
INTRODUCTION
The bidirectional communication between cells and the microenvironment is critical not only for normal tissue homeostasis [1], but also in disordered tissues. In particular, interactions among premalignant lesion cells, circumjacent stromal cells, and infiltrated immune cells represent a powerful relationship that can significantly modify the development of potentially malignant disorders [2, 3]. In 1970s, Noone and co-workers first described the relationship between the prognosis of oral squamous cell carcinoma (OSCC), a common malignancy, and T lymphocytes within the cancer microenvironment [4]. Since then, the microenvironment has been characterized in disordered tissues, including cancer and oral potentially malignant disorders (OPMD) to identify and develop novel therapies to treat OPMD and to prevent OPMD from acquiring cancer characteristics. The term “OPMD” has been used to broadly describe clinical presentations that may transform into oral carcinoma, including Oral Erythroplakia (OE), Oral Leukoplakia (OLK), Oral Lichen Planus (OLP), Oral Submucous Fibrosis (OSMF), and Actinic Keratosis [5, 6]. In the case of leukoplakia, the progress from leukoplakia to OSCC is histologically classified as mild, moderate, or severe [7]. While its progression results from heterogeneities, including aberrant mutations, there is a growing appreciation that the progression is also influenced by the microenvironment [2, 3]. As the environmental conditions keep evolving and oncogenic signals are generated during the progression from normal tissues to OPMD and subsequently to OSCC, the microenvironment also changes [8, 9]. These underscore the need to discuss the changes in the OPMD microenvironment that is dynamic. How OPMD lesional cells communicate with the microenvironment bidirectionally and promote the process in their own niche needs to be deciphered.
In this review, we summarize the current literature on the roles of different stromal cells during the progression from OPMD to OSCC and metastasis, which provides new insights on how to prevent, repress, or even reverse the progression towards malignancy. Since the microenvironment is extensive and complex and changes occur at each phase of the differentiation process from OPMD to OSCC, we chose to discuss the specific aspect of the microenvironment during the progression from OPMD to OSCC. We summarize the evidence that supports the breadth of communication within OPMD, whereby tissue cells can not only promote the progression from OPMD to OSCC, but also reverse it.
Clinical association between inflammation, OPMD, and OSCC incidence
The etiology of OPMD is multifactorial. Tobacco and alcohol are considered major risk factors, but the role of the microenvironment is increasingly recognized as being significant in OSCC development [8, 9]. In order to visualize the abstract concept of the microenvironment, we summarized direct pieces of clinical evidence. A deregulated microenvironment affects the progress from OPMD to OSCC. More specifically, when subjected to carcinogenic factors (for example, tobacco and alcohol), the oral mucosa may induce OPMD, and lymphocyte populations may change secondarily [10]. OPMD with chronic inflammation generally supports high OSCC incidence [2, 11]. It has been reported that chronic inflammation can instigate cell proliferation and activity, which induces irreversible DNA damage [12]. Cytokines released through chronic inflammatory OLK promote carcinogenesis that, in turn, instigates the inflammatory response, forming a cyclic progression [2]. In addition, immune response failure can induce OPMD and OSCC [13–18].
Clinical association between infection and OPMD
Oral inflammation such as human papilloma virus (HPV) infection may be associated with OPMD, indicating that a deregulated microenvironment can affect the occurrence of OPMD. For instance, HPV infection is highly prevalent in OPMD. In some studies on OPMD, HPV was detected in potentially malignant lesions [19–26]. A recent meta-analysis of 4580 specimens showed that the prevalence of HPV in normal oral mucosa, non-dysplastic leukoplakia, dysplastic leukoplakia, and in other OPMD and OSCC is 10%, 20.2%, 26.2%, and 46.5%, respectively [27]. They also detected the likelihood of HPV and showed that it is 2 to 3 times greater in OPMD than in the normal oral mucosa, while it is 4 to 5 times greater in OSCC [27]. Moreover, some reports implicated high-risk HPV genotypes, particularly HPV16 and/or 6 positive, as the most prevalent in OPMD and OSCC [28–30]. The prevalence of HPV16 and/or 6 and 18 is 33% and 14%, respectively [31]. The effect of oral Candida in OPMD is another well-known example [32]. For example, the prevalence of Candida infection in OLK, erosive OLP, and OSF is 47%, 44.29%, and 36.67%, respectively [33–35]. Other infections may be associated with OPMD such as Helicobacter pylori [36], HCV [37], and fungal hyphae [38], and the infection rate is about 90% [36], 23% [37], and over 25% [38], respectively.
Clinical association between OPMD and OSCC
Chronic inflamed OPMD generally exhibits high OSCC incidence, indicating that a deregulated microenvironment can affect the occurrence of cancer [2]. For example, there is a strong association between OSCC risk and OPMD (OLK and OSMF) [11]. Although OE is less common than OLK, it exhibits a higher risk for malignant transformation; more than 90% of these red lesions show severe epithelial dysplasia or in situ carcinoma [39]. As for OLK, cancer occurs in 0.13% to 34% of patients per year [40]. For OSMF, the rate of malignant transformation ranges from 4% to 8% [41].
Clinical association between OPMD with infection and OSCC
The presence of infection in OPMD may be associated with carcinomatous transformation, suggesting that a deregulated microenvironment can affect the progression of carcinomatous transformation. For instance, HPV infection in OPMD is linked to the progression of oncogenesis [28, 31, 42]. Proliferative verrucous leucoplakia, a rare and particularly aggressive form of OLK, is thought to have the strongest relationship with HPV infection [42]. This clinical disorder has a high potential for malignant transformation with a 90% malignant evolution to OSCC [43]. Regarding the effect of oral Candida in the development of OPMD into cancer, Candida infection may affect dysplasia of the epithelia and malignant transformation of OPMD [32, 44]. Candida-infected OLK showed a higher rate of malignant transformation on follow-up [32, 45].
Clinical association between impaired immune responses and high OSCC incidence
Impaired immune responses can induce OPMD and OSCC. In an analysis of 122,993 patients with AIDS, the observed incidence of both AIDS-related cancers (For example, non-Hodgkin lymphoma) and non–AIDS-related cancers (For example, cancers of the tongue) were higher [13]. These results were confirmed in other studies [14–17, 46]. There is also evidence showing that adequate immune function may protect OPMD from acquiring malignant characteristics [47, 48]. In some cases of viral infections, the adequate immune system may exert its protective function either by limiting the outgrowth of virus-transformed cells or OSCC cells that require the continued expression of viral proteins for their survival, or by curbing viral replication at an earlier stage, which is called “tumorigenic hit” [47]. This contrasts with studies supporting the pro-tumorigenic functions of inflammation [49].
Alternative therapeutic opportunities
These clinical phenomena indicate new challenges in our understanding of the functions of inflammation and suggest new treatment strategies, including reeducating immune cells in OPMD and OSCC. In such a complex environment, therapeutic effects depend on the reeducation of the stroma, which has a strong influence on the progression from OPMD to OSCC. For example, whereas macrophages can be stimulated or educated by the Th2-derived anti-inflammatory cytokines, IL-4 and IL-13, and exhibit pro-tumorigenic effects [50–52], they can also be re-educated by various pharmaceutical drugs to exhibit opposite functions [53–57]. Thus, we could take advantage of the plasticity of the OPMD stroma by re-educating cells to prevent OSCC occurrence rather than by simply ablating stromal cells.
