compared with bortezomib, MLN9708 is orally bioactive, has a shorter proteasome dissociation half-life and improved pharmacokinetics, and has low rates of peripheral neuropathy [14]. Upon exposure to aqueous solutions or plasma, MLN9708 immediately hydrolyzes to its biologically active boronic acid form MLN2238 (ixazomib). Ixazomib inhibits cell growth and induces apoptosis in multiple myeloma cells resistant to conventional therapies and bortezomib. Ixazomib-triggered multiple myeloma cell death has been shown to be associated with activation of caspases, activation of the p53 pathway, induction of endoplasmic reticulum stress response proteins, inhibition of NF-κB, and upregulation of miR33b [15, 16]. Several clinical trials have shown promise for ixazomib, both as a single drug and in combination with dexamethasone, in patients with relapsed/refractory multiple myeloma [17, 18]. The potential efficacy of ixazomib for treatment of refractory/relapsed DLBCL, including DHL, remains unclear.
Our purpose in the present study was to examine the antitumor activity and biological effects of ixazomib in both in vitro and in vivo models of refractory/relapsed DLBCL and DHL.
RESULTS
In vitro ixazomib sensitivity in patient-derived DLBCL cell lines
To evaluate the antitumor efficacy of ixazomib in human DLBCL, we first examined the in vitro effects of the drug in 28 representative DLBCL cell lines, 18 GCB and 10 non-GCB, using concentration-dependent, 72 h viability assays. Both GCB and non-GCB DLBCL cell lines showed modest responses to ixazomib, with IC50 values ranging from 21 to 200 nmol/L (nM) (Figure 1A; see Supplementary Figure 1 for concentration-response curves). The MZ and RC cell lines were most responsive to the drug, with IC50 values of 21 and 40 nM, respectively. The IC50 values of ixazomib in all DLBCL cell lines were then compared with those of other proteasome inhibitors such as bortezomib and carfilzomib. The average IC50 for ixazomib (120 nM) was 14-fold higher than that of bortezomib (average 8.6 nM) and 8.8-fold higher than that of carfilzomib (average 13.5 nM; Figure 1B). Response to ixazomib did not differ significantly between GCB and non-GCB cell lines (p = 0.6052; Figure 1C). Four DLBCL cell lines carried the MYC and BCL2 translocations and met criteria for DHL [19–22]. Eight cell lines expressed both MYC and BCL-2 proteins, measured by RPPA analysis (Table 1) and therefore met the criteria for double expressor lymphoma (DEL), and 9 cell lines carried the p53 gene mutations (Table 1). There was no significant difference in ixazomib IC50 values between the DH/DEL group and the non-DH/DEL groups (p = 0.5288; Figure 1D), nor was there a difference between the mutant p53 and wild-type p53 groups (p = 0.6416; Figure 1E), suggesting that ixazomib is also effective in poor responder DLBCL groups. Furthermore, there was no significant difference in ixazomib sensitivity between the doxorubicin-sensitive and the doxorubicin-resistant cell lines (p = 0.4295; Figure 1F), suggest that ixazomib is a promising therapeutic agent that has the potential to overcome treatment resistance in DLBCL. Since ixazomib effectively inhibits DLBCL cell viability, we next examined whether the drug can induce apoptosis and/or cell cycle arrest in two representative DLBCL cell lines (RC and MZ), shown to be highly sensitive to ixazomib. Cells treated with ixazomib showed both time- (24 and 48 h) and concentration-dependent increases in apoptosis (Figure 1G; see Supplementary Figure 2 for detailed histograms). Furthermore, cells treated with ixazomib accumulated in sub-G0/G1 and underwent G2M cell cycle arrest (Figure 1H).
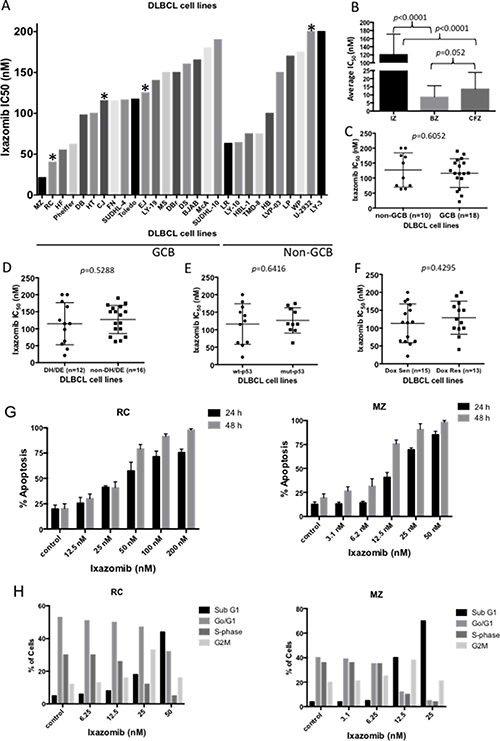
Figure 1: In vitro antitumor efficacy of the proteasome inhibitor ixazomib in DLBCL. (A) The effect of ixazomib in a concentration-dependent manner for 72 h on viability of 28 representative DLBCL cell lines (10 non-GCB subtype and 18 GCB subtype) was assessed. The IC50 value for each cell line was calculated and plotted. * denotes double-hit DLBCL cell lines. (B) The average IC50 value of ixazomib (IZ) in all DLBCL cell lines was compared with those of other proteasome inhibitors such as bortezomib (BZ) and carfilzomib (CFZ). The average IC50 of ixazomib (120 nM) was 14-fold higher than that of bortezomib (8.6 nM) and 8.8-fold higher than that of carfilzomib (13.5 nM). (C) Comparison of ixazomib IC50 values for non-GCB (n = 10) and GCB (n = 18) DLBCL subtypes revealed no significant difference. (D–F) Comparison of ixazomib IC50 values for double hit/double expressor lymphoma (DH/DEL) vs. non-DH/DEL groups (D), wild-type p53 vs. mutant p53 groups (E), and doxorubicin-sensitive vs. doxorubicin-resistant groups (F) revealed no significant differences. (G) Ixazomib-sensitive DLBCL cell lines RC (left panel) and MZ (right panel) were treated with ixazomib at various concentrations for 24 or 48 h, followed by annexin V staining/FACS analysis for apoptosis identification. (H) RC (left panel) and MZ (right panel) cells were treated with ixazomib at various doses for 24 h and were then subjected to cell cycle analysis. Error bars indicate the standard error of the mean.
