
INTRODUCTION
Glioblastoma
Glioblastomas (GB), classified by the World Health Organization as Grade IV-Diffuse Glioma [1], are the most frequent and lethal malignant primary adult brain tumor [2]. The current standard of care for newly diagnosed - or de novo - GB (~90–95% of GB) includes maximal surgical resection and fractionated radiotherapy (30 × 2 Gy) with concomitant Temozolomide, also called the Stupp regimen [3]. However, prognosis remains extremely poor, with a median overall survival (OS) of 14–15 months [2]. A major molecular prognostic factor identified in GB is IDH1/2 mutations, a benefic prognosis factor that closely concerns secondary GB, which progress from low-grade diffuse astrocytoma or anaplastic astrocytoma (~5–10% of GB) [1]. Another well-identified prognosis factor is the methylation status of the O6-alkylguanine DNA methyltransferase (MGMT) gene, encoding a DNA-repair enzyme for Temozolomide lesions. According to studies, ~35–45% of wild-type IDH GB present a MGMT promoter methylation, associated to a better prognosis [4]. Recent studies have also highlighted new prognosis factors in GB, such as TERT promoter mutations (~70–75% of de novo GB, worse prognosis factor), histone H3F3A K27 and G34 mutations (~5% of adult GB), ATRX mutations and a positive glioma-CpG island methylator phenotype (G-CIMP), a benefic prognosis factor closely associated to secondary IDH mutant GB [4, 5]. TP53 mutations (observed in 27% and 81% of IDH-wild type and IDH-mutant GB, respectively [1]) and EGFR amplification (~40–50% of GB) and/or mutations, such as EGFR variant III, appear to be quite frequent in GB but do not seem to be associated to a worse outcome in GB patients [6]. Besides these molecular considerations, prompt relapses experienced by patients may be explained by the aggressiveness of GB, prone to invade surrounding brain tissue [2]. GB are also highly angiogenic, radio/chemoresistant and characterized by a strong cellular heterogeneity. Notably, a cancer cell subpopulation, called GB-initiating cells (GIC) or stem-like cells, appears to be particularly responsible for tumor maintenance and recurrence, as they can recapitulate the heterogeneity of the original brain tumor in orthotopically-xenografted nude mice [7].
GIC are characterized by their ability to self-renew in vitro (as neurosphere 3D structures) and in vivo, their marked expression of stem markers (CD133, Nestin, Olig2, Sox2, Nanog, BMI1, A2B5, ITGA6, L1CAM…), their multipotent aptitude to differentiate into neuronal, astrocytic or oligodendrocytic lineages, and a higher tumorigenic potential in orthotopically xenografted athymic nude mice [7]. Of note, it appears that the GIC subpopulation is strongly heterogeneous and could not be defined by the expression of an unique marker, but rather by a combination of markers. For example, it was previously shown that CD133-negative GB cells could also be identified as tumorigenic GIC [8]. Furthermore, GIC are particularly aggressive, invasive, radio/chemoresistant [7, 9] and display high autophagic capacities that may participate to these resistance processes [10]. GIC participate to angiogenesis via pro-angiogenic factor synthesis, cooperation with endothelial cells (EC), vasculogenesis by vasculogenic mimicry formation and transdifferentiation into pericytes or EC (for review [11]). GIC can also induce immunosuppression, notably via cytokines secretion [12]. Moreover, several signals and stresses, such as hypoxia [13], specific growth factors [14, 15], viral infections [16], chemotherapy [17] and ionizing radiations [9] contribute to the generation/amplification of GIC population from more differentiated GB cells, a process, which can evoke a possible cancer cell dedifferentiation or reprogramming [18]. Collectively these properties (Figure 1) suggest that targeting GIC represents a therapeutic interest in GB. To this end, blockade of specific pathways identified in GIC maintenance and functions (Notch, Sonic Hedgehog/Wnt, Akt…) may be a valuable strategy [7], but could lack efficiency due to compensatory pathways. Another alternative to eradicate GIC could be the targeting of their niches. Actually, GIC preferentially reside in perivascular and necro/hypoxic niches where they closely interact with the microenvironment [19]. These interactions with microenvironment elements, like stromal cells or extracellular matrix (ECM) components, seem to be critical for GIC maintenance, notably through metabolic and/or epigenetic modifications [19, 20] and could constitute a putative target.
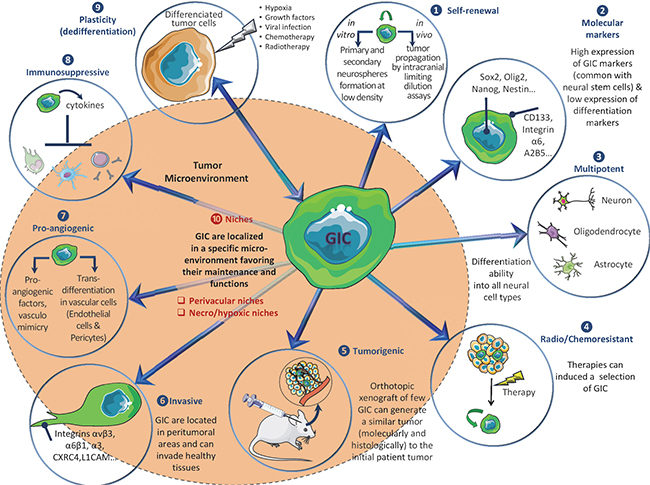
Figure 1: Overview of main GIC properties.
Integrins
Key components of the dialogue between cells and the microenvironment, integrins (ITG) are composed of two non-covalently associated α and β subunits, which are characterized by a large extracellular domain, a short transmembrane domain and a small intracellular non-catalytic cytoplasmic tail [21]. To date, 18 α and 8 β subunits have been identified in humans and form at least 24 unique heterodimers. Each α/β combination determines specific integrin binding ability and functions. Structural characteristics and ligands of these heterodimeric glycoproteins have already been reviewed [22] and are summarized in Figure 2. These receptors play a role in the regulation of cell adhesion to ECM proteins or cell surface proteins (immune cells, platelets…) [22]. They are central regulators, which act as transmembrane links between extracellular contacts and intracellular cytoskeleton via a bidirectional signalling. First, upon extracellular ligands binding, integrins cluster in the membrane and transduce intracellular signals through their cytoplasmic domain (mostly via β subunit) by activation of kinases (Focal Adhesion Kinase (FAK), Integrin-Linked Kinase (ILK)…) or Rho-GTPases. Integrins can then activate pathways leading to gene transcription to sustain proliferation, survival, differentiation and migration (outside-in signalling) [22, 23]. Second, cytoplasmic proteins can also modulate integrins extracellular affinity for their ligands (inside-out signalling). In addition, integrins also bind cytoskeleton proteins (α-actinin, tensin, vinculin, talin, paxillin, intermediate filaments…) and, then, are involved in structural cell functions. All these characteristics (Figure 2) may confer a high degree of complexity and flexibility in integrin-linked cell functions and signalling pathways, since several distinct integrins can be expressed on specific cells and, depending on context generate dramatically different responses. As a consequence, integrin-pathways alterations have also been linked to several pathologies, such as auto-immune and thrombotic diseases, ischemic brain injury, inflammation, fibrosis and cancer.
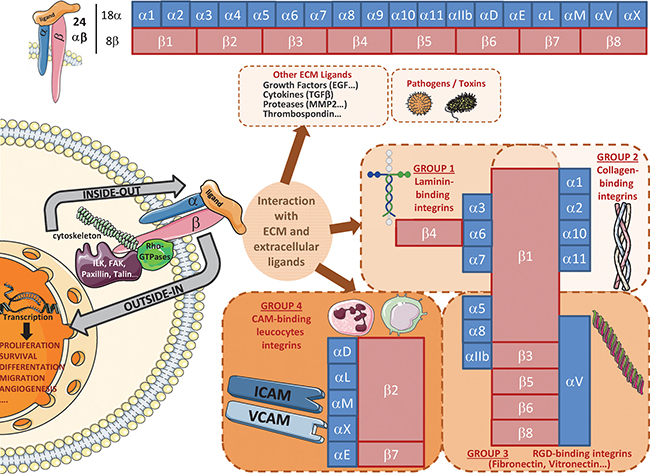
Figure 2: Overview of main integrin heterodimers and ligands.
Integrins in cancer
Integrins are rarely mutated in tumors. They do not act as oncogenes but may cooperate with oncogenes to favor tumorigenesis, either by particular expression and/or localization in tumor cells compared to normal cells, by post-translational modifications or via integrin recycling. Indeed, in several cancers, modifications of the integrin pattern are often associated with tumor progression. For example, α6β4, α5β1, αvβ6, and αvβ3 are upregulated in different tumor types, including GB [24, 25], and correlated with poor patient survival. However, integrin expression can also be decreased in tumors (e.g. α2β1 in breast cancer, α6β4/α6β1 in esophageal carcinoma), but still in favor of cancer progression [23, 24]. Altered integrins expression has also been reported in different models of carcinogenesis induction [24]. Moreover, post-translational modifications (glycosylation/sialylation, citrullination and carbamylation) modulate integrin conformation, affinity and functions and play a role in cancer progression [26, 27]. Finally, increased integrin recycling is associated with invasive and metastatic processes in cancer cells [28].
All these modulations affect integrins functions. First they are involved in invasion (and metastasis) as they directly provide the traction necessary for cell migration (for review [24]). They also participate in ECM remodeling through protease regulation and localization at migration front. Subsequently, integrins regulate tumor cell proliferation and survival through ECM-direct binding and/or intracellular signalling [24]. Furthermore, integrins interact and crosstalk with other cell surface receptors, like growth factor receptors (for review [29]). Finally, integrins are expressed by various tumor microenvironment cells (EC, pericytes, fibroblasts, immune cells, platelets…) and allow cell interactions to promote tumorigenesis. Integrin involvement in angiogenesis is particularly well described [30], as well as their role in epithelial to mesenchymal transition (EMT) which often mediates the acquisition of a migratory and/or invasive phenotype in cancer cells. Even if GB cannot be considered as tumors of epithelial origin, EMT can occur in response to treatment [31]. For example, α5β1 integrin was shown to mediate EMT in GB cells [32]. Consequently, integrins contribute to cancer progression and future studies have to identify the roles of these regulators precisely.
Due to their involvement in tumor cell functions, their crosstalk with numerous pathways and their localization at the cell membrane, integrins represent promising targets for cancer treatment. Consequently, different integrin inhibitors are under clinical investigations, notably in GB (detailed below). However, disappointing results in a phase III-study [33] forced researchers to reappraise the roles of the different integrins in GB, notably their specific expression according to tumor heterogeneity. In this review, we summarized integrin expression patterns and characteristics in GB and particularly concentrated on tumor molecular and cellular heterogeneity, notably regarding GIC. We chose to specifically focus on newly diagnosed GB and to not address here lower grades diffuse gliomas. We also discussed the roles of these adhesion molecules in GB progression and made an overview of therapeutic options.
ROLES OF INTEGRINS IN GLIOBLASTOMA
Integrins expression in glioblastoma
αvβ3 and αvβ5 were first identified as attractive therapeutic targets in GB [34, 35]. These integrins are not expressed on normal brain cells [35] but specifically on both tumor-associated EC and GB cells [35]. Their ECM ligands, vitronectin [34] and fibronectin [36], are also upregulated in GB. Currently, αvβ3/αvβ5 and α6β1, a laminin-interacting integrin also overexpressed in GB [37], are the most studied in GB but others may play major roles. Indeed, comparative immunohistochemistry (IHC) staining of integrins in GB versus control tissues reveal an overexpression of α2, α3, α4, α5, α6 and β1 [38], as well as of β3/αvβ3, αvβ5 and αvβ8 [39]. Of note, β8 and α5β1 were particularly observed in peri-necrotic and perivascular areas [40].
Although several integrins are overexpressed in GB, links between integrin expression and patient overall survival (OS) were scarcely explored. α3, α5, α7, αvβ3, αvβ5 and αvβ8 expression levels were positively associated with higher glioma grades [35, 39, 41–43]. αvβ3 or α3β1 overexpressions at the protein level were also correlated with poor GB prognosis [25, 43]. Consequently we conducted a statistical analysis (detailed in supplemental data) based on TCGA Affymetrix data [44] to determine GB patient OS according to major integrin expression. We focused on a group of 184 newly diagnosed primary GB solely treated with standard chemoradiotherapy [3]. In this particular population, overexpression (based on an upper threshold of the third quartile) of β1, β3, α3, α5 and αv, but neither β5, β4, α6 nor α7, was significantly associated with OS decrease, whether through univariate or multivariate analysis (Figure 3). α5 and α3 overexpressions were previously associated to worse prognosis in GB (n = 127, TCGA dataset and high-grade glioma cohorts, for α5 [42, 45]) or grade III/IV glioma patients (n = 68, immunohistochemistry data, for α3 [43]). It was also recently showed that β1 may be downregulated at both protein and RNA levels in GB patients with OS > 23 months (n = 14/26) [46]. However, α6, hypothesized to be associated with decreased OS in all-grade glioma [47] and in three-fold (or more) overexpressing GB (n = 7/193 patients, REMBRANDT dataset [48]), fails to show a similar pattern in our analysis. Similarly, no difference could be highlighted for β4, shown to be associated with GB worse prognosis (n = 393, TCGA dataset) [49]. Lower expression of α7 integrin, which was recently identified as a new functional marker in GB (probably as a heterodimer with β1), was also correlated with better prognosis outcomes in TCGA GB patients (n = 595) and in three additional independent GB cohorts [41]. However, our analysis failed to highlight such survival gain in α7-low patients. These disparities for α6, α7 and β4 could be linked to the fact that we restrained our analysis to a homogenous primary GB patients subgroup treated with standard chemoradiation.