Progression from OPMD to OSCC; disruption of tissue homeostasis
From a number of clinical associations discussed above, it is clear that the progression from OPMD to OSCC is indeed educated by an abnormal immunoenvironment. During the progression from OPMD to OSCC, the coordinated interactions between inflammatory cells that are present in epithelial tissues are disordered as the process occurs and, in turn, the tissue microenvironment regulates the carcinogenic process (Figures 1, 2, and Supplementary Table 1) [58]. We will first discuss that the stroma is hijacked for its own benefit at primary sites to escape from immune attack and to undergo the carcinogenic process to differentiate into OSCC, with a focus on the role of macrophages, myeloid-derived suppressor cells, dendritic cells, T cells, cancer-associated fibroblasts, mesenchymal stem cells, and mast cells.
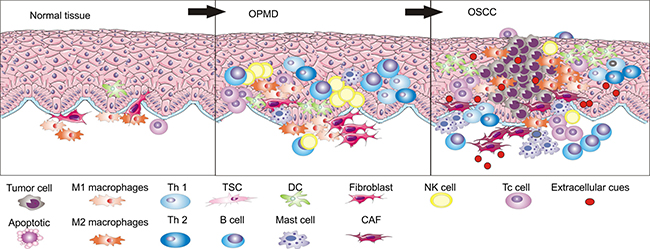
Figure 1: During the development process from normal epithelia to OSCC through OPMD, immune cell populations are different in number and types.
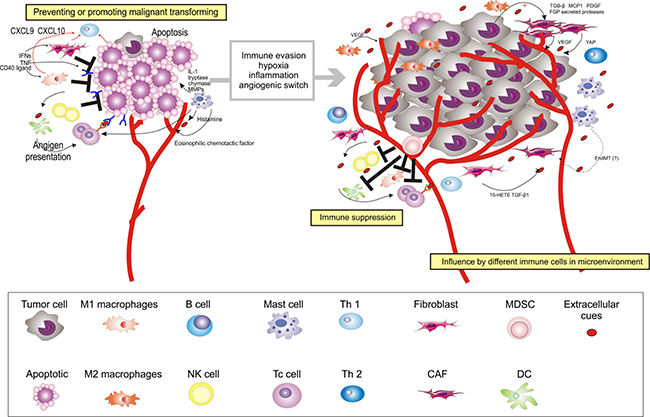
Figure 2: Multiple stromal cell types converge to support the development of OSCC. After circumventing cell-intrinsic mechanisms of apoptosis, cancer cells are subjected to elimination pressure by the immune system. Tumor cell-specific antigens play a role during this process; they are recognized by cytotoxic immune cells, leading to their destruction. Fibroblasts, macrophages, NK cells, and Th1 cells within the microenvironment also contribute to the growth-suppressive state. Chemokines (for example, CXCL9 and CXCL10) can be induced by IFNs (IFN-gamma) and TNF or CD40 ligand, and recruit Th1 cells. After recruiting Th1 cells, IFNs (IFN-gamma) could further switch the macrophage phenotype in the oral premalignant lesions to M1 macrophages. However, fibroblasts, macrophages, and Th1 cells may later become educated by the cancer to acquire pro-tumorigenic functions. NK cells may later decrease in the microenvironment of OSCC. For instance, M2 macrophages support diverse phenotypes during the process from OPMD to OSCC, including acquisition of the malignant phenotype and angiogenesis, by stimulating IFN and secreting growth factors (for example, VEGF). As OPMD acquire a malignant phenotype, immunosuppressor cells (For example, MDSCs) are mobilized into the circulation to disrupt immune surveillance through multiple mechanisms, including, but not limited to, disruption of antigen presentation by DCs, inhibition of Tc cells, altering M1 macrophage polarization, and inhibition of NK cell cytotoxicity. CAFs, which become generated by cell factor (For example, TGF-β1, 15-HETE, and FSP1) activated by growth factors and cytokines (For example, TGF-β, MCP1, PDGF, and FGF), and secrete factors to support carcinogenesis (For example, VEGF). MCs aggravate the progression from OPMD to OSCC by secreting mediators, including IL-1, tryptase, chymase, MMPs, histamine, and eosinophilic chemotactic factor. In addition to cellular conditions, several extracellular properties contribute to the development of malignancy, including immune evasion, hypoxia, inflammation, and angiogenic switch. EndMT, endothelial-to-mesenchymal transition; Ag, antigen; TNF, tumor necrosis factor.
Macrophage plasticity contributes to the differentiation of OPMD into OSCC
Macrophages are essential for all animal lives. They have the ability of phagocytizing pathogens, repairing tissue, and maintaining tissue integrity [59]. They are differentiated from progenitor cells from the fetal yolk sac [60, 61], can renew by themselves [62], or are derived from the bone marrow and spleen [63]. Although they have long been regarded as important immune cells during the immune response, there is a growing appreciation that macrophages play a role in supporting multiple aspects of OPMD [59, 64]. Perhaps the most notable is that macrophages in OPMD present the M1 phenotype [64]. Indeed, a study on OLK demonstrated that the number of M1 macrophages is increased and showed a positive relationship between Th1 cells and M1 macrophages [64].
When facing different physiological conditions, macrophages can alter their polarization state functionally (Supplementary Table 1). At the extremes of their phenotypic continuum [65], macrophages can be divided into ‘Inhibit’ type macrophages (M1) and ‘Heal’ type macrophages (called M2) [59, 66, 67]. Classically activated M1 macrophages can rapidly phagocytize pathogens, inhibit cell proliferation, cause tissues damage, and act as pro-inflammatory cells [59, 67, 68], while the alternatively activated M2 macrophages can promote cell proliferation, tissue repair, tissue integrity maintenance, and act as anti-inflammatory cells [59, 67, 68]. Thus, the M1 and M2 phenotypes demonstrate the two major activities of macrophages and form a counterbalanced system. In order to be a threat to the organism from within and without, macrophages are constantly and rapidly changing their physiology [66, 69, 70]. This fluid circumstance is called macrophage plasticity [65, 66, 69, 70]. Here, we should note that, although the classification is accepted by most people and is really useful, it is somewhat too simple, as the complexity of macrophage activation has not been fully represented [65].
Currently, the functional role of macrophages in the progression from OPMD to OSCC is unclear. Environmental conditions such as the persistent type I interferons (IFN-alpha and IFN-beta)-stimulating microenvironment may mediate this transition from the premalignant to the malignant lesion [71]. By cancer immunoediting, cancer cells can acquire resistance to the antitumor responses of IFNs (IFN-alpha and IFN-beta) in the continuously stimulating environment [71]. Premalignant epithelial cells are genetically altered and are continuously stimulated by carcinogens. In this persistent stimulating environment, epithelial cells, fibroblasts, and macrophages may produce chemokines, which can recruit Th1 cells [64, 72, 73]. Some studies suggested that the expression of these chemokines can be induced by IFNs (IFN-gamma and IFN-alpha) and tumor necrosis factor (TNF) or CD40 ligand [74, 75]. These chemokines (CXCL9 and CXCL10) can be induced by IFNs (IFN-gamma and IFN-alpha), which mediate the recruitment of Th1 cells [72, 73, 76]. Indeed, it has been reported that CXCL9 is expressed in the subepithelial lesion of OLK. After recruiting Th1 cells, IFNs (IFN-gamma and IFN-alpha) could further switch the macrophage phenotype in the oral premalignant lesions to M1 macrophages [77–83]. In fact, using a mouse tumor model, a study showed that IFN-induced M1 macrophages play an important role during cancer immunoediting [84]. Furthermore, it has also been suggested that densities of M1 macrophages in tumor islets may be associated with the extended survival of patients [85, 86].