Table 1: Characterization of DLBCL cell lines
DLBCL Cell lines |
Subtype |
STR DNA Finger- Printing |
p53 status |
BCL2/MYC |
IZ IC50(nM) |
Doxo IC50(ng/mL) |
|
|---|---|---|---|---|---|---|---|
1 |
OCI-LY10 |
ABC |
Yes |
wt |
+/− |
64 |
12.23 |
2 |
OCI-LY3 |
ABC |
Yes |
wt |
+/+ |
200 |
8.45 |
3 |
SUDHL-4 |
GCB |
Yes |
mut |
+/− |
116 |
15.62 |
4 |
HF |
GCB |
Yes |
wt |
+/+ |
55 |
8.87 |
5 |
McA |
GCB |
Yes |
wt |
+/+ |
180 |
8.49 |
6 |
LR |
ABC |
Yes |
wt |
+/+ |
63 |
14.8 |
7 |
MZ |
GCB |
Yes |
wt |
+/+ |
21 |
10.4 |
8 |
FN |
GCB |
Yes |
wt |
+/+ |
115 |
12.8 |
9 |
DBr |
GCB |
Yes |
wt |
−/− |
150 |
30.67 |
10 |
WP |
ABC |
Yes |
mut |
−/− |
175 |
45.03 |
11 |
HB |
ABC |
Yes |
mut |
−/+ |
100 |
55.06 |
12 |
CJ |
GCB (DHL) |
Yes |
mut |
+/+ |
115 |
134.1 |
13 |
LP |
ABC |
Yes |
mut |
−/− |
170 |
115.2 |
14 |
MS |
GCB |
Yes |
mut |
+/− |
150 |
97.1 |
15 |
DS |
GCB |
Yes |
mut |
−/− |
160 |
122.2 |
16 |
EJ |
GCB (DHL) |
Yes |
wt |
+/+ |
125 |
65.2 |
17 |
Toledo |
GCB |
Yes |
wt |
+/− |
117 |
12 |
18 |
Pfeiffer |
GCB |
Yes |
mut |
−/− |
62 |
50 |
19 |
OCI-LY19 |
GCB |
Yes |
wt |
+/+ |
140 |
20 |
20 |
BJAB |
GCB |
Yes |
wt |
−/+ |
165 |
22 |
21 |
U2932 |
ABC (DHL) |
Yes |
ND |
+/+ |
200 |
75 |
22 |
RC |
GCB (DHL) |
yes |
ND |
+/+ |
40 |
95 |
23 |
LVP-03 |
PM-BCL |
yes |
ND |
−/− |
150 |
98 |
24 |
HBL-1 |
ABC |
Yes |
ND |
+/− |
75 |
65 |
25 |
TMD-8 |
ABC |
Yes |
ND |
−/+ |
75 |
20 |
26 |
DB |
GCB |
Yes |
mut |
+/+ |
98 |
60 |
27 |
HT |
GCB |
Yes |
ND |
−/− |
100 |
85 |
28 |
SUDHL-10 |
GCB |
Yes |
ND |
+/− |
190 |
120 |
ABC and GCB-DLBCL subtypes of the cell lines were previously characterized. All cell lines were authenticated using short tandem repeat DNA fingerprinting at the Characterized Cell Line Core Facility at The University of Texas MD Anderson Cancer Center. BCL-2 and MYC protein expressions were determined by reverse-phase protein array and validated by Western blotting. All cells were collected at log phase before protein purification. The cut-offs were determined by log2 protein expression level, values below zero were considered negative and above zero were considered positive. Ixazomib (IZ) and Doxorubicin (Dox) IC50 values for each cell line were determined using a dose-dependent 72 h viability assay. For Doxorubicin, cell lines with IC50 values below 50 ng/ml were considered “Dox Sensitive” and cell lines with IC50 values of 50 ng/ml and above were considered “Dox Resistant”. ABC, activated-like B cell; GCB, germinal center B cell; DH, double hit; PM-BCL, plasmablastic B cell lymphoma; STR, short tandem repeat; wt, wild type; mut, mutant; ND, no data; IZ, ixazomib; Doxo, doxorubicin.
In vitro effects of ixazomib in primary DLBCL cells and in vivo efficacy of ixazomib in a DHL xeno-transplant NOD SCID gamma (NSG) mouse model
To assess the effects of ixazomib on the viability of fresh primary human DLBCL cells, lymphoma cells were isolated from a panel of 10 DLBCL patients who had been exposed to various standard therapies (Table 2). The primary cells were treated with ixazomib in a concentration-dependent manner and the observed median IC50 values ranged from 36–200 nM (Figure 2A and Table 2), similar to the range found in DLBCL cell lines in vitro. Notably, the similar range of drug concentrations of ixazomib did not affect proliferation of resting PBMCs from three healthy volunteers (Figure 2A). These results indicate that ixazomib selectively inhibits proliferation of primary DLBCL cells without affecting the growth of normal lymphocytes. To determine whether the in vitro effects of ixazomib on DLBCL cells translate to an in vivo setting, we evaluated the ability of ixazomib to suppress tumor growth in vivo in a DHL xeno-transplant model established in NSG mice. Treatment of tumor-bearing mice with ixazomib 3.5 mg/kg or 7 mg/kg body weight orally twice a week for 2 weeks resulted in significant reduction of tumor burden (Figure 2B). The magnitude and durability of the response increased with the dose; treatment with 7 mg/kg conferred a TGImax of 64% (p < 0.001). Due to the aggressiveness of the tumor in the control group, the experiment was terminated after the fifth treatment and all mice in the control and ixazomib 3.5 mg/kg treatment groups were sacrificed. Mice in the ixazomib 7 mg/kg treatment group were also sacrificed within the next 10 days due to excessive tumor burden. A representative tumor tissue sectioned from an ixazomib-treated mouse show increased caspase-3 activation in comparison to a tumor tissue sectioned from a control untreated mouse, indicative of cells undergoing apoptosis in vivo after ixazomib treatment (Figure 2C). In addition, more cells were found in the mitotic phase of the cell cycle in control vs. ixazomib-treated mice (Figure 2C and Supplementary Figure 3), further validating our in vitro findings showing ixazomib induces cell cycle arrest.
Table 2: Patients with refractory DLBCL
DLBCLPatient |
Sex |
Age |
COO |
Treatment |
Response |
Status |
Sample source |
OS (months) |
IZ IC50nM |
|---|---|---|---|---|---|---|---|---|---|
PT-1 |
M |
46.9 |
Non-GCB |
RICE, splenectomy |
progression |
Dead |
Fluid |
6.5 |
59.2 |
PT-2 |
M |
82.5 |
Non-GCB |
R-CHOP |
relapse and progression |
Dead |
PB |
13.5 |
57.9 |
PT-3 |
F |
48.2 |
Non-GCB |
R-ESHAP, hyper- CVAD, auto SCT |
relapse and progression |
Dead |
Fluid |
14.8 |
35.9 |
PT-4 |
F |
75.6 |
Non-GCB |
R-CHOP, R-ESHAP, Rituxan+MTX +cytarabine, COPP, radiaRon, Revlimid |
partial remission and progression |
Dead |
Apheresis |
40.7 |
122 |
PT-5 |
M |
55.5 |
GCB |
CHOP, hyper-CVAD, RICE, RDAP |
relapse and progression |
Dead |
PB |
14.4 |
200 |
PT-6 |
M |
47.4 |
Non-GCB |
CHOP, DA-EPOCH, ICE, auto SCT, hyper- CVAD |
partial remission and progression |
Dead |
Apheresis |
29.2 |
201 |
PT-7 |
M |
38.7 |
GCB |
R-CHOP, RICE |
progression |
Dead |
Fluid |
8.2 |
132 |
PT-8 |
F |
69.0 |
GCB |
R-hyper-CVAD, R- EPOCH, RICE, ritumimab_revlimid |
progression |
Dead |
Fluid |
49.3 |
60.8 |
PT-9 |
F |
68.2 |
GCB |
R-CHOP, R-GEMOX, benamusRne +rituximab |
progression |
Dead |
Apheresis |
38.1 |
44.5 |
PT-10 |
M |
63.5 |
GCB |
R-CHOP, RICE, auto SCT, R-GEMOX |
relapse and progression |
Dead |
PB |
10.2 |
75 |
Dx, diagnosis; COO, cell of origin; OS, overall survival; IZ, ixazomib; NA, not available; GCB, germinal center B cell; RICE: rituximab, ifosfamide, carboplatin, etopiside; VTEP, R-CHOP: rituximab-cyclophophamide, hydroxydaunorubicin, oncovin, prednisone; R-ESHAP: rituximab-etoposide, solu-medrol-methylprednisolone, high-dose Ara-C-cytarabine, platinol; CVAD: cyclophosphamide, vincristine, doxorubicin, dexamethasone; DA-EPOCH: dose adjusted-etoposide, prednisolone, oncovin, cyclophosphamide, hydroxydaunorubicin; R-GEMOX: rituximab-gemcitabine, oxaliplatin; SCT, stem cell transplant; PB, peripheral blood.
