
INTRODUCTION
The normal epithelial-adenoma-carcinoma axis was proposed thirty years ago by Vogelstein and Fearon [1]. The genetic changes involve the initial loss or mutation of APC/β-catenin genes for the formation of aberrant crypts foci, mutations of KRAS/BRAF to advanced adenoma, and other acquired mutations for transformation to carcinoma [2]. The tumorigenesis took several decades. Epidemiology studies have demonstrated an average 2% cancer transformation rate of polyps every year [3] that result in a 5~6% lifetime probability of developing into colorectal cancer in an average-risk individual at 68 years of age [2]. The central theme of this theory is the necessity of unstable chromosomes by the loss of APC function in the first stage and the accumulation of sequentially acquired mutations to exert tumor characteristics in a time-dependent manner. However, in our previous study, APC was not the most frequently mutated gene in 50 colorectal (CRC) tumors, nor did it have the highest mutated allelic frequency within one tumor [4]. Instead, TP53 gained the highest mutation rate (46%) among 20 frequently aberrant genes in CRC and almost had the highest alleice frequency if multiple gene mutations were found within an individual tumor. This observation challenged the dogma for the essential presence of an APC mutation and raised the possibility of multiple different mutation pathways that determine cancer transformation.
To elucidate the role of each gene during tumorigenesis, it is warranted to identify the mutation profiles of each stage of pre-cancerous neoplasia. Recently, Sievers et al. [5] surveyed the genetic landscapes of 48 small (< 9 mm) resected polyps from 36 asymptomatic patients after a follow-up by routine CT scan for 1–3 years. They concluded that there was no significant correlation between the genetic profile and the adenoma growth rate. However, they found multiple subclones with lower allelic frequency of driver mutations coexisted early in small polyps and might cause malignancy transformation till it acquired additional driver mutations or received non-genetic changes. The results imply the importance of qualitative and quantitative measurement of tumor genome. In this study, we conduct a retrospective cohort study to investigate the mutation patterns of 53 archived polyp tissues with histologically proven findings from patients with hyperplasia to villous adenoma by performing next generation sequencing (NGS) with a 50-gene panel. The platform is the same as the one we used in our previous study for 50 CRC fresh tumor tissues. Therefore, we can parallel the data from the two studies to observe the changes in the altered somatic gene pattern from the initiation of cell proliferation, the pre-cancerous stage, to the early and late stages of cancer in a cross-sectional view. This study may reveal how a polyp evolves into cancer through multiple steps of genome alteration and reestablish the proliferation tree map by quantitating the relative allelic frequency of each mutated gene within one tumor.
However, mutation patterns from cross-sectional studies are affected by individual host genomic background and unaccountable environmental factors of the study cohorts. The best way to investigate the contribution of each gene in cancer formation should be the longitudinal follow-up of a single polyp from a benign state to malignancy. As it is difficult to monitor the evolution of the cancer genome in that way, we collected six pairs of synchronous polyp and tumor tissues from 6 individual patients to focus on the differences in the gene spectrum of pre-cancer and cancer lesions within the same individual. All the information can help us elucidate the dominant gene and pathway paving the road to cancer.
RESULTS
General patient characteristics and germline variants in patients with polyps compared to those in CRC patients
Table 1 summarizes the clinical features of 53 patients with polyps. The number of patients with non-neoplastic polyps (NNP, n = 27) was equal to the number with neoplastic polyps (NP, n = 26). Of the enrolled participants, 45% had more than one polyp, and they were diagnosed at an average of 58 years old. Figure 1A shows nine (17%, 9/53) patients who were identified as carrying germline variants. Among them, two had diagnoses of NNP, and the remaining seven patients had advanced polyps. Table 2 summarizes the germline variants found in the polyps and CRC groups. In patients with polyps, 5 germline variants were detected in the three most common CRC-susceptible genes, APC, MLH-1 and SMAD4. The tolerant APC variants V1125A and V1352A were represented as germline mutation hotspots (9%, 5/53). MLH1 V143D and R144C were predicted as damaging mutations, and one rare SMAD4 N316S variant was discovered in one patient. On the other hand, the damaging CDH1 T340A, damaging NRAS G138R and tolerated MLH-1 R148Q variant were exclusively present in the cancer group. Among the eight cancer-predisposing germline mutations, all were minor alleles in the Taiwanese except for the MLH1 V143D mutation, which had a 6.0% frequency in the general Taiwanese population.
Table 1: Clinical characteristics of 53 patients with polyps
Clinical Features |
Patient no (%) |
|
|---|---|---|
Sex |
Male |
40 (75%) |
Female |
13 (25%) |
|
Age |
Mean (range) |
58 (26–75) |
Pathology classification |
Non-neoplasia polyps |
27 |
Hyperplasia |
5 (9%) |
|
Adenomatous |
11 (21%) |
|
Tubular adenoma < 1 cm |
11 (21%) |
|
Neoplastic polyps |
26 |
|
Tubular adenoma ≥ 1 cm |
5 (9%) |
|
Tubulovillous |
13 (25%) |
|
Villous adenoma |
8 (15%) |
|
Patients with > 1 polyps (%) |
24 (45%) |
|
Patients with synchronous cancer (%) |
6$ (11%) |
|
Polyps location |
Right* |
15 (28%) |
Left** |
28 (53%) |
|
Rectum |
10 (19%) |
|
Treatment |
Polypectomy removed |
53 (100%) |
$: 6 patients provided tumor FFPE specimen for comparison of mutation patterns with those in paired polyps.
