
INTRODUCTION
Telomerase reverse transcriptase (TERT) encodes the catalytic subunit of telomerase, a ribonucleoprotein responsible for maintaining telomere length of chromosomes which play an integral role in cell immortality. Using linkage analysis and high-throughput sequencing, Horn et al. [1] reported somatic mutations in 74% of metastatic melanoma human cell lines, 85% of metastatic melanoma tumor tissues and 33% of primary melanomas. These mutations are the result of a cytidine to thymidine transition in the promoter of the TERT gene, at chromosome 5, 1,295,228 C>T and 1,295,250 C>T, hereafter termed C228T and C250T. These mutations create a putative consensus ETS (E26 transformation-specific) /ternary complex factor binding motif (GGAA/T), which is associated with an increase in TERT expression [1, 2]. The presence of these mutations in cutaneous melanoma is associated with fast growing melanomas [3] and poor prognosis [4]. The co-existence of TERT promoter mutations with BRAF or NRAS mutations (in 55% of cases) is associated with poor disease-free and melanoma-specific survival [5]. TERT promoter mutations occur frequently in a number of other cancers: 80–90% of glioblastoma multiforme, 60% of hepatocellular carcinoma, 60% of bladder cancer, 70% of basal cell carcinoma, 50% of cutaneous squamous cell carcinoma and up to 30% of thyroid cancers [6-11] and are associated with aggressive disease in thyroid carcinoma [12], glioblastoma [13], neuroblastoma [14] and renal cell carcinoma [15]. Therefore, it is of significant clinical benefit to develop a non-invasive and sensitive test that determines the TERT promoter mutation status in cancer patients.
Molecular profiling of tumors to aid cancer prognosis and to identify actionable therapeutic targets has become routine practice in clinical oncology. Whilst tumor tissue samples are typically used for mutation analysis, access to the tumor for biopsy, and the quality and quantity of the sample may hinder detection, particularly when methods with limited sensitivity are employed. Commonly used methods include Sanger sequencing, melting curve analysis and pyrosequencing which have limits of sensitivity of 15%-20%, 10% and 5% respectively [16]. More recently, tumor related aberrations have been determined in plasma cell free DNA (cfDNA) [17-22]. This is referred to as “liquid biopsy”, a relatively non-invasive test that can be performed regularly and provides information from the sum of all tumors at any one time point. It is, therefore, a valuable biomarker for monitoring disease progression and response to therapy [19, 23].
Whilst a variety of methods have been used to detect mutations from circulating tumor DNA (ctDNA), Hindson et al., have shown droplet digital PCR (ddPCR) to be a highly sensitive platform, enabling absolute quantitation of mutant BRAF down to 0.001% allelic fraction [24]. Various studies have since shown the utility of testing mutant BRAF in plasma of melanoma patients using ddPCR [18, 23, 25-27]. In particular, our laboratory has demonstrated that ctDNA analysis allows tracking of patient response to therapy and resistance acquisition [23]. Given the high prevalence of the TERT promoter mutations C228T and C250T in cutaneous melanoma [5, 28], their addition to existing tests for detection of mutant BRAF and NRAS will allow monitoring of most melanoma patients using ddPCR. Furthermore, it has been shown that concurrence of mutations in the TERT promoter with BRAF or NRAS mutations predispose patients to fast growing and aggressive disease, thus detection of multiple mutations including mutant TERT could serve as a prognostic marker.
We report here on the development of a ddPCR probe based assay to simultaneously detect the TERT promoter mutations C250T and C228T. One probe binds the wild-type sequence overlapping position C228, while a second probe binds the mutant sequence resulting from C228T or C250T mutations, as both mutations reconstitute the putative ETS binding site (Figure 1). First, we tested the concordance of this assay for the detection of TERT promoter mutations in 39 melanoma cell lines relative to Sanger sequencing, and in 22 plasma samples relative to patient matched tumor tissue. We also determined the sensitivity and specificity of this assay for the detection of TERT promoter mutations using plasma derived cfDNA from 56 melanoma patients and 56 healthy controls.
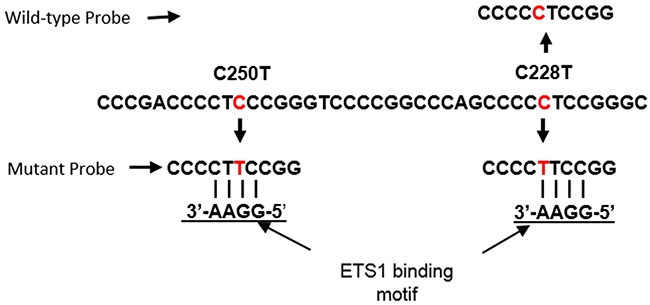
Figure 1: Location of ddPCR assay probes relative to ETS1 binding motifs generated by the C228T and C250T TERT promoter mutations. Probes for the identification of wild-type and mutant sequences are indicated. Both mutant sites are detected by the same probe.
RESULTS
The designed primer sets were tested for amplification of the genomic region of interest by end-point PCR. Amplification conditions were optimized by testing a range of annealing temperatures (55-61ºC). As shown in Figure 2A, the primers failed to amplify the required fragment in the absence of Q-solution (Qiagen). Optimal amplification was achieved in the presence of Q-solution between 61-64ºC (Figure 2B). The PCR fragment obtained was subjected to Sanger sequencing to confirm its specificity.