Recent evidence indicates a change to the M2 macrophage phenotype during the progression from OPMD to OSCC. A study using the WHO histopathological classification of OLK described that there was a significant increase in M2 macrophages during the development of dysplasia [64]. Other studies using pathological graded OSCC cells suggested that the number of M2 macrophages increased based on the histopathological grade [87]. Although the exact function of these cells in OLK and OSCC is unknown, evidence suggests that they may correlate with angiogenesis, OPMD malignant potential, and OSCC progression. In fact, some researchers reported that macrophage accumulation correlates with the expression of vascular endothelial growth factor (VEGF) and the density of microvessels [88, 89]. They also suggested that angiogenesis does not depend on the number of macrophages, but on their predominant phenotype [90]. Later, some studies on distinct phenotypes of macrophages showed increased infiltration of M2 polarized macrophages in early stage OSCC and OSCC with nodal lymphogenic metastasis [87, 88, 90, 91].
Taken together, these findings suggest that macrophage plasticity contributes to the progression from OPMD to OSCC. Such results support the potential of re-educating macrophages to prevent OPMD from acquiring cancer characteristics as a potential therapeutic strategy and to evaluate the prognosis of the patients.
The interaction between dendritic cells and T cells shapes dysplastic cells
Apart from macrophage plasticity, immunosurveillance mechanisms have been described to suppress malignancy [92]. However, in an equilibrium phase, dendritic cells and T cells can interact and sculpt premalignant lesion to malignant.
Dendritic cells (DCs), potent antigen-presenting cells, can induce a T cell response [93]. DCs trap, process, and present antigens to naive or memory T cell in the context of the major histocompatibility complex class I pathway (MHC class I) [94, 95]. The presentation of antigens by DCs, which is restricted by the MHC class I, to CD8+ T cells generates specific cytolytic effector T cells. This is a critical step in adaptive immune response [96] (Figure 3). Dendritic Langerhans cells (LCs) is a subset of DCs present in oral mucosal linings. They can provide immunosurveillance to tissue compartments [94]. The interaction between LCs and T cells, which can select the type and direct the immune response, is crucial [94]. Given their crucial regulatory roles in directing the immune response, it is not surprising that DCs affect the progression from OPMD to OSCC. In OLK tissue, increased numbers of LCs and CD8+ T cells correlate with leukoplakia with dysplasia, whereas, in OSCC, the number of dendritic LCs are increased compared to that in leukoplakia with dysplasia [58]. A review indicated that the immune system, especially the interaction between DCs and T cells, and dysplasia cells or early tumor cell formation have three phases (elimination, equilibrium, and escape) [97]. In the early phase, the immune system can eliminate the damaged cells [98]. Altered self-expression by keratinocytes causes the recruitment of DCs [58]. The presence of dysplasia could result in influx of inflammation that can enhance the interplay between LCs and T cells [58]. In an equilibrium phase, by iteratively selecting and/or promoting the generation of tumor cell variants, the immune system increases the cell capacity to survive an immune attack [97, 99]. In fact, OLK with dysplasia may be an example in which proliferation of dysplastic cells and activation of the immune system are in an equilibrium phase [99]. In the escape phase, tumor, which has been sculpted by the immune system expands in an uncontrolled manner, where OPMD acquires malignant characteristics [99]. Interestingly, a recent study indicated that programmed death ligand 1 (PD-L1)-expressing dysplastic epithelial cells in OPMD may evade the host immune system [100]. Programmed death 1 [PD-1]/PD-L1 pathway, an important inhibitory checkpoint, has been highlighted recently [101]. By activating this pathway, adaptive immune resistance helps establishing an immunosuppressive microenvironment, which is one of the major mechanisms of tumor escape in the equilibrium phase [100–102]. These findings highlight potential therapeutic strategies where re-educating DCs in the equilibrium phase might prevent premalignant lesions from acquiring malignant characteristics.
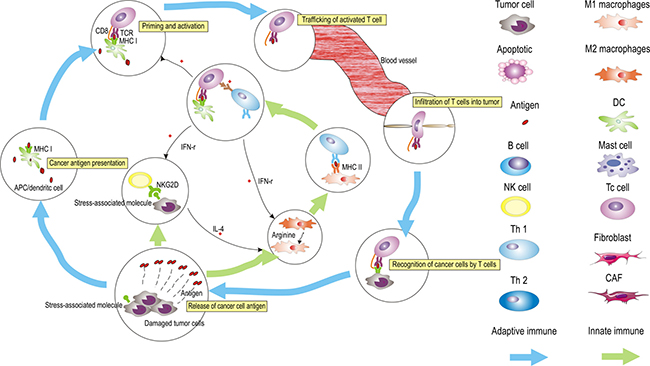
Figure 3: The generation of tumor immunity is a cyclic process that can be self-propagating, leading to the accumulation of immuno-stimulatory factors that, in principle, should amplify and broaden adaptive immune responses. This cycle can be divided into six major steps, starting with the release of antigens from the cancer cells and ending with the killing of cancer cells, which can be regulated by innate immune cells (NK cells, macrophages, and Th1 cells). The process is described above, with the primary cell types involved. APCs, antigen-presenting cells.
Myeloid-derived suppressor cells suppress immune surveillance
During the malignant progression from OPMD to OSCC, it is critical for cells to evade and suppress the host immune system (Figure 2), which can be achieved by inhibiting various effector immune cells or by stimulating immunosuppressive cells. It is generally agreed that myeloid-derived suppressor cells (MDSCs) are involved in immune evasion [103]. They may comprise aberrant cell populations that appear during the progression from OPMD to OSCC.
MDSCs have been functionally described as immunosuppressive, immature myeloid cells that inhibit T cell proliferative responses, antibody production, and CTL induction [104]. During tumorigenesis, MDSCs are educated, infiltrate, promote tumor vascularization, and disrupt major mechanisms of immunosurveillance by directing progenitor of DCs, activating T cells, altering M1 macrophage polarization, and inhibiting NK cell cytotoxicity [105–109]. Recent studies showed that chronic inflammation can induce MDSCs accumulation [110, 111]. When subjected to inflammation, the oral mucosa may induce OPMD, and OPMD usually gets secondarily inflamed [2, 11]. Thus, we believe that MDSCs promote the malignant progression from OPMD to OSCC. Since MDSCs include mixed subpopulations with a great diversity of plasticities and maturities and can differentiate into different types of cells, MDSCs are attractive for the design of therapies that aim at remodeling the immunosuppressive activity of MDSCs.