*Right side including polyps located in cecum, ascending, transverse and hepatic flexure.
**Left side including polyps located in sigmoid, descending and splenic flexure.
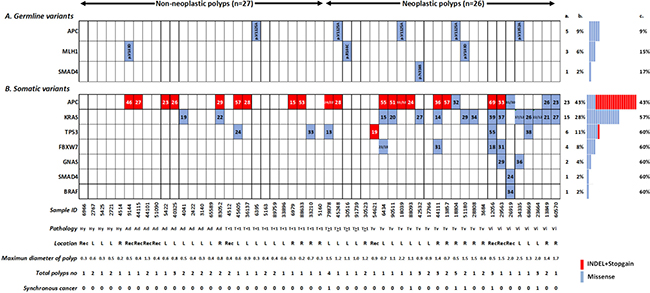
Figure 1: Distribution of 5 germline variants in 3 cancer-susceptible genes (A) and 40 somatic variants in 7 genes (B) in 53 polyp tissues. Sample order is lined up by pathology finding and lesion location. The color and number in the square cell represents the variant type (red denotes an indel or nonsense mutation, and blue denotes missense mutation) and variant allelic frequency. More than one number in one cell indicates multiple mutations in the same gene. Columns a and b denote the number and percentage of samples altered per gene; column c denotes the accumulated percentage of samples with mutated genes. The patient information denoted at the bottom of the figure includes the sample ID, histological classification, location, maximum diameters of the polyp (cm), number of polyps and whether synchronous cancer was present (1 stands for yes). “Hy” = Hyperplasia; “Ad” = adenomatous; “T < 1” = tubular size < 1 cm; “T ≥ 1” = tubular size ≥ 1 cm; “Tv” = tubulovillous; ”Vi” = villous; “Re” = rectum; “R” = right side; and “L” = left side.
Table 2: Germline mutation list and frequency comparison found in 53 polyps patients and 50 colorectal cancer patients
Gene |
Locus |
Exon/CDS change |
A. A. change |
SIFT prediction |
% in polyps |
% in CRC* |
Allelic frequency in Taiwanese** |
|---|---|---|---|---|---|---|---|
APC |
chr5:112174665 |
Exon15/c.T3374C |
p.V1125A |
Tolerated |
7.5%(4/53) |
2% (1/50) |
0.7% |
chr5:112175346 |
Exon15/c.T4055C |
p.V1352A |
Tolerated |
1.9% (1/53) |
2% (1/50) |
0.0% |
|
MLH1 |
chr3:37067240 |
Exon11/c.T428A |
p.V143D |
Damaging |
3.8% (2/53) |
0% (0/50) |
6.0% |
chr3:37067242 |
Exon11/c.C430T |
p.R144C |
Damaging |
1.9% (1/53) |
0% (0/50) |
1.3% |
|
chr3:37067255 |
Exon11/c.G443A |
p.R148Q |
Tolerated |
0.0% (0/53) |
2% (1/50) |
0.0% |
|
SMAD4 |
chr18:48586278 |
Exon8/c.A947G |
p.N316S |
Tolerated |
1.9% (1/53) |
0% (0/50) |
0.0% |
CDH1 |
chr16:68846047 |
Exon8/c.A1018G |
p.T340A |
Damaging |
0.0% (0/53) |
6% (3/50) |
0.7% |
NRAS |
chr1:115252228 |
Exon4/c.G412A |
p.G138R |
Damaging |
0.0% (0/53) |
2% (1/50) |
0.0% |
Total |
17% (9/53) |
12% (6/50) |
*Data have been published in previous study (P-Y Chang et al., Oncotarget, 2016).
**Data were collected from 150 healthy Taiwanese people with age larger than 70 years old by whole genome sequencing.
Somatic variants in patients with polyps
After the subtraction of inherited variants, somatic mutations in each FFPE polyp sample were identified. APC (43%), followed by KRAS (28%) and TP53 (11%), are the most frequently mutated genes and those three genes cover 60% of the polyp patients (Figure 1B). In sum, a total of 40 somatic variants in 7 genes in the polyps group were identified and confirmed by Sanger sequencing (variant information was listed in Table 3). Regarding the variant effect, 20 missense mutations (50%), 4 indel mutations (10%), and 16 nonsense mutations (40%) were identified. Among these, 3 (7.5%) variants, APC S1344*, APC F1500fs and GNAS R186C, were novel and were not reported in the COSMIC database. Missense mutations of in codons 12 and 13 of KRAS (including G12V, G12D, and G13D) were the most frequently observed variants in the samples (23%, 12/53). Indel and nonsense mutations, which can lead to truncated proteins, were distributed mostly in the APC and TP53 genes.