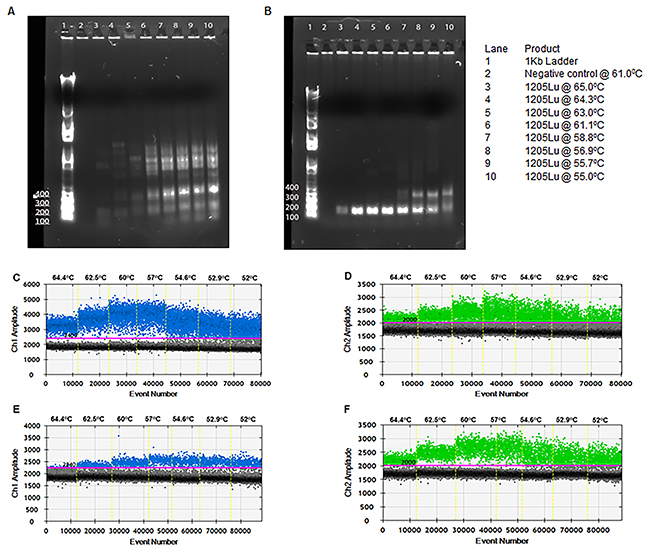
Figure 2: Optimization of ddPCR for detection of TERT promoter mutations. PCR fragments from cell line 1205Lu amplified at varying temperatures without (A) and with (B) “Q solution”. gDNA of cell lines 1205Lu-C228T (C and D) and UACC62-C250T (E and F) were used as template for the TERT ddPCR at varying annealing temperatures. FAM signal from mutant probe binding to C228T (C) or C250T (E). HEX signal from binding of wild-type probe (D and F).
Next, droplet digital PCRs were performed at a gradient of annealing temperatures from 52°C to 65°C for the detection of the C228T mutation in gDNA from 1205Lu cells (Figure 2C and 2D) and the C250T mutation in gDNA from UACC62 cells (Figure 2E and 2F). Optimal droplet segregation was observed at 57°C. Hereafter all ddPCR assays were performed with an annealing/extension temperature of 57°C.
To evaluate the quantitative linearity and the limit of detection (LOD) of the ddPCR assay, serial dilutions of mutant gDNA from cell lines 1205Lu (C228T mutant) and UACC62 (C250T mutant) were mixed in a background of wild-type human genomic DNA to achieve a final concentration of gDNA of 20 ng/μL (Figure 3), with each dilution tested in 8 replicates. At 0% mutant DNA, we identified that a maximum of 2 false positive droplets were observed in some of the 8 replicates, with an average of 0.068 ± 0.049%. Therefore, the lower LOD was defined at 0.17%, the percentage false positives detectable at two standard deviations over mean background [29].
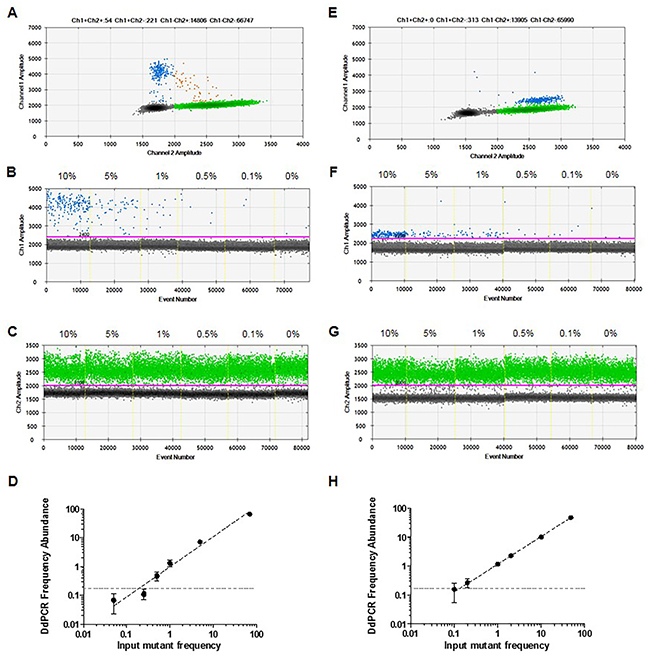
Figure 3: Detection of TERT promoter mutations in the presence of homologous wild-type DNA. Serial dilutions of DNA from mutant cell lines 1205Lu – C228T (A-D) and UACC62 – C250T (E-H) were prepared in a constant background of wild-type human genomic DNA. 2D plots of ddPCR read out at 10% of mutant DNA (A and E). 1D plots indicating mutant (B and F) and wild-type (C and G) DNA detection. Analytical sensitivity (LOD) of the assay (D and H). Obtained frequency abundances and standard deviations were plotted versus expected mutant frequencies based on input. The LOD, defined as 2 SD over the mean frequency abundance obtained at 0% when only wild-type DNA was used as input, was indicated as dashed lines in both graphs.
To validate the assay, we tested 39 cell lines with known TERT promoter mutant or wild-type status (Table 1). We confirmed detection of the C228T and/or the C250T TERT promoter mutation in only those cell lines identified as positive for these two mutations, while those previously identified as wild-type showed no positivity for TERT DNA mutations by ddPCR. Cell lines that harbored an alternative TERT mutation other than C228T or C250T showed as wild-type in our assay. In addition, the C250T mutation was not detected in cell line C021, due to the presence of a C253T single nucleotide polymorphism in the probe binding site (Supplementary Figure 1). Simultaneous C250T and C253T mutations have been reported in 2% of melanoma cells lines [1].
