
INTRODUCTION
Angiogenesis is the formation of new blood vessels that sprouting from the pre-existing primitive vessels and occurs during normal development, reproduction and tissue repairing [1]. However, excessive angiogenesis is related with some disease states, such as cancer [2], diabetic retinopathy [3], preeclampsia [4] and so on. The proliferation and migration of endothelial cells into the extracellular matrix towards the angiogenic factors play central roles in the process of angiogenesis. Targeting ECs to prevent dysfunction or inhibit pathological angiogenesis is potentially beneficial for a wide variety of diseases, and the current studies focus primarily on growth factors, receptors and signaling molecules. Of the growth factors involved in the angiogenesis, vascular endothelial growth factor (VEGF) is a key regulator. VEGF family currently includes six known members: VEGF-A through E and placental growth factor. VEGF-A is most prevalent and consists of five isoforms of which VEGF165 is the predominant molecular species [5]. VEGF induced by hypoxia [6], initiates the process of angiogenesis, via the activation of two receptor tyrosine kinases (RTKs), VEGF receptor-1 (VEGFR1) and VEGF receptor-2 (VEGFR-2)[7]. There are three core signaling pathways on the downstream of VEGFR2. They are the PI3K (phosphatidylinositol 3-kinase)–Akt (Protein Kinase B) pathway, which promotes cell survival and vascular permeability, the Raf-MEK (mitogen-activated or extracellular signal-regulated protein kinase kinase)-MAPK (mitogen-activated protein kinase) pathway, which activates cell proliferation, and the Src-FAK (focal adhesion kinase) pathway, which increases cell motility [8]. MicroRNAs also involved in the VEGF pathways. MiR-26a serveds as a critical regulator of VEGF-mediated angiogenesis through directly targeting Nogo-B receptor (NgBR) in endothelial cells [9]. MiR-29a lowered VEGF production and angiogenic activities in synovial fibroblasts through targeting the 3'-UTR of VEGF, thus miR-29a deficiency exacerbated synovitis pathogenesis in the end-stage OA knees [10]. Pleiotropic action of miR-21 induced the expression of pERK, HIF-1α, and VEGF in the high glucose condition by simultaneously targeting SPRY1, SMAD7, and PTEN in ARPE-19 cells [11].
Targeting endothelial cell metabolism to treat angiogenesis and endothelial cell dysfunction is becoming a novel therapeutic strategy. Despite their close proximity to oxygenated blood, ECs rely on glycolysis instead of oxidative metabolism for adenosine triphosphate (ATP) production [12]. The loss of the glycolytic enzyme PFKFB3 (fructose-2, 6-biphosphatase 3) in ECs impaired vessel formation [13]. In contrast to cardiac myocytes and other cell types, energy requirements in the endothelial cells are relatively low, so mitochondria in ECs play a vital role in signaling cellular responses to environmental cues [14], including biogenesis, dynamics, mitophagy, ROS production, calcium homeostasis, regulated cell death, and heme synthesis. But the effects of VEGF on mitochondria are still unknown.
To better understand the VEGF pathways will help us evaluate the mechanism of angiogenesis and provide new targets for therapy. In this present paper, we evaluated the effects of VEGF on the proliferation, migration, cell cycle and angiogenesis of endothelial cells in vitro. We also explored the mechanism of VEGF based on the mitochondria and expected to find the new targets for angiogenic vascular disorders.
RESULTS
VEGF significantly promoted the proliferation, migration, angiogenesis and cell cycle of endothelial cells
To investigate the effects of VEGF on endothelial cells, HUVECs were treated with VEGF at different concentrations in vitro. As measured by MTT assay, VEGF significantly promoted the proliferation of HUVECs at the concentration of 20 nM (Figure 1A). HUVECs also showed significant increased migration in response to 20 nM VEGF. The increased migration rate is nearly two fold of the control (Figure 1B). As expected, ECs showed a reduced of cells in G1-phase and an accumulation of cells in S and G2 phase, correlating with the increased proliferation in response to VEGF (Figure 1C). We used a matrigel angiogenesis assay to explore the effects of VEGF on the in vitro tube formation and found that VEGF alone promoted the ability of HUVECs to form tube-like structures in vitro (Figure 1D).