Cancer-associated fibroblasts elicit pro-malignant functions in OPMD
Several important factors are involved in the malignant transformation of OPMD to OSCC such as angiogenesis, lymphangiogenesis, genetic alterations, and mutational factors [112, 113]. These changes can be achieved by activation of fibroblasts.
Fibroblasts are stromal components with diverse functions. They can deposit extracellular matrix (ECM), regulate the differentiation of connective tissue, modulate immune responses, and mediate homeostasis [114]. Cancer-associated fibroblasts (CAFs), also called peritumoral fibroblasts and myofibroblasts (MFs), are activated fibroblasts [115], similar to fibroblasts associated with wound healing [114]. In the OPMD and OSCC microenvironment, CAFs are present in abnormally high numbers that increase as the disease progresses from OPMD to OSCC and are different from normal fibroblasts [41, 116]. In general, CAFs express high levels of alpha-smooth muscle actin (a-SMA), p53, podoplanin, CD10, FAP, fibroblast specific protein 1 (FSP1), MMPs, tenascin-C, and PDGFR a/b and low levels of caveolin-1 (Cav-1) and cytokeratin [114]. In a clinical experiment, CAFs were positively detected from 0% no MFs to 21–40% MFs in OLK epithelial tissue, but not in the normal epithelium [41, 116–118]. Similarly, in OSF, the frequencies of CAFs were 100% and 86%, respectively [41, 119]. In OSCC tissue, the number of CAFs is interestingly high [41, 116, 120, 121]. As discussed above, the rate of malignant transformation of OSF is higher than that of OLK, which corresponds to the frequency of CAFs in OSF and OSCC. Another interesting study continuously observed CAF expression during the process of differentiation from normal tissue to OPMD to OSCC [116]. CAFs were not expressed in normal tissue and mild epithelial dysplasia, but expressed in severe epithelial dysplasia and OSCC [116]. These changes indicate that CAFs should be considered a cell type with potent effects on the process of differentiation from normal tissue to OPMD to OSCC, which further strengthens the “tissue organization field theory” [8, 122]. It emphasizes that mutations in epithelial/stromal cells and disturbance between stromal-epithelial interactions may be important for tumorigenesis [8, 122].
How CAFs are generated during the progression from OPMD to OSCC remains unclear. Some studies suggested that they arise from the transition of fibroblasts into myofibroblasts. Currently, the leading signal that mediates the phenotypic conversion is the transforming growth factor-β [120, 123–126]. Further in vitro studies suggested that 15-hydroxyeicosatetraenoic acid (15-HETE) converted fibroblasts into CAFs by the transforming growth factor (TGF)-β1-mediated FGF-2 signaling pathway [127, 128]. Such conversion is associated with the emergence of the mesenchymal marker, fibroblast-specific protein-1 (FSP1) and the down-regulation of CD31/PECAM [126]. Other studies pointed to another possible origin from endothelial-to-mesenchymal transition (EMT), vascular smooth muscle cells have different differentiation potentials to generate mesenchymal cells [120, 129–131]. Mouse experiments showed that CAFs can be generated from EMT [126]. Tumor-associated endothelial cells are not only a source for CAFs, but also promote the generation of CAFs since they can dedifferentiate to generate cells expressing CAF markers [132].
CAFs are raised and activated by growth factors and cytokines in the milieu such as TGF-β, monocyte chemotactic protein 1 (MCP1), platelet-derived growth factor (PDGF), fibroblast growth factor (FGF), and secreted proteases [120, 125].
Later, studies showed the importance of the YAP transcription factor for CAFs to remodel the microenvironment and to support carcinogenesis [133]. In turn, they also can regulate factors that re-educate the cytoskeleton and modulate matrix stiffness. This effect can induce a feedback loop to induce YAP expression.
Once CAFs are activated, they will secrete a large number of growth and proinflammatory factors. These factors play an important role in supporting carcinogenesis. For example, VEGF can permeate the vascular system and induce angiogenesis [134, 135]. Thus, the interdependency of multiple cell types may be disturbed.
Impairment of mesenchymal stem cells is related to the progression from OPMD to OSCC
Mesenchymal stem cells (MSCs), identified in the epithelium and primarily in the basal layer [136–139], can differentiate into three germ layers and demonstrate multi-potent trans-differentiation [140, 141]. In the microenvironment of OPMD tissue, MSCs present an imbalance between regenerative and metabolic self-regulatory functions. For example, in OLK tissues, the self-renewal ability was enhanced, while the potential for trans-differentiation was decreased, compared with that of MSCs in normal tissues [142]. Indeed, during the progression of OLK, collagen IV (Col IV) was decreased and matrix metalloproteinases-9 (MMP-9) was increased in MSCs [142, 143]. Previous studies suggested that Col IV continuity was destroyed and MMP-9 positive cells were increasing in the zone adjacent to fragmented basement membranes in epithelial dysplasia and cancer [144–146]. Therefore, impairment of MSCs may be critical to destroy the basement membrane in the mucosa of OPMD and OSCC.
Why the capacity for multi-potent differentiation was greatly decreased in MSCs and concurrent with the progression from OPMD and OSCC remains unclear. The impairment of MSCs may be associated with EMT. The abnormal epithelia such as epithelial dysplasia and OSCC epithelia may interfere with EMT and produce abnormal MSCs presenting with malfunctions [142]. In addition, inflammation can reduce the multi-potent differentiation capacity of MSCs [147]. Indeed, high levels of inflammatory factors are detected in OPMD tissues [148–151].
Abnormal MSCs produced in OPMD are weaker in regulating inflammation [150]. Previous studies showed that MSCs can immunosuppress the proliferative responses of T cells via active IDO [150, 152]. However, MSCs show low immunogenicity and immunomodulatory functions in OLP [150]. In an in vitro study, MSCs not only immunosuppressed the proliferation of T cells directly, but also inducted T cells with a CD4+CD25+Foxp3+ regulatory phenotype [153]. All these effects contribute to the generation of an immunosuppressive environment.
“Piece meal degranulation” of mast cells aggravates the progression from OPMD to OSCC
Mast cells (MCs), also called mastocytes and labrocytes, are large connective tissue cells, distributed preferentially in the capillaries and containing many granules rich in histamine and heparin [154]. The granules in their cytoplasm are basophilic and may obscure the nucleus [154]. In the microenvironment of oral premalignant lesions, MCs are aberrantly increased and present different phenotypes. For example, in a study comparing the number, morphology, and topographical distribution of MCs in OPMD (including OLK, OSF, and OSCC), the number of typical (TMCs), atypical (AMCs), and granular mast cells (GMCs) increased in cases with mild, moderate, and severe inflammation [155].
“Piece meal degranulation” is the characteristic trait of MCs, whereby MCs secretes certain mediators by selecting cell secretion pathways [156, 157]. The secreted mediators in OPMD play various roles in the malignant process [155]. It includes TNF-alpha, interleukin-1, heparin, histamine, chymase, bFGF, VEGF, TGF-β, IL- 8, Colony Stimulating Factor (CSF), FGF, Nerve Growth Factor (NGF), TGF, PDGF, and Stem Cell Factor (SCF) [155–159].