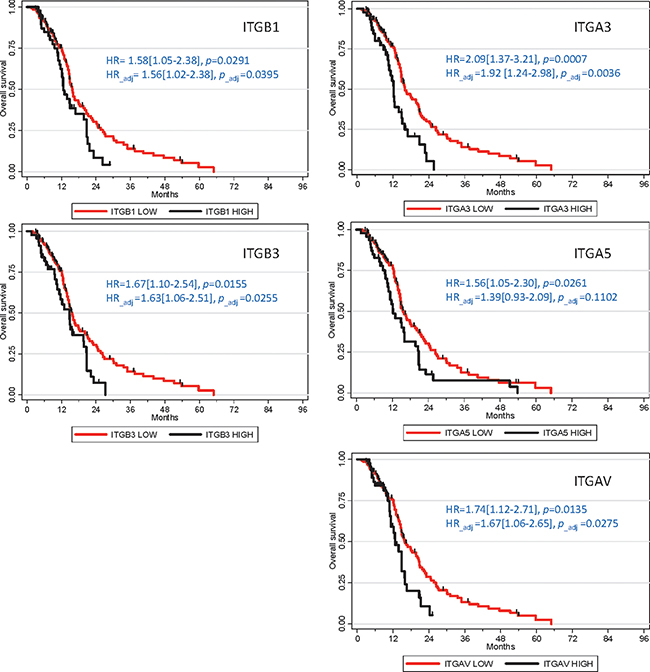
Figure 3: Integrins overexpression association with poor prognosis in GB patients. Kaplan-Meier survival plots were established using TCGA Affymetrix dataset (n = 184). Hazard ratios (HR) and p-values were given for univariate analyses. Adjusted HR and p-values were calculated for multivariate analyses, in relation to other prognostic clinical covariates (Age, G-CIMP status and Karnofsky score).
Whereas several works studied the expression of integrins at the RNA level in GB patient cohorts, data of their expression at the protein level are scarce in such cohorts. CORE (n = 224) and CENTRIC (n = 274) clinical trial cohorts were explored for αvβ3, αvβ5 and αvβ8 staining by IHC and showed that αvβ3 is mainly expressed by GB endothelial cells, contrary to αvβ8 which is expressed almost exclusively by GB tumor cells. Of note, αvβ5 is expressed by both cell populations [50]. These results confirm those found in an independent cohort of 324 patients for which 147~160 GB samples were stained by IHC [39]. Proteomic analyses also demonstrated that αv integrins are overexpressed in GB endothelial cells compared to physiological endothelial cells (10 GB samples) [51] and that sialylated β8 integrin is upregulated in GB samples in comparison with control adult astrocytes [27]. Using the Human Protein Atlas resource portal [52], we also noticed that α3, αv, β1, β4, β5 and β8 integrins may be overexpressed in high grade glioma patient samples compared to control cortex tissues (IHC data), confirming previous studies [38, 39, 43].
These data show that several integrins are overexpressed in GB and are associated with worse GB prognosis, indicating that they could play a role in GB progression and that specific targeting of overexpressed-integrins could be of therapeutic interest in GB.
Integrins and glioblastoma molecular subtypes
Several integrins are overexpressed in GB [35, 38–40, 42, 43, 49] but these results have to be analyzed in the light of GB molecular heterogeneity. Indeed, based on RNA and genomic profiling using TCGA databases, GB were classified by Verhaak et al. in four subtypes: Pro-Neural, Neural, Classical and Mesenchymal [53]. Philips et al. also classified GB of a different dataset in three subtypes (Pro-Neural, Proliferative and Mesenchymal), with Proliferative GB corresponding to TCGA Neural and Classic groups (40–50% of GB). In addition to this genomic heterogeneity between GB patients, it was also demonstrated that intratumoral heterogeneity, with all four subtypes being represented, can occur within a same GB tumor area, either between spatially distinct fragments [54] or even at the single-cell level [55]. Each subtype is characterized by specific genetic alterations. The Proliferative signature is characterized by EGFR amplification and stem marker overexpression (Nestin, Notch and Hedgehog). Within this group, it was originally described that Neural GB overexpress neuronal markers (e.g. NEFL). Pro-Neural subtype (5–20%) is associated with TP53 mutation, PI3K-pathway overactivation, PDGFRa amplification and Olig2/Sox2 stem markers overexpression. A small IDH1-mutants subpopulation was also exclusively found in Pro-Neural GB. Mesenchymal subtype (35–50%) is characterized by several deregulated pathways (TNF/NFκB, MET, YKL40, CXCR4, TGF-β, CD44…), a very invasive and angiogenic phenotype and a strong aggressiveness [56]. Interestingly, single-cell analysis demonstrated that a notable stemness signature was associated to Pro-Neural and Classic cell subtypes, and underrepresented in the Mesenchymal subtype [55]. In line with this, Mesenchymal GB were shown to contain a more progenitor type of GIC, contrary to Pro-Neural tumors containing a more stem GIC type [56, 57]. Of note, major studies recently classified GB by combining RNA, genomic and DNA methylation profiles [58, 59]. This new classification into six different clusters (LGm1 to LGm6) showed that primary GB (IDH wild type) can be classified into three subsets, with LGm4 being enriched in Classic-like GB, LGm5 in Mesenchymal-like GB and LGm6 in Pro-neural and Mesenchymal GB [59]. The Neural group, although represented in those three subsets, failed to show any specificity for any of these new subtypes. Finally, the newest study of the Verhaak group was designed to characterize the transcriptional heterogeneity of IDH wild-type GB by only using genes solely expressed by tumor cells and not by tumor-associated cells [60]. For this purpose, they combined RNA-sequencing data of (i) ~600 single cells isolated from 8 GB, (ii) 37 paired GB bulk tumors and their in vitro derived-neurospheres and (iii) matching microdissected tissues of both GB tumor cores and leading edges. They concluded that IDH wild-type GB can be classified into three subgroups (Pro-neural, Classic and Mesenchymal) and that the previously described Neural phenotype could be non-tumor specific [60].
Regarding integrin, very little was known, to our knowledge, about their differential expression among the different GB subtypes, neither in the different subsets defined by RNA profiling nor in the new LGm clusters. We then used publically available TCGA data [44] to performed a statistical analysis (see supplemental data) of the major integrins expression in all primary GB (n=500) and showed that most of them (β1, β4, β5, β8, α3, α5, α6, α7, αv) are significantly overexpressed in GB compared to normal brain (Figure 4, Supplementary Figure 1 and Supplementary Table 1A). However, contrary to numerous studies showing β3 transcript overexpression in GB [35, 39], this particular integrin did not appear to be overexpressed in GB for this specific dataset. We then analyzed an additional TCGA dataset, based on RNA-seq profiling of n = 149 primary GB and found that β3 is indeed overexpressed in GB (Supplementary Table 1B), as in the REMBRANDT dataset [39]. In addition, all these integrins but three (β8, α6 and α7) show a stronger overexpression in Mesenchymal GB compared to other subtypes and a lower overexpression in Pro-Neural GB (β3 excepted) (Figure 4 and Supplementary Figure 2). However, β8, α6 and α7 display a distinct pattern, with a higher overexpression in Classic GB compared to Mesenchymal and Pro-Neural GB (Figure 4), confirming some recent data on α7 [41]. In line with this, α7 overexpression was shown to especially correlate with poor prognosis outcome in Pro-neural GB patients (TCGA dataset) [41]. In support to our analysis, ITGB8 and ITGA7 were previously described as genes related to the Classic subtype signature [55]. Moreover, ITGB1 was defined as a top gene of the Mesenchymal subtype signature [60]. Of note, the last study by Verhaak’s group highlighted that Mesenchymal GB are enriched in tumor associated microglial cells, as the macrophage marker ITGAM (CD11b) appeared, among others, to be specifically overexpressed in this subtype [60].
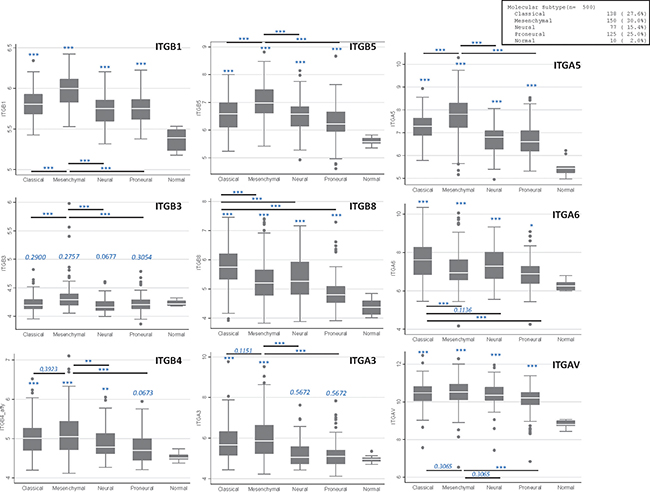
Figure 4: Main integrins expression in GB samples according to molecular subtypes. Plots were established using TCGA Affymetrix dataset (n = 500). *p ≤ 0.05; ** p ≤ 0.01; ***p ≤ 0.001. When not stated, values are compared to the “normal” subgroup.
According to our analyses, two different integrin expression patterns were then highlighted according to GB subtypes: Mesenchymal GB show a global integrin overexpression compared to other subtypes, apart from β8 and α6, mainly overexpressed in Classic GB. These specificities could be of interest in clinic to target selective integrins according to their overexpression in particular GB subtypes.
As an additional and relevant critical aspect of GB heterogeneity in clinic, it would be of major interest to decipher whether or not integrin expression profile could vary over time between primary GB and the associated recurrent tumors. Several studies were recently undertaken to assess this longitudinal transcriptomic heterogeneity in GB [60, 61]. It is noteworthy that the genomic and transcriptomic analyses of recurrent tumors revealed in these GB the presence of some exclusive LTBP4 (latent TGF-β binding protein 4) mutations associated to its overexpression [61]. As further highlighted in the present review, LTBPs, some key components of the latent TGFβ complex, appeared to be required for TGFβ activation in response to the binding of specific αv integrins (mainly αvβ6 and αvβ8) to the latent TGFβ complex. So, recurrent GB may present a deregulation of the TGFβ pathway, known to sustain GB aggressiveness, and it would be of interest to explore the role of the related αv integrins in this deregulation.
Integrins in glioblastoma proliferation, migration and invasion
Integrins sustain tumor cells proliferation, notably via their association with ECM components and maintenance of cell adhesion to substrate [24]. Few studies were conducted in GB. α5β1, which reduces GB cell proliferation when inhibited [62], acts through its interaction with Anosmin-1 [63] or Rap1A Rho-GTPase [64]. β4 interaction with its extracellular ligand netrin-4 also sustained GB cell proliferation [49].
However, integrins contribution to adhesive, migratory and invasive behaviors has been extensively studied in GB. First, αvβ3/αvβ5 encourage GB cell migration/invasion by direct adhesion to ECM (via fibronectin, vitronectin, osteopontin or periostin) [36, 65, 66] and then activate intracellular signalling pathways. For example, they trigger FAK, which controls cytoskeletal organization, force generation and survival, or activate additional pathways (Shc/MAP-Kinases, Rho-GTPases, Src Family Kinases) [23, 24, 67]. αvβ3 promotes GB invasion via the activation of MMP-2 at the plasma membrane and the subsequent degradation of complex ECM [24]. Finally, αvβ3/αvβ5 inhibition in murine models reduces GB cell migration/invasion [68]. Besides αvβ3/αvβ5, α6β1 was also associated with a stronger invasive phenotype. Its expression in U87 GB cell line enhances cells invasion in vitro and in vivo [37], as well as in GIC (our unpublished data). α6β1 cooperation with ERK and N-cadherin could support this effect [69]. Recently, β1 (associated with α3, α5 or α9) [45, 62, 70–72] and β8 [73, 74] have also been involved in GB cells migration/invasion, even if mechanisms are not well known yet. α5β1 could act through its interaction with MMP-2 [75] and αvβ8 through the modulation of Rho-GTPases activities (via RhoGDI1) [73].