Table 3: Forty somatic variants distributed on 7 genes in 53 polyps tissues
Gene name (RefSeq ID) |
Nucleotide Change |
Amino Acid Change |
Variant effect |
Patient no |
|---|---|---|---|---|
TP53 (NM_000546) |
c.C321A |
COSM45040_p.Y107* |
stopgain |
1 |
c.A641G |
COSM43687_p.H214R |
nonsynonymous missense |
1 |
|
c.A659G |
COSM10758_p.Y220C |
nonsynonymous missense |
1 |
|
c.A701G |
COSM10725_p.Y234C |
nonsynonymous missense |
1 |
|
c.G731A |
COSM10883_p.G244D |
nonsynonymous missense |
1 |
|
c.G733A |
COSM6932_p.G245S |
nonsynonymous missense |
1 |
|
KRAS (NM_033360) |
c.G34A |
COSM517_p.G12S |
nonsynonymous missense |
1 |
c.G34T |
COSM516_p.G12C |
nonsynonymous missense |
1 |
|
c.G35A |
COSM521_p.G12D |
nonsynonymous missense |
5 |
|
c.G35T |
COSM520_p.G12V |
nonsynonymous missense |
3 |
|
c.G38A |
COSM532_p.G13D |
nonsynonymous missense |
4 |
|
c.A183C |
COSM554_p.Q61H |
nonsynonymous missense |
2 |
|
c.G436A |
COSM19404_p.A146T |
nonsynonymous missense |
1 |
|
APC (NM_000038) |
c.C2626T |
COSM18852_p.R876* |
stopgain |
2 |
c.C3340T |
COSM13125_p.R1114* |
stopgain |
1 |
|
c.C3367T |
COSM1432255_p.Q1123* |
stopgain |
1 |
|
c.G3856T |
COSM18772_p.E1286* |
stopgain |
2 |
|
c.C3871T |
COSM19072_p.Q1291* |
stopgain |
2 |
|
c.C3880T |
COSM18960_p.Q1294* |
stopgain |
1 |
|
c.3894_3895delTG |
COSM18980 p.S1298fs |
frameshift |
1 |
|
c.C3907T |
COSM13728_p.Q1303* |
stopgain |
1 |
|
c.G3916T |
COSM18760_p.E1306* |
stopgain |
2 |
|
c.C4031G |
p.S1344* |
stopgain |
1 |
|
c.G4033T |
COSM18759_p.E1345* |
stopgain |
1 |
|
c.4060delT |
COSM292626_p.S1355fs*60 |
frameshift |
1 |
|
c.C4067G |
COSM18779_p.S1356* |
stopgain |
1 |
|
c.G4120T |
COSM19085_p.E1374* |
stopgain |
1 |
|
c.C4285T |
COSM18836_p.Q1429* |
stopgain |
1 |
|
c.C4348T |
COSM13127_p.R1450* |
stopgain |
4 |
|
c.4385_4388delAGAG |
COSM1432412_p.E1464fs*8 |
frameshift |
1 |
|
c.4498delT |
p.F1500fs |
frameshift |
1 |
|
c.G4660T |
COSM33818_p.E1554* |
stopgain |
1 |
|
SMAD4 (NM_005359) |
c.G1082A |
COSM14122_p.R361H |
nonsynonymous missense |
1 |
FBXW7 (NM_018315) |
c.C1153T |
COSM170725_p.R385C |
nonsynonymous missense |
1 |
c.G1196A |
COSM94297_p.R399Q |
nonsynonymous missense |
1 |
|
c.G1196T |
COSM99619_p.R399L |
nonsynonymous missense |
1 |
|
c.C1273T |
COSM74637_p.R425C |
nonsynonymous missense |
1 |
|
c.C1505T |
COSM295016_p.S502L |
nonsynonymous missense |
1 |
|
BRAF (NM_004333) |
c.T1799A |
COSM476_p.V600E |
nonsynonymous missense |
1 |
GNAS (NM_001077489) |
c.C556T |
p.R186C |
nonsynonymous missense |
2 |
Two novel variants on APC gene and one variants on GNAS gene which are not recorded in the COSMIC database are shaded. Nomenclature of variants follows the guideline of Human Genome Variation Society. *stands for stop codon.
Gradually acquired somatic mutations in polyps and CRC tissue samples and their correlation with clinicopathological factors
For better clarification of the molecular features alone with the development from adenoma-to-cancer with various clinicopathological factors, we enrolled our previous study results for cross-sectional comparison of the genetic alteration (somatic mutation profiles of 50 CRC patients are in Supplementary Figure 1). Four subgroups were stratified: NNP, NP, CRC early stage and CRC late stage.
First, the average numbers of somatic variants in the four subgroups were 0.5, 1.7, 2.1 and 2.3 (Figure 2A). Figure 2B summarized the mutation rate of the top 5 mutated genes in different stages of neoplasm. It was clear that with the exception of APC with the same high mutation rate from the initiation stage to the end stage of cancer, the KRAS, TP53, PIK3CA and SMAD4 gene mutation rates were elevated when malignancy developed (p < 0.05), and a significant increase of the positive rate on TP53 and PIK3CA from NNP, NP, early stage and late stage of carcinoma (7%, 15%, 33.3%, and 65% for TP53, p < 0.001; 0%, 0%, 23.3%, and 25% for PIK3CA, p = 0.002) were also observed. Figure 2C further demonstrated the individual gene mutation rate with the location. The statistical results indicated no particular tendency in the mutation pattern at different locations of the large intestine lumen.