Table 1: Validation of C228T and C250T TERT promoter mutation detection in melanoma cell lines
Cell line |
Sanger sequencing |
ddPCR |
|---|---|---|
C024 |
wt |
wt |
C055 |
wt |
wt |
C092 |
wt |
wt |
C096 |
wt |
wt |
HGA |
wt |
wt |
C022 |
C228T |
C228T |
C037 |
C228T |
C228T |
C058 |
C228T |
C228T |
D41 |
C228T |
C228T |
MM409 |
C228T |
C228T |
D22 |
C228T |
C228T |
MM473 |
C228T |
C228T |
A06 |
C228Ta |
C228Ta |
C076 |
C228Ta |
C228Ta |
MM455 |
C228Ta |
C228Ta |
1205Lu |
C228Ta |
C228Ta |
A15 |
C250T |
C250T |
A14 |
C250T |
C250T |
C002 |
C250T |
C250T |
MM537 |
C250T |
C250T |
SKMEL13 |
C250T |
C250T |
MM386 |
C250T |
C250T |
D01 |
C250T |
C250T |
MM229 |
C250Ta |
C250Ta |
MM253 |
C250Ta |
C250Ta |
MM266 |
C250Ta |
C250Ta |
C001 |
C250Ta |
C250Ta |
C045 |
C250Ta |
C250Ta |
D40 |
C250Ta |
C250Ta |
UACC62 |
C250Ta |
C250Ta |
MM396 |
C227T/C228T |
wt |
A07 |
C227T/C228T |
wt |
C054 |
C227T/C228T |
wt |
C062 |
C227T/C228T |
wt |
C057 |
C241T/C242T |
wt |
C108 |
C241T/C242T |
wt |
D28 |
C241T/C242T |
wt |
SKMEL5 |
C241T/C242T |
wt |
C021 |
C250Tb |
wt |
aHomozygous
bC021 carried an additional C253T polymorphism.
Tumor tissue samples from 22 stage IV (AJCC) metastatic melanoma patients were tested for C228T and C250T TERT promoter mutations by ddPCR using the TERT assay (Table 2). As reported in the literature [4, 28, 30], most tumor tissues tested harbored at least one of these mutations (68%, n=15); 11 harbored the C228T mutation and 4 harbored the C250T mutation. No tissue samples were found to contain both TERT promoter mutations.
Table 2: Detection of TERT promoter mutations in ctDNA and paired tumor tissue
Tumor Tissue |
|||
Plasma ctDNA |
+ |
− |
Total |
+ |
8 |
0 |
8 |
− |
7 |
7 |
14 |
Total |
15 |
7 |
22 |
Plasma derived cfDNA from these 22 patients were also tested for TERT promoter mutations. These plasma samples were collected from patients with active metastatic disease prior to any systemic therapeutic intervention. Overall, the concordance rate between tumor tissue and plasma testing was 68% (15/22). No patient was positive for a TERT promoter mutation in plasma and negative in its corresponding tumor tissue (100% specificity). Of 15 plasmas from patients with confirmed TERT promoter positive tumors, 8 were identified as positive for the same mutation, whereas 7 cases were positive in the tissue but negative in the plasma sample (Table 2). Thus, the sensitivity of our TERT C228T/C250T mutation detection in plasma was estimated as 53% (95% CI 27%-79%). In a cox regression analysis, patients with detectable ctDNA at baseline (n=8) had a significantly shorter progression free survival (PFS) compared to patients that had no detectable ctDNA (n=7) (p=0.028, Hazard ratio: 4.48 (CI, 1.18-17.06) (Figure 4A).
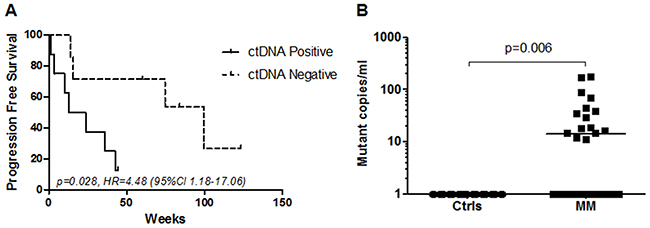
Figure 4: Detection of TERT promoter mutations in plasma. (A) Kaplan-Meier plots of PFS probabilities of patients with detectable (n=8) and undetectable (n=7) ctDNA levels at baseline. Cox regression p-value, Hazard ratio (HR) and confidence interval (CI) are indicated. (B) Copies of mutant DNA per mL of plasma were significantly higher in metastatic melanoma patients (MM) (N=56) compared to healthy controls (Ctrls) (N=56). P=0.006, Mann-Whitney U-test.
To further demonstrate the detection rate of TERT ctDNA in metastatic melanoma we tested 56 plasma samples from randomly selected stage IV (AJCC) melanoma patients (mean age 65 years, ranging from 35 to 85 years) with known BRAF but unknown TERT mutational status and compared this to 56 plasma samples from healthy individuals (mean age 51 years, ranging from 24 to 81 years). The TERT ddPCR assay detected a statistically significant difference in the copies of mutant TERT ctDNA in plasma from metastatic melanoma patients relative to those from healthy controls (p=0.006, Figure 4). We found TERT mutant DNA in 11 of 38 BRAF wild-type and in 4 of the 18 BRAF V600E/K patients. The number of TERT promoter copies per mL of plasma detected in the melanoma patient cohort varied from 11.2 to 176 copies per mL (Figure 4). No TERT promoter mutant DNA was detected in any of the 56 healthy control plasmas. Based on these results the assay specificity was estimated as 100% (95% CI 94%-100%).