Consequently, targeting specific integrins in GB could reduce tumor invasion and aggressiveness.
Integrins in glioblastoma angiogenesis
Knockout-strategies demonstrated the role of integrins in tumor angiogenesis [30]. In GB, characterized by a high vascularization and an overactive angiogenesis depending on VEGF and bFGF [76], this process is notably mediated by αvβ3, αvβ5, β1 and αvβ8. EC-expressed αvβ3/αvβ5 can provide survival signals and traction for invading cells, two mechanisms necessary to angiogenesis [34, 35]. αvβ3- and αvβ5-associated angiogenesis are respectively dependent on tumor cell-secreted bFGF/TNFα and VEGF through an amplification loop leading to αvβ3/αvβ5 overexpression on EC [77]. Overexpressed αvβ3/αvβ5 mediate in turn adhesive interactions with ECM proteins (vitronectin, fibronectin, fibrinogen, osteopontin, von Willebrand factor). In cooperation with bFGF/VEGF, αvβ3/αvβ5 also activate signalling pathways (FAK/ILK, PI3K/Akt and SDF1-CXCR4 [78]) that promote EC proliferation, survival and migration [30]. Regarding PI3K/Akt, a recent study associates this pathway with Plexin-B1/Rho/αvβ3 and Serine/arginine Protein Kinase-1 [79]. Moreover, migration can be improved by αvβ3 interaction with pleiotrophin (a secreted heparin-binding cytokine) on GB cells and EC [80] and with the cell adhesion molecule L1CAM on GIC [81]. Besides αvβ3/αvβ5, β1 is also involved in GB angiogenesis [76]. α5β1, similarly to αvβ3, is also upregulated by FGF on tumor endothelium during angiogenesis and promotes EC survival and migration [82]. β1 can be associated with other α-subunits, like α9, to support EC-mediated angiogenesis in GB [83]. α4β1 is also involved in pericyte-dependent angiogenesis to favor blood vessel maturation/stabilization [30]. Finally, αvβ8, overexpressed on GB cells [35, 40, 74], is involved in vessel formation/remodeling in GB through an autocrine TGF-β-dependent differential control of angiogenesis [74].
To summarize, several integrins, notably the αv and β1 families, appear to be interesting targets in GB to reduce angiogenesis in these highly vascularized brain tumors.
Integrins in glioblastoma survival and resistance to therapy
Depending on the context, integrins can either enhance cell survival or apoptosis, as they constantly challenge their microenvironment to regulate the death/survival balance. However, cancer cells deregulate this balance to promote survival. These different mechanisms, i.e. integrin ligation-related activation of pro-survival signals and unligated-integrin effects on both Integrin Mediated cell Death (IMD) or Anoikis (cell detachment-induced apoptotic death) were previously reviewed [24]. Briefly, IMD, a Caspase 8-dependent apoptotic process activated by unligated-integrins [24], can be deficient in tumor cells to promote survival [84]. In glioma, integrin inhibition can also induce a TGFβ-dependent anoikis [85]. Finally, in GB, both αvβ3 and the cytoskeleton regulatory kinase PAK4 were recently highlighted to mediate evasion of a p21-dependent senescence [86].
Moreover, chemo/radiotherapy exposure was associated with alterations of integrin-linked pathways in GB. First, irradiation up-regulated αvβ3 in U87 cells [87]. Second, α2β1, α3β1 and α5β1 were overexpressed in multi-drug resistant human glioma cells [88]. These results suggest, as for other cancers, an important role of integrins in GB resistance after chemo/radiotherapy. Cell adhesion to ECM, particularly through integrin ligation, is known to confer resistance in tumors, either to chemotherapy (cell-adhesion-mediated drug resistance, CAM-DR), or to irradiation (cell-adhesion-mediated radioresistance, CAM-RR).
Regarding CAM-DR, β1 and β4 integrins can confer Temozolomide-resistance in GB cells [89, 90]. An α5β1/p53 interconnection was notably demonstrated to modulate two anti-apoptotic proteins: Phosphoprotein enriched in astrocytes 15 (PEA-15) and Survivin. α5β1-overexpressing GB may then benefit from targeted-therapies associating integrin antagonists and survivin repressors [91]. Another putative and more general mechanism could be through integrin interaction with Periostin, an ECM protein recently highlighted in anti-angiogenic therapy resistance in glioma models [92]. Finally, as the high GB intratumoral heterogeneity (depicted above and in [54, 55]) could have major clinical implications in resistance to treatments, it is noteworthy that a recent single-cell clonal analysis of a GB patient sample by RNA-sequencing highlighted that TMZ-resistant clones displayed an up-regulation of αv integrin and a downregulation of α7 integrin compared to TMZ-sensitive clones [93].
CAM-RR is more documented in GB. U87 and SF763 GB cells, which express αvβ3 and αvβ5, respectively, were shown to be radioresistant through an ILK/RhoB pathway. Specific αvβ3/αvβ5 inhibition by Cilengitide or ILK blockade led to a significant in vitro radiosensitization [94]. In vivo, U251 cells-xenografted rats treated with Cilengitide and radiotherapy displayed a survival advantage compared to radiotherapy alone [95]. This radiosensitization was associated with an increase of radiation-induced apoptosis [95]. Furthermore, EC radiosensitization can also induce GB cell radiosensitization in vitro and in vivo [95]. So αvβ3/αvβ5 blockade could lead to radiosensitization potentiation by targeting both endothelial and tumor cells. Another integrin involved in GB CAM-RR is β1, through the activation of a pro-survival pathway transduced by Akt, paxillin, p130Cas and JNK [96]. β1 and JNK co-inhibition was recently shown to potently radiosensitize GB-initiating cells in vitro and in vivo [97]. ERK may also be involved since this MAP-kinase participates to chemoresistance in glioma cells [89]. Then, several studies imply integrins in GB radioresistance. Growing evidence demonstrated that induction of survival pathways (PI3K/Akt, NFKB, Bcl2, JNK…) or inhibition of pro-apoptotic pathways (p53), most often through FAK activation, could be involved [23, 98, 99]. The anti-apoptotic survivin, induced by irradiation [100], can also modulate the radiotherapy-induced mitotic U87 cell death through an ILK/HIF-1α/survivin pathway [101]. Besides ILK/FAK, other focal adhesion proteins such as PINCH1 and ILKAP contribute to GB radioresistance [102]. Among potential mechanisms in GB, integrins could also act by cell cycle modulation [103], since irradiation-induced cell cycle arrest is potentiated by α6β4/laminin-5 adhesion in prostate cancer [104]. Moreover, IMD resistance might also occur after radiotherapy. Indeed, as irradiation induces adhesion of breast cancer cells to laminin, fibronectin and collagen [105], it could be hypothesized that such process impairs IMD. Finally, integrins cooperation with growth factor pathways could also mediate CAM-RR in GB, as concurrent inhibition of β1/EGFR can radiosensitize tumor cells [106].
Altogether, these observations suggest that several integrins, particularly αvβ3/αvβ5/β1/β4, are interesting candidates in GB to be combined to ionizing radiations for radiosensitizing strategies. However, most of these studies were conducted on adherent GB cell lines and this could limit the significance of these resistance pathways in clinic. To better recapitulate the behavior of GB cells in vivo, other in vitro GB models could be more appropriate, and notably the GIC-enriched neurosphere 3D cell model.
Integrins in glioblastoma-initiating cells
Integrins were recently highlighted in cancer stem cell (CSC) biology, as they could represent specific CSC biomarkers and/or participate to CSC phenotype and functions [107]. In GB, α6 was shown to be preferentially and strongly expressed by GIC compared to differentiated cells [48]. Furthermore, α6High-expressing GIC present higher proliferation, stronger neurospheres-forming ability in vitro and tumorigenesis in vivo compared to α6Low GIC [48]. α6 also regulates GIC invasion process by cooperation with N-cadherin and ERK pathway [69]. α3 integrin was also shown to be overexpressed in GIC and to promote invasion [71]. Of note, the cell surface tetraspanin family member CD151 was recently found as a novel regulator of GIC by interacting with α6 and α3 [108]. Recently, a monoclonal antibody screening identified α7 integrin as a functional GIC marker. Indeed, α7 appeared to sustain the stem characteristics, the laminin-linked invasion ability and the tumorigenic capacity of this GIC subpopulation [41]. Besides these three α integrins, β1 integrin, even if not specifically expressed by GIC, can form a complex at the GIC membrane with astrocytes-secreted Connective Tissue Growth Factor and tyrosine kinase receptor type A and activate NFκB-driven invasion [109]. Another argument linking integrins with GIC biology is that particular integrins, belonging notably to the αv-family, can activate TGF-β pathway [110]. Indeed, TGF-β was described as a stemness gatekeeper in glioma [111] and TGF-β receptor antagonists target GIC and reduce tumorigenesis and radioresistance [112]. Finally, besides integrins, the junctional adhesion molecule-A may also promote GIC self-renewal [47].
In addition, recent studies also highlighted the differential expression of integrins in GIC according to their molecular subtypes. Indeed, even if molecular classifications were originally set up on GB whole tissues, GIC can also be classified according to their transcriptional subtypes. In that way, it was shown that GIC can be clustered in two major subgroups with distinct functional and molecular properties [56, 57, 113, 114]. The first one corresponds to a more stem-like GIC group, characterized by CD133 overexpression, an increased rate of asymmetric division, a higher invasive phenotype in xenografted-mice and a significant enrichment in Pro-Neural genes. The second group matches with a progenitor-like GIC type, corresponding to more adherent cells/neurospheres, a decrease in stem marker expression (Olig2, CD133), a trend toward a decreased tumorigenic ability in orthotopically-xenografted mice and an enrichment in Mesenchymal genes. These two subgroups were confirmed in two studies realized either in 48 different patient-derived GIC cell lines [115] or at the intratumoral level in a spectrum of GIC clones derived from the same tumor [116]. In the study by Xie and colleagues, an additional GIC cluster, corresponding to the Classic subtype, was highlighted, the Neural subtype being underrepresented (n = 3/48) [115]. Pro-Neural GIC clones or cell lines were found to be more sensitive to chemo- and radiotherapies, to have a higher proliferation rate and to give a higher rate of macroscopic tumors in xenografted mice [115, 116]. Regarding the correlation of GIC subtypes with survival prognosis in GB patients or in xenografted-mice, no clear trend could be defined since opposite results were obtained according to the studies, with a worse survival either for Mesenchymal GIC [56, 117] or for Pro-Neural GIC [115, 116].
This molecular and functional heterogeneity between GIC clones/cell lines was shown to be associated with differential integrin expression between GIC subgroups. Indeed, integrin α4 appeared to be downregulated in Pro-Neural stem-like GIC [114]. Integrins β1 and α5 were demonstrated to be overexpressed in Mesenchymal progenitor-like GIC at the transcriptional level [116, 117]. β1 overexpression was also confirmed at the protein level in Mesenchymal GIC clones [116], as well as the Mesenchymal marker CD44, known to bind and activate α5β1 in cancer cells [118]. This suggests an important role for α5β1 integrin in Mesenchymal GIC. Of note, this Mesenchymal GIC subtype is characterized by a down-regulation of miR-9-3p compared to Pro-Neural GIC [119]. As ITGB1 is a known target inhibited by miR-9-3p in cancer cells [120], this could contribute to the overexpression of β1 in Mesenchymal GIC. Finally, we used the Human Glioblastoma Cell Culture (HGCC) resource portal (www.hgcc.se, detailed in [115]) to compare the expression of the main integrins in the 48 GIC cell lines according to their molecular subtypes. We noticed, as we observed in the TCGA dataset analysis detailed above, that integrins α3, α4, α5, αv, β1, β3, β4 and β5 were overexpressed in the Mesenchymal GIC compared to the Pro-Neural GIC cell lines. All these integrins, except β5, were also downregulated in the Classic GIC in comparison with Mesenchymal GIC. On the contrary, α6 failed to show any significant variations between the different subgroups. β8, as observed in the TCGA Affymetrix dataset, is overexpressed in the Classic GIC subtype compared to Pro-Neural/Mesenchymal GIC cell lines. Altogether, it can be postulated that most of the integrins with a role in GB are preferentially expressed in Mesenchymal GB cells and GIC compared to Classic/Pro-Neural subtypes, with the notable exception of α6 and β8, preferentially expressed in the Classic GB cells.
Besides this molecular heterogeneity between GIC populations, it was also shown that anatomical location of the biopsy has an impact on integrin expression in GIC. Indeed, GIC generated from the GB peritumoral area displayed a higher migratory and invasive phenotype compared to GIC from the tumor mass. This invasive capacity was correlated to the overexpression of integrin β3 in peritumoral GIC and inhibition of αvβ3 blocked this invasive phenotype [121].