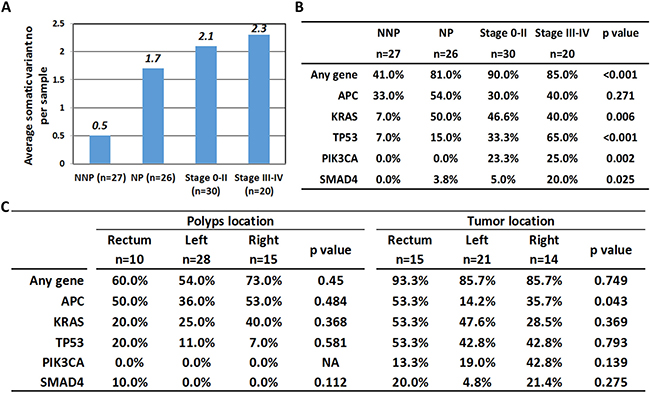
Figure 2: Correlation of gene mutation number and mutation rate with clinicopathological factors. One hundred three samples were divided into 4 categories. NNP: non-neoplastic polyps, NP: neoplastic polyps, stage 0–II: colorectal cancer stage 0–II and stage III–IV: CRC with stage III–IV. (A) Comparison of average somatic variant number in 4 subgroups. (B) Comparison of mutation rate of top five mutated genes. (C) Correlation of mutation rate of top five mutated genes with lesion location in polyps and CRC groups.
Second, Supplementary Figure 2 illustrated the spatial distribution of the variants in the 5 most frequently mutated genes in the polyps and CRC tissues. In overview, variants in each gene were located in the well-known mutation areas regardless of whether they were derived from polyps or tumor tissues. For example, all mutations on the APC gene were distributed in the β-catenin-binding domain, and the variants in the TP53 gene were located in the DNA-binding domain. KRAS mutations from adenomas and CRC occurred exclusively in codons 12, 13, 60, 61 and 146. Unexpectedly, no PIK3CA mutation was found in the 53 polyp tissues, in which mutations in Exon 9 and Exon 20 are frequently reported in CRC. The similar mutation locations in each gene in both groups indicates the continuity of the deleterious genome from adenoma to carcinoma.
Distinct mutation profiles of synchronous polyps and tumors in one individual
Figure 3 demonstrates the comparison of the mutation patterns of paired polyp and carcinoma tissues in six patients. Figure 3A includes three patients (patient 1–3) whose polyps and cancer located at different sites within the large intestine. The results indicate that completely different mutation alleles could exist in distant lesions. For example, patient 1 has one tubulovillous polyp located in the descending intestine with an APC E1374* mutation. However, their stage II carcinoma located in the rectum presented KRAS G12V and PIK3CA H1047R, two different mutations. In patient 2, although KRAS mutations were found in both the polyp and carcinoma, the mutation alleles were G12D and G12C in the polyp and G13D in the carcinoma, which all have been confirmed by Sanger sequencing. The same finding was observed in patient 3. APC and TP53 were mutated in both neoplasms with non-consistent mutation points. Figure 3B includes two patients with polyps and tumors that existed at the same ascending area. These two patients had germline mutations in MLH1 and APC respectively. After excluding the germline mutation, distinct molecular features were also observed in paired lesions, even when they were located in the same area. Figure 3C demonstrates only one concordant pattern of two lesions at the same location within one patient in our study. In summary, independent mutation patterns were observed in synchronous lesions within one individual. However, the TP53 and PIK3CA somatic mutations were found to be present in the majority of tumor tissues but not in the polyp lesions.
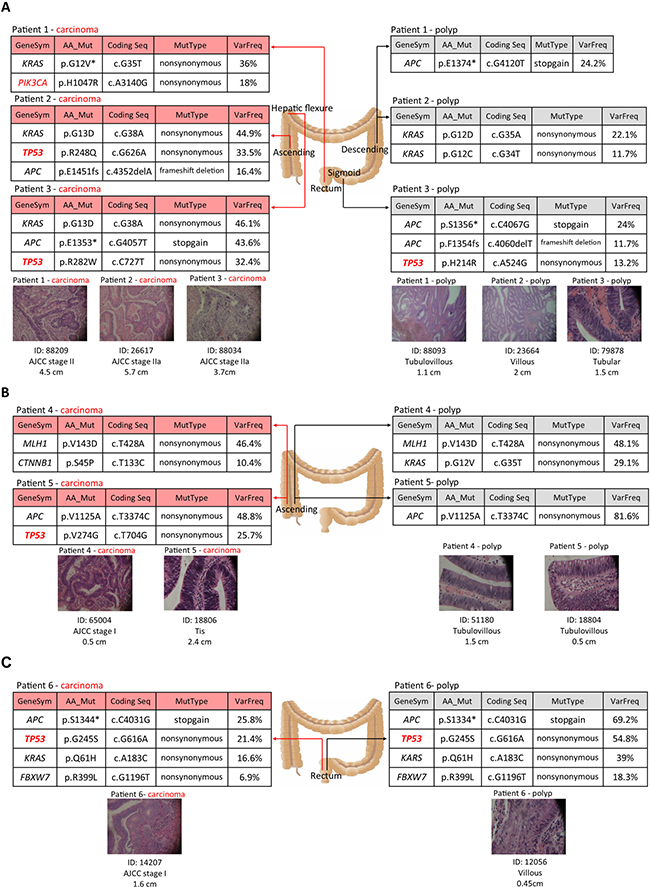
Figure 3: Distinct molecular features of synchronous polyps and cancer in six individuals. (A) Mutation profiles found in three patients with polyps and cancer at different locations. (B) Mutation profiles in two patients with polyps and cancer at the same location and with germline mutations. (C) Mutation patterns in one patient with polyp and cancer at the same location. The histology of each lesion is shown by the hematoxylin and eosin stained slide with ×40 magnification. The mutation details of each neoplasm are summarized as a table.