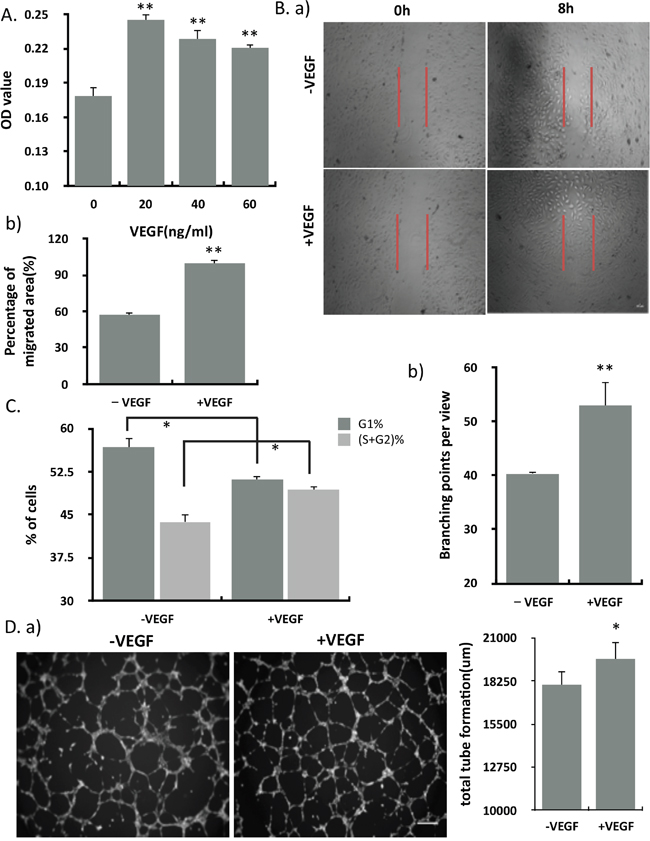
Figure 1: VEGF significantly promoted the proliferation, migration, angiogenesis and cell cycle of endothelial cells. (A) The proliferation of endothelial cells treated with different concentration of VEGF (n=12). (B) Migration of HUVEC in the absence and presence of VEGF (20 ng/ml) was assessed by wound healing assay. a) Representative micrographs of wound healing assays at 0 hrs and 8 hrs after creating a wound field. Scale bar, 200um. b) Quantitative assessment of percentage of cells migrating into the wound field (n=3). (C) PI staining followed by flow cytometry was used to assess cell cycle. Cell cycle phase length was calculated from the percentage of cells present in each cell cycle phase (n=3). (D) ECs were seeded on extracellular matrigel and exposed to VEGF treatments. a) Representative bright field micrographs of EC matrigel angiogenesis after 5h of VEGF treatment. b) Quantitative analysis of the number of branching points and total tube length (n=3). Scale bar, 100um. Results represented mean ± SEM of n independent experiments. **P<0.01, *P<0.05, 1-way ANOVA.
Mitochondrial function was enhanced in response to VEGF
ECs relay on glycolysis rather than on oxidative phosphorylation in the vessel sprouting. To explore which mechanism VEGF promotes ECs, we found that mitochondrial oxidative respiration was enhanced in ECs treated with VEGF (Figure 2A). Intracellular ATP levels increased (Figure 2B) and the ROS production decreased (Figure 2D) in VEGF-treated ECs. Moreover, the genes of ROS defence system, including catalase and glutathione peroxidase (GPX1), whose expression both increased in the VEGF stimulation (Figure 2C). Overall, VEGF protected endothelial cells by enhancing mitochondrial function.