To conclude, specific integrins, such as α6, seem to contribute to GIC identification and/or functions. Moreover, the GIC heterogeneity, either at the molecular or anatomical levels, between GB patients or even between GIC clones of a same patient, is associated with a differential integrin expression. Notably, β1 integrin displayed an important overexpression in Mesenchymal GIC subtypes. Consequently, targeting these specific integrins preferentially expressed in GIC or in certain GIC subtypes might represent a potent way to alter GIC stemness and tumorigenicity in order to improve GB treatment.
Integrins in glioblastoma microenvironment and niches
Tumor microenvironment modifications contribute to cancer aggressiveness and recurrence. GB, characterized by a specific and tightly regulated microenvironment, are among the most vascularized and hypoxic tumors [19]. Cell membrane-expressed integrins can interact and modulate GB specific microenvironment via pro-migratory and pro-invasive properties (previously reviewed [24] and described above), hypoxic signalling/conditions, growth factor pathways, immune system and GIC maintenance within their niches. These mechanisms are detailed below and Figure 5.
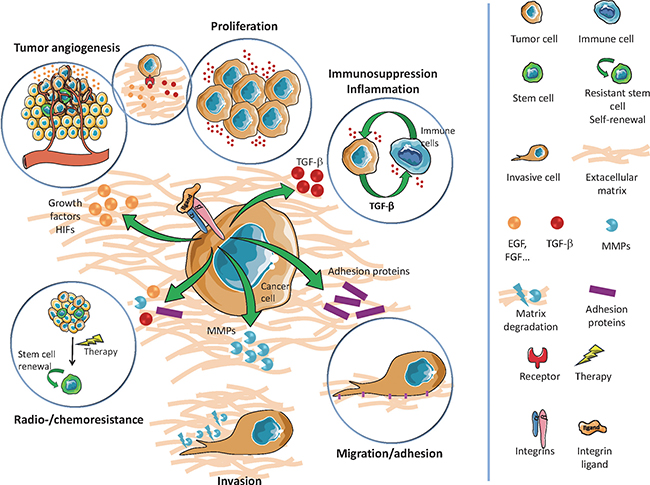
Figure 5: Overview of integrin interactions with glioblastoma microenvironment and niches
Hypoxia occurs in growing tumors when distant or abnormal tumor vasculature cannot provide blood supply. In GB, oxygen concentration decreases to 0.1–2% (2–10% in healthy brain). Hypoxia-signalling pathway, via hypoxia inducible factors (HIF-1/HIF-2), is then activated to promote, among others, angiogenesis. In different tumor types, hypoxia could up-regulate integrins (α6, αvβ3), which contribute to hypoxia cell adaptation [122, 123]. In GB, integrins are part of this hypoxia-signalling pathway and strongly support GB angiogenesis. For instance, α6-overexpressing U87 cells form bigger tumors than wild-type U87 in immunodeficient mice due to improved vascularisation [37]. Moreover, EGFRvIII/β3 integrin complexes were identified as promoter of GB progression in hypoxic environment [124]. Additionally, our laboratory demonstrated that αvβ3/αvβ5 were overexpressed in hypoxic U87 and SF763 GB cells, leading to FAK activation and subsequent RhoB/GSK-3-dependent HIF-1 stabilization [125]. In this study, the αvβ3/αvβ5-inhibitor Cilengitide, known to induce tumor regression via angiogenesis blockade and tumor growth inhibition [68], significantly decreased in vitro HIF-1 expression under hypoxia. Additionally, siRNA-mediated β3 inhibition led in vivo to an oxygenation of U87 xenografts [125]. It was also demonstrated in U87 that the T3 thyroid hormone, through iodothyronine receptor domains on αvβ3 and a Src/PI3K/ERK pathway, could enhance cell proliferation and HIF-1 expression [126]. Finally, in U251 cells, melatonin, a potent antioxidant, was shown to reduce cell migration/invasion in hypoxia through oxidative stress modulation and inhibition of the αvβ3/FAK/Proline-rich tyrosine Kinase 2 pathway [127]. Concerning integrin-mediated downstream pathways, and particularly ILK, two small-molecule ILK inhibitors (QLT0254/QLT0267) led in vitro to U87 cell cycle arrest and apoptosis, associated with AKT inhibition and VEGF secretion. In vivo, QLT0267 reduced U87 tumor growth by downregulating HIF-1 and VEGF, suggesting that ILK contribute to GB angiogenic process [128]. Consequently, specific integrins and associated-downstream pathways participate to hypoxia signalling in GB and then could impact on tumor aggressiveness.
As previously described, integrins cooperate with growth factors and/or their receptors and these pathways extensively cross talk. We focus here on TGF-β, involved in multiple cancer processes (for review [111]). TGF-β is secreted in a latent form consisting of a complex of three proteins: TGF-β (isoforms 1/2/3), inhibitor LAPs (latency-associated protein) and ECM-binding protein LTBPs (latent TGF-β binding proteins). To be activated, TGF-β needs to be liberated from latent complex, e.g. via conformational change or proteolysis. Interestingly, LAPs of TGF-β1 and TGF-β3 contain an integrin-binding site (RGD, arginine-glycine-aspartic acid sequence) and RGD-binding integrins (αv family, particularly αvβ6/αvβ8) can activate latent TGF-β through direct binding [110]. TGF-β is often overexpressed in GB and associated with tumor initiation and progression, as it promotes proliferation, invasion, angiogenesis, stemness, resistance and immune suppression [111]. Notably, inhibition of TGF-β pathway, up-regulated in glioma after irradiation [129], radiosensitizes GB cells [130]. Regarding the specific signalling, TGF-β binds to its receptors TGF-β-R-I/II/III and activates the canonical (SMAD proteins) and non-canonical (MAPK, PI3K/Akt, NF-κB, RhoGTPases…) pathways [111]. A recent study also demonstrates a network between integrins, aryl hydrocarbon receptor and TGF-β [131]. So αv integrins, through TGF-β activation, can promote GB progression.
Finally, integrin involvement in microenvironment may also depend on immune response modulation. Indeed, integrins regulate the recruitment of myeloid cells [22] and lead to immunosuppression or inflammation in favor of tumor progression, as exemplified for α4β1 [132]. In GB, a proteomic secretome analysis highlighted that osteopontin (interacting with αvβ3/αvβ5/αvβ6/αvβ1) and lactadherin (interacting with αvβ3/αvβ5) are able to induce M2 microglia reprogramming via an integrin/FAK/PI3K pathway [133]. M2 tumor-associated macrophages are recruited through association of their αvβ3 integrins with Periostin to favor tumor growth [134]. Another mechanism of integrin-mediated immunosuppression could involve TGF-β activation [110]. Depending on tumor context, GB or microglial cells-secreted TGF-β mediates immunosuppression either via the inhibition of natural killer cells, the IL-2R-mediated downregulation of proliferative signals in human T cells or the generation of immunosuppressive regulatory T cells [111].
Furthermore, all these microenvironment-related mechanisms can be transposed to GIC niches. GIC subpopulation is enriched in perivascular and hypoxic niches, which support their stemness and protect them from chemo/radiotherapies (for reviews [7, 19]). Moreover, hypoxia, HIFs and HIF-regulated genes play key role in GIC self-renewal and differentiation capacities compared to normal neural progenitors but also in GB cell dedifferentiation into GIC [9, 135]. Consequently, by modulating hypoxia and HIFs, integrins might regulate GIC characteristics, GIC differentiation and GB cell dedifferentiation. Additionally, integrins were previously described as key regulators to maintain stemness (see above) [48, 71]. Stem cell niches are indeed critical to preserve stemness, and integrins, by interacting with this particular local microenvironment, may provide signals allowing stem cells maintenance, depending or not on ECM interactions. Additionally, integrins could also enhance GIC properties via cooperation with growth factor receptors [24] and modulation of tumor immunity [12].
Integrin-targeting in glioblastoma
Integrins, localized at the cell surface, represent attractive targets for GB treatment. Some integrins, like αvβ5, seems also quite selective to tumor cells, then limiting toxicity on healthy tissues. Diverse integrin-targeting agents (antibodies, peptidic/peptidomimetic antagonists and other small molecules) were therefore placed into clinical development and clinical trials, previously reviewed [23, 30] and updated in Table 1, were then set-up in different tumor types, including GB.
Table 1: Integrins targeting agents in cancer, including GB
MOLECULE |
COMMERCIAL NAME |
COMPANY |
TYPE |
TARGET |
DISEASE |
TRIAL STATUS |
REFERENCES |
|---|---|---|---|---|---|---|---|
GLPG0187 |
x |
Galapagos SASU (France) |
Integrin receptor antagonist |
Broad spectrum integrins (αvβ1, αvβ3, αvβ5, αvβ6, αvβ8 and α5β1) |
Advanced solid tumours |
Phase I |
[141] |
EMD121974 |
Cilengitide |
EMD pharm. |
Cyclicized RGD- containing pentapeptide |
αvβ3 and αvβ5 |
Renal cell carcinoma, colon cancer, GB, melanoma, refractory advanced solid tumours, AML |
Phase III |
[142] |
MEDI-522 |
Etaracizumab |
Medimmune Inc. (USA) |
IgG1 humanized monoclonal antibody |
αvβ3 |
Melanoma, prostate/colon/ thyroid cancer |
Phase II |
[143] |
M200 |
Volociximab |
Protein design labs (USA) |
Chimeric monoclonal antibody |
α5β1 |
Renal cell carcinoma, melanoma, NSCLC, pancreatic cancer |
Phase II |
[144] |
PF0460541 |
x |
Pfizer (USA) |
IgG1 humanized monoclonal antibody |
α5β1 |
Solid tumours |
Phase I |
[145] |
ATN-161 |
x |
Attenuon, LLC (USA) |
Fibronectin like pentapeptide peptidomimetic |
α5β1 |
Advanced solid tumours |
Phases I/II |
[146] |
CNTO 95 |
Intetumumab |
Centocor (USA) |
Humanized monoclonal antibody |
αv |
Refractory advanced solid tumours |
Phase II |
[147] |
EMD525797 |
x |
Merk (Germany) |
Humanized monoclonal antibody |
αv |
Colorectal carcinoma and prostate |
Phase II |
[148] |
E7820 |
x |
Eisai medical research (USA) |
Aromatic sulfonamide derivative |
α2 |
Colorectal carcinoma |
Phase II |
[149] |
OS2966 |
x |
Oncosynergy (USA) |
monoclonal antibody |
β1 |
GB |
Phase I |
Planned in 2017 [150] |
αvβ3/αvβ5, the most studied integrins in GB, are involved in tumor progression and angiogenesis. The main developed molecule to target αvβ3/αvβ5 is Cilengitide (EMD121974). This cyclized RGD pentapeptide potently blocks αvβ3/αvβ5 activation and preclinical studies demonstrated its efficacy in vitro and in vivo in GB (for review [68]). Consequently, this molecule entered into phase I/II clinical trials in GB, in association with radio/chemotherapies, first in recurrent and then in newly diagnosed GB. These trials, previously reviewed [68] and updated in Table 2, showed interesting clinical responses with a good tolerance and clinical benefice on progression free survival (PFS) as well as on OS (Table 2). The CENTRIC (phase III) and CORE (phase II) clinical trials were then designed to evaluate Cilengitide efficacy in GB patients with methylated or unmethylated MGMT gene promoter, respectively. However, these studies failed to achieve expected results, with no OS improvement (Table 2). Several reasons were evoked and notably the fact that Cilengitide short half-life could lead to a weak systemic concentration and an additional difficulty to reach its targets [136]. The absence of an adapted biomarker to identify responding GB patients was also mentioned. A retrospective study showed that αvβ3/αvβ5 expression did not correlate with an improved OS in CENTRIC patients and that only an overexpressed αvβ3 in tumor cells, but not endothelial cells, of CORE cohort patients may be associated to a better outcome in Cilengitide-treated arm (Table 2). Moreover, GIC, the most resistant cells within the tumoral mass, express low αvβ3/αvβ5 levels [35]. So, it could be hypothesized that Cilengitide could not conveniently target GIC, one of the major reservoirs for GB recurrence. However, even if Cilengitide did not improve OS and will not be further developed as an anticancer drug, integrins still remains under consideration in GB and other solid tumors (Table 1).