DISCUSSION
The majority of colorectal cancers progress from polyps. In Taiwan, 25.8% of the average-risk population at age ≥ 50 years had polyps, and 22% of them were advanced neoplasms [6]. Synchronous polyps, which means two or more polyps developed simultaneously, are also prevalent in polyps population (~25% [7, 8] and 45% in this study). According to the results of a large meta-analysis, individuals who performed polypectomy and found having synchronous polyps will have an odds ratio of 2.47 (95% CI, 1.74–3.50) to have a metachronous polyp in the future and an odds ratio of 5.15 (95% CI, 2.02–13.14) to have a metachronous CRC if the initial polyp was severe dysplasia [9]. This phenomenon echoes the observation that the causes of polyp production and progression to cancer are contributed to the genetic nature of cancer-susceptible individuals (20%) and environmental insults (80%) [10]. If individuals had the cancer-prone genome and were constantly under a cancer triggering environment, then they were at high risk to develop polyps or cancer repeatedly. Therefore, for cancer prevention purposes, we need a biomarker to identify individuals at high risk of having polyps (e.g. germline mutations), individuals with polyps at high risk to develop to cancer (e.g. specific somatic mutations).
Molecular alterations have been thoroughly investigated in CRC by high throughput sequencing platforms [11, 12]. However, few studies investigated the molecular background of adenomas and only screened in limited clinical specimen. Zhou et al. used the exome capture sequencing method to investigate the genetic alterations in only one pair of a tubular adenoma and adenocarcinoma that were 4 cm apart [13]. Kang et al. reported the intratumor heterogeneity by thoroughly measuring the point mutations, chromosome copy number and methylation in opposite sides of a 6 cm adenoma [14]. Another two studies focus the genome of adenomas specific in 36 patients with 48 small-sized polyps [5] and in 12 patients of familial adenomatous polyposis [15]. In the present study, we collected 53 polyps from 53 patients with pathology from non-neoplastic to neoplastic findings and combined the results from 50 CRC tissues in our previous study to comprehensively compare the germline and somatic mutation profiles by the same NGS cancer panel.
For germline mutations, our results reveal a 17% of mutation rate in cancer-susceptibility genes in the polyps group (Table 2). Most of the variants had significantly higher allelic frequencies than those in the general Taiwanese population and damaging effects on the protein function according to the SIFT prediction. APC is an autosomal dominant gene with 100% penetrance. Variants in the Wnt/β-Catenin pathway may contribute to colorectal cancer development [16]. In particular, the APC V1125A variant in this study has been reported previously in a Taiwanese CRC cohort study [17]. On the other hand, any kind of MLH1 mutation carriers are reported to have an estimated 34% risk for males and 36% risk for females to develop into colorectal cancer at age 70 years [18]. In sum, the findings of this retrospective polyps study combined with the novel CDH1 and NRAS germline mutations found in the CRC groups emphasize the existence of the Taiwanese-specific genome and provided candidate genes for cancer risk assessment in the general population.
For somatic mutations, the number of mutated genes and the mutation rates of specific genes significantly increased from non-neoplastic polyps to the late stage of carcinoma (Figure 2), which demonstrates that cancer might form when the acquired mutations reach the threshold. However, is the total number of mutated genes or the mutation of specific genes vital to cancer formation? By quantitative evaluation of the intra-tumor allelic frequency, we can anticipate the evolutionary history from adenoma to carcinoma. Although the allelic frequency is dependent on the percentage of tumor content in the original sample [12], the individual allelic frequency of multiple mutants occurring within one sample can still provide information about how subclones carrying different mutations evolved. In 53 polyp tissues, 15 samples had two or more somatic mutations, and nine of them had the highest mutant percentage in the APC gene. This phenomenon fits Volgestein’s theory in which cells with an APC mutation at the initial stage acquired new mutations such as KRAS and TP53 in the original clone when neoplasm grows (Supplementary Figure 3A). However, in the CRC group, the gene with the highest mutation frequency within one tumor is TP53 which suggests that multiple alternative pathways other than APC mutations are involved [19]. Subclones that were not necessarily derived from the original APC-mutated clone may carry the essential driver mutations or defective pathways, such as TP53 or PIK3CA, and gain a growth advantage with neoplasm progression (Supplementary Figure 3B).
The p53 protein induces G1 cell-cycle arrest and facilitates DNA repair prior to a cell committing to the process of DNA replication. The fact of p53 inactivation as a critical rate-limiting event can be evident by introducing a human TP53 knock-in mouse model (Hupki mouse) [20]. Concordant with our finding, the elevated TP53 mutation rate in CRC but not adenoma and its pathological role in CRC has been reported by other groups [21–23]. On the other hand, PIK3CA mutation is associated with phosphorylated AKT expression to decrease apoptosis and increase tumor invasion [24]. Activating mutations in PIK3CA are present in many cancer types, including colorectal cancer, with a 10–30% mutation rate [25]. However, PIK3CA mutation is uncommon in polyps. Vicki L.J. Whitehall et al. surveyed 426 colorectal polyps, and PIK3CA mutations were only present in 4 tubulovillous adenomas and 1 tubulovillous adenoma; no PIK3CA mutations were found in non-neoplastic polyps [26]. In our studies, 0% of PIK3CA mutations could be found in 53 polyp tissues, but 24% were found in the CRC group. More interestingly, PIK3CA and TP53 mutations were mutually exclusive in CRC patients (Supplementary Figure 1) and contributed to a total of 68% of the mutation rate (34/50). This result demonstrated the crucial roles of these two genes in tumorigenesis.