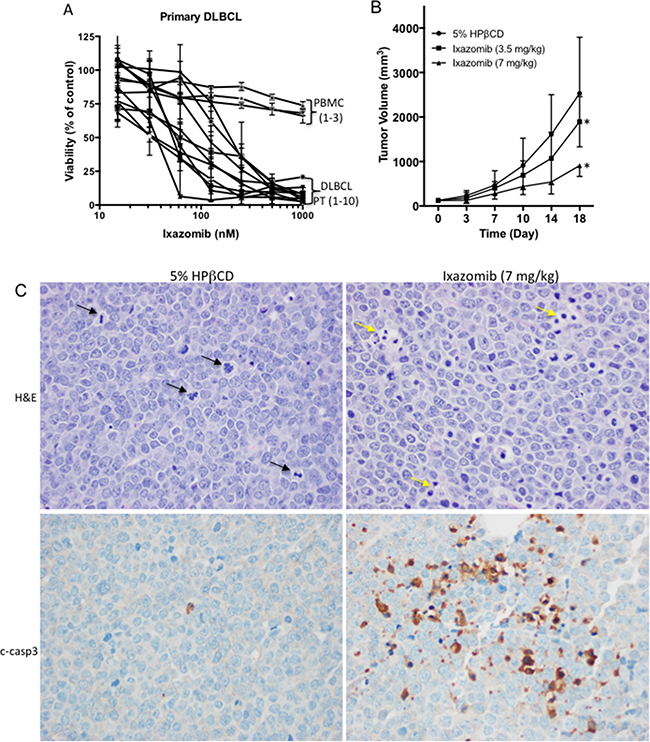
Figure 2: In vitro effects of ixazomib in primary DLBCL cells and in vivo efficacy of ixazomib in a DHL xeno-transplant mouse model. (A) The effect of ixazomib at various doses for 72 h was assessed by viability assay in primary DLBCL cells from 10 representative patients and peripheral blood mononuclear cells (PBMC) isolated from 3 healthy donors. (B) The effects of ixazomib on tumor growth in NOD-SCID mice with established RC tumors are shown. The graph describes the change in tumor volume as a function of the time after initiation of treatment. Each point on the curves represents the mean of 10 tumors. Error bars depict the SEMs. Ixazomib was administered orally twice a week. A 5% solution of HPβCD was used as the vehicle control. *p-value < 0.001. (C) Tumor tissues from control and ixazomib-treated mice were fixed, sectioned, and stained for H&E (top panels) or cleaved caspase-3 (bottom panels). 40×; black arrows pointing to mitotic cells; yellow arrows pointing to apoptotic bodies.
Correlation between proteasomal activities and ixazomib sensitivity in DLBCL
To determine whether proteasome activity can predict ixazomib sensitivity, we utilized the ProCISE assay to quantify proteasome subunit occupancy in 24 representative DLBCL cell lines (16 GCB and 8 non-GCB). In general, constitutive (c)-proteasome and immuno (i)-proteasome activity were detected in most of the DLBCL cell lines, although i-proteasome activity was low in some cell lines (Supplementary Figures 4 and 5). The sensitivity trend of ixazomib positively correlated with activity of the beta2 subunit of the c-proteasome in all DLBCL cell lines, where cell lines with lower beta2 subunit is more sensitive to ixazomib treatments (Table 3). However, when the cell lines were stratified into GCB and non-GCB subsets, in ixazomib-treated cells the GCB subset showed a trend toward a positive correlation for the c-proteasome subunits and a negative correlation for the i-proteasome subunits, particularly MECL-1 (Figure 3A). In non-GCB cells, there was a trend for positive correlation with i-proteasome subunits (Figure 3B). These results suggest that GCB cells with higher i-proteasome subunit activity are more sensitive to ixazomib, whereas non-GCB cells with higher i-proteasome subunit activity are more resistant to the drug. Since i-proteasome subunits can be induced by interferon-gamma (IFN-γ), we selected three DLBCL cell lines that were deficient or had low expression of both the LMP-2 and MECL-1 subunits and stimulated them with IFN-γ. After 48 h of IFN-γ exposure, the LMP-2 and MECL-1 subunits were induced (Figure 3C) and the cells were sensitized to ixazomib (Figure 3D).
Table 3: Proteasomal activity vs. ixazomib sensitivity in DLBCL
All |
Correlation Coefficient (r) |
p value |
|---|---|---|
Beta 1 |
0.2009 |
0.3467 |
Beta 2 |
0.402 |
0.0515 |
Beta 5 |
0.1037 |
0.6298 |
MECL1 |
0.04508 |
0.8343 |
LMP2 |
0.1211 |
0.5728 |
LMP-7 |
0.1874 |
0.3806 |
GCB |
Correlation Coefficient (r) |
p value |
Beta 1 |
0.2613 |
0.3284 |
Beta 2 |
0.4833 |
0.0579 |
Beta 5 |
0.3827 |
0.1435 |
MECL1 |
−0.3999 |
0.1248 |
LMP2 |
−0.1473 |
0.5861 |
LMP-7 |
−0.05122 |
0.8506 |
Non-GCB |
Correlation Coefficient (r) |
p value |
Beta 1 |
0.04858 |
0.9091 |
Beta 2 |
0.2867 |
0.4912 |
Beta 5 |
−0.2903 |
0.4855 |
MECL1 |
0.3886 |
0.3413 |
LMP2 |
0.5883 |
0.1251 |
LMP-7 |
0.5191 |
0.1874 |
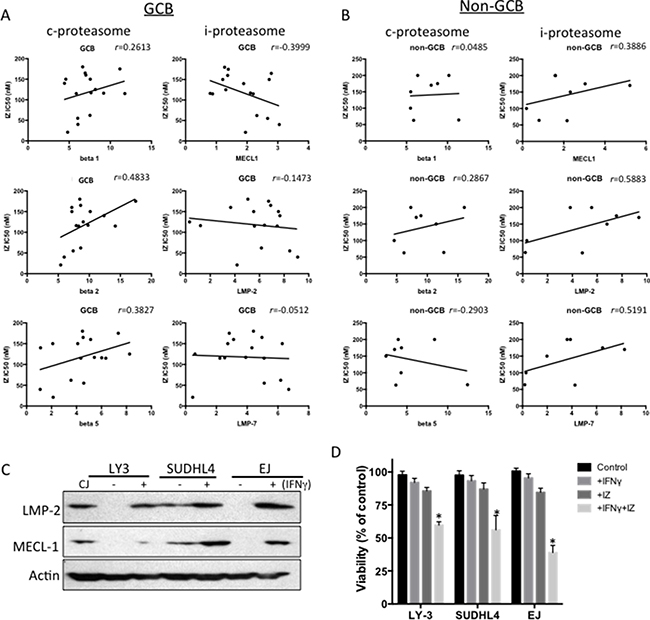
Figure 3: Correlation between proteasomal activities and ixazomib sensitivity in DLBCL. (A, B) Proteasomal activities were assessed by the ProCISE assay and plotted against the corresponding ixazomib IC50 for GCB (A) and non-GCB (B) DLBCL cell lines. Correlation coefficients were determined by the Pearson rank correlation and one-tailed t-test. (C) Three DLBCL cell lines (OCI-LY3, SUDHL-4, and EJ) with low/negative activity of i-proteasome subunits LMP2 and MECL-1 were stimulated with IFN-γ for 48 h and the cell extracts were subjected to western blotting for LMP-1 and MECL-1. Actin was used as a loading control. The CJ cell line was used as a positive control. (D) The same three DLBCL cell lines (OCI-LY3, SUDHL-4, and EJ) with low/negative activity of i-proteasome subunits LMP2 and MECL-1 were stimulated with IFN-γ (100 units/mL) for 48 h and then treated with ixazomib (IZ; IC25 concentrations) for another 48 h and cell viability was assessed.*p-value < 0.05.