Table 2: Summary of clinical trials using cilengitide in GB
TRIAL NAME |
PHASE |
PATIENTS |
TUMOR TYPE |
CILENGITIDE DOSE + TREATMENTS |
OUTCOMES (OS in months, PFS at 6 months) |
REFERENCES |
|---|---|---|---|---|---|---|
NABTT9911 |
I |
51 |
Recurrent malignant glioma |
Single agent 120 to 2400 mg/m2 2×/week |
OS: 5.6 |
[151] |
PBTC-012 |
I |
31 |
Recurrent pediatric brain tumours |
Single agent 120 to 2400 mg/m2 2×/week |
Complete response: 1 |
[152] |
NCT00979862 |
I |
45 |
Recurrent GB |
Cilengitide 2000 mg/m2 2×/week |
OS : 6.5 |
[153] |
CILENT-0902 |
I |
40 |
Children With Diffuse Intrinsic Pontine Glioma |
Radiotherapy + |
Completed |
No published results |
EMD-009 |
II |
81 |
Recurrent GB |
Single agent 500 or 2000 mg/m2 2×/week |
500mg: OS: 6.5; PFS-6: 10% |
[154] |
EMD-010 |
I/IIa |
52 |
Newly diagnosed GB |
Standard treatment + |
OS : 16.1 |
[155] |
NABTC03-02 |
II |
30 |
Recurrent GB |
Single agent 500 vs 2000 mg/m2 3 days before surgery and then 2000 mg/m2 2×/week |
PFS-6: 12% |
[156] |
NABTT0306 |
II |
112 |
Newly diagnosed GB |
Standard treatment + |
500mg: OS: 17.4 |
[157] |
ACNS0621 |
II |
30 |
Recurrent or refractory pediatric brain tumours |
Single agent 1800 mg/m2 2×/week |
Stable disease: 1 |
[158] |
CORE NCT00813943 |
II |
264 |
Newly diagnosed GB (unmethylated MGMT) |
Standard treatment |
Cilengitide 2x/wk : OS : 16.3 |
|
EXCENTRIC |
II |
48 |
Newly diagnosed GB (unmethylated MGMT) |
Standard treatment + Cilengitide 2000 mg/m² 2×/week + |
OS : 14.5 |
[160] |
CENTRIC NCT00689221 |
III |
504 |
Newly diagnosed GB (methylated MGMT) |
Standard treatment + 2000 mg/m2 2×/week |
OS : 26.3 |
[33] |
NCT01517776 |
II |
Recurrent High-grade Gliomas |
TMZ 75 mg/m²/d |
Terminated. |
||
NCT01044225 |
II |
Newly Diagnosed GB (unmethylated MGMT) |
Standard treatment + Cilengitide 2000 mg/m² 2×/week |
Terminated. |
Besides being attractive therapeutic targets, integrins may offer several other clinical perspectives. First, they represent interesting prognosis biomarkers. For example, αvβ3 and α3β1 high expression was associated with poor prognosis in GB patients [25, 43]. Second, their extracellular part could be recognized using non-invasive imaging systems. Such tools may be useful for diagnosis, for selection of responding patients and for the follow-up of anti-integrin treatment efficacy. Interestingly, 18F-fluciclatide-labeled αvβ3/αvβ5 integrins are able to provide clinical information and guide patient care in GB [137]. Other examples are DA364, a RGD-cyclic probe allowing GB detection by near-infrared fluorescence imaging [138], or a new 131I-labeled RGD-cyclic peptide dimer for αvβ3 imaging [139]. Third, considering a specific integrins pattern on tumor cells compared to normal cells, integrins targeting to deliver therapeutics seems another promising application in GB treatment. Indeed, they could serve as delivering target to specifically supply tumor cells with chemotherapy, immunotherapy, radionucleotide, gene therapy agents or even nanoparticles. This could improve treatment efficacy but also reduce toxicity. For example, an anti-αvβ3 monoclonal antibody (Abegrin) associated with a radioimmunotherapeutic agent reduce tumor volume of orthotopic GB cells [140].
CONCLUSIONS
Due to their involvement in GB radio/chemoresistance and progression (Table 3), integrins appear of great interest in GB treatment, either as targeted therapies, drug-delivering vectors or diagnostic tools for tumor imaging. Considering the strong GB heterogeneity, future preclinical and clinical studies have to focus on the particular integrins expression pattern within GB microenvironment. It is indeed crucial to identify the integrin profiles of specific subpopulations, notably GIC, and to correlate them to cell radio/chemosensitivity. Another challenge is to establish specific integrin signatures for each molecular GB subtype. These research axes represent key steps towards treatment personalization in GB and outcome improvement.
Table 3: Summary of integrins involved in GB
INTEGRINS |
ROLES IN GB |
REFERENCES |
|---|---|---|
αvβ3 |
Migration |
|
Invasion |
||
Angiogenesis |
||
Survival |
[86] |
|
Therapy resistance |
||
Prognostic marker |
[25] |
|
αvβ4 |
Proliferation |
[49] |
Therapy resistance |
[90] |
|
αvβ5 |
Migration, Invasion |
|
Angiogenesis |
[77] |
|
Therapy resistance |
||
αvβ8 |
Invasion |
[73] |
Angiogenesis |
[74] |
|
α3β1 |
Migration, Invasion |
|
Stemness |
[71] |
|
Prognostic marker |
[43] |
|
α5β1 |
Proliferation |
|
Migration |
[82] |
|
Invasion |
||
Survival |
||
Therapy resistance |
[42] |
|
α6β1 |
Invasion |
|
Stemness |
[48] |
|
α9β1 |
Migration |
[72] |
Angiogenesis |
[83] |
ACKNOWLEDGMENTS AND FUNDING
We want to acknowledge the Institut Claudius Regaud and GRICR, INSERM, the Canceropôle Grand Sud-Ouest (A.L), the Association pour la Recherche sur les Tumeurs Cérébrales (A.L) and the THE program from Plan Cancer 2016 (MoGlimaging). L.M and S.B were supported by a PhD fellowship from Plan Cancer/ITMO, P.D by a government PhD fellowship and J.S by a PhD fellowship from Ligue Nationale Contre le Cancer.
CONFLICTS OF INTEREST
None.
REFERENCES
1. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016; 131:803–20.
2. Brandes AA, Tosoni A, Franceschi E, Reni M, Gatta G, Vecht C. Glioblastoma in adults. Crit Rev Oncol Hematol. 2008; 67:139–52.
3. Weller M, van den Bent M, Hopkins K, Tonn JC, Stupp R, Falini A, Cohen-Jonathan-Moyal E, Frappaz D, Henriksson R, Balana C, Chinot O, Ram Z, Reifenberger G, et al. EANO guideline for the diagnosis and treatment of anaplastic gliomas and glioblastoma. Lancet Oncology. 2014; 15:e395–403.
4. Karsy M, Guan J, Cohen AL, Jensen RL, Colman H. New Molecular Considerations for Glioma: IDH, ATRX, BRAF, TERT, H3 K27M. Curr Neurol Neurosci Rep. 2017; 17:19.
5. Aldape K, Zadeh G, Mansouri S, Reifenberger G, von Deimling A. Glioblastoma: pathology, molecular mechanisms and markers. Acta Neuropathol. 2015; 129:829–48.
6. van den Bent MJ, Weller M, Wen PY, Kros JM, Aldape K, Chang S. A clinical perspective on the 2016 WHO brain tumor classification and routine molecular diagnostics. Neuro Oncol. 2017.
7. Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN. Cancer stem cells in glioblastoma. Genes Dev. 2015; 29:1203–17.
8. Clement V, Dutoit V, Marino D, Dietrich PY, Radovanovic I. Limits of CD133 as a marker of glioma self-renewing cells. Int J Cancer. 2009; 125:244–8.
9. Dahan P, Martinez Gala J, Delmas C, Monferran S, Malric L, Zentkowski D, Lubrano V, Toulas C, Cohen-Jonathan Moyal E, Lemarie A. Ionizing radiations sustain glioblastoma cell dedifferentiation to a stem-like phenotype through survivin: possible involvement in radioresistance. Cell Death Dis. 2014; 5:e1543.
10. Koukourakis MI, Mitrakas AG, Giatromanolaki A. Therapeutic interactions of autophagy with radiation and temozolomide in glioblastoma: evidence and issues to resolve. Br J Cancer. 2016; 114:485–96.
11. Guelfi S, Duffau H, Bauchet L, Rothhut B, Hugnot JP. Vascular Transdifferentiation in the CNS: A Focus on Neural and Glioblastoma Stem-Like Cells. Stem Cells Int. 2016; 2016:2759403.
12. Wu A, Wei J, Kong LY, Wang Y, Priebe W, Qiao W, Sawaya R, Heimberger AB. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. 2010; 12:1113–25.
13. Heddleston JM, Li Z, McLendon RE, Hjelmeland AB, Rich JN. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle. 2009; 8:3274–84.
14. Fessler E, Borovski T, Medema JP. Endothelial cells induce cancer stem cell features in differentiated glioblastoma cells via bFGF. Mol Cancer. 2015; 14:157.
15. Li Y, Li A, Glas M, Lal B, Ying M, Sang Y, Xia S, Trageser D, Guerrero-Cazares H, Eberhart CG, Quinones-Hinojosa A, Scheffler B, Laterra J. c-Met signaling induces a reprogramming network and supports the glioblastoma stem-like phenotype. Proc Natl Acad Sci USA. 2011; 108:9951–6.
16. Fornara O, Bartek J, Jr., Rahbar A, Odeberg J, Khan Z, Peredo I, Hamerlik P, Bartek J, Stragliotto G, Landazuri N, Soderberg-Naucler C. Cytomegalovirus infection induces a stem cell phenotype in human primary glioblastoma cells: prognostic significance and biological impact. Cell Death Differ. 2016.
17. Auffinger B, Tobias AL, Han Y, Lee G, Guo D, Dey M, Lesniak MS, Ahmed AU. Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Cell Death Differ. 2014; 21:1119–31.
18. Safa AR, Saadatzadeh MR, Cohen-Gadol AA, Pollok KE, Bijangi-Vishehsaraei K. Glioblastoma stem cells (GSCs) epigenetic plasticity and interconversion between differentiated non-GSCs and GSCs. Genes Dis. 2015; 2:152–63.
19. Lathia JD, Heddleston JM, Venere M, Rich JN. Deadly teamwork: neural cancer stem cells and the tumor microenvironment. Cell Stem Cell. 2011; 8:482–5.
20. Mack SC, Hubert CG, Miller TE, Taylor MD, Rich JN. An epigenetic gateway to brain tumor cell identity. Nat Neurosci. 2016; 19:10–9.
21. Humphries MJ. Integrin structure. Biochem Soc Trans. 2000; 28:311–39.
22. Takada Y, Ye X, Simon S. The integrins. Genome Biol. 2007; 8:215.
23. Hehlgans S, Haase M, Cordes N. Signalling via integrins: implications for cell survival and anticancer strategies. Biochim Biophys Acta. 2007; 1775:163–80.
24. Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010; 10:9–22.
25. Ducassou A, Uro-Coste E, Verrelle P, Filleron T, Benouaich-Amiel A, Lubrano V, Sol JC, Delisle MB, Favre G, Ken S, Laprie A, De Porre P, Toulas C, et al. alphavbeta3 Integrin and Fibroblast growth factor receptor 1 (FGFR1): Prognostic factors in a phase I-II clinical trial associating continuous administration of Tipifarnib with radiotherapy for patients with newly diagnosed glioblastoma. Eur J Cancer. 2013; 49:2161–9.
26. Ata R, Antonescu CN. Integrins and Cell Metabolism: An Intimate Relationship Impacting Cancer. Int J Mol Sci. 2017; 18.
27. Autelitano F, Loyaux D, Roudieres S, Deon C, Guette F, Fabre P, Ping Q, Wang S, Auvergne R, Badarinarayana V, Smith M, Guillemot JC, Goldman SA, et al. Identification of novel tumor-associated cell surface sialoglycoproteins in human glioblastoma tumors using quantitative proteomics. PLoS One. 2014; 9:e110316.
28. Onodera Y, Nam JM, Sabe H. Intracellular trafficking of integrins in cancer cells. Pharmacol Ther. 2013; 140:1–9.
29. Streuli CH, Akhtar N. Signal co-operation between integrins and other receptor systems. Biochem J. 2009; 418:491–506.
30. Avraamides CJ, Garmy-Susini B, Varner JA. Integrins in angiogenesis and lymphangiogenesis. Nat Rev Cancer. 2008; 8:604–17.
31. Kast RE, Skuli N, Karpel-Massler G, Frosina G, Ryken T, Halatsch ME. Blocking epithelial-to-mesenchymal transition in glioblastoma with a sextet of repurposed drugs: the EIS regimen. Oncotarget. 2017.
32. Renner G, Noulet F, Mercier MC, Choulier L, Etienne-Selloum N, Gies JP, Lehmann M, Lelong-Rebel I, Martin S, Dontenwill M. Expression/activation of alpha5beta1 integrin is linked to the beta-catenin signaling pathway to drive migration in glioma cells. Oncotarget. 2016; 7:62194–62207. https://doi.org/10.18632/oncotarget.11552.