“Piece meal degranulation” of Mast cells aggravates the progression from OLK to OSCC. Interleukin-1, which is released by stimulated MCs, contributes to increased epithelial proliferation [160]. Histamine could facilitate the antigen to connective tissue by increasing the permeability of the mucous [160]. Similarly, studies concluded that the number of MCs increases as the differentiation from normal tissue to OLK with high-grade dysplasia and may be associated with angiogenesis [161–164]. Thus, it can be used to determine disease progression.
“Piece meal degranulation” of Mast cells aggravates the process of OSF. Similar to the number in OLK, MCs are more abundant in OSF than in the normal buccal mucosa [164, 165]. Interleukin-1, which is released by stimulated MCs, increases the fibroblastic response, which could cause the production of type-1 collagen and fibronectin. Histamine could contribute to vesicle formation and symptoms of itching sensation and submucosal edema of OSF. Eosinophilic chemotactic factor released by MCs increases the permeability of the vascellum, since eosinophils are sometimes a part of inflammatory cell infiltration [166] Archana Yadav et al. showed that tryptase and chymase are critical in the pathogenesis of OSF and its malignant transformation [167]. Interestingly, MCs also contribute to increased epithelial proliferation as in OLK [168].
“Piece meal degranulation” of Mast cells also aggravates the process of OLP. The number of MCs was elevated in the OLP tissues when compared with the normal tissue [164, 169]. MCs can interact with T-cells and contribute to the OLP progression in different phases of OLP [170]. It acts as a destructor of the basement membrane [170]. For example, TNF-alpha can cause the elevation of matrix metalloproteinases (collagenase), which can destruct the basement membrane and increase the expression of adhesion molecules (E-selectin and ICAM) [170]. The characteristic trafficking of lymphocytes observed in OLP is also attributed to “Piece meal degranulation” of MCs. The vasopermeable function of histamine leads to submucosal edema and proliferation of T-cells. The antigen induced T-cells cause the degeneration of basal cells, and apoptosis of keratinocytes. Thus, characteristic Civatte bodies can be detected in OLP [170].
All these studies highlight an alternative therapeutic approach by which blocking MCs’ pro-tumor mediators might prevent OPMD from acquiring cancer characteristics.
Extracellular cues influence the progression of OPMD to OSCC
Beyond the effects of immune cells in the progression from OPMD to OSCC, the ECM has the capacity to influence the progression of OPMD. In fact, the ECM composition is a major predictor of the acquisition of a malignant behavior by OPMD. Potentially malignant oral lesions with high expression of FGF-2 and its receptors, FGFR-2 and FGFR-3, in the microenvironment are associated with the progression to OSCC [128]. OLK with high expression of tenascins and MMP-2, ECM glycoproteins, may predict the malignant potential of tobacco-associated OLK [171, 172]. Evidence also indicates that VEGFR-2 and MMP-9 protein levels are associated with epithelial dysplasia grading [173]. It has also been suggested that Nuclear factor-κB (NF-κB)-dependent cytokines (such as TNF-alpha, IL-1, IL-6, and IL-8) increase in saliva and OPMD tissues [174–177]. The NF-κB signaling pathway is critical in carcinogenesis, chemo-resistance, and protection from apoptosis in head and neck cancers [178–181]. It can mediate signal regulatory protein α (SIRPα), a cell-surface protein expressed on macrophages, to control macrophage plasticity [182]. Accumulating studies indicated that these cytokines may play critical roles in carcinogenesis and are associated with vessel density and worsened outcomes [175–177, 183]. Proteomics showed that distinct extracellular cues come from distinct cells, called the ‘matrisome’ [184]. All these data suggest that the surrounding and infiltrating extracellular cues may provide new opportunities to prevent OPMD from acquiring malignant characteristics.
Therapeutic strategies to remodel the microenvironment
Directly targeting various aspects of the cells in the OPMD tissue is the main therapeutic strategy against OPMD. However, the cells in the OPMD microenvironment are genetically stable compared to tissue lesional cells and, thus, are likely to be less susceptible to therapeutic resistance. Moreover, increasing studies indicated heterogeneity at different levels in OPMD lesional cells [185], suggesting that reeducating the microenvironment cells may become a new choice (Figure 4). Since, in the microenvironment, the cells have the paradoxical capacity of both promoting and impairing the progression from OPMD to OSCC, this plasticity must be taking into account when designing novel therapeutic strategies. One should think of manipulating and reprograming the cells in the microenvironment rather than to simply destruct or deplete the lesional cells. Although increasing immunotherapies generate exciting effects in cancer clinical trials [186–189], there is few examples of such cell manipulation and re-education approach to clinically prevent OPMD from acquiring OSCC characteristics. As the cells in the microenvironment change as OPMD progresses to OSCC, immunotherapies that can re-educate the microenvironment components may provide a new strategy to prevent OSCC.
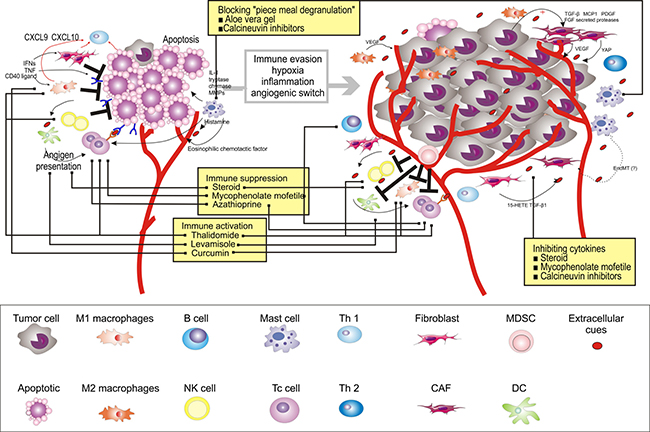
Figure 4: Therapeutic strategies to re-educate the OPMD (using OLP for as an example) microenvironment. Multiple strategies to target the microenvironment are currently tested in clinical trials, as indicated here, and referenced throughout the review. Immune activation, marked by stimulating T cells (e.g., thalidomide and levamisole), M1 (e.g., thalidomide and curcumin), and NK cells (e.g., thalidomide), is also a promising avenue of therapeutic intervention. Sequestration of cytokines within the microenvironment, in particular T cells (e.g., steroid, mycophenolate mofetil, and Azathioprine) and B cells (e.g., steroid and mycophenolate mofetil), can be achieved by inhibiting key cytokine pathways or NF-κB signaling pathway (e.g., steroid, mycophenolate mofetil, and calcineurin inhibitors). Alternatively, “piece meal degranulation” of mast cells can be blocked by using Aloe Vera gel and calcineurin inhibitors.