Proteomic analysis of ixazomib-treated DLBCL cells
To further gain mechanistic insights into the response of DLBCL to ixazomib, we utilized a proteomics approach via RPPA in two representative DLBCL cell lines that were shown to be highly sensitive to ixazomib. RC and MZ cells were treated with ixazomib at IC75 concentrations at different time intervals (12 and 24 h), and then proteins were extracted and purified from control and treated cells and cellular protein level changes were measured across a set of 285 antibodies. As shown in the “subtracted” heat maps (Figure 4A), a total of 71 proteins were altered by a log2 fold change of > 0.4 in at lease one comparison with IZ-treated RC and MZ cells. Of the 71 proteins, 41 (58%) were found overlapping, either up (20)- or down (21)- regulated in both RC and MZ cells treated with IZ (Figure 4B); these proteins are involved in various cellular processes, including apoptosis (caspases 3,7 and bax), DNA damage (γH2AX, Histone 3), growth/survival signaling (AKT, MCL-1, Stat5a), cell cycle regulation (p16INK4a), metabolism (Mitochondria/Hexokinase II), and tumor repressor (Merlin) (Figure 4B). There were some variations between the two cell lines. Nine proteins were found induced in RC cells after IZ treatment but were found down-regulated in MZ cells after IZ treatment. On the other hand, 20 proteins were found induced in MZ cells but were found down-regulated in IZ-treated RC cells. Using Western blot analysis, we confirmed that ixazomib induces apoptosis through activation of caspases 3 and 7 and cleavage of PARP (Figure 4C). Induction of γH2AX by ixazomib in both RC and MZ cells (Figure 4C) suggests that the drug can induce DNA damage in DLBCL cells.
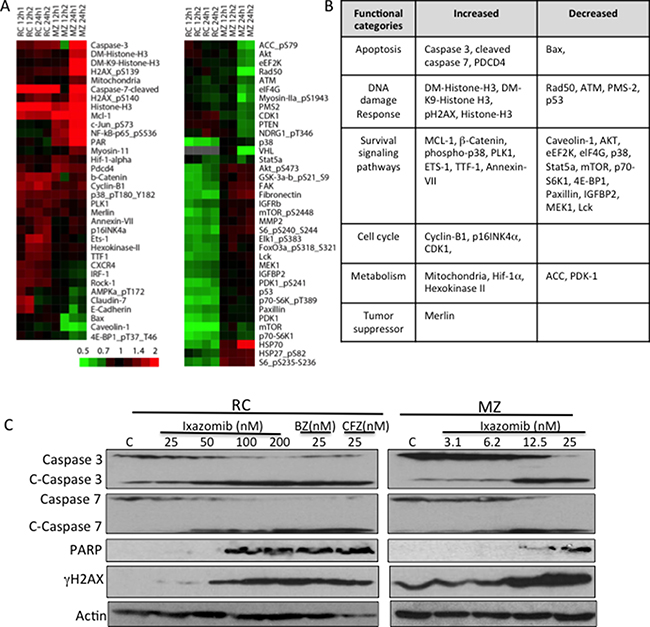
Figure 4: Proteomic analysis in control and ixazomib-treated DLBCL cells. Ixazomib-sensitive RC and MZ DLBCL cells were treated with ixazomib (IZ; at IC75 concentrations, 50 nM and 30 nM, respectively) for 12 or 24 h. Protein extracts from treated cells were subjected to reverse-phase protein array (RPPA) analysis. (A) “Subtracted” heat maps of RC and MZ lines for antibodies whose RPPA log2 values changed with IZ treatment by at least 0.4 from control in at least 4 of the 8 time points. The color bar shows the fold-change from control. (B) Table showing the overlapped increased and decreased proteins in both RC and MZ cells after ixazomib treatment. (C) RC and MZ cells were treated with various concentrations of ixazomib. RC cells treated with bortezomib (BZ) and carfilzomib (CFZ) were used as controls. Protein extracts from treated cells were subjected to western blotting for caspase 3, caspase 7, cleaved PARP, γH2AX, and actin (loading control).
Synergism of ixazomib with inhibition of the DNA damage repair pathway through CHK2 to induce robust DNA damage and apoptosis
Since ixazomib was shown to induce a robust DNA damage response in DLBCL cells, we evaluated whether the DNA damage repair (DDR) pathways were induced in ixazomib-treated DLBCL cells. As shown in Figure 5A, pATM and pATR proteins were inhibited and pCHK2 and pCHK1 were significantly induced in cells treated with ixazomib. Since CHK2 shows a more significant increased in both cell lines tested, we examine whether blocking ixazomib-induced pCHK2 would lead to a more robust antitumor response in DLBCL cells. As expected, ixazomib in combination with a CHK2 inhibitor (CHK2i) showed synergistic activity in DLBCL cell lines that were more sensitive to ixazomib (RC, MZ, and HT), but not in cells that were more resistant to ixazomib (U-2932) (Figure 5B; see Supplementary Figure 4 for detailed synergy calculations). Similar results were obtained in primary DLBCL cells from two patients, one sensitive (PT-9) and one resistant (PT-5) to ixazomib (Figure 5C). CHK2i inhibited ixazomib-induced CHK2 phosphorylation in ixazomib-sensitive RC and PT-9 cells, but not in ixazomib-resistant U-2932 cells (Figure 5D). Similarly, combined treatment with ixazomib and CHK2i caused a striking increase in γH2AX (DNA damage) in DLBCL cell line (RC) and primary (PT-9) cells but not in U-2932 cells (Figure 5D), and increase in cells undergoing apoptosis as compared with either agent alone (Figure 5E). Furthermore, these results were independently confirmed using siRNA-mediated knock-down of CHK2 in RC and U-2932 cells, which shows that CHK2 silencing sensitized ixazomib-sensitive cells (RC) but not ixazomib-resistant cells (U-2932) (Figure 5F). These findings confirm that co-administration of a CHK2i markedly potentiates DNA damage in DLBCL cells exposed to ixazomib and that this event precedes induction of extensive apoptosis.