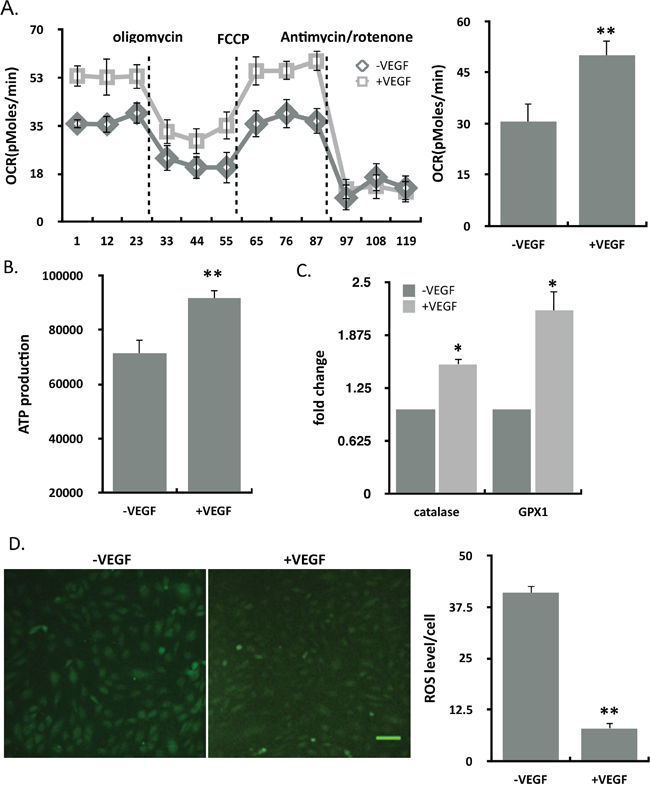
Figure 2: VEGF enhanced the mitochondrial function. (A) Oxygen consumption rate (OCR) in control and VEGF-treated ECs using by seahorse. Statistics of FCCP-coupled OCR was shown in the graph (n=10). (B) Intracellular ATP levels in control and VEGF-treated ECs (n=16). (C) Catalase and GPX1 mRNA were analysed by means of quantitative RT-PCR. Relative expression values of the VEGF-untreated cells were taken as 1.0 (n=4). (D) ROS staining was performed using DCFH-DA in ECs in the absence and presence of VEGF. Left: Representative images of ROS staining; right: Statistics of ROS fluorescence intensity in per cell (n=8). Scale bar, 100um. Results represented mean ± SEM of n independent experiments. **P<0.01, *P<0.05, 1-way ANOVA.
VEGF activated mTOR signaling pathway
mTOR is a central regulator of cellular growth and inhibition of mTOR by Rapamycin decreased oxygen consumption and mitochondrial capacity. To determine the molecular basis for VEGF effects on the mitochondria of ECs, we investigated the role of mTOR signaling in the VEGF stimulation. VEGF could activate the phosphorylation of the ribosomal S6 protein in ECs, a best-characterised mTOR substrate, while expression of total S6 had no change (Figure 3A). Rapamycin, the mTOR inhibitor, could inhibit the increasing of P-S6 (Figure 3B) and the proliferation induced by VEGF (Figure 3C), accompanying with an accumulation of cells in G1 and reduced S+G2 phase (Figure 3D). It was concluded that VEGF activated mTOR signaling pathway.
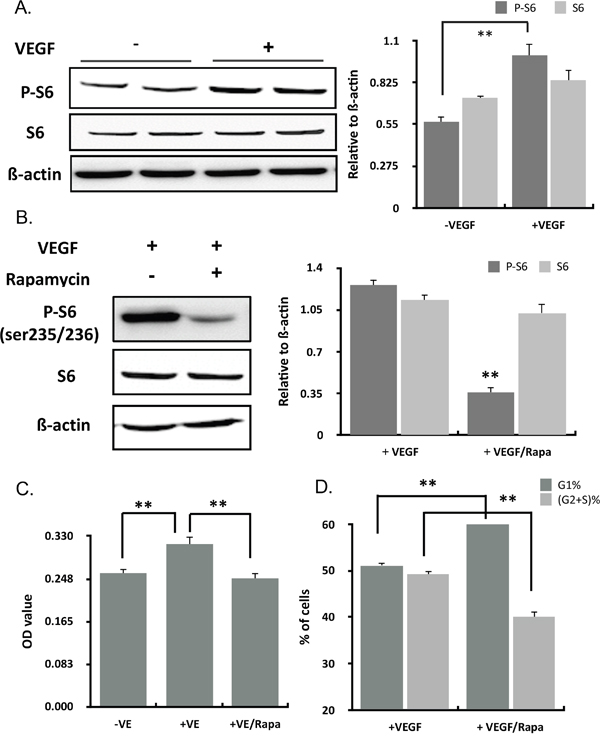
Figure 3: VEGF activated mTOR signaling pathway. (A) HUVECs cultured in M199 medium with 5%FBS were induced with VEGF for 24 hours. P-S6 and S6 were determined by western blots. (B) HUVECs cultured in M199 medium with 5%FBS were induced with VEGF and rapamycin for 24 hours. P-S6 and S6 were determined by western blots. (C) Cell proliferation in cultured HUVECs treated with VEGF and the inhibitor of mTOR signaling, rapamycin (50 nM) for 24 hours (n=12). (D) Cell cycle in cultured HUVECs treated with VEGF and an inhibitor of mTOR signaling, rapamycin (50 nM) for 24h determining by PI staining (n=3).