Various therapies aimed at re-educating the immune system, many of which currently focus on patients with OLP given their high numbers of lymphocytes [190]. Thalidomide is an immune-stimulant that can inhibit TNF-alpha and co-stimulate T cells, M1, and NK cells [191]. In the clinical trial for Thalidomide in patients with OLP, significant resolution of lesion and improvement in pain was observed [192]. Several immune-stimulants have also been used. For example, levamisole, an anthelminthic drug, presents an immune-modulating effect by potentiating the activity of interleukins, interferons, and T-cell-mediated immunity [193]. Another immune-therapy success story involves a different approach, using curcumin to activate M1 [194]. Interestingly, studies demonstrated that it is effective for the management of OLP with no side effects [195]. Taken together, these studies suggest that activation of normal immune cell functionality may provide optimal benefits to the patients; however, these trials are still at an early stage with small sample sizes, and the mechanisms underlying the efficacy of these drugs remain unclear. Moreover, the evaluation of the long-term impact of these drugs on the patients’ safety will take years.
Further emerging examples of microenvironment-directed therapies that do not focus on target cell depletion include sequestration of cytokines or chemokines [196]. Among the cell types and signaling molecules discussed here, strategies that inhibit key cytokine pathways or NF-κB signaling pathway are being investigated in clinical trials [193, 195, 197–214] (Figure 4). For example, steroids, the first-line treatment for OLP, may inhibit the production of cytokines and reduce the number of immune cells [195, 197–212, 215]. Several drugs have been combined with steroids to further improve its clinical efficacy [193]. For example, Azathioprine, a synthetic purine analog, was used in combination or in sequence with a steroid to treat patients with OLP [193]. It has been reported to interfere with T-cell activation. Similarly, mycophenolate mofetil has potent cytostatic effects on B cells and T cells and, thus, inhibits chemokines [214]. In addition, calcineurin inhibitors, namely cyclosporine, tacrolimus, and pimecrolimus, exert immunosuppressive effect by inhibiting cytokine expression [213]. They can also prevent mast cells from releasing cytokines that mediate inflammation [213]. Similarly, Aloe Vera gel, a type of nutraceutical, was used in patients with OLP. The Allianceae family inhibits antigen antibody-mediated release of histamine and leukotriene from mast cells [216]. Four RCTs have been reported, of which three reported significantly higher overall clinical improvement [216–219]. It is plausible that microenvironment-targeted therapies should aim at manipulating and reprograming the cells in the microenvironment rather than simply destructing or depleting the lesional cells.
CONCLUSIONS
The literature summarized in this review is encouraging for the OPMD microenvironment field and introduces new concepts and potential therapeutic strategies to reprogram the cells in the microenvironment. Nonetheless, challenges come with advances, how to identify and re-educate the cells and extracellular cues that are increasingly complex and interconnected with other components in the microenvironment remains unclear. Indeed, given that the OPMD microenvironment and mechanisms are not clear and broadly diverse during the progression from OPMD to OSCC, insights into the underlying mechanisms, how to deal with unsure response to current standard-of-care therapies by a diversity of cells will be a new challenge in this field. Another unclear question in the area is that cells in the microenvironment may be programed and sculpted by specific component(s) of OPMD and how this, in turn, results in stromal cell diversity. Additional points need be considered, including determining which patients to target, which classic therapies to combine with microenvironment-cell-targeted agents, whether intrinsic or acquired resistance like in cancer exists, and how to overcome it. However, from the studies summarized in this review, we now have a set of tools to solve these problems. For example, in order to choose specific therapies to different patients, we can select patients by analyzing the entire cells and the subset of cells in the OPMD microenvironment rather than by simply analyzing individual cells. Additionally, OPMD cell–directed agents will have to be combined with immunotherapies in a manner that allows researchers to identify the interactions between stromal cells and chemotherapies and targeted agents. Looking ahead, perhaps the most encouraging idea is that remodeling dysfunctional cells in the OPMD microenvironment could yield striking results in preventing OPMD from acquiring OSCC characteristics, as evidenced by the accumulating studies in the OPMD microenvironment field.
ACKNOWLEDGMENTS
This review was supported by the Sichuan University.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This work was supported by grants from the Nonprofit Industry Research Specific Fund of National Health and Family Planning Commission of China (No. 201502018), the National Natural Science Foundation of China (Nos 81470747, 81520108009, 81621062, 81200791, 81472533, 81102060, and 81270040), and 111 Project of MOE (B14038), China. The funding agencies had no role in the study design, the collection, analysis, or interpretation of data, the writing of the report, or the decision to submit the article for publication.
REFERENCES
1. Liu J, Mao JJ, Chen L. Epithelial-mesenchymal interactions as a working concept for oral mucosa regeneration. Tissue Eng Part B Rev. 2011; 17:25–31.
2. Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002; 420:860–867.
3. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008; 454:436–444.
4. Noone RB, Bonner H Jr, Raymond S, Brown AS, Graham WP 3rd, Lehr HB. Lymph node metastases in oral carcinoma. A correlation of histopathology with survival. Plast Reconstr Surg. 1974; 53:158–166.
5. Warnakulasuriya S, Johnson NW, van der Waal I. Nomenclature and classification of potentially malignant disorders of the oral mucosa. J Oral Pathol Med. 2007; 36:575–580.
6. Walsh T, Liu JL, Brocklehurst P, Glenny AM, Lingen M, Kerr AR, Ogden G, Warnakulasuriya S, Scully C. Clinical assessment to screen for the detection of oral cavity cancer and potentially malignant disorders in apparently healthy adults. Cochrane Database Syst Rev. 2013; CD010173.
7. Liu SC, Klein-Szanto AJ. Markers of proliferation in normal and leukoplakic oral epithelia. Oral Oncol. 2000; 36:145–151.
8. Beacham DA, Cukierman E. Stromagenesis: the changing face of fibroblastic microenvironments during tumor progression. Semin Cancer Biol. 2005; 15:329–341.
9. Amatangelo MD, Bassi DE, Klein-Szanto AJ, Cukierman E. Stroma-derived three-dimensional matrices are necessary and sufficient to promote desmoplastic differentiation of normal fibroblasts. Am J Pathol. 2005; 167:475–488.
10. Yeh CY, Lin CL, Chang MC, Chen HM, Kok SH, Chang SH, Kuo YS, Hahn LJ, Chan CP, Lee JJ, Jeng JH. Differences in oral habit and lymphocyte subpopulation affect malignant transformation of patients with oral precancer. J Formos Med Assoc. 2016; 115:263–268.
11. Li S, Lee YC, Li Q, Chen CJ, Hsu WL, Lou PJ, Zhu C, Pan J, Shen H, Ma H, Cai L, He B, Wang Y, et al. Oral lesions, chronic diseases and the risk of head and neck cancer. Oral Oncol. 2015; 51:1082–1087.
12. Rai B, Kaur J, Jacobs R, Singh J. Possible action mechanism for curcumin in pre-cancerous lesions based on serum and salivary markers of oxidative stress. J Oral Sci. 2010; 52:251–256.
13. Gallagher B, Wang Z, Schymura MJ, Kahn A, Fordyce EJ. Cancer incidence in New York State acquired immunodeficiency syndrome patients. Am J Epidemiol. 2001; 154:544–556.
14. Chiao EY, Krown SE. Update on non-acquired immunodeficiency syndrome-defining malignancies. Curr Opin Oncol. 2003; 15:389–397.
15. Singh B, Sabin S, Rofim O, Shaha A, Har-El G, Lucente FE. Alterations in head and neck cancer occurring in HIV-infected patients—results of a pilot, longitudinal, prospective study. Acta Oncol. 1999; 38:1047–1050.