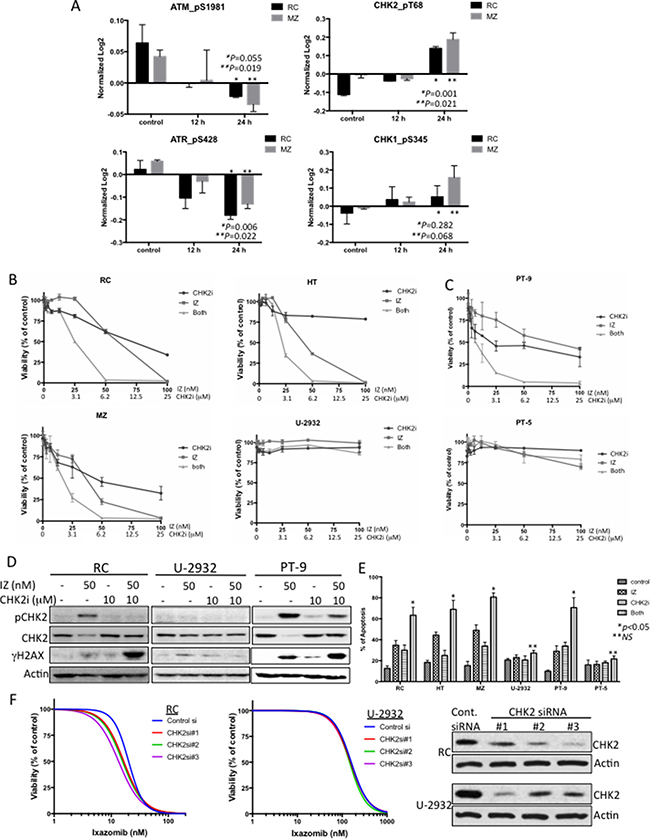
Figure 5: Synergism of ixazomib with inhibition of the DNA damage repair pathway through CHK2 to induce robust DNA damage and apoptosis. (A) Protein level changes of pATM, pATR, pCHK1 and pCHK2 in control and IZ-treated RC (black bars) and MZ (gray bars) cells for 12 and 24 h. Data for these proteins did not showed up in the heat maps because they were below the 0.4-cutoff threshold. (B) Representative ixazomib-sensitive (RC, MZ, and HT) and ixazomib-resistant (U-2932) DLBCL cell lines were co-treated with ixazomib (IZ) and a CHK2 inhibitor (CHK2i) at concentration-dependent manner for 72 h and cell viability was assessed. Data from two independent experiments performed in triplicate are shown. (C) Primary DLBCL cells from two representative patients (PT-9 [ixazomib-sensitive] and PT-5 [ixazomib-resistant]) were co-treated with ixazomib and CHK2i at various doses for 48 h, and cell viability was assessed. Data from two independent experiments performed in triplicate are shown. (D) RC, U-2932, and primary DLBCL (PT-9) cells were treated with ixazomib (50 nM) alone, CHK2i (10 μM) alone, or combination of both drugs for 24 h. Protein extracts from the cells were subjected to western blot analysis to detect pCHK2, CHK2, γH2AX (a DNA damage marker), and actin (protein loading control). (E) Representative ixazomib-sensitive (RC, MZ, HT, and PT-9) and ixazomib-resistant (U-2932 and PT-5) DLBCL cells were treated with ixazomib (50 nM) alone, CHK2i (10 μM) alone, or combination of both drugs for 24 h. Apoptotic cells were detected by annexin V/propidium iodide staining. Data shown represent means ± SD from three independent experiments. (F) RC and U-2932 cells were transiently transfected with control or CHK2 siRNA. At 48 h post-transfection, protein purified from transfected cells was subjected to western blotting for CHK2 and actin (loading control). Small portion of the transfected cells were treated with increasing concentrations of ixazomib for an additional 72 h and viability was assessed. Data are expressed as means ± standard deviation of at least 2 independent experiments.
DISCUSSION
Although earlier studies have shown that the novel proteasome inhibitor ixazomib has in vitro and vitro antitumor activity in DLBCL, its efficacy in high-risk DLBCL, including DHL, has yet to be examined [14, 23]. In the present study, we further show the following: (i) ixazomib can effectively kill refractory/relapsed GCB and non-GCB DLBCL cells, as well as DHL cells in in vitro and in vivo models; (ii) ixazomib has low toxicity in normal PBMCs; (iii) ixazomib sensitivity correlates with immunoproteasome activity, particularly in the GCB subtype of DLBCL; (iv) ixazomib induces DNA damage and apoptosis in DLBCL cells; and (v) a CHK2 inhibitor suppresses ixazomib-induced CHK2 phosphorylation, resulting in a synergistic inhibition of cell growth and DNA damage.
Bortezomib was the first proteasome inhibitor that was approved by the United States Food and Drug Administration for the treatment of patients with multiple myeloma and relapsed mantle cell lymphoma [24, 25]. Dunleavy and colleagues showed that single agent bortezomib has minimal activity in DLBCL, but enhances the activity of standard chemotherapy regimens in patients with ABC (non-GCB) subtype of DLBCL [26]. Therefore, patients with relapsed/refractory non-GCB DLBCL may benefit from treatment with a combination of proteasome inhibitors and other chemotherapy regimen [26, 27]. Here, we show that ixazomib is active in many DLBCL cell lines of both GCB and non-GCB subtype. In addition, we show that ixazomib is active against MYC/BCL2 DHL, MYC/BCL2 DEL cell lines, mutant p53 DLBCL cell lines, and doxorubicin-resistant DLBCL cell lines. These results show that ixazomib is also active in high-risk DLBCL groups, extending beyond subtypes tested in previous studies [14, 28]. Furthermore, ixazomib inhibited viability of fresh primary DLBCL cells derived from 10 patients with treatment-refractory disease, but had little effect on normal PBMCs, indicating that the drug is effective but safe for DLBCL patients.
In this study we have shown that the IC50 of ixazomib is significantly higher than that of other proteasome inhibitors such as bortezomib and carfilzomib. This difference may be due to ixazomib having a shorter proteasome dissociation half-life than bortezomib, [29] so that a higher concentration of ixazomib is needed to attain an equivalent effect. Clinical pharmacokinetic studies estimated a plasma Cmax of 150 nM from once-weekly orally administered ixazomib, [30] higher than the average IC50 of 120 nM in our study. Our finding that ixazomib was very effective in a DHL xeno-transplant mouse model, significantly decreasing tumor burden, suggest that this drug may be promising for the treatment of patients with DHL, a group of patients with a treatment resistant tumor and very poor prognosis, as well as patients with refractory DLBCL overall. This observation is also in line with reports that ixazomib had significantly less severe side effects than bortezomib and displayed a manageable safety profile in clinical trials of heavily pretreated patients who had refractory myeloma [17, 18]. The mechanism(s) for ixazomib effectiveness in high risk DLBCL subtypes such as DHL is still unknown, but could possibly be due to higher immunoproteasome activity in these cells, as our study shows that ixzomib treatments were more responsive to cells with higher immunoproteasome subunit MECL1 activity, particularly in the GCB-DLBCL subtype. Further studies are required to validate these findings.
In an RPPA analysis performed on two representative cell lines, ixazomib consistently induced DNA damage through upregulation of the DNA damage marker γH2AX. To mitigate DNA damage, ixazomib-treated cells also activated DNA damage response (DDR) pathways by activating CHK1/CHK2 pathway. Two major signaling pathways drive DDR, ATM/CHK2 for DNA double-strand breaks and ATR/CHK1 for single-stranded DNA breaks. Inactivation of the DDR pathways has been shown to efficiently sensitize malignant cells to radiotherapy and chemotherapy [31, 32]. In addition, therapeutic targeting of CHK2 in the DDR pathway has shown to be feasible in DLBCL [33, 34]. Interestingly, our findings show that while pCHK2 was induced in ixazomib-treated cells, the level of phosorylated ATM was inhibited, suggesting that the activation of CHK2, and probably CHK1, is independent of ATM and ATR activation, respectively. This could be due to stimulation by different kinases, such as DNA-PK, which is known to activate CHK2 independent of ATM, [35, 36] or protein accumulation due to proteasomal inhibition, as the turnover of many of these DNA damage-associated proteins is controlled by the 26S proteasome [37]. Our results also show that a CHK2 inhibitor blocked ixazomib-induced CHK2, synergistically inhibiting cell growth, inducing apoptosis, and increasing DNA damage, confirming that abrogation of the CHK2 DDR in ixazomib-treated cells will lead subsequently to more robust DNA damage. These findings suggest that targeting CHK2, and probably CHK1, in combination with proteasome inhibition could be an innovative strategy to overcome chemoresistance in patients with DLBCL [38].