DISCUSSION
In the present study, we found that VEGF significant stimulated endothelial cells proliferation, migration, cell cycle and tube formation in vitro. We further investigated the mechanism and found that mitochondrial functions were enhanced induced by VEGF. Mitochondria in endothelial cells primarily function in signaling cellular responses to environmental cues not to produce ATP [14]. Mitochondria dysfunction was induced by many diseases in the endothelium, thus interventions that restore mitochondria function might be protective [15].
Our results showed that mitochondrial oxidative respiration and intracellular ATP levels increased in response to VEGF. The ROS production decreased, while the genes of ROS defence system, including catalase and glutathione peroxidase (GPX1), whose expression both increased in the VEGF stimulation. VEGF activated mTOR signaling pathway to promote the mitochondria functions which may become a potential regulator of mitochondria function. Rapamycin, the inhibitor of mTOR pathway could inhibit the proliferation and cell cycle induced by VEGF. mTOR signaling pathway regulates cell metabolism and energy homeostasis [16] and positively regulates PGC-1α expression [17], a coactivator that controls mitochondrial biogenesis. PGC-1α could enhance oxidative phosphorylation, mitochondrial biogenesis and the oxygen consumption rate in cancer cells [18]. ROS accumulation in cancer cells induces expression of PGC-1å/ß to promote detoxification through direct induction of superoxide dismutase 2 (SOD2), catalase and glutathione peroxidase [19].
It has been reported that one class of anti-angiogenic agents used in treating cancer directly inhibit endothelial cells function and induce endothelial cells death by targeting mitochondria. 10-(6’-plastoquinonyl) decyltriphenylphosphonium (SkQ1) is a new antioxidant with the highest mitochondria membrane penetrating ability and it could modulate the tumor angiogenesis [20]. Vitamin E analogues are first recognized for their selective toxicity to cancer cells. Vitamin E analogues a-TOS and a-TEA bind to Complex II in the inner-mitochondrial membrane, leading to displacement of ubiquinone and the generation of reactive oxygen species [21]. Paclitaxel (Taxol) is potent chemotherapeutic used in the treatment of ovarian, lung and breast malignancies. It could directly interact with isolated mitochondria, initiating the MPTP and inducing cytochrome C release [22]. Thus mitochondria are the focal point for a variety of angiogenic signals [23].
In this paper, in vivo experiments were lacking. To develop robust combinatory cancer hallmark–based gene signature sets (CSS sets) that more accurately predict prognosis [24], so we believe that it would be a good idea to take the RNA-seq or microarray to identify if the expression of the VEGF and the related mitochondrial genes could predict cancer recurrence. What’s more, the effects of other growth factors, such as VEGF-E and TGF-ß need to be explored in the further.
In summary, our findings provide the evidence that mitochondria play a dominant role in the stimulation of VEGF. The mitochondrial functions were enhanced. This is the first time to demonstrate the mechanism of VEGF signaling pathway based on the mitochondria. It is of significant importance and may provide new mechanistic insights into angiogenic vascular disorders.
MATERIALS AND METHODS
Cell culture and chemicals
Human umbilical vein endothelial cells (HUVECs) were purchased from PromoCell (Germany). HUVECs were grown in endothelial cells medium (Promocell) and maintained at 37°C and 5% CO2. Cells were used within five passages. The information of drugs were shown in the below table.