16. Wistuba II, Behrens C, Gazdar AF. Pathogenesis of non-AIDS-defining cancers: a review. AIDS Patient Care STDS. 1999; 13:415–426.
17. Deeken JF, Tjen AL, Rudek MA, Okuliar C, Young M, Little RF, Dezube BJ. The rising challenge of non-AIDS-defining cancers in HIV-infected patients. Clin Infect Dis. 2012; 55:1228–1235.
18. Remick SC. Non-AIDS-defining cancers. Hematol Oncol Clin North Am. 1996; 10:1203–1213.
19. D'Costa J, Saranath D, Dedhia P, Sanghvi V, Mehta AR. Detection of HPV-16 genome in human oral cancers and potentially malignant lesions from India. Oral Oncol. 1998; 34:413–420.
20. Campisi G, Giovannelli L, Arico P, Lama A, Di Liberto C, Ammatuna P, D'Angelo M. HPV DNA in clinically different variants of oral leukoplakia and lichen planus. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2004; 98:705–711.
21. Furrer VE, Benitez MB, Furnes M, Lanfranchi HE, Modesti NM. Biopsy vs. superficial scraping: detection of human papillomavirus 6, 11, 16, and 18 in potentially malignant and malignant oral lesions. J Oral Pathol Med. 2006; 35:338–344.
22. Reed SG, Wahlquist AE. Adults With Oral High-risk Human Papillomavirus (HPV) and/or Smoking History Have a Higher Risk for Clinically Diagnosed Oral Premalignant Lesions. J Evid Based Dent Pract. 2015; 15:134–136.
23. Gupta S. Role of human papillomavirus in oral squamous cell carcinoma and oral potentially malignant disorders: A review of the literature. Indian J Dent. 2015; 6:91–98.
24. Dalla Torre D, Burtscher D, Edlinger M, Solder E, Widschwendter A, Rasse M, Puelacher W. Comparison of the prevalence of human papilloma virus infection in histopathologically confirmed premalignant oral lesions and healthy oral mucosa by brush smear detection. Oral Surg Oral Med Oral Pathol Oral Radiol. 2015; 119:333–339.
25. Llamas-Martinez S, Esparza-Gomez G, Campo-Trapero J, Cancela-Rodriguez P, Bascones-Martinez A, Moreno-Lopez LA, Garcia-Nunez JA, Cerero-Lapiedra R. Genotypic determination by PCR-RFLP of human papillomavirus in normal oral mucosa, oral leukoplakia and oral squamous cell carcinoma samples in Madrid (Spain). Anticancer Res. 2008; 28:3733–3741.
26. Nawandar DM, Wang A, Makielski K, Lee D, Ma S, Barlow E, Reusch J, Jiang R, Wille CK, Greenspan D, Greenspan JS, Mertz JE, Hutt-Fletcher L, et al. Differentiation-Dependent KLF4 Expression Promotes Lytic Epstein-Barr Virus Infection in Epithelial Cells. PLoS Pathog. 2015; 11:e1005195.
27. Miller CS, Johnstone BM. Human papillomavirus as a risk factor for oral squamous cell carcinoma: a meta-analysis, 1982–1997. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2001; 91:622–635.
28. Ostwald C, Rutsatz K, Schweder J, Schmidt W, Gundlach K, Barten M. Human papillomavirus 6/11, 16 and 18 in oral carcinomas and benign oral lesions. Med Microbiol Immunol. 2003; 192:145–148.
29. Acay RR, dos Santos E, de Sousa SO. Correlation between c-Jun and human papillomavirus in oral premalignant and malignant lesions. Oral Oncol. 2008; 44:698–702.
30. Fregonesi PA, Teresa DB, Duarte RA, Neto CB, de Oliveira MR, Soares CP. p16(INK4A) immunohistochemical overexpression in premalignant and malignant oral lesions infected with human papillomavirus. J Histochem Cytochem. 2003; 51:1291–1297.
31. Elamin F, Steingrimsdottir H, Wanakulasuriya S, Johnson N, Tavassoli M. Prevalence of human papillomavirus infection in premalignant and malignant lesions of the oral cavity in U.K. subjects: a novel method of detection. Oral Oncol. 1998; 34:191–197.
32. Sankari SL, Gayathri K, Balachander N, Malathi L. Candida in potentially malignant oral disorders. J Pharm Bioallied Sci. 2015; 7:S162–164.
33. Dilhari A, Weerasekera MM, Siriwardhana A, Maheshika O, Gunasekara C, Karunathilaka S, Nagahawatte A, Fernando N. Candida infection in oral leukoplakia: an unperceived public health problem. Acta Odontol Scand. 2016; 74:565–569.
34. Zeng X, Hou X, Wang Z, Jiang L, Xiong C, Zhou M, Chen Q. Carriage rate and virulence attributes of oral Candida albicans isolates from patients with oral lichen planus: a study in an ethnic Chinese cohort. Mycoses. 2009; 52:161–165.
35. Kamat MS, Vanaki SS, Puranik RS, Puranik SR, Kaur R. Oral Candida carriage, quantification, and species characterization in oral submucous fibrosis patients and healthy individuals. J Investig Clin Dent. 2011; 2:275–279.
36. Kazanowska-Dygdala M, Dus I, Radwan-Oczko M. The presence of Helicobacter pylori in oral cavities of patients with leukoplakia and oral lichen planus. J Appl Oral Sci. 2016; 24:18–23.
37. Bagan JV, Ramon C, Gonzalez L, Diago M, Milian MA, Cors R, Lloria E, Cardona F, Jimenez Y. Preliminary investigation of the association of oral lichen planus and hepatitis C. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1998; 85:532–536.
38. Hongal BP, Kulkarni VV, Deshmukh RS, Joshi PS, Karande PP, Shroff AS. Prevalence of fungal hyphae in potentially malignant lesions and conditions-does its occurrence play a role in epithelial dysplasia? J Oral Maxillofac Pathol. 2015; 19:10–17.
39. van der Waal I. Potentially malignant disorders of the oral and oropharyngeal mucosa; terminology, classification and present concepts of management. Oral Oncol. 2009; 45:317–323.
40. Anderson A, Ishak N. Marked variation in malignant transformation rates of oral leukoplakia. Evid Based Dent. 2015; 16:102–103.
41. Gupta K, Metgud R, Gupta J. Evaluation of stromal myofibroblasts in oral leukoplakia, oral submucous fibrosis, and oral squamous cell carcinoma--an immunohistochemical study. J Cancer Res Ther. 2015; 11:893–898.
42. Campisi G, Panzarella V, Giuliani M, Lajolo C, Di Fede O, Falaschini S, Di Liberto C, Scully C, Lo Muzio L. Human papillomavirus: its identity and controversial role in oral oncogenesis, premalignant and malignant lesions (review). Int J Oncol. 2007; 30:813–823.
43. Palefsky JM, Silverman S, Abdelsalaam M, Daniels TE, Greenspan JS. Association between Proliferative Verrucous Leukoplakia and Infection with Human Papillomavirus Type-16. J Oral Pathol Med. 1995; 24:193–197.