Taken together, our results demonstrate the preclinical efficacy and biological effects of ixazomib in in vitro and in vivo DLBCL models. To the best of our knowledge, this is the first report showing that ixazomib is an effective proteasome inhibitor active in high-risk DLBC, including DHL, and this drug therefore has the potential to become an effective agent in the treatment of patients with DLBCL refractory to standard therapy. Our data provide pre-clinical evidence to support the clinical development of ixazomib, probably in combination with novel check-point kinase inhibitors, for the treatment of patients with DLBCL, particularly refractory DLBCL.
MATERIALS AND METHODS
Cells and reagents
DLBCL cell lines MS, DS, DBr, JM (McA), FN, EJ, HF, HB, MZ, LR, CJ, LP, WP, LVP-03, and RC were established in our laboratory and were characterized and described previously [19, 39]. The Pfeifer and BJAB DLBCL cell lines were purchased from ATCC (Manassas, VA). The DLBCL cell lines U-2932, OCI-LY19, Toledo, SUDHL-4, SUDHL-10, HBL-1, TMD-8, DB, HT, OCI-LY10, and OCI-LY3 were obtained from Drs. Michael Rosenblum and Eric Davis (UT MD Anderson Cancer Center). All cell lines were routinely tested for mycoplasma using a Myco Tect kit (Invitrogen, Carlsbad, CA) and were validated by short tandem repeat DNA fingerprinting at the Characterized Cell Line Core Facility at The University of Texas MD Anderson Cancer Center. Stocks of authenticated cell lines were stored in liquid nitrogen for future use, and all cell lines used in the studies described here were from these authenticated stocks and were not passage more than six months from the time of thawing.
Primary DLBCL cells were isolated from samples obtained from patients through a protocol approved by the Institutional Review Board at MD Anderson Cancer Center. Normal peripheral blood mononuclear cells (PBMCs) were isolated from blood samples from healthy volunteers using the human B-cell enrichment cocktail from StemCell Technologies (Vancouver, BC, Canada) and used as lineage/cellular controls. Informed consent was obtained from all donors. The cells were cultured in RPMI medium (Gibco, Rockville, MD) containing 15% fetal calf serum (Gibco) and 1% penicillin/streptomycin (Hyclone, Logan, UT).
MLN9708 (ixazomib citrate, ixazomib) and bortezomib drug stocks were provided by Millennium Pharmaceuticals, Inc., a subsidiary of Takeda Pharmaceutical Company Limited (Cambridge, MA). Carfilzomib was provided by Amgen (Thousand Oaks, CA). CHK2 inhibitor was purchased from Sigma Aldrich (St. Louis, MO).
Antibodies and siRNA
Monoclonal and polyclonal antibodies specific for the following molecules were used: γ-H2AX, caspase 3, c-caspase 3, caspase 7, c-caspase 7, CHK2, and pCHK2 (Cell Signaling Technology, Danvers, MA); LMP-2, MECL-1, and PARP (Santa Cruz Biotechnology, Santa Cruz, CA). Pre-designed and validated CHK2 siRNA (S22119, S22120, S22121) and control siRNA were purchased from ThermoFisher Scientific (Waltham, MA).
Viability assays
Cells from representative DLBCL cell lines were plated at 5,000 cells per well in 384-well plates. The cells were incubated for 72 hours (h) in 20 μL medium containing 15% fetal bovine serum and the indicated drug at various concentrations or dimethylsulfoxide (DMSO) as control. Viability was assessed with the Celltiter-Glo Luminescent Cell Viability Assay according to the manufacturer’s instructions (Promega, Madison, WI). To determine the half maximal inhibitory concentration (IC50), six to eight concentrations in a sequence of two-fold increases were chosen for each cell line so that the IC50 point was approximately in the middle of the concentration range. The experiments were performed at least two times independently; in every experiment, each concentration was tested in triplicate samples.
Western blotting, apoptosis, cell cycle analysis, and transient transfection methods were performed as previously described [40, 41].
In vivo ixazomib treatment in a xeno-transplant DHL mouse model
All animal experiments were reviewed and approved by the MD Anderson Institutional Animal Care and Use Committee (IACUC; protocol #: 00001480-RN00). Six-week-old female severe combined immunodeficient (SCID) NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ mice were purchased from Jackson Laboratories (Bar Harbor, ME) and housed under specific pathogen-free conditions at the SCID Mouse Barrier Facility at MD Anderson. A recently established DHL cell line, RC, was used for the in vivo study [21]. RC cells (1 × 107 in 100 μL phosphate-buffered saline solution [PBS]) were injected intraperitoneally into the right abdomen of each mouse. After 3 weeks, when the tumors were palpable (approximately 125 mm3), mice were randomly divided into three groups (n = 10/group), and the drug treatment was started that day (day 0). Mice were treated with ixazomib (3.5 mg/kg or 7 mg/kg body weight) or with vehicle control (5% 2-hydroxypropyl-β-cyclodextrin (HPβCD)), as previously described, [23] in a 100-μL volume via oral gavage twice a week (Monday and Thursday) for 2 weeks. Ixazomib was formulated for oral dosing in 5% HPβCD. Electronic calipers were used to measure the length and width of each tumor twice a week. Tumor volume was calculated by applying the following equation: tumor volume = length × width2/2. Maximum tumor growth inhibition (TGImax) was calculated as the greatest treatment response using the following equation: TGImax = (1 − mean tumor volume of the treated group/mean tumor volume of the vehicle control group) × 100. The animals were sacrificed when tumor reached protocol limit, ~2,500 mm3. Tumors were removed and the animal carcasses disposed of according to IACUC guidelines.
ProCISE assay
The proteasome constitutive/immunoproteasome subunit enzyme-liked immunosorbent (ProCISE) assay was carried out in 24 representative DLBCL cell lines (16 GCB and 8 non-GCB), using a previously published methodology [42].
Reverse-phase protein array
Reverse-phase protein arrays (RPPA) were analyzed and antibodies validated by the RPPA Core Facility at MD Anderson [43]. Total protein lysates were prepared by resuspending cells in lysis buffer (1% Triton X-100, 50 mM HEPES [pH 7.4], 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 100 mM NaF, 10 mM NaPPi, 10% glycerol, 1 mM PMSF, 1 mM Na3VO4, and 10 μg/mL aprotinin). Protein lysates were adjusted to a 1 μg/μL concentration, and a serial dilution of five concentrations was printed on nitrocellulose-coated slides (Grace Bio-Labs, Bend, OR) with 10% of the samples replicated for quality control (2470 Arrayer; Aushon Biosystems, Billerica, MA). Immunostaining was performed using a DakoCytomation-catalyzed system with the diaminobenzidine colorimetric reaction (Dako, Carpenteria, CA). Slides were scanned on a flatbed scanner to produce 16-bit tiff images. Spot intensities were analyzed and quantified using an Array-Pro Analyzer (Meyer Instruments, Houston, TX) to generate spot signal intensities. Overall, 285 unique antibodies and 4 secondary antibody negative controls were analyzed. The Gene Cluster 3.0 (http://cluster2.software.informer.com/3.0/) software was used for data analysis, and the heat map was created by Java TreeView (http://rana.lbl.gov/EisenSoftware.htm) software.