Name |
Corporation |
SKU |
|---|---|---|
DCFH-DA |
Sigma |
D6883 |
rapamycin |
Sigma |
V900930 |
MTT |
Sigma |
M2128 |
matrigel |
BD |
356230 |
CellTiter-Glo® Luminescent Cell Viability Assay |
Promega |
G7570 |
anti-Phospho-S6 Ribosomal Protein |
Cell Signaling |
#2211 |
anti-S6 Ribosomal Protein (5G10) |
Cell Signaling |
#2217 |
anti-ß-actin |
Beijing TDY Biotech LTD |
#M009 |
HRP-conjugated secondary mouse antibody |
Beijing TDY Biotech LTD |
#E009 |
HRP-conjugated secondary rabbit antibody |
Beijing TDY Biotech LTD |
#E011 |
endothelial cells medium |
PromoCell |
C-22010 |
TransStart Green qPCR Supermix |
Transgene |
AQ101-02 |
HUVEC |
PromoCell |
C-12206 |
Cell proliferation assay
HUVECs were seeded into 96-well cell culture plates and allowed them to adhere for 12h. After incubation with various concentration of VEGF for 24h, 10ul of MTT was added to each well for 3h incubation. Subsequently, cells were dissolved by 150 mL of DMSO, mixed and measured the absorbance (A) by Multiskan (Thermo) at 562 nm.
Scratch wound assay
HUVECs were deprived of serum for 12h and scratched with a sterile P200 pipette tip according to a paradigm previously described [25]. After removal of the debris by repeated washes, cells were subjected to VEGF treatment and scratch wound closure was monitored by phase microscopy capturing images of the same field with a 4X objective at 0h and 8h. The percentage of migrated area was calculated with “Image J” software.
PI staining for cell cycle
HUVECs were pretreated with VEGF for 24h. Cells were trypsinized and fixed by -80°C 70% ethanol for 24 hours. And then cells were centrifuged and stained with PI buffer in dark at 37°C for 30min. Cell cycle was evaluated by flow cytometry according to the manufacturer’s protocol (BD PharMingen, San Diego, CA, USA). PI buffer(200μL) including: 160μL 0.1% Triton X-100/PBS, 20μL 0.5 mg/ml PI, 20μL 10 mg/ml RNase A.
In vitro angiogenesis assay
Angiogenic potential was assessed by the spontaneous formation of capillary-like structures by HUVECs on growth factor–reduced Matrigel. HUVECs (1×104 cells/ well) were seeded in 96-well matrigel-coated plates using completed medium. After 1 hours, cells were stimulated with 20 nM VEGF. And after 4-5 hours, cells were observed with a Nikon inverted microscope and images recorded and analysed by using Image Pro-Plus image analysis software (Media Cybernetics).
Measurement of oxygen consumption rate
Cellular OXPHOS was monitored using the Seahorse Bioscience Extracellular Flux Analyzer (XF24, Seahorse Bioscience Inc., North Billerica, MA, USA) by measuring the OCR (indicative of respiration). Briefly, 3000-5000 cells were seeded in 24-well plates designed for XF24 in 150μL of appropriate growth media and incubated overnight, then with different treatments. Before measurements, cells were washed with unbuffered media once, then immersed in 675μL unbuffered media and incubated in the absence of CO2 for 1h at 37°C. OCR was then measured in a typical 8-min cycle of mix (2-4 min), dwell (2 min) and measure (2-4 min) as recommended by Seahorse Bioscience.
Intracellular ATP content measurements
CellTiter-Glo Luminescent Cell Viability Assay was used according to the manufacturer’s instructions. Luminescence signal was measured using synergy HT fluorometer (Bio Tek, Winooski, VT, USA). The values were normalized to control for each corresponding point.
Measurements of intracellular reactive oxygen species
Intracellular ROS was quantified by a laser confocal microscope using the fluorescent probe, 2’, 7’-dichlorodihydrofluorescein diacetate (DCFH-DA). HUVECs were incubated in phenol red-free media supplemented with 10μM DCFH-DA in dark for 20 min at 37°C. They were then washed twice with PBS and immediately observed.
Quantitative reverse transcription PCR
Quantitative reverse transcription PCR (qRT-PCR) was carried out using the following oligonucleotides and PCR conditions: 2 minutes at 95°C (20 seconds at 95°C, 20 seconds at the annealing temp, and 30 seconds at 72°C) × 35, and 10 minutes at 72°C. Primers were as follows: ß-actin, forward: 5′-CATCACTATTGGCAACGAG-C-3′, reverse: 5′-ACGCAGCTCAGTAACAGTCC-3′; catalase, forward: 5′-TGGAGCTGGTAACCCAGTAGG-3′, reverse: 5′-CCTTTGCCTTGGAGTATTTGGTA-3′;GPX1, forward: 5′-CAGTCGGTGTATGCCTTCTCG-3′, reverse: 5′-GAGGGACGCCACATTCTCG-3′.