DISCUSSION
Here we describe and validate a method to detect the two most common TERT promoter mutations found in melanoma tumors using ddPCR. TERT promoter mutations occur in melanoma as frequently as [4], or more frequently [5] than BRAF mutations, and yet mainly BRAF mutant specific cfDNA is being used to monitor melanoma patients for response to therapy and disease progression [26]. The inclusion of TERT promoter mutations within ctDNA for monitoring would increase the number of patients for whom ctDNA could be used to determine disease status, particularly amongst BRAF and NRAS wild-type melanoma patients. This will enable large studies on the clinical utility of ctDNA monitoring to provide evidence of the efficacy of this marker for determining disease progression, to inform cessation of ineffective therapies [23, 27] and to guide alternative therapy.
Our assay allowed for detection of mutant TERT in biologically relevant samples, such as FFPE tumor DNA and plasma of metastatic melanoma patients at high specificity. Using cell line derived DNA, we optimized the assay to detect as little as 0.17% mutant TERT DNA in dilutions of wild-type DNA. This is significantly lower than limits of detection reported for other mutation detection platforms such as allele specific PCR at 1% [31] and pyrosequencing at 5%, melting curve analysis at 10% and Sanger sequencing at 20% [16]. While we and others have shown ddPCR to detect BRAF mutant fraction as low as 0.001% [24, 32], we were unable to achieve this sensitivity with the TERT assay developed here, possibly due to the highly GC rich area of the promoter region of this gene, resulting in background signal and limited segregation of positive and negative droplets. In fact, during the development of this assay, multiple primers, probes and amplification conditions were tested without success. The conditions detailed here, including the addition of LNA at the specific nucleotides and the use of Q-solution in the amplification mix, were indispensable for successful amplification.
We validated the assay in terms of accuracy and reliability by showing 97.4% concordance with the genotype of 39 melanoma cell lines. Of the cell lines analyzed that harbored either a C228T or C250T mutation, 14 were heterozygous and 9 homozygous. A major limitation of our assay is that it cannot detect other TERT promoter mutations and it can be affected by SNPs within the probe binding sites. This was apparent by the results obtained from 9 cell lines with known TERT promoter dinucleotide mutations C227T/C228T and C241T/C242T, which have been reported to exist in 5.2% and 10.4% of primary melanomas respectively [1]. Similarly, a negative result was reported for cell line CO12 which harbors a C253T SNP on the probe binding site. Further development of ddPCR assays to detect these other TERT promoter mutations [5] would ensure that a maximum number of patients could be monitored. In addition and given that SNPs in this region can also affect patient prognosis [5], germline sequence analysis should be performed complementary to the analysis of TERT promoter somatic mutations.
It is notable that all patients with TERT promoter mutations in plasma had corresponding mutations in matched tumor tissue and as such no false positive plasma samples were detected. High concordance between mutational profiles in plasma ctDNA and matched tumor tissue have been reported in several studies from patients with melanoma [18, 25, 26], breast cancer [20, 33, 34], non-small cell lung cancer [35, 36] and colorectal cancer [17, 20, 37]. In our study, 7 patients with TERT promoter positive tumors had no detectable TERT promoter mutations in matched plasma samples. This is similar to the findings by Lee et al, who detected ctDNA in 53% of patients prior to treatment initiation [38]. The lack of detectable ctDNA in a subset of patients may be explained by the pathophysiology of the tumor or its metastasis, as ctDNA concentration has been correlated with tumor size [38-40], metastatic spread or disease burden [25, 38, 41], tumor vascularization [42] and site of metastasis [20]. A retrospective analysis of PFS in this group of patients revealed a significant difference between patients with negative and positive ctDNA results. This further supports previous findings that low or undetectable level of ctDNA is a predictor of long term treatment benefit [18, 23, 25, 26, 38].
Previous studies have reported detection rates for BRAF V600E mutations in plasma of metastatic patients at 76 to 84.3% [25, 26] and for BRAF V600K at 81 to 89% [18, 26]. In other cancers, Bettegowda et al. [20] identified mutant ctDNA in 75% of patients with a variety of cancers including ovarian, breast, bladder, gastroesophageal and colorectal cancers. Considering our detection rates of TERT promoter mutations in ctDNA are lower (53%) than these reports, it would be necessary for this investigation to be conducted in a larger cohort controlling for tumor burden, metastatic sites and mutation variety. Nevertheless, our TERT promoter mutation assay allowed ctDNA detection in 11 of 38 BRAF wild-type tumors. Thus, our assay may facilitate ctDNA monitoring on BRAF wild-type cases, most of which will receive immunotherapy as a first line of treatment.