33. Stupp R, Hegi ME, Gorlia T, Erridge SC, Perry J, Hong YK, Aldape KD, Lhermitte B, Pietsch T, Grujicic D, Steinbach JP, Wick W, Tarnawski R, et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071–22072 study): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014.
34. Gladson CL, Wilcox JN, Sanders L, Gillespie GY, Cheresh DA. Cerebral microenvironment influences expression of the vitronectin gene in astrocytic tumors. J Cell Sci. 1995; 108:947–56.
35. Roth P, Silginer M, Goodman SL, Hasenbach K, Thies S, Maurer G, Schraml P, Tabatabai G, Moch H, Tritschler I, Weller M. Integrin control of the transforming growth factor-beta pathway in glioblastoma. Brain. 2013; 136:564–76.
36. Serres E, Debarbieux F, Stanchi F, Maggiorella L, Grall D, Turchi L, Burel-Vandenbos F, Figarella-Branger D, Virolle T, Rougon G, Van Obberghen-Schilling E. Fibronectin expression in glioblastomas promotes cell cohesion, collective invasion of basement membrane in vitro and orthotopic tumor growth in mice. Oncogene. 2014; 33:3451–62.
37. Delamarre E, Taboubi S, Mathieu S, Berenguer C, Rigot V, Lissitzky JC, Figarella-Branger D, Ouafik L, Luis J. Expression of integrin alpha6beta1 enhances tumorigenesis in glioma cells. Am J Pathol. 2009; 175:844–55.
38. Gingras MC, Roussel E, Bruner JM, Branch CD, Moser RP. Comparison of cell adhesion molecule expression between glioblastoma multiforme and autologous normal brain tissue. J Neuroimmunol. 1995; 57:143–53.
39. Schittenhelm J, Schwab EI, Sperveslage J, Tatagiba M, Meyermann R, Fend F, Goodman SL, Sipos B. Longitudinal expression analysis of alphav integrins in human gliomas reveals upregulation of integrin alphavbeta3 as a negative prognostic factor. J Neuropathol Exp Neurol. 2013; 72:194–210.
40. Riemenschneider MJ, Mueller W, Betensky RA, Mohapatra G, Louis DN. In situ analysis of integrin and growth factor receptor signaling pathways in human glioblastomas suggests overlapping relationships with focal adhesion kinase activation. Am J Pathol. 2005; 167:1379–87.
41. Haas TL, Sciuto MR, Brunetto L, Valvo C, Signore M, Fiori ME, di Martino S, Giannetti S, Morgante L, Boe A, Patrizii M, Warnken U, Schnolzer M, et al. Integrin alpha7 Is a Functional Marker and Potential Therapeutic Target in Glioblastoma. Cell Stem Cell. 2017; 21:35–50 e9.
42. Janouskova H, Maglott A, Leger DY, Bossert C, Noulet F, Guerin E, Guenot D, Pinel S, Chastagner P, Plenat F, Entz-Werle N, Lehmann-Che J, Godet J, et al. Integrin alpha5beta1 plays a critical role in resistance to temozolomide by interfering with the p53 pathway in high-grade glioma. Cancer Res. 2012; 72:3463–70.
43. Zhou P, Erfani S, Liu Z, Jia C, Chen Y, Xu B, Deng X, Alfaro JE, Chen L, Napier D, Lu M, Huang JA, Liu C, et al. CD151-alpha3beta1 integrin complexes are prognostic markers of glioblastoma and cooperate with EGFR to drive tumor cell motility and invasion. Oncotarget. 2015; 6:29675–93. https://doi.org/10.18632/oncotarget.4896.
44. Cancer Genome Atlas Research N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008; 455:1061–8.
45. Mallawaaratchy DM, Buckland ME, McDonald KL, Li CC, Ly L, Sykes EK, Christopherson RI, Kaufman KL. Membrane proteome analysis of glioblastoma cell invasion. J Neuropathol Exp Neurol. 2015; 74:425–41.
46. Virga J, Bognar L, Hortobagyi T, Zahuczky G, Csosz E, Kallo G, Toth J, Hutoczki G, Remenyi-Puskar J, Steiner L, Klekner A. Prognostic Role of the Expression of Invasion-Related Molecules in Glioblastoma. J Neurol Surg A Cent Eur Neurosurg. 2017; 78:12–9.
47. Lathia JD, Li M, Sinyuk M, Alvarado AG, Flavahan WA, Stoltz K, Rosager AM, Hale J, Hitomi M, Gallagher J, Wu Q, Martin J, Vidal JG, et al. High-throughput flow cytometry screening reveals a role for junctional adhesion molecule a as a cancer stem cell maintenance factor. Cell Rep. 2014; 6:117–29.
48. Lathia JD, Gallagher J, Heddleston JM, Wang J, Eyler CE, Macswords J, Wu Q, Vasanji A, McLendon RE, Hjelmeland AB, Rich JN. Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem Cell. 2010; 6:421–32.
49. Hu Y, Ylivinkka I, Chen P, Li L, Hautaniemi S, Nyman TA, Keski-Oja J, Hyytiainen M. Netrin-4 promotes glioblastoma cell proliferation through integrin beta4 signaling. Neoplasia. 2012; 14:219–27.
50. Weller M, Nabors LB, Gorlia T, Leske H, Rushing E, Bady P, Hicking C, Perry J, Hong YK, Roth P, Wick W, Goodman SL, Hegi ME, et al. Cilengitide in newly diagnosed glioblastoma: biomarker expression and outcome. Oncotarget. 2016; 7:15018–32. https://doi.org/10.18632/oncotarget.7588.
51. Mustafa DA, Dekker LJ, Stingl C, Kremer A, Stoop M, Sillevis Smitt PA, Kros JM, Luider TM. A proteome comparison between physiological angiogenesis and angiogenesis in glioblastoma. Mol Cell Proteomics. 2012; 11:M111 008466.
52. Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, Olsson I, Edlund K, Lundberg E, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015; 347:1260419.
53. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O’Kelly M, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010; 17:98–110.
54. Sottoriva A, Spiteri I, Piccirillo SG, Touloumis A, Collins VP, Marioni JC, Curtis C, Watts C, Tavare S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci USA. 2013; 110:4009–14.
55. Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, Louis DN, Rozenblatt-Rosen O, Suva ML, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014; 344:1396–401.
56. Cusulin C, Chesnelong C, Bose P, Bilenky M, Kopciuk K, Chan JA, Cairncross JG, Jones SJ, Marra MA, Luchman HA, Weiss S. Precursor States of Brain Tumor Initiating Cell Lines Are Predictive of Survival in Xenografts and Associated with Glioblastoma Subtypes. Stem Cell Reports. 2015; 5:1–9.
57. Gunther HS, Schmidt NO, Phillips HS, Kemming D, Kharbanda S, Soriano R, Modrusan Z, Meissner H, Westphal M, Lamszus K. Glioblastoma-derived stem cell-enriched cultures form distinct subgroups according to molecular and phenotypic criteria. Oncogene. 2008; 27:2897–909.
58. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, Beroukhim R, Bernard B, Wu CJ, et al. The somatic genomic landscape of glioblastoma. Cell. 2013; 155:462–77.
59. Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, Morozova O, Newton Y, Radenbaugh A, Pagnotta SM, Anjum S, Wang J, Manyam G, et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell. 2016; 164:550–63.
60. Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L, deCarvalho AC, Lyu S, Li P, Li Y, Barthel F, Cho HJ, Lin YH, et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell. 2017; 32:42–56 e6.
61. Wang J, Cazzato E, Ladewig E, Frattini V, Rosenbloom DI, Zairis S, Abate F, Liu Z, Elliott O, Shin YJ, Lee JK, Lee IH, Park WY, et al. Clonal evolution of glioblastoma under therapy. Nat Genet. 2016; 48:768–76.
62. Maglott A, Bartik P, Cosgun S, Klotz P, Ronde P, Fuhrmann G, Takeda K, Martin S, Dontenwill M. The small alpha5beta1 integrin antagonist, SJ749, reduces proliferation and clonogenicity of human astrocytoma cells. Cancer Res. 2006; 66:6002–7.
63. Choy CT, Kim H, Lee JY, Williams DM, Palethorpe D, Fellows G, Wright AJ, Laing K, Bridges LR, Howe FA, Kim SH. Anosmin-1 contributes to brain tumor malignancy through integrin signal pathways. Endocr Relat Cancer. 2014; 21:85–99.
64. Sayyah J, Bartakova A, Nogal N, Quilliam LA, Stupack DG, Brown JH. The Ras-related protein, Rap1A, mediates thrombin-stimulated, integrin-dependent glioblastoma cell proliferation and tumor growth. J Biol Chem. 2014; 289:17689–98.
65. Ding Q, Stewart J, Jr., Prince CW, Chang PL, Trikha M, Han X, Grammer JR, Gladson CL. Promotion of malignant astrocytoma cell migration by osteopontin expressed in the normal brain: differences in integrin signaling during cell adhesion to osteopontin versus vitronectin. Cancer Res. 2002; 62:5336–43.
66. Mikheev AM, Mikheeva SA, Trister AD, Tokita MJ, Emerson SN, Parada CA, Born DE, Carnemolla B, Frankel S, Kim DH, Oxford RG, Kosai Y, Tozer-Fink KR, et al. Periostin is a novel therapeutic target that predicts and regulates glioma malignancy. Neuro Oncol. 2015; 17:372–82.
67. Lawson CD, Burridge K. The on-off relationship of Rho and Rac during integrin-mediated adhesion and cell migration. Small GTPases. 2014; 5:e27958.
68. Scaringi C, Minniti G, Caporello P, Enrici RM. Integrin inhibitor cilengitide for the treatment of glioblastoma: a brief overview of current clinical results. Anticancer Res. 2012; 32:4213–23.
69. Velpula KK, Rehman AA, Chelluboina B, Dasari VR, Gondi CS, Rao JS, Veeravalli KK. Glioma stem cell invasion through regulation of the interconnected ERK, integrin alpha6 and N-cadherin signaling pathway. Cell Signal. 2012; 24:2076–84.
70. Kawataki T, Yamane T, Naganuma H, Rousselle P, Anduren I, Tryggvason K, Patarroyo M. Laminin isoforms and their integrin receptors in glioma cell migration and invasiveness: Evidence for a role of alpha5-laminin(s) and alpha3beta1 integrin. Exp Cell Res. 2007; 313:3819–31.
71. Nakada M, Nambu E, Furuyama N, Yoshida Y, Takino T, Hayashi Y, Sato H, Sai Y, Tsuji T, Miyamoto KI, Hirao A, Hamada JI. Integrin alpha3 is overexpressed in glioma stem-like cells and promotes invasion. Br J Cancer. 2013; 108:2516–24.
72. Veeravalli KK, Ponnala S, Chetty C, Tsung AJ, Gujrati M, Rao JS. Integrin alpha9beta1-mediated cell migration in glioblastoma via SSAT and Kir4.2 potassium channel pathway. Cell Signal. 2012; 24:272–81.
73. Reyes SB, Narayanan AS, Lee HS, Tchaicha JH, Aldape KD, Lang FF, Tolias KF, McCarty JH. alphavbeta8 integrin interacts with RhoGDI1 to regulate Rac1 and Cdc42 activation and drive glioblastoma cell invasion. Mol Biol Cell. 2013; 24:474–82.
74. Tchaicha JH, Reyes SB, Shin J, Hossain MG, Lang FF, McCarty JH. Glioblastoma angiogenesis and tumor cell invasiveness are differentially regulated by beta8 integrin. Cancer Res. 2011; 71:6371–81.
75. Kesanakurti D, Chetty C, Dinh DH, Gujrati M, Rao JS. Role of MMP-2 in the regulation of IL-6/Stat3 survival signaling via interaction with alpha5beta1 integrin in glioma. Oncogene. 2013; 32:327–40.
76. Zhang M, Ye G, Li J, Wang Y. Recent advance in molecular angiogenesis in glioblastoma: the challenge and hope for anti-angiogenic therapy. Brain Tumor Pathol. 2015; 32:229–36.
77. Friedlander M, Brooks PC, Shaffer RW, Kincaid CM, Varner JA, Cheresh DA. Definition of two angiogenic pathways by distinct alpha v integrins. Science. 1995; 270:1500–2.
78. Maddirela DR, Kesanakurti D, Gujrati M, Rao JS. MMP-2 suppression abrogates irradiation-induced microtubule formation in endothelial cells by inhibiting alphavbeta3-mediated SDF-1/CXCR4 signaling. Int J Oncol. 2013; 42:1279–88.
79. Chang Y, Li L, Zhang L, Guo X, Feng Z, Zhou J, Zhou S, Feng G, Han F, Huang W, Yang J, Huang F. Plexin-B1 indirectly affects glioma invasiveness and angiogenesis by regulating the RhoA/alphavbeta3 signaling pathway and SRPK1. Tumour Biol. 2016; 37:11225–36.