Western blots analysis
Cell lysate proteins were separated by SDS-PAGE (10% gels) and transferred to nitrocellulose membranes. Membranes were blocked in Tris-buffered saline with 0.05% Tween 20 (TBST) containing 5% nonfat dry milk powder for 1 hour. Western blots were probed with primary antibodies for 1 hour, washed 3 times with TBST, and then incubated with the appropriate secondary antibodies for 30 minutes. Membranes were then washed with TBST 3 times, before developing with SuperSignal West Dura chemiluminescent substrate. The following antibodies were used: rabbit anti-Phospho-S6 Ribosomal Protein (Ser235/236) (Cell Signaling, #2211, 1:1000), rabbit anti-S6 Ribosomal Protein (5G10) (Cell Signaling, #2217, 1:1000), mouse anti-ß-actin (Beijing TDY Biotech LTD, #M009, 1:5000), HRP-conjugated secondary mouse (Beijing TDY Biotech LTD, #E009, 1:5000) and rabbit antibodies (Beijing TDY Biotech LTD, #E011, 1:5000).
Statistical analyses
All data were derived from ≥3 independent experiments. Statistical analyses were conducted using SPSS version 19.0 (IBM SPSS, Armonk, NY, USA). Values were calculated as mean ± standard error of the mean. Significant differences between the groups were determined using one-way analysis of variance test. A p-value P<0.05 was considered to indicate a statistically significant difference.
Abbreviations
VEGF: vascular endothelial growth factor; ECs: endothelial cells; HUVECs: Human umbilical vein endothelial cells; ROS: reactive oxygen species; GPX1: glutathione peroxidase (GPX1); mTOR: mammalian target of rapamycinm; ATP: adenosine triphosphate; PFKFB3: fructose-2, 6-biphosphatase 3.
Author contributions
Dongqing Guo designed and performed the experiments, and wrote the manuscript text. Qiyan Wang, Chun Li and Yong Wangmodified the manuscript. Xing Chen conceived the experiments, analyzed the data, modified the manuscript. All authors reviewed the manuscript.
ACKNOWLEDGMENTS
This work is supported by the research grants by Yong Wang (National Natural Science Foundation of China, No.81673712), Xing Chen (National Natural Science Foundation of China, No.61772531 and 11631014), and Dongqing Guo (Fundamental Research Funds of Beijing University of Chinese Medicine, No.2017-JYB-JS-017).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Folkman J. Seminars in Medicine of the Beth Israel Hospital, Boston. Clinical applications of research on angiogenesis. N Engl J Med. 1995; 333:1757-1763.
2. Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996; 86:353-364.
3. Aiello LP, Avery RL, Arrigg PG, Keyt BA, Jampel HD, Shah ST, Pasquale LR, Thieme H, Iwamoto MA, Park JE, Nguyen HV, Aiello LM, Ferrara N, King GL. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N Engl J Med. 1994; 331:1480-1487.
4. Wang K, Ahmad S, Cai M, Rennie J, Fujisawa T, Crispi F, Baily J, Miller MR, Cudmore M, Hadoke PW, Wang R, Gratacos E, Buhimschi IA, et al. Dysregulation of hydrogen sulfide producing enzyme cystathionine gamma-lyase contributes to maternal hypertension and placental abnormalities in preeclampsia. Circulation. 2013; 127:2514-2522.
5. Kaigler D, Wang Z, Horger K, Mooney DJ, Krebsbach PH. VEGF scaffolds enhance angiogenesis and bone regeneration in irradiated osseous defects. J Bone Miner Res. 2006; 21:735-744.
6. Finkenzeller G, Technau A, Marme D. Hypoxia-induced transcription of the vascular endothelial growth factor gene is independent of functional AP-1 transcription factor. Biochem Biophys Res Commun. 1995; 208:432-439.