44. Rehani S, Rao NN, Rao A, Carnelio S, Ramakrishnaiah SH, Prakash PY. Spectrophotometric analysis of the expression of secreted aspartyl proteinases from Candida in leukoplakia and oral squamous cell carcinoma. J Oral Sci. 2011; 53:421–425.
45. Russell C, Jones JH. The histology of prolonged candidal infection of the rat's tongue. J Oral Pathol. 1975; 4:330–339.
46. Mitsuyasu RT. Non-AIDS-defining cancers. Top Antivir Med. 2014; 22:660–665.
47. Schulz TF. Cancer and viral infections in immunocompromised individuals. Int J Cancer. 2009; 125:1755–1763.
48. Vajdic CM, van Leeuwen MT. Cancer incidence and risk factors after solid organ transplantation. Int J Cancer. 2009; 125:1747–1754.
49. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010; 140:883–899.
50. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002; 23:549–555.
51. Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003; 3:23–35.
52. Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med. 1992; 176:287–292.
53. Cook J, Hagemann T. Tumour-associated macrophages and cancer. Curr Opin Pharmacol. 2013; 13:595–601.
54. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V, Setty M, Leslie CS, Oei Y, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013; 19:1264–1272.
55. Hagemann T, Lawrence T, McNeish I, Charles KA, Kulbe H, Thompson RG, Robinson SC, Balkwill FR. "Re-educating" tumor-associated macrophages by targeting NF-kappaB. J Exp Med. 2008; 205:1261–1268.
56. Wang HW, Joyce JA. Alternative activation of tumor-associated macrophages by IL-4: priming for protumoral functions. Cell Cycle. 2010; 9:4824–4835.
57. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010; 11:889–896.
58. Ohman J, Magnusson B, Telemo E, Jontell M, Hasseus B. Langerhans cells and T cells sense cell dysplasia in oral leukoplakias and oral squamous cell carcinomas--evidence for immunosurveillance. Scand J Immunol. 2012; 76:39–48.
59. Mills CD, Ley K. M1 and M2 macrophages: the chicken and the egg of immunity. J Innate Immun. 2014; 6:716–726.
60. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, Samokhvalov IM, Merad M. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010; 330:841–845.
61. Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, Prinz M, Wu B, Jacobsen SE, Pollard JW, Frampton J, Liu KJ, Geissmann F. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012; 336:86–90.
62. Jenkins SJ, Ruckerl D, Cook PC, Jones LH, Finkelman FD, van Rooijen N, MacDonald AS, Allen JE. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science. 2011; 332:1284–1288.
63. Varol C, Yona S, Jung S. Origins and tissue-context-dependent fates of blood monocytes. Immunol Cell Biol. 2009; 87:30–38.
64. Mori K, Haraguchi S, Hiori M, Shimada J, Ohmori Y. Tumor-associated macrophages in oral premalignant lesions coexpress CD163 and STAT1 in a Th1-dominated microenvironment. BMC Cancer. 2015; 15:573.
65. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008; 8:958–969.
66. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. 2000; 164:6166–6173.
67. Mills CD. M1 and M2 Macrophages: Oracles of Health and Disease. Crit Rev Immunol. 2012; 32:463–488.
68. Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009; 9:239–252.
69. Mills CD. Macrophage arginine metabolism to ornithine/urea or nitric oxide/citrulline: a life or death issue. Crit Rev Immunol. 2001; 21:399–425.
70. Mills CD, Shearer J, Evans R, Caldwell MD. Macrophage arginine metabolism and the inhibition or stimulation of cancer. J Immunol. 1992; 149:2709–2714.
71. Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol. 2006; 6:836–848.
72. Ohmori Y, Hamilton TA. Cell type and stimulus specific regulation of chemokine gene expression. Biochem Biophys Res Commun. 1994; 198:590–596.
73. Hiroi M, Ohmori Y. Constitutive nuclear factor κB activity is required to elicit interferon-gamma-induced expression of chemokine CXC ligand 9 (CXCL9) and CXCL10 in human tumour cell lines. Biochem J. 2003; 376:393–402.
74. Ohmori Y, Hamilton TA. The interferon-stimulated response element and a kappa B site mediate synergistic induction of murine IP-10 gene transcription by IFN-gamma and TNF-alpha. J Immunol. 1995; 154:5235–5244.
75. Altenburg A, Baldus SE, Smola H, Pfister H, Hess S. CD40 ligand-CD40 interaction induces chemokines in cervical carcinoma cells in synergism with IFN-gamma. J Immunol. 1999; 162:4140–4147.
76. Loetscher M, Gerber B, Loetscher P, Jones SA, Piali L, Clark-Lewis I, Baggiolini M, Moser B. Chemokine receptor specific for IP10 and mig: structure, function, and expression in activated T-lymphocytes. J Exp Med. 1996; 184:963–969.
77. Hsieh CS, Macatonia SE, O'Garra A, Murphy KM. T cell genetic background determines default T helper phenotype development in vitro. J Exp Med. 1995; 181:713–721.
78. O'Garra A, Murphy K. Role of cytokines in determining T-lymphocyte function. Curr Opin Immunol. 1994; 6:458–466.
79. Murphy KM, Reiner SL. The lineage decisions of helper T cells. Nat Rev Immunol. 2002; 2:933–944.
80. Mantovani A, Locati M. Orchestration of macrophage polarization. Blood. 2009; 114:3135–3136.
81. Gobert AP, Wilson KT. Editorial: Orchestration of macrophage polarization by polyamines. J Leukoc Biol. 2012; 91:677–679.
82. Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010; 32:593–604.
83. Varin A, Gordon S. Alternative activation of macrophages: immune function and cellular biology. Immunobiology. 2009; 214:630–641.
84. O'Sullivan T, Saddawi-Konefka R, Vermi W, Koebel CM, Arthur C, White JM, Uppaluri R, Andrews DM, Ngiow SF, Teng MW, Smyth MJ, Schreiber RD, Bui JD. Cancer immunoediting by the innate immune system in the absence of adaptive immunity. J Exp Med. 2012; 209:1869–1882.
85. Ma J, Liu L, Che G, Yu N, Dai F, You Z. The M1 form of tumor-associated macrophages in non-small cell lung cancer is positively associated with survival time. BMC Cancer. 2010; 10:112.
86. Ohri CM, Shikotra A, Green RH, Waller DA, Bradding P. Macrophages within NSCLC tumour islets are predominantly of a cytotoxic M1 phenotype associated with extended survival. Eur Respir J. 2009; 33:118–126.
87. Mori K, Hiroi M, Shimada J, Ohmori Y. Infiltration of m2 tumor-associated macrophages in oral squamous cell carcinoma correlates with tumor malignancy. Cancers (Basel). 2011; 3:3726–3739.
88. Boas DS, Takiya CM, Gurgel CA, Cabral MG, Santos JN. Tumor-infiltrating macrophage and microvessel density in oral squamous cell carcinoma. Braz Dent J. 2013; 24:194–199.
89. El-Rouby DH. Association of macrophages with angiogenesis in oral verrucous and squamous cell carcinomas. J Oral Pathol Med. 2010; 39:559–564.
90. Freedland SJ, Williams CD, Masko EM