For investigating the development of polyps and cancer under the same genetic background, the genomes of six pairs of synchronous polyps and tumors were compared. Independent molecular features and a right-sided tumor tendency of synchronous neoplasm within one individual were observed. Recent molecular pathological epidemiology (MPE) theory provokes a concept that the molecular features and behavior of tumor cells are influenced by host immunity and inflammation [27], as well as the interaction of various molecules [24, 28] and exposures to the local microbiome [29]. Concordant with our finding, Wheeler et al. recently reported on the distinct genomic profiling of 6 synchronous CRCs in one patient with Lynch syndrome. This result demonstrates that each lesion had a unique pattern of affected genes [30]. The continuous interaction of a growing neoplasm with the surrounding environment results in the tumor heterogeneity nature and the unpredictable development of any given lesion, especially when more than one polyp existed in one patient. However, we found that TP53 or PIK3CA mutation always appeared in the carcinoma counterpart but not in the polyp site, except for one pair at a very adjacent location (patient 6 in Figure 3). The same finding has been reported by Haley’ s group [12]. They investigated the genomic profiles of 5 paired adenoma and invasive carcinoma tissues and found that PIK3CA mutations were only present in two of five carcinoma tissues and not in the adenoma. This result implies that the detection of the presence of TP53 or PIK3CA mutations may serve as a predictor for being prone to the development of cancer.
In conclusion, this is the first comprehensive investigation of both polyps and the CRC genome using a next generation sequencing platform. The paired genome comparison of synchronous polyps and cancers also provides direct evidence of the impact of local environment and highlights the importance of acquiring and expanding subclones with TP53 or PIK3CA mutations. The vital role of genetic alteration in the TP53 and PI3K pathway provides the possibility for designing a better screening strategy and targeted treatment to stop the tumorigenesis process before cancer transformation.
MATERIALS AND METHODS
Study patients
Polyp patients
Fifty-three participants who received polypectomy to remove polyps in a national cancer screening program at Chang Gung Memorial Hospital in Taiwan were enrolled between 2013 and 2015. Resected polyps were classified according to World Health Organization criteria and general consensus [31]. Histological findings were reported by experienced pathologists. The classification was divided into six categories: hyperplasia, adenomatous, tubular adenoma with largest diameter < 1 cm, tubular adenoma with largest diameter ≥ 1 cm, tubulovillous adenoma and villous adenoma. The former three classifications were grouped as non-neoplasia polyps (NNP), and the last three were defined as advanced or neoplastic polyps (NP). Clinicopathological factors, including age, sex, and the anatomic subsite of lesions in the intestinal lumen, were recorded at enrollment. The polyp location was classified into 3 parts: right side (tumors at the cecum, ascending colon, hepatic flexure, and transverse colon); left side (tumors at the splenic flexure, descending colon, and sigmoid colon); and the rectum. Twenty-four patients were found to have more than one polyp, and 6 patients had synchronous cancer. The tumors were surgically removed and pathologically confirmed. All patients were provided with a form of written informed consent, and the study was approved by the Institutional Review Board of Chang Gung Memorial Hospital (101-4609A3; 102-5224B).
Colorectal cancer patients
Fifty patients with untreated CRC were enrolled, and their fresh tumor and adjacent normal tissues had been investigated for somatic and germline mutations in our previous study [4]. The pathological classification of the tumor stage ranged from AJCC stage 0 to stage IV, with 30 early stage cases (stage 0–II) and 20 late stage cases (stage III–IV).
Controls
Taiwanese genome reference data were obtained from 150 healthy controls by whole genome sequencing. All controls had no family history of cancer and remained healthy at the age of 70 years old when they provide the blood samples (See Acknowledgement).
Sample preparation and routine laboratory test
Fifty-three formalin-fixed paraffin-embedded (FFPE) polyps and 6 synchronous FFPE tumor blocks were submitted to the laboratory of anatomic pathology for inspection. One hematoxylin & eosin (H&E) slide of each sample was used for reviewing neoplastic contents and photorecorded by pathologists. The other 3–5 unstained slides of 10 mm-thick sections with cellularity larger than 20% were collected and subjected to the following DNA extraction. For the confirmation of germline mutations, EDTA blood samples were simultaneously collected from 53 patients. Genomic DNA from PBMCs were extracted using a DNeasy Blood and Tissue Kit (Qiagen) and stored at −80°C until use.
DNA extraction procedure for FFPE sections
At least three slides of 10 μm thick FFPE tissue were added to the Covaris microTUBE (COVARIS). The samples were processed by using the Focused-ultrasonicator Covaris S220 to dissociate the paraffin and rehydrate the tissue. The extraction procedures followed the manufacturer’s instructions.