Nagore and colleagues [5] have shown that melanoma patients harboring these specific TERT promoter mutations, in combination with BRAF/NRAS mutations within their tumor tissue, have a significantly shorter disease free survival than patients without this combination. In fact, Li et al., have shown that TERT promoter mutations are key downstream targets of the RAS-ERK pathway for malignant progression of BRAF mutant melanomas [43]. Furthermore, Akincilar et al. [44] have shown that TERT transcription is driven by mediation of long-range chromatin interaction and enrichment of active histone marks through the recruitment of GABPA to mutant TERT promoters, specifically C228T and C250T. These authors have consequently suggested that inhibitors could be designed to hinder TERT transcription in cancer cells with these mutations. As such, routine genetic testing of melanoma patients for TERT promoter mutations in addition to mutant BRAF and NRAS would be clinically beneficial.
TERT promoter mutations have been identified in numerous other cancers such as thyroid, bladder, hepatocellular cancer and malignant glioblastoma [6-8]. Consequently, the assay described here may allow ctDNA monitoring in multiple other malignancies. However, the assay would require validation for each of these cancers.
In conclusion, we report on the development of a ddPCR assay for the detection of two common TERT promoter mutations in cell lines, tumor tissue and ctDNA. Our results suggest that the TERT ddPCR assay could prove useful as a companion diagnostic to predict treatment benefit and to monitor response in melanoma patients and could be extended to other malignancies.
MATERIALS AND METHODS
Ethics
This study was approved by the Human Ethics Committees at Edith Cowan University (No. 11543) and Sir Charles Gairdner Hospital (No.2013-246).
Genomic DNA extraction
Genomic DNA (gDNA) with known TERT promoter mutations was obtained from melanoma cell lines 1205Lu (Wistar Institute) and UACC62 (National Cancer Institute) to be used as positive controls. In addition, gDNA was extracted from 39 melanoma cell lines from the QIMR Berghofer Medical Research Institute [45]. Wild-type gDNA was obtained from the white blood cell pellets collected from 4 mL whole blood from one healthy control. DNA was isolated using the QIAamp DNA Mini Kit (Qiagen, Australia) as per the manufacturer’s instructions. gDNA was eluted in AE buffer (Qiagen) and stored at 4°C until further processing.
Plasma sample preparation
Blood samples were collected from American Joint Committee on Cancer (AJCC) stage IV melanoma patients, prior to initiation of any systemic therapy, into EDTA vacutainer tubes and stored at 4°C. Plasma was separated within 24 hours by centrifugation at 1600 g for 10 minutes, followed by a second centrifugation at 2000 g for 10 minutes, then stored at -80°C until extraction.
DNA extraction from plasma
cfDNA was isolated from 5 mL of plasma from healthy donors and AJCC stage IV metastatic melanoma patients using the QIAamp Circulating Nucleic Acid Kit (Qiagen) as per the manufacturer’s instructions. cfDNA was eluted in 40 μl AVE buffer (Qiagen) and stored at -80°C until ctDNA quantification.
DNA extraction from FFPE tissue
Following review and macrodissection by an experienced pathologist, genomic DNA (gDNA) was extracted from 10 x 5μm unstained sections of FFPE tissue using the QIAamp DNA mini kit (Qiagen) as per the manufacturer’s instructions. Only FFPE tissues stored at room temperature, for less than 7 years were used. The DNA concentration and purity was determined using the NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE) and Qubit 2.0 Fluorometer (Life Technologies, USA) instruments.
PCR
The following primers were used to amplify a 163bp product incorporating both hotspot mutations (C228T and C250T) in the TERT promoter region: 5’- AGCGCTGCCTGAAACTCG -3’ (forward) and 5’- CCTGCCCCTTCACCTTCCAG -3’ (reverse). Primers were synthesized by GeneWorks (Thebarton, SA, Australia). For optimization of the PCR amplification of TERT promoter mutations, we first performed end point PCRs containing, 1 x ddPCR supermix (Bio-Rad), 900 nM of each primer and 50 ng of template gDNA, with and without 1 x Q solution (Qiagen). Amplifications were performed using the following cycling conditions: 1 cycle of 95°C for 15 minutes, 40 cycles of 95°C for 30 seconds and a range of temperatures from 55°C to 65°C for 30 seconds, followed by 68°C for 30 seconds and 1 cycle of 68°C for 10 minutes. PCR products of 163bp were detected by gel electrophoresis on a 1% agarose gel in Tris-acetate-EDTA (TAE) buffer containing SYBR® Safe DNA Gel Stain (Life Technologies).
Droplet digital PCR
A probe was designed to detect both C228T and C250T mutation as both mutations result in the same sequencing string (Figure 1). Due to the short size of the probe, Locked Nucleic Acid (LNA) bases were introduced on the bases indicated with a “+” (TERT Mut:/56-FAM/CCC+C+T+T+CCGG/3IABkFQ/). A second probe was designed to recognize the C228 loci, also containing LNA bases, (TERT WT, /5HEX/CCCC+C+T+CCGG/3IABkFQ/). Probes were custom synthesized by Integrated DNA Technologies (IDT). Amplifications were performed in a 20 μL reaction containing 1 x ddPCR Supermix for Probes (No dUTP, Bio-Rad), 1x Q solution (Qiagen), 250 nM of each probe and 900 nM of each primer plus template.
Droplets were generated using the Automatic Droplet generator QX200 AutoDG (Bio-Rad). Amplifications were performed using the following cycling conditions: 1 cycle of 95°C (2.5C/s ramp) for 10 minutes, 40 cycles of 94°C (2.5C/s ramp) for 30 seconds and 57°C for 1 minute, followed by 1 cycle of 98°C (2.5C/s ramp) for 10 minutes. Annealing/extension temperature was optimized using temperature gradients from 52°C to 65°C. The sample was held at 4°C until further processing. Droplets were analyzed through a QX200 droplet reader (Bio-Rad). QuantaSoft analysis software (Bio-Rad) was used to acquire and analyze data.