80. Mikelis C, Sfaelou E, Koutsioumpa M, Kieffer N, Papadimitriou E. Integrin alpha(v)beta(3) is a pleiotrophin receptor required for pleiotrophin-induced endothelial cell migration through receptor protein tyrosine phosphatase beta/zeta. FASEB J. 2009; 23:1459–69.
81. Burgett ME, Lathia JD, Roth P, Nowacki AS, Galileo DS, Pugacheva E, Huang P, Vasanji A, Li M, Byzova T, Mikkelsen T, Bao S, Rich JN, et al. Direct contact with perivascular tumor cells enhances integrin alphavbeta3 signaling and migration of endothelial cells. Oncotarget. 2016; 7:43852–67. https://doi.org/10.18632/oncotarget.9700.
82. Lee HS, Oh SJ, Lee KH, Lee YS, Ko E, Kim KE, Kim HC, Kim S, Song PH, Kim YI, Kim C, Han S. Gln-362 of angiopoietin-2 mediates migration of tumor and endothelial cells through association with alpha5beta1 integrin. J Biol Chem. 2014; 289:31330–40.
83. Walsh EM, Kim R, Del Valle L, Weaver M, Sheffield J, Lazarovici P, Marcinkiewicz C. Importance of interaction between nerve growth factor and alpha9beta1 integrin in glial tumor angiogenesis. Neuro Oncol. 2012; 14:890–901.
84. Stupack DG, Teitz T, Potter MD, Mikolon D, Houghton PJ, Kidd VJ, Lahti JM, Cheresh DA. Potentiation of neuroblastoma metastasis by loss of caspase-8. Nature. 2006; 439:95–9.
85. Silginer M, Weller M, Ziegler U, Roth P. Integrin inhibition promotes atypical anoikis in glioma cells. Cell Death Dis. 2014; 5:e1012.
86. Franovic A, Elliott KC, Seguin L, Camargo MF, Weis SM, Cheresh DA. Glioblastomas require integrin alphavbeta3/PAK4 signaling to escape senescence. Cancer Res. 2015; 75:4466–73.
87. Wild-Bode C, Weller M, Rimner A, Dichgans J, Wick W. Sublethal irradiation promotes migration and invasiveness of glioma cells: implications for radiotherapy of human glioblastoma. Cancer Res. 2001; 61:2744–50.
88. Hikawa T, Mori T, Abe T, Hori S. The ability in adhesion and invasion of drug-resistant human glioma cells. J Exp Clin Cancer Res. 2000; 19:357–62.
89. Han S, Li Z, Master LM, Master ZW, Wu A. Exogenous IGFBP-2 promotes proliferation, invasion, and chemoresistance to temozolomide in glioma cells via the integrin beta1-ERK pathway. Br J Cancer. 2014; 111:1400–9.
90. Li L, Hu Y, Ylivinkka I, Li H, Chen P, Keski-Oja J, Hyytiainen M. NETRIN-4 protects glioblastoma cells FROM temozolomide induced senescence. PLoS One. 2013; 8:e80363.
91. Renner G, Janouskova H, Noulet F, Koenig V, Guerin E, Bar S, Nuesch J, Rechenmacher F, Neubauer S, Kessler H, Blandin AF, Choulier L, Etienne-Selloum N, et al. Integrin alpha5beta1 and p53 convergent pathways in the control of anti-apoptotic proteins PEA-15 and survivin in high-grade glioma. Cell Death Differ. 2016; 23:640–53.
92. Park SY, Piao Y, Jeong KJ, Dong J, de Groot JF. Periostin (POSTN) Regulates Tumor Resistance to Antiangiogenic Therapy in Glioma Models. Mol Cancer Ther. 2016; 15:2187–97.
93. Meyer M, Reimand J, Lan X, Head R, Zhu X, Kushida M, Bayani J, Pressey JC, Lionel AC, Clarke ID, Cusimano M, Squire JA, Scherer SW, et al. Single cell-derived clonal analysis of human glioblastoma links functional and genomic heterogeneity. Proc Natl Acad Sci USA. 2015; 112:851–6.
94. Monferran S, Skuli N, Delmas C, Favre G, Bonnet J, Cohen-Jonathan-Moyal E, Toulas C. Alphavbeta3 and alphavbeta5 integrins control glioma cell response to ionising radiation through ILK and RhoB. Int J Cancer. 2008; 123:357–64.
95. Mikkelsen T, Brodie C, Finniss S, Berens ME, Rennert JL, Nelson K, Lemke N, Brown SL, Hahn D, Neuteboom B, Goodman SL. Radiation sensitization of glioblastoma by cilengitide has unanticipated schedule-dependency. Int J Cancer. 2009; 124:2719–27.
96. Cordes N, Seidler J, Durzok R, Geinitz H, Brakebusch C. beta1-integrin-mediated signaling essentially contributes to cell survival after radiation-induced genotoxic injury. Oncogene. 2006; 25:1378–90.
97. Vehlow A, Klapproth E, Storch K, Dickreuter E, Seifert M, Dietrich A, Butof R, Temme A, Cordes N. Adhesion- and stress-related adaptation of glioma radiochemoresistance is circumvented by beta1 integrin/JNK co-targeting. Oncotarget. 2017; 8:49224–49237. https://doi.org/10.18632/oncotarget.17480.
98. Lim ST, Chen XL, Lim Y, Hanson DA, Vo TT, Howerton K, Larocque N, Fisher SJ, Schlaepfer DD, Ilic D. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol Cell. 2008; 29:9–22.
99. Matter ML, Ruoslahti E. A signaling pathway from the alpha5beta1 and alpha(v)beta3 integrins that elevates bcl-2 transcription. J Biol Chem. 2001; 276:27757–63.
100. Guvenc H, Pavlyukov MS, Joshi K, Kurt H, Banasavadi-Siddegowda YK, Mao P, Hong C, Yamada R, Kwon CH, Bhasin D, Chettiar S, Kitange G, Park IH, et al. Impairment of glioma stem cell survival and growth by a novel inhibitor for Survivin-Ran protein complex. Clin Cancer Res. 2013; 19:631–42.
101. Lanvin O, Monferran S, Delmas C, Couderc B, Toulas C, Cohen-Jonathan-Moyal E. Radiation-induced mitotic cell death and glioblastoma radioresistance: a new regulating pathway controlled by integrin-linked kinase, hypoxia-inducible factor 1 alpha and survivin in U87 cells. Eur J Cancer. 2013; 49:2884–91.
102. Hausmann C, Temme A, Cordes N, Eke I. ILKAP, ILK and PINCH1 control cell survival of p53-wildtype glioblastoma cells after irradiation. Oncotarget. 2015; 6:34592–605. https://doi.org/10.18632/oncotarget.5423.
103. Wang P, Ballestrem C, Streuli CH. The C terminus of talin links integrins to cell cycle progression. J Cell Biol. 2011; 195:499–513.
104. Kremer CL, Schmelz M, Cress AE. Integrin-dependent amplification of the G2 arrest induced by ionizing radiation. Prostate. 2006; 66:88–96.
105. Lee SH, Cheng H, Yuan Y, Wu S. Regulation of ionizing radiation-induced adhesion of breast cancer cells to fibronectin by alpha5beta1 integrin. Radiat Res. 2014; 181:650–8.
106. Eke I, Zscheppang K, Dickreuter E, Hickmann L, Mazzeo E, Unger K, Krause M, Cordes N. Simultaneous beta1 integrin-EGFR targeting and radiosensitization of human head and neck cancer. J Natl Cancer Inst. 2015; 107.
107. Seguin L, Desgrosellier JS, Weis SM, Cheresh DA. Integrins and cancer: regulators of cancer stemness, metastasis, and drug resistance. Trends Cell Biol. 2015; 25: 234–40.
108. Tilghman J, Schiapparelli P, Lal B, Ying M, Quinones-Hinojosa A, Xia S, Laterra J. Regulation of Glioblastoma Tumor-Propagating Cells by the Integrin Partner Tetraspanin CD151. Neoplasia. 2016; 18:185–98.
109. Edwards LA, Woolard K, Son MJ, Li A, Lee J, Ene C, Mantey SA, Maric D, Song H, Belova G, Jensen RT, Zhang W, Fine HA. Effect of brain- and tumor-derived connective tissue growth factor on glioma invasion. J Natl Cancer Inst. 2011; 103:1162–78.
110. Munger JS, Sheppard D. Cross talk among TGF-beta signaling pathways, integrins, and the extracellular matrix. Cold Spring Harb Perspect Biol. 2011; 3:a005017.
111. Joseph JV, Balasubramaniyan V, Walenkamp A, Kruyt FA. TGF-beta as a therapeutic target in high grade gliomas - promises and challenges. Biochem Pharmacol. 2013; 85:478–85.
112. Hardee ME, Marciscano AE, Medina-Ramirez CM, Zagzag D, Narayana A, Lonning SM, Barcellos-Hoff MH. Resistance of glioblastoma-initiating cells to radiation mediated by the tumor microenvironment can be abolished by inhibiting transforming growth factor-beta. Cancer Res. 2012; 72:4119–29.
113. Chen R, Nishimura MC, Bumbaca SM, Kharbanda S, Forrest WF, Kasman IM, Greve JM, Soriano RH, Gilmour LL, Rivers CS, Modrusan Z, Nacu S, Guerrero S, et al. A hierarchy of self-renewing tumor-initiating cell types in glioblastoma. Cancer Cell. 2010; 17:362–75.
114. Schulte A, Gunther HS, Phillips HS, Kemming D, Martens T, Kharbanda S, Soriano RH, Modrusan Z, Zapf S, Westphal M, Lamszus K. A distinct subset of glioma cell lines with stem cell-like properties reflects the transcriptional phenotype of glioblastomas and overexpresses CXCR4 as therapeutic target. Glia. 2011; 59:590–602.
115. Xie Y, Bergstrom T, Jiang Y, Johansson P, Marinescu VD, Lindberg N, Segerman A, Wicher G, Niklasson M, Baskaran S, Sreedharan S, Everlien I, Kastemar M, et al. The Human Glioblastoma Cell Culture Resource: Validated Cell Models Representing All Molecular Subtypes. EBioMedicine. 2015; 2:1351–63.
116. Segerman A, Niklasson M, Haglund C, Bergstrom T, Jarvius M, Xie Y, Westermark A, Sonmez D, Hermansson A, Kastemar M, Naimaie-Ali Z, Nyberg F, Berglund M, et al. Clonal Variation in Drug and Radiation Response among Glioma-Initiating Cells Is Linked to Proneural-Mesenchymal Transition. Cell Rep. 2016; 17:2994–3009.
117. Marziali G, Signore M, Buccarelli M, Grande S, Palma A, Biffoni M, Rosi A, D’Alessandris QG, Martini M, Larocca LM, De Maria R, Pallini R, Ricci-Vitiani L. Metabolic/Proteomic Signature Defines Two Glioblastoma Subtypes With Different Clinical Outcome. Sci Rep. 2016; 6: 21557.
118. McFarlane S, McFarlane C, Montgomery N, Hill A, Waugh DJ. CD44-mediated activation of alpha5beta1-integrin, cortactin and paxillin signaling underpins adhesion of basal-like breast cancer cells to endothelium and fibronectin-enriched matrices. Oncotarget. 2015; 6:36762–73. https://doi.org/10.18632/oncotarget.5461.
119. Marziali G, Buccarelli M, Giuliani A, Ilari R, Grande S, Palma A, D’Alessandris QG, Martini M, Biffoni M, Pallini R, Ricci-Vitiani L. A three-microRNA signature identifies two subtypes of glioblastoma patients with different clinical outcomes. Mol Oncol. 2017.
120. Zawistowski JS, Nakamura K, Parker JS, Granger DA, Golitz BT, Johnson GL. MicroRNA 9-3p targets beta1 integrin to sensitize claudin-low breast cancer cells to MEK inhibition. Mol Cell Biol. 2013; 33:2260–74.
121. Ruiz-Ontanon P, Orgaz JL, Aldaz B, Elosegui-Artola A, Martino J, Berciano MT, Montero JA, Grande L, Nogueira L, Diaz-Moralli S, Esparis-Ogando A, Vazquez-Barquero A, Lafarga M, et al. Cellular plasticity confers migratory and invasive advantages to a population of glioblastoma-initiating cells that infiltrate peritumoral tissue. Stem Cells. 2013; 31:1075–85.
122. Brooks DL, Schwab LP, Krutilina R, Parke DN, Sethuraman A, Hoogewijs D, Schorg A, Gotwald L, Fan M, Wenger RH, Seagroves TN. ITGA6 is directly regulated by hypoxia-inducible factors and enriches for cancer stem cell activity and invasion in metastatic breast cancer models. Mol Cancer. 2016; 15:26.
123. Cowden Dahl KD, Robertson SE, Weaver VM, Simon MC. Hypoxia-inducible factor regulates alphavbeta3 integrin cell surface expression. Mol Biol Cell. 2005; 16:1901–12.