Next generation sequencing by ampliseq cancer hotspot panel
All tissue samples were subjected to the AmpliSeq Cancer Hotspot Panel Version 2 (Thermal Fisher). The panel specifically targets 50 cancer-related genes which harbors 2855 COSMIC [32] hotspots. The AmpliSeq Library was prepared according to the manufacturer’s protocol. For the detailed procedures, please refer to our previous study [4].
Germline mutation detection
Mutations found in FFPE tissue samples will be further tested by Sanger sequencing in paired blood genomic DNA to exclude germline mutations. Germline mutations are defined as a gene alteration found in both the tissue and blood DNA with an allelic frequency of approximately 50%.
Bioinformatics analysis
Mutations located in exonic regions that were nonsynonymous changes causing amino acid alterations with allelic frequencies less than 3% in Asian populations were filtered in. To further eliminate high-artifact background from the fragmented FFPE DNA, only those mutations with a frequency of > 10% and variant coverage of > 30 were considered candidate variants for further analysis. Variants with amino acid changes were further examined for whether they were potentially damaging by using the Sorting Intolerant From Tolerant (SIFT) software, which can predict the possible impact of an amino acid substitution on the structure and function of a protein [33]. SIFT calculates the conservation value and scales the probability for each position. The SIFT score ranges from 0.0 (damaging) to 1.0 (tolerated).
Statistical analysis
Descriptive statistics were summarized in percentages. Between-group comparisons were conducted using the chi-square test for each marker. A P value less than 0.05 (2-tailed) was considered statistically significant. All statistical tests were conducted using PASW Statistics 18. The protein domain structure and distribution of variants in specific proteins were plotted using DOG Version 1.0 (http://dog.biocuckoo.org).
Author contributions
JJL developed the concept, initiated the study, and revised the manuscript. PYC designed the experiments and wrote the manuscript. SCC, MCW, NCC, WHL and HLL performed the experiments and organized the figures and tables. SH and YHW aided in the retrieval of the FFPE samples and reviewed the section morphology. JSC and WST collected the clinical samples, collected patient information, and obtained consent from the enrolled patients.
ACKNOWLEDGMENTS
The frequency of the eight germline variants in the Taiwanese population were kindly provided by the Whole-Genome Research Core Laboratory of Human Diseases at Chang Gung Medical Foundation Chang Gung Memorial Hospital, Keelung. We thank Professor Swei Hsueh in the Department of Anatomy Pathology, Chang Gung Memorial Hospital for providing academic support in reviewing the histopathology section.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
GRANT SUPPORT
This study was funded by Chang Gung Memorial Hospital (CMRPG3C1371-3, and PMRPG3D0031-4).
REFERENCES
1. Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988; 319:525–532.
2. Grady WM, Carethers JM. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology. 2008; 135:1079–1099.
3. Stryker SJ, Wolff BG, Culp CE, Libbe SD, Ilstrup DM, MacCarty RL. Natural history of untreated colonic polyps. Gastroenterology. 1987; 93:1009–1013.
4. Chang PY, Chen JS, Chang NC, Chang SC, Wang MC, Tsai SH, Wen YH, Tsai WS, Chan EC, Lu JJ. NRAS germline variant G138R and multiple rare somatic mutations on APC in colorectal cancer patients in Taiwan by next generation sequencing. Oncotarget. 2016; 7:37566–37580. http://doi.org/10.18632/oncotarget.8885.
5. Sievers CK, Zou LS, Pickhardt PJ, Matkowskyj KA, Albrecht DM, Clipson L, Bacher JW, Pooler BD, Moawad FJ, Cash BD, Reichelderfer M, Vo TN, Newton MA, et al. Subclonal diversity arises early even in small colorectal tumours and contributes to differential growth fates. Gut. 2016. [Epub ahead of print].
6. Chiu HM, Lin JT, Chen CC, Lee YC, Liao WC, Liang JT, Shun CT, Wang HP, Wu MS. Prevalence and characteristics of nonpolypoid colorectal neoplasm in an asymptomatic and average-risk Chinese population. Clin Gastroenterol Hepatol. 2009; 7:463–470.
7. Eshghi MJ, Fatemi R, Hashemy A, Aldulaimi D, Khodadoostan M. A retrospective study of patients with colorectal polyps. Gastroenterol Hepatol Bed Bench. 2011; 4:17–22.
8. Demetriades H, Kanellos I, Blouhos K, Tsachalis T, Vasiliadis K, Pramateftakis MG, Betsis D. Synchronous polyps in patients with colorectal cancer. Tech Coloproctol. 2004; 8:s72–75.
9. Jayasekara H, Reece JC, Buchanan DD, Ahnen DJ, Parry S, Jenkins MA, Win AK. Risk factors for metachronous colorectal cancer or polyp: A systematic review and meta-analysis. J Gastroenterol Hepatol. 2017; 32:301–326.
10. Abdel-Rahman WM, Peltomaki P. Molecular basis and diagnostics of hereditary colorectal cancers. Ann Med. 2004; 36:379–388.
11. The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012; 487:330–337.
12. Haley L, Tseng LH, Zheng G, Dudley J, Anderson DA, Azad NS, Gocke CD, Eshleman JR, Lin MT. Performance characteristics of next-generation sequencing in clinical mutation detection of colorectal cancers. Mod Pathol. 2015; 28:1390–1399.