To evaluate the LOD of our TERT ddPCR assay, gDNA from cell lines 1205Lu (C228T) or UACC62 (C250T) were serially diluted into normal human DNA obtained from white blood cells of healthy controls to achieve from 100% to 0% mutant alleles. Each dilution was tested in a series of eight repetitions all completed in one run.
Cell lines with known C228T and C250T TERT promoter mutations, as well as cell lines wild-type for both mutations (as determined by Sanger sequencing) were used to validate the assay. The reaction mix was prepared as above using 50 ng of gDNA as template.
For plasma ctDNA analysis, 5 μL of cfDNA (maximum template volume possible) was added per reaction irrespective of the cfDNA concentration. Each run included a non-template control, gDNA from a healthy control and gDNA from the cell lines containing the TERT mutations: 1205Lu (C228T) and UACC62 (C250T). Only samples with more than two positive droplets were considered positive. The number of mutated DNA copies per 20 μl reaction was extrapolated to calculate copies per mL using the following equation:
Copies/mL of plasma = C*EV/TV/PV.
PV = Volume of plasma used for cfDNA extraction (ml)
EV = Volume in which cfDNA was eluted (μl)
TV = Volume of cfDNA added to the PCR reaction (μl)
C = copies/20μl (data derived from QuantaSoft).
Statistical analysis
Sensitivity and specificity of the assay was calculated using a contingency table analyzed using a Fisher’s exact test. Comparison between ctDNA concentrations in patient and control samples were performed using the non-parametric Mann-Whitney U-test. A Cox proportional hazards regression analysis was performed to examine association of ctDNA detection with PFS. Statistical analyses were performed using Statistical Package for Social Sciences for Window version 22 (SPSS, Chicago, IL) and plotted using GraphPad Prism version 5.
Author contributions
All authors contributed to the work presented in this study and reviewed the final manuscript. ACM designed the study, performed experiments, analyzed data and wrote the manuscript. LC, MP, TG, CR performed experiments and analyzed data. BA analyzed samples and provided expert advice. MAK, TMM and MM recruited and clinically assessed patients and provided clinical information. ALP and NKH supplied material, genetic data and provided expert advice. MZ and ESG designed experiments, analyzed data, supervised the study and wrote the paper.
ACKNOWLEDGMENTS
The authors thank all the participants, patients and healthy volunteers, for their assistance with the study. We also would like to thank Ms Sarah Wickham for her assistance with data collection.
CONFLICTS OF INTEREST
No author has any conflicts of interest to declare.
GRANT SUPPORT
ACM has received financial support through an “Australian Government Research Training Program” scholarship and the Edith Cowan University “Inspiring Minds” scholarship. ESG is supported by a fellowship from the Cancer Research Trust. This study was funded by a Western Australia Cancer Council grant (1100249) and Perpetual/Ramaciotti Health Investment grant to ESG; National Health and Medical Research (NHMRC) grants (1046711 and 1119791) and Department of Health WA grant to MZ; and Spinnaker Health Research Foundation grant to MAK. NKH is supported by an uncoupled NHMRC research fellowship (1117663).
REFERENCES
1. Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K. TERT promoter mutations in familial and sporadic melanoma. Science. 2013; 339:959-61.
2. Kumar R, Heidenreich B, Hosen I, Rachakonda S, Hemminki K. Patterns of Telomerase reverse transcriptase (TERT) promoter mutations in melanoma and bladder cancer. Cancer Res. 2014; 74:S559.
3. Nagore E, Heidenreich B, Requena C, García-Casado Z, Martorell-Calatayud A, Pont-Sanjuan V, Jimenez-Sanchez AI, Kumar R. TERT promoter mutations associate with fast-growing melanoma. Pigment Cell Melanoma Res. 2016; 29:236–38.
4. Griewank KG, Murali R, Puig-Butille JA, Schilling B, Livingstone E, Potrony M, Carrera C, Schimming T, Möller I, Schwamborn M, Sucker A, Hillen U, Badenas C, et al. TERT promoter mutation status as an independent prognostic factor in cutaneous melanoma. J Natl Cancer Inst. 2014; 106:dju246.
5. Nagore E, Heidenreich B, Rachakonda S, Garcia-Casado Z, Requena C, Soriano V, Frank C, Traves V, Quecedo E, Sanjuan-Gimenez J, Hemminki K, Landi MT, Kumar R. TERT promoter mutations in melanoma survival. Int J Cancer. 2016; 139:75–84.
6. Liu T, Wang N, Cao J, Sofiadis A, Dinets A, Zedenius J, Larsson C, Xu D. The age-and shorter telomere-dependent TERT promoter mutation in follicular thyroid cell-derived carcinomas. Oncogene. 2014; 33:4978-84.
7. Liu X, Wu G, Shan Y, Hartmann C, Von Deimling A, Xing M. Highly prevalent TERT promoter mutations in bladder cancer and glioblastoma. Cell Cycle. 2013; 12:1637-8.