Statistical analysis
The IC50 values were calculated for each cell line using CalcuSyn software (Biosoft, Cambridge, MA). The Pearson rank correlation coefficient was used to evaluate the correlation between proteasome inhibitor IC50 values and proteasome enzymatic activity. Asymptotic t approximation was used for computation of its p-value. Relative protein levels for each sample in the RPPA analysis were determined by interpolation of each dilution curve from the “standard curve” using R package SuperCurve. All data points were normalized for protein loading and transformed to linear values for bar graphing. Normalized linear values were transformed to log2 values and then median-centered for hierarchical cluster analysis and for heatmap generation. The heatmap was generated in Cluster 3.0 (http://cluster2.software.informer.com/3.0/) as a hierarchical cluster using Pearson correlation and a center metric, visualized in Treeview (http://rana.lbl.gov/EisenSoftware.htm), and presented in a high-resolution .bmp format. Significance in the experiments for TGImax was determined by the Student t-test and Wilcoxon rank sum test, respectively. Significance for complete response was determined by a two-tailed Fisher exact test. P-values of < 0.05 is considered statistically significant.
Abbreviations
DLBCL: diffuse large B cell lymphoma; DHL: double-hit lymphoma; RPPA: revers-phase protein array; CHK2: check-point kinase 2; R-CHOP: rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone; GCB: germinal center B-cell; DMSO: dimethylsulfoxide; SCID: severe combined immunodeficient; DEL: double expressor lymphoma; DDR: DNA damage repair.
Author contributions
WL and LVP conceived and designed the experiments. WL, JC, AT, CR, LL, and LVP performed the data analysis and interpretation. SZ and CS performed the bioinformatics analysis. RED analyzed the RPPA data. WL, KHY, JW, RED, SH, LJM, RJF, and LVP were involved in the manuscript preparation. All authors read and approved the final manuscript.
ACKNOWLEDGMENTS AND FUNDING
This work was supported financially by grants from the National Natural Science Foundation of China (No. 31100944), the Natural Science Foundation of Jiangsu Province, China (No. BK2009107), and the Research Fund for the Visiting Scholar Program by the China Scholarship Council. The Characterized Cell Line Core and the Flow Cytometry and Cellular Imaging Core Facilities at The University of Texas MD Anderson Cancer Center are funded by the U.S. National Cancer Institute through a Cancer Center Support Grant (P30CA016672). We thank Kathryn Hale for editing assistance on the manuscript.
CONFLICTS OF INTEREST
LVP received research funding from Takeda Pharmaceuticals. No other potential conflicts of interest were disclosed.
REFERENCES
1. Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW, editor. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Fourth Edition. Lyon, France: IARC; 2008.
2. Johnson NA, Savage KJ, Ludkovski O, Ben-Neriah S, Woods R, Steidl C, Dyer MJ, Siebert R, Kuruvilla J, Klasa R, Connors JM, Gascoyne RD, Horsman DE. Lymphomas with concurrent BCL2 and MYC translocations: the critical factors associated with survival. Blood. 2009; 114:2273–9.
3. Hu S, Xu-Monette ZY, Tzankov A, Green T, Wu L, Balasubramanyam A, Liu WM, Visco C, Li Y, Miranda RN, Montes-Moreno S, Dybkaer K, Chiu A, et al. MYC/BCL2 protein coexpression contributes to the inferior survival of activated B-cell subtype of diffuse large B-cell lymphoma and demonstrates high-risk gene expression signatures: a report from The International DLBCL Rituximab-CHOP Consortium Program. Blood. 2013; 121:4021–31.
4. Johnson NA, Slack GW, Savage KJ, Connors JM, Ben-Neriah S, Rogic S, Scott DW, Tan KL, Steidl C, Sehn LH, Chan WC, Iqbal J, Meyer PN, et al. Concurrent expression of MYC, BCL2 in diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol. 2012; 30:3452–9.
5. Green TM, Nielsen O, de Stricker K, Xu-Monette ZY, Young KH, Moller MB. High levels of nuclear MYC protein predict the presence of MYC rearrangement in diffuse large B-cell lymphoma. Am J Surg Pathol. 2012; 36:612–9.
6. Oki Y, Noorani M, Lin P, Davis RE, Neelapu SS, Ma L, Ahmed M, Rodriguez MA, Hagemeister FB, Fowler N, Wang M, Fanale MA, Nastoupil L, et al. Double hit lymphoma: the MD Anderson Cancer Center clinical experience. Br J Haematol. 2014; 166:891–901.
7. Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003; 426:895–9.
8. Adams J. The proteasome: a suitable antineoplastic target. Nat Rev Cancer. 2004; 4:349–60.
9. Ocio EM, Mateos MV, San-Miguel JF. Novel agents derived from the currently approved treatments for MM: novel proteasome inhibitors and novel IMIDs. Exp Opin Investig Drugs. 2012; 21:1075–87.
10. Wang M, Han XH, Zhang L, Yang J, Qian JF, Shi YK, Kwak LW, Romaguera J, Yi Q. Bortezomib is synergistic with rituximab and cyclophosphamide in inducing apoptosis of mantle cell lymphoma cells in vitro and in vivo. Leukemia. 2008; 22:179–85.
11. Suh KS, Goy A. Bortezomib in mantle cell lymphoma. Future Oncol. 2008; 4:149–68.
12. Goy A, Bernstein SH, Kahl BS, Djulbegovic B, Robertson MJ, de Vos S, Epner E, Krishnan A, Leonard JP, Lonial S, Nasta S, O'Connor OA, Shi H, et al. Bortezomib in patients with relapsed or refractory mantle cell lymphoma: updated time-to-event analyses of the multicenter phase 2 PINNACLE study. Ann Oncol. 2009; 20:520–5.
13. Fisher RI, Bernstein SH, Kahl BS, Djulbegovic B, Robertson MJ, de Vos S, Epner E, Krishnan A, Leonard JP, Lonial S, Stadtmauer EA, O'Connor OA, Shi H, et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol. 2006; 24:4867–74.
14. Kupperman E, Lee EC, Cao Y, Bannerman B, Fitzgerald M, Berger A, Yu J, Yang Y, Hales P, Bruzzese F, Liu J, Blank J, Garcia K, et al. Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res. 2010; 70:1970–80.
15. Chauhan D, Tian Z, Zhou B, Kuhn D, Orlowski R, Raje N, Richardson P, Anderson KC. In vitro and in vivo selective antitumor activity of a novel orally bioavailable proteasome inhibitor MLN9708 against multiple myeloma cells. Clin Cancer Res. 2011; 17:5311–21.
16. Tian Z, Zhao JJ, Tai YT, Amin SB, Hu Y, Berger AJ, Richardson P, Chauhan D, Anderson KC. Investigational agent MLN9708/2238 targets tumor-suppressor miR33b in MM cells. Blood. 2012; 120:3958–67.
17. Kumar SK, Bensinger WI, Zimmerman TM, Reeder CB, Berenson JR, Berg D, Hui AM, Gupta N, Di Bacco A, Yu J, Shou Y, Niesvizky R. Phase 1 study of weekly dosing with the investigational oral proteasome inhibitor ixazomib in relapsed/refractory multiple myeloma. Blood. 2014; 124:1047–55.