7. Shibuya M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis: A Crucial Target for Anti- and Pro-Angiogenic Therapies. Genes Cancer. 2011; 2:1097-1105. doi: 10.1177/1947601911423031.
8. Claesson-Welsh L. VEGF receptor signal transduction - A brief update. Vascul Pharmacol. 2016; 86:14-17.
9. Jo HN, Kang H, Lee A, Choi J, Chang W, Lee MS, Kim J. Endothelial miR-26a regulates VEGF-Nogo-B receptor-mediated angiogenesis. BMB Rep. 2017.
10. Ko JY, Lee MS, Lian WS, Weng WT, Sun YC, Chen YS, Wang FS. MicroRNA-29a Counteracts Synovitis in Knee Osteoarthritis Pathogenesis by Targeting VEGF. Sci Rep. 2017; 7:3584.
11. Haque R, Iuvone PM, He L, Su K, Choi C, Ngo A, Gokhale S, Aseem M, Park D. The MicroRNA-21 signaling pathway is involved in prorenin receptor (PRR) -induced VEGF expression in ARPE-19 cells under a hyperglycemic condition. Mol Vis. 2017; 23:251-262.
12. Parra-Bonilla G, Alvarez DF, Al-Mehdi AB, Alexeyev M, Stevens T. Critical role for lactate dehydrogenase A in aerobic glycolysis that sustains pulmonary microvascular endothelial cell proliferation. Am J Physiol-Lung C. 2010; 299:L513-L522.
13. De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, Quaegebeur A, Ghesquiere B, Cauwenberghs S, Eelen G, Phng LK, Betz I, Tembuyser B, et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell. 2013; 154:651-663.
14. Darley-Usmar V. The powerhouse takes control of the cell; the role of mitochondria in signal transduction. Free Radic Biol Med. 2004; 37:753-754.
15. Smith RA, Hartley RC, Cocheme HM, Murphy MP. Mitochondrial pharmacology. Trends Pharmacol Sci. 2012; 33:341-352.
16. Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017; 168:960-976.
17. Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature. 2007; 450:736-740.
18. LeBleu VS, O'Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, de Carvalho FM, Damascena A, Domingos Chinen LT, Rocha RM, Asara JM, Kalluri R. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol. 2014; 16:992-1003, 1001-1015.
19. Hu J, Hwang SS, Liesa M, Gan B, Sahin E, Jaskelioff M, Ding Z, Ying H, Boutin AT, Zhang H, Johnson S, Ivanova E, Kost-Alimova M, et al. Antitelomerase therapy provokes ALT and mitochondrial adaptive mechanisms in cancer. Cell. 2012; 148:651-663.
20. Bazhin AV, Yang Y, D'Haese JG, Werner J, Philippov PP, Karakhanova S. The novel mitochondria-targeted antioxidant SkQ1 modulates angiogenesis and inflammatory micromilieu in a murine orthotopic model of pancreatic cancer. Int J Cancer. 2016; 139:130-139.
21. Neuzil J, Tomasetti M, Mellick AS, Alleva R, Salvatore BA, Birringer M, Fariss MW. Vitamin E analogues: a new class of inducers of apoptosis with selective anti-cancer effects. Curr Cancer Drug Targets. 2004; 4:355-372.
22. Andre N, Braguer D, Brasseur G, Goncalves A, Lemesle-Meunier D, Guise S, Jordan MA, Briand C. Paclitaxel induces release of cytochrome c from mitochondria isolated from human neuroblastoma cells'. Cancer Res. 2000; 60:5349-5353.
23. Park D, Dilda PJ. Mitochondria as targets in angiogenesis inhibition. Mol Aspects Med. 2010; 31:113-131.
24. Gao S, Tibiche C, Zou J, Zaman N, Trifiro M, O'Connor-McCourt M, Wang E. Identification and Construction of Combinatory Cancer Hallmark-Based Gene Signature Sets to Predict Recurrence and Chemotherapy Benefit in Stage II Colorectal Cancer. JAMA Oncol. 2016; 2:37-45.
25. Sinagra T, Merlo S, Spampinato SF, Pasquale RD, Sortino MA. High mobility group box 1 contributes to wound healing induced by inhibition of dipeptidylpeptidase 4 in cultured keratinocytes. Frontiers in pharmacology. 2015; 6:126.