8. Borah S, Xi L, Zaug AJ, Powell NM, Dancik GM, Cohen SB, Costello JC, Theodorescu D, Cech TR. TERT promoter mutations and telomerase reactivation in urothelial cancer. Science. 2015; 347:1006-10.
9. Vinagre J, Almeida A, Pópulo H, Batista R, Lyra J, Pinto V, Coelho R, Celestino R, Prazeres H, Lima L, Melo M, da Rocha AG, Preto A, et al. Frequency of TERT promoter mutations in human cancers. Nat Commun. 2013; 4:2185.
10. Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA Jr, Friedman AH, Friedman H, Gallia GL, Giovanella BC, Grollman AP, He TC, He Y, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci USA. 2013; 110:6021–26.
11. Nault JC, Mallet M, Pilati C, Calderaro J, Bioulac-Sage P, Laurent C, Laurent A, Cherqui D, Balabaud C, Zucman-Rossi J. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat Commun. 2013; 4:2218.
12. Yin DT, Yu K, Lu RQ, Li X, Xu J, Lei M, Li H, Wang Y, Liu Z. Clinicopathological significance of TERT promoter mutation in papillary thyroid carcinomas: a systematic review and meta-analysis. Clin Endocrinol (Oxf). 2016; 85:299–305.
13. Huse JT. TERT promoter mutation designates biologically aggressive primary glioblastoma. Neuro-oncol. 2015; 17:5–6.
14. Simon M, Hosen I, Gousias K, Rachakonda S, Heidenreich B, Gessi M, Schramm J, Hemminki K, Waha A, Kumar R. TERT promoter mutations: a novel independent prognostic factor in primary glioblastomas. Neuro-oncol. 2015; 17:45-52.
15. Wang K, Liu T, Liu L, Liu J, Liu C, Wang C, Ge N, Ren H, Yan K, Hu S, Björkholm M, Fan Y, Xu D. TERT promoter mutations in renal cell carcinomas and upper tract urothelial carcinomas. Oncotarget. 2014; 5:1829–36. https://doi.org/10.18632/oncotarget.1829.
16. Tsiatis AC, Norris-Kirby A, Rich RG, Hafez MJ, Gocke CD, Eshleman JR, Murphy KM. Comparison of Sanger sequencing, pyrosequencing, and melting curve analysis for the detection of KRAS mutations: diagnostic and clinical implications. J Mol Diagn. 2010; 12:425-32.
17. Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, Thornton K, Agrawal N, Sokoll L, Szabo SA, Kinzler KW, Vogelstein B, Diaz LA Jr. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008; 14:985–90.
18. Ascierto PA, Minor D, Ribas A, Lebbe C, O’Hagan A, Arya N, Guckert M, Schadendorf D, Kefford RF, Grob JJ, Hamid O, Amaravadi R, Simeone E, et al. Phase II trial (BREAK-2) of the BRAF inhibitor dabrafenib (GSK2118436) in patients with metastatic melanoma. J Clin Oncol. 2013; 31:3205–11.
19. Heitzer E, Ulz P, Geigl JB. Circulating tumor DNA as a liquid biopsy for cancer. Clin Chem. 2015; 61:112-23.
20. Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM, Antonarakis ES, Azad NS, Bardelli A, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014; 6:224ra24.
21. Tsao SC, Weiss J, Hudson C, Christophi C, Cebon J, Behren A, Dobrovic A. Monitoring response to therapy in melanoma by quantifying circulating tumour DNA with droplet digital PCR for BRAF and NRAS mutations. Sci Rep. 2015; 5:11198.
22. Chang GA, Tadepalli JS, Shao Y, Zhang Y, Weiss S, Robinson E, Spittle C, Furtado M, Shelton DN, Karlin-Neumann G, Pavlick A, Osman I, Polsky D. Sensitivity of plasma BRAFmutant and NRASmutant cell-free DNA assays to detect metastatic melanoma in patients with low RECIST scores and non-RECIST disease progression. Mol Oncol. 2016; 10:157–65.
23. Gray ES, Rizos H, Reid AL, Boyd SC, Pereira MR, Lo J, Tembe V, Freeman J, Lee JH, Scolyer RA, Siew K, Lomma C, Cooper A, et al. Circulating tumor DNA to monitor treatment response and detect acquired resistance in patients with metastatic melanoma. Oncotarget. 2015; 6:42008–18. https://doi.org/10.18632/oncotarget.5788.
24. Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, Bright IJ, Lucero MY, Hiddessen AL, Legler TC, Kitano TK, Hodel MR, Petersen JF, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011; 83:8604–10.
25. Sanmamed MF, Fernández-Landázuri S, Rodríguez C, Zárate R, Lozano MD, Zubiri L, Perez-Gracia JL, Martín-Algarra S, González A. Quantitative cell-free circulating BRAFV600E mutation analysis by use of droplet digital PCR in the follow-up of patients with melanoma being treated with BRAF inhibitors. Clin Chem. 2015; 61:297-304.
26. Santiago-Walker A, Gagnon R, Mazumdar J, Casey M, Long GV, Schadendorf D, Flaherty K, Kefford R, Hauschild A, Hwu P. Correlation of BRAF mutation status in circulating-free DNA and tumor and association with clinical outcome across four BRAFi and MEKi clinical trials. Clin Cancer Res. 2016; 22:567–74.