18. Richardson PG, Baz R, Wang M, Jakubowiak AJ, Laubach JP, Harvey RD, Talpaz M, Berg D, Liu G, Yu J, Gupta N, Di Bacco A, Hui AM, et al. Phase 1 study of twice-weekly ixazomib, an oral proteasome inhibitor, in relapsed/refractory multiple myeloma patients. Blood. 2014; 124:1038–46.
19. Ford RJ, Goodacre A, Ramirez I, Mehta SR, Cabanillas F. Establishment and characterization of human B-cell lymphoma cell lines using B-cell growth factor. Blood. 1990; 75:1311–8.
20. Goy A, Ramdas L, Remache YK, Gu J, Fayad L, Hayes KJ, Coombes KR, Barkoh BA, Katz R, Ford R, Cabanillas F, Gilles F. Establishment and characterization by gene expression profiling of a new diffuse large B-cell lymphoma cell line, EJ-1, carrying t(14;18) and t(8;14) translocations. Lab Invest. 2003; 83:913–6.
21. Pham LV, Lu G, Tamayo AT, Chen J, Challagundla P, Jorgensen JL, Medeiros LJ, Ford RJ. Establishment and characterization of a novel MYC/BCL2 "double-hit" diffuse large B cell lymphoma cell line, RC. J Hematol Oncol. 2015; 8:121.
22. Quentmeier H, Amini RM, Berglund M, Dirks WG, Ehrentraut S, Geffers R, Macleod RA, Nagel S, Romani J, Scherr M, Zaborski M, Drexler HG. U-2932: two clones in one cell line, a tool for the study of clonal evolution. Leukemia. 2013; 27:1155–64.
23. Lee EC, Fitzgerald M, Bannerman B, Donelan J, Bano K, Terkelsen J, Bradley DP, Subakan O, Silva MD, Liu R, Pickard M, Li Z, Tayber O, et al. Antitumor activity of the investigational proteasome inhibitor MLN9708 in mouse models of B-cell and plasma cell malignancies. Clin Cancer Res. 2011; 17:7313–23.
24. Arkwright R, Pham TM, Zonder JA, Dou QP. The preclinical discovery and development of bortezomib for the treatment of mantle cell lymphoma. Exp Op Drug Disc. 2017; 12:225–35.
25. Kane RC, Bross PF, Farrell AT, Pazdur R. Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. The Oncologist. 2003; 8:508–13.
26. Dunleavy K, Pittaluga S, Czuczman MS, Dave SS, Wright G, Grant N, Shovlin M, Jaffe ES, Janik JE, Staudt LM, Wilson WH. Differential efficacy of bortezomib plus chemotherapy within molecular subtypes of diffuse large B-cell lymphoma. Blood. 2009; 113:6069–76.
27. Bose P, Batalo MS, Holkova B, Grant S. Bortezomib for the treatment of non-Hodgkin's lymphoma. Exp Opin Phamacother. 2014; 15:2443–59.
28. Lee EC, Fitzgerald M, Bannerman B, Donelan J, Bano K, Terkelsen J, Bradley DP, Subakan O, Silva MD, Liu R, Pickard M, Li Z, Tayber O, et al. Antitumor activity of the investigational proteasome inhibitor MLN9708 in mouse models of B-cell and plasma cell malignancies. Clin Cancer Res.17:7313–23.
29. Dick LR, Fleming PE. Building on bortezomib: second-generation proteasome inhibitors as anti-cancer therapy. Drug Discov Today. 2010; 15:243–9.
30. Gupta N, Diderichsen PM, Hanley MJ, Berg D, van de Velde H, Harvey RD, Venkatakrishnan K. Population Pharmacokinetic Analysis of Ixazomib, an Oral Proteasome Inhibitor, Including Data from the Phase III TOURMALINE-MM1 Study to Inform Labelling. Clin Pharmacokinet. 2017; 56:1355–1368.
31. Manic G, Obrist F, Sistigu A, Vitale I. Trial Watch: Targeting ATM-CHK2 and ATR-CHK1 pathways for anticancer therapy. Mol Cell Oncol. 2015; 2:e1012976.
32. Matt S, Hofmann TG. The DNA damage-induced cell death response: a roadmap to kill cancer cells. Cell Mol Life Sci. 2016; 73:2829–50.
33. Dai B, Zhao XF, Mazan-Mamczarz K, Hagner P, Corl S, Bahassi el M, Lu S, Stambrook PJ, Shapiro P, Gartenhaus RB. Functional and molecular interactions between ERK, CHK2 in diffuse large B-cell lymphoma. Nat Commun. 2011; 2:402.
34. Derenzini E, Agostinelli C, Imbrogno E, Iacobucci I, Casadei B, Brighenti E, Righi S, Fuligni F, Ghelli Luserna Di Rora A, Ferrari A, Martinelli G, Pileri S, Zinzani PL. Constitutive activation of the DNA damage response pathway as a novel therapeutic target in diffuse large B-cell lymphoma. Oncotarget. 2015; 6:6553–69. https://doi.org/10.18632/oncotarget.2720.
35. Li J, Stern DF. DNA damage regulates Chk2 association with chromatin. J Biol Chem. 2005; 280:37948–56.
36. Li J, Stern DF. Regulation of CHK2 by DNA-dependent protein kinase. J Biol Chem. 2005; 280:12041–50.
37. Choudhury A, Cuddihy A, Bristow RG. Radiation and new molecular agents part I: targeting ATM-ATR checkpoints, DNA repair, and the proteasome. Sem Rad Oncol. 2006; 16:51–8.
38. Ravi D, Beheshti A, Abermil N, Passero F, Sharma J, Coyle M, Kritharis A, Kandela I, Hlatky L, Sitkovsky MV, Mazar A, Gartenhaus RB, Evens AM. Proteasomal Inhibition by Ixazomib Induces CHK1 and MYC-Dependent Cell Death in T-cell and Hodgkin Lymphoma. Cancer Res. 2016; 76:3319–31.
39. Pham LV, Fu L, Tamayo AT, Bueso-Ramos C, Drakos E, Vega F, Medeiros LJ, Ford RJ. Constitutive BR3 receptor signaling in diffuse, large B-cell lymphomas stabilizes nuclear factor-kappaB-inducing kinase while activating both canonical and alternative nuclear factor-kappaB pathways. Blood. 2011; 117:200–10.
40. Pham LV, Tamayo AT, Li C, Bornmann W, Priebe W, Ford RJ. Degrasyn potentiates the antitumor effects of bortezomib in mantle cell lymphoma cells in vitro and in vivo: therapeutic implications. Mol Cancer Ther. 2010; 9:2026–36.
41. Pham LV, Tamayo AT, Yoshimura LC, Lo P, Ford RJ. Inhibition of constitutive NF-kappa B activation in mantle cell lymphoma B cells leads to induction of cell cycle arrest and apoptosis. J Immunol. 2003; 171:88–95.
42. Parlati F, Lee SJ, Aujay M, Suzuki E, Levitsky K, Lorens JB, Micklem DR, Ruurs P, Sylvain C, Lu Y, Shenk KD, Bennett MK. Carfilzomib can induce tumor cell death through selective inhibition of the chymotrypsin-like activity of the proteasome. Blood. 2009; 114:3439–47.
43. Byers LA, Wang J, Nilsson MB, Fujimoto J, Saintigny P, Yordy J, Giri U, Peyton M, Fan YH, Diao L, Masrorpour F, Shen L, Liu W, et al. Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer Discov. 2012; 2:798–811.