13. Zhou D, Yang L, Zheng L, Ge W, Li D, Zhang Y, Hu X, Gao Z, Xu J, Huang Y, Hu H, Zhang H, Zhang H, et al. Exome capture sequencing of adenoma reveals genetic alterations in multiple cellular pathways at the early stage of colorectal tumorigenesis. PLoS One. 2013; 8:e53310.
14. Kang H, Salomon MP, Sottoriva A, Zhao J, Toy M, Press MF, Curtis C, Marjoram P, Siegmund K, Shibata D. Many private mutations originate from the first few divisions of a human colorectal adenoma. J Pathol. 2015; 237:355–362.
15. Borras E, San Lucas FA, Chang K, Zhou R, Masand G, Fowler J, Mork ME, You YN, Taggart MW, McAllister F, Jones DA, Davies GE, Edelmann W, et al. Genomic Landscape of Colorectal Mucosa and Adenomas. Cancer Prev Res (Phila). 2016; 9:417–427.
16. Hildebrandt MA, Reyes ME, Lin M, He Y, Nguyen SV, Hawk ET, Wu X. Germline Genetic Variants in the Wnt/beta-Catenin Pathway as Predictors of Colorectal Cancer Risk. Cancer Epidemiol Biomarkers Prev. 2016; 25:540–546.
17. Chen SP, Tsai ST, Jao SW, Huang YL, Chao YC, Chen YL, Wu CC, Lin SZ, Harn HJ. Single nucleotide polymorphisms of the APC gene and colorectal cancer risk: a case-control study in Taiwan. BMC Cancer. 2006; 6:83.
18. Dowty JG, Win AK, Buchanan DD, Lindor NM, Macrae FA, Clendenning M, Antill YC, Thibodeau SN, Casey G, Gallinger S, Marchand LL, Newcomb PA, Haile RW, et al. Cancer risks for MLH1 and MSH2 mutation carriers. Hum Mutat. 2013; 34:490–497.
19. Smith G, Carey FA, Beattie J, Wilkie MJV, Lightfoot TJ, Coxhead J, Garner RC, Steele RJC, Wolf CR. Mutations in APC, Kirsten-ras, and p53--alternative genetic pathways to colorectal cancer. Proc Natl Acad Sci USA. 2002; 99: 9433–9438.
20. Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010; 2:a001008.
21. Iacopetta B. TP53 mutation in colorectal cancer. Hum Mutat. 2003; 21:271–276.
22. Yan WF, Wu G, Sun PC, Qiu D. P53 mutations occur more commonly than KRAS mutations in colorectal adenoma. Int J Clin Exp Med. 2015; 8:1370–1375.
23. Li XL, Zhou J, Chen ZR, Chng WJ. P53 mutations in colorectal cancer - molecular pathogenesis and pharmacological reactivation. World J Gastroenterol. 2015; 21:84–93.
24. Ogino S, Lochhead P, Giovannucci E, Meyerhardt JA, Fuchs CS, Chan AT. Discovery of colorectal cancer PIK3CA mutation as potential predictive biomarker: power and promise of molecular pathological epidemiology. Oncogene. 2014; 33:2949–2955.
25. Rosty C, Young JP, Walsh MD, Clendenning M, Sanderson K, Walters RJ, Parry S, Jenkins MA, Win AK, Southey MC, Hopper JL, Giles GG, Williamson EJ, et al. PIK3CA activating mutation in colorectal carcinoma: associations with molecular features and survival. PLoS One. 2013; 8:e65479.
26. Whitehall VL, Rickman C, Bond CE, Ramsnes I, Greco SA, Umapathy A, McKeone D, Faleiro RJ, Buttenshaw RL, Worthley DL, Nayler S, Zhao ZZ, Montgomery GW, et al. Oncogenic PIK3CA mutations in colorectal cancers and polyps. Int J Cancer. 2012; 131:813–820.
27. Cereda M, Gambardella G, Benedetti L, Iannelli F, Patel D, Basso G, Guerra RF, Mourikis TP, Puccio I, Sinha S, Laghi L, Spencer J, Rodriguez-Justo M, et al. Patients with genetically heterogeneous synchronous colorectal cancer carry rare damaging germline mutations in immune-related genes. Nat Commun. 2016; 7:12072.
28. Collisson EA, Cho RJ, Gray JW. What are we learning from the cancer genome? Nat Rev Clin Oncol. 2012; 9:621–630.
29. Keku TO, Dulal S, Deveaux A, Jovov B, Han X. The gastrointestinal microbiota and colorectal cancer. Am J Physiol Gastrointest Liver Physiol. 2015; 308:G351–363.
30. Wheeler SR, Shi C, Holt JA, Vnencak-Jones CL. Mutation profiles of synchronous colorectal cancers from a patient with Lynch syndrome suggest distinct oncogenic pathways. J Gastrointest Oncol. 2016; 7:E64–71.
31. Colucci PM, Yale SH, Rall CJ. Colorectal polyps. Clin Med Res. 2003; 1:261–262.
32. COSMIC v81: Catalogue Of Somatic Mutations In Cancer. http://cancersangeracuk/cancergenome/projects/cosmic/.
33. Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GL, Edwards KJ, Day IN, Gaunt TR. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat. 2013; 34:57–65.