27. Girotti MR, Gremel G, Lee R, Galvani E, Rothwell D, Viros A, Mandal AK, Lim KH, Saturno G, Furney SJ, Baenke F, Pedersen M, Rogan J, et al. Application of Sequencing, Liquid Biopsies, and Patient-Derived Xenografts for Personalized Medicine in Melanoma. Cancer Discov. 2016; 6:286–99.
28. Heidenreich B, Nagore E, Rachakonda PS, Garcia-Casado Z, Requena C, Traves V, Becker J, Soufir N, Hemminki K, Kumar R. Telomerase reverse transcriptase promoter mutations in primary cutaneous melanoma. Nat Commun. 2014; 5:3401.
29. Armbruster DA, Pry T. Limit of blank, limit of detection and limit of quantitation. Clin Biochem Rev. 2008; 29:S49–52.
30. Hayward NK, Wilmott JS, Waddell N, Johansson PA, Field MA, Nones K, Patch AM, Kakavand H, Alexandrov LB, Burke H, Jakrot V, Kazakoff S, Holmes O, et al. Whole-genome landscapes of major melanoma subtypes. Nature. 2017; 545:175-80.
31. Gonzalez de Castro D, Angulo B, Gomez B, Mair D, Martinez R, Suarez-Gauthier A, Shieh F, Velez M, Brophy VH, Lawrence HJ, Lopez-Rios F. A comparison of three methods for detecting KRAS mutations in formalin-fixed colorectal cancer specimens. Br J Cancer. 2012; 107:345–51.
32. Reid AL, Freeman JB, Millward M, Ziman M, Gray ES. Detection of BRAF-V600E and V600K in melanoma circulating tumour cells by droplet digital PCR. Clin Biochem. 2015; 48:999–1002.
33. Higgins MJ, Jelovac D, Barnathan E, Blair B, Slater S, Powers P, Zorzi J, Jeter SC, Oliver GR, Fetting J, Emens L, Riley C, Stearns V, et al. Detection of tumor PIK3CA status in metastatic breast cancer using peripheral blood. Clin Cancer Res. 2012; 18:3462–69.
34. Dawson SJ, Tsui DW, Murtaza M, Biggs H, Rueda OM, Chin SF, Dunning MJ, Gale D, Forshew T, Mahler-Araujo B, Rajan S, Humphray S, Becq J, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013; 368:1199–209.
35. Newman AM, Bratman SV, To J, Wynne JF, Eclov NC, Modlin LA, Liu CL, Neal JW, Wakelee HA, Merritt RE, Shrager JB, Loo BW Jr, Alizadeh AA, Diehn M. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014; 20:548–54.
36. Narayan A, Carriero NJ, Gettinger SN, Kluytenaar J, Kozak KR, Yock TI, Muscato NE, Ugarelli P, Decker RH, Patel AA. Ultrasensitive measurement of hotspot mutations in tumor DNA in blood using error-suppressed multiplexed deep sequencing. Cancer Res. 2012; 72:3492-8.
37. Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M, Siravegna G, Bencardino K, Cercek A, Chen CT, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012; 486:532–6.
38. Lee JH, Long GV, Boyd S, Lo S, Menzies AM, Tembe V, Guminski A, Jakrot V, Scolyer RA, Mann GJ, Kefford RF, Carlino MS, Rizos H. Circulating tumour DNA predicts response to anti-PD1 antibodies in metastatic melanoma. Ann Oncol. 2017; 28:1130–36.
39. Thierry AR, Mouliere F, Gongora C, Ollier J, Robert B, Ychou M, Del Rio M, Molina F. Origin and quantification of circulating DNA in mice with human colorectal cancer xenografts. Nucleic Acids Res. 2010; 38:6159-75.
40. Kamat AA, Bischoff FZ, Dang D, Baldwin MF, Han LY, Lin YG, Merritt WM, Landen CN Jr, Lu C, Gershenson DM, Simpson JL, Sood AK. Circulating cell-free DNA: a novel biomarker for response to therapy in ovarian carcinoma. Cancer Biol Ther. 2006; 5:1369–74.
41. Parkinson CA, Gale D, Piskorz A, Hodgkin C, Biggs H, Addley H, Moyle P, Freeman S, Sala E, Hosking K, Gounaris I, Jimenez-Linan M, Earl HM, et al. Circulating tumour DNA carrying patient-specific mutations in TP53 as an early response biomarker in relapsed high grade serous ovarian cancer. J Clin Oncol. 2016; 34:e23040.
42. Thierry AR, El Messaoudi S, Gahan PB, Anker P, Stroun M. Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev. 2016; 35:347–76.
43. Li Y, Cheng HS, Chng WJ, Tergaonkar V. Activation of mutant TERT promoter by RAS-ERK signaling is a key step in malignant progression of BRAF-mutant human melanomas. Proc Natl Acad Sci USA. 2016; 113:14402–7.
44. Akıncılar SC, Khattar E, Boon PL, Unal B, Fullwood MJ, Tergaonkar V. Long-range chromatin interactions drive mutant TERT promoter activation. Cancer Discov. 2016; 6:1276–91.
45. O’Rourke MG, Johnson M, Lanagan C, See J, Yang J, Bell JR, Slater GJ, Kerr BM, Crowe B, Purdie DM, Elliott SL, Ellem KA, Schmidt CW. Durable complete clinical responses in a phase I/II trial using an autologous melanoma cell/dendritic cell vaccine. Cancer Immunol Immunother. 2003; 52:387–95.