124. Liu Z, Han L, Dong Y, Tan Y, Li Y, Zhao M, Xie H, Ju H, Wang H, Zhao Y, Zheng Q, Wang Q, Su J, et al. EGFRvIII/integrin beta3 interaction in hypoxic and vitronectinenriching microenvironment promote GBM progression and metastasis. Oncotarget. 2016; 7:4680–94. https://doi.org/10.18632/oncotarget.6730.
125. Skuli N, Monferran S, Delmas C, Favre G, Bonnet J, Toulas C, Cohen-Jonathan Moyal E. Alphavbeta3/alphavbeta5 integrins-FAK-RhoB: a novel pathway for hypoxia regulation in glioblastoma. Cancer Res. 2009; 69:3308–16.
126. Lin HY, Sun M, Tang HY, Lin C, Luidens MK, Mousa SA, Incerpi S, Drusano GL, Davis FB, Davis PJ. L-Thyroxine vs. 3,5,3′-triiodo-L-thyronine and cell proliferation: activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase. Am J Physiol Cell Physiol. 2009; 296:C980–91.
127. Xu CS, Wang ZF, Huang XD, Dai LM, Cao CJ, Li ZQ. Involvement of ROS-alpha v beta 3 integrin-FAK/Pyk2 in the inhibitory effect of melatonin on U251 glioma cell migration and invasion under hypoxia. J Transl Med. 2015; 13:95.
128. Edwards LA, Woo J, Huxham LA, Verreault M, Dragowska WH, Chiu G, Rajput A, Kyle AH, Kalra J, Yapp D, Yan H, Minchinton AI, Huntsman D, et al. Suppression of VEGF secretion and changes in glioblastoma multiforme microenvironment by inhibition of integrin-linked kinase (ILK). Mol Cancer Ther. 2008; 7:59–70.
129. Satoh E, Naganuma H, Sasaki A, Nagasaka M, Ogata H, Nukui H. Effect of irradiation on transforming growth factor-beta secretion by malignant glioma cells. J Neurooncol. 1997; 33:195–200.
130. Zhang M, Kleber S, Rohrich M, Timke C, Han N, Tuettenberg J, Martin-Villalba A, Debus J, Peschke P, Wirkner U, Lahn M, Huber PE. Blockade of TGF-beta signaling by the TGFbetaR-I kinase inhibitor LY2109761 enhances radiation response and prolongs survival in glioblastoma. Cancer Res. 2011; 71:7155–67.
131. Silginer M, Burghardt I, Gramatzki D, Bunse L, Leske H, Rushing EJ, Hao N, Platten M, Weller M, Roth P. The aryl hydrocarbon receptor links integrin signaling to the TGF-beta pathway. Oncogene. 2016; 35:3260–71.
132. Schmid MC, Franco I, Kang SW, Hirsch E, Quilliam LA, Varner JA. PI3-kinase gamma promotes Rap1a-mediated activation of myeloid cell integrin alpha4beta1, leading to tumor inflammation and growth. PLoS One. 2013; 8:e60226.
133. Ellert-Miklaszewska A, Wisniewski P, Kijewska M, Gajdanowicz P, Pszczolkowska D, Przanowski P, Dabrowski M, Maleszewska M, Kaminska B. Tumour-processed osteopontin and lactadherin drive the protumorigenic reprogramming of microglia and glioma progression. Oncogene. 2016; 35:6366–77.
134. Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J, Wu L, Sloan AE, McLendon RE, Li X, Rich JN, Bao S. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol. 2015; 17:170–82.
135. Qiang L, Wu T, Zhang HW, Lu N, Hu R, Wang YJ, Zhao L, Chen FH, Wang XT, You QD, Guo QL. HIF-1 alpha is critical for hypoxia-mediated maintenance of glioblastoma stem cells by activating Notch signaling pathway. Cell Death Differ. 2012; 19:284–94.
136. Tucci M, Stucci S, Silvestris F. Does cilengitide deserve another chance? Lancet Oncology. 2014; 15:E584–E5.
137. Battle MR, Goggi JL, Allen L, Barnett J, Morrison MS. Monitoring tumor response to antiangiogenic sunitinib therapy with 18F-fluciclatide, an 18F-labeled alphaVbeta3-integrin and alphaV beta5-integrin imaging agent. J Nucl Med. 2011; 52:424–30.
138. Lanzardo S, Conti L, Brioschi C, Bartolomeo MP, Arosio D, Belvisi L, Manzoni L, Maiocchi A, Maisano F, Forni G. A new optical imaging probe targeting alphaVbeta3 integrin in glioblastoma xenografts. Contrast Media Mol Imaging. 2011; 6:449–58.
139. Ma H, Hao P, Zhang L, Ma C, Yan P, Wang RF, Zhang CL. A new cyclic RGD peptide dimer for integrin alphavbeta3 imaging. Eur Rev Med Pharmacol Sci. 2016; 20:613–9.
140. Veeravagu A, Liu Z, Niu G, Chen K, Jia B, Cai W, Jin C, Hsu AR, Connolly AJ, Tse V, Wang F, Chen X. Integrin alphavbeta3-targeted radioimmunotherapy of glioblastoma multiforme. Clin Cancer Res. 2008; 14:7330–9.
141. Cirkel GA, Kerklaan BM, Vanhoutte F, Van der Aa A, Lorenzon G, Namour F, Pujuguet P, Darquenne S, de Vos FY, Snijders TJ, Voest EE, Schellens JH, Lolkema MP. A dose escalating phase I study of GLPG0187, a broad spectrum integrin receptor antagonist, in adult patients with progressive high-grade glioma and other advanced solid malignancies. Invest New Drugs. 2016; 34:184–92.
142. Reardon DA, Neyns B, Weller M, Tonn JC, Nabors LB, Stupp R. Cilengitide: an RGD pentapeptide alphanubeta3 and alphanubeta5 integrin inhibitor in development for glioblastoma and other malignancies. Future Oncol. 2011; 7:339–54.
143. Hersey P, Sosman J, O’Day S, Richards J, Bedikian A, Gonzalez R, Sharfman W, Weber R, Logan T, Kirkwood JM. A phase II, randomized, open-label study evaluating the antitumor activity of MEDI-522, a humanized monoclonal antibody directed against the human alpha v beta 3 (avb3) integrin, ± dacarbazine (DTIC) in patients with metastatic melanoma (MM) ASCO Annual Meeting. 2005; 23:Abstract 16S.
144. Almokadem S, Belani CP. Volociximab in cancer. Expert Opin Biol Ther. 2012; 12:251–7.
145. Mateo J, Berlin J, de Bono JS, Cohen RB, Keedy V, Mugundu G, Zhang L, Abbattista A, Davis C, Gallo Stampino C, Borghaei H. A first-in-human study of the anti-alpha5beta1 integrin monoclonal antibody PF-04605412 administered intravenously to patients with advanced solid tumors. Cancer Chemother Pharmacol. 2014; 74:1039–46.
146. Donate F, Parry GC, Shaked Y, Hensley H, Guan X, Beck I, Tel-Tsur Z, Plunkett ML, Manuia M, Shaw DE, Kerbel RS, Mazar AP. Pharmacology of the novel antiangiogenic peptide ATN-161 (Ac-PHSCN-NH2): observation of a U-shaped dose-response curve in several preclinical models of angiogenesis and tumor growth. Clin Cancer Res. 2008; 14:2137–44.
147. O’Day S, Pavlick A, Loquai C, Lawson D, Gutzmer R, Richards J, Schadendorf D, Thompson JA, Gonzalez R, Trefzer U, Mohr P, Ottensmeier C, Chao D, et al. A randomised, phase II study of intetumumab, an anti-alphav-integrin mAb, alone and with dacarbazine in stage IV melanoma. Br J Cancer. 2011; 105:346–52.
148. Wirth M, Heidenreich A, Gschwend JE, Gil T, Zastrow S, Laniado M, Gerloff J, Zuhlsdorf M, Mordenti G, Uhl W, Lannert H. A multicenter phase 1 study of EMD 525797 (DI17E6), a novel humanized monoclonal antibody targeting alphav integrins, in progressive castration-resistant prostate cancer with bone metastases after chemotherapy. Eur Urol. 2014; 65:897–904.
149. Semba T, Funahashi Y, Ono N, Yamamoto Y, Sugi NH, Asada M, Yoshimatsu K, Wakabayashi T. An angiogenesis inhibitor E7820 shows broad-spectrum tumor growth inhibition in a xenograft model: possible value of integrin alpha2 on platelets as a biological marker. Clin Cancer Res. 2004; 10:1430–8.
150. Carbonell WS, DeLay M, Jahangiri A, Park CC, Aghi MK. beta1 integrin targeting potentiates antiangiogenic therapy and inhibits the growth of bevacizumab-resistant glioblastoma. Cancer Res. 2013; 73:3145–54.
151. Nabors LB, Mikkelsen T, Rosenfeld SS, Hochberg F, Akella NS, Fisher JD, Cloud GA, Zhang Y, Carson K, Wittemer SM, Colevas AD, Grossman SA. Phase I and correlative biology study of cilengitide in patients with recurrent malignant glioma. J Clin Oncol. 2007; 25:1651–7.
152. MacDonald TJ, Stewart CF, Kocak M, Goldman S, Ellenbogen RG, Phillips P, Lafond D, Poussaint TY, Kieran MW, Boyett JM, Kun LE. Phase I clinical trial of cilengitide in children with refractory brain tumors: Pediatric Brain Tumor Consortium Study PBTC-012. J Clin Oncol. 2008; 26:919–24.
153. Gerstner ER, Ye X, Duda DG, Levine MA, Mikkelsen T, Kaley TJ, Olson JJ, Nabors BL, Ahluwalia MS, Wen PY, Jain RK, Batchelor TT, Grossman S. A phase I study of cediranib in combination with cilengitide in patients with recurrent glioblastoma. Neuro Oncol. 2015; 17:1386–92.
154. Reardon DA, Fink KL, Mikkelsen T, Cloughesy TF, O’Neill A, Plotkin S, Glantz M, Ravin P, Raizer JJ, Rich KM, Schiff D, Shapiro WR, Burdette-Radoux S, et al. Randomized phase II study of cilengitide, an integrin-targeting arginine-glycine-aspartic acid peptide, in recurrent glioblastoma multiforme. J Clin Oncol. 2008; 26:5610–7.
155. Stupp R, Hegi ME, Neyns B, Goldbrunner R, Schlegel U, Clement PM, Grabenbauer GG, Ochsenbein AF, Simon M, Dietrich PY, Pietsch T, Hicking C, Tonn JC, et al. Phase I/IIa study of cilengitide and temozolomide with concomitant radiotherapy followed by cilengitide and temozolomide maintenance therapy in patients with newly diagnosed glioblastoma. J Clin Oncol. 2010; 28:2712–8.
156. Gilbert MR, Kuhn J, Lamborn KR, Lieberman F, Wen PY, Mehta M, Cloughesy T, Lassman AB, Deangelis LM, Chang S, Prados M. Cilengitide in patients with recurrent glioblastoma: the results of NABTC 03–02, a phase II trial with measures of treatment delivery. J Neurooncol. 2012; 106:147–53.
157. Nabors LB, Mikkelsen T, Hegi ME, Ye X, Batchelor T, Lesser G, Peereboom D, Rosenfeld MR, Olsen J, Brem S, Fisher JD, Grossman SA. A safety run-in and randomized phase 2 study of cilengitide combined with chemoradiation for newly diagnosed glioblastoma (NABTT 0306). Cancer. 2012; 118:5601–7.
158. MacDonald TJ, Vezina G, Stewart CF, Turner D, Pierson CR, Chen L, Pollack IF, Gajjar A, Kieran MW. Phase II study of cilengitide in the treatment of refractory or relapsed high-grade gliomas in children: a report from the Children’s Oncology Group. Neuro Oncol. 2013; 15:1438–44.
159. Nabors LB, Fink KL, Mikkelsen T, Grujicic D, Tarnawski R, Nam do H, Mazurkiewicz M, Salacz M, Ashby L, Zagonel V, Depenni R, Perry JR, Hicking C, et al. Two cilengitide regimens in combination with standard treatment for patients with newly diagnosed glioblastoma and unmethylated MGMT gene promoter: results of the open-label, controlled, randomized phase II CORE study. Neuro Oncol. 2015; 17:708–17.
160. Khasraw M, Lee A, McCowatt S, Kerestes Z, Buyse ME, Back M, Kichenadasse G, Ackland S, Wheeler H. Cilengitide with metronomic temozolomide, procarbazine, and standard radiotherapy in patients with glioblastoma and unmethylated MGMT gene promoter in ExCentric, an open-label phase II trial. J Neurooncol. 2016; 128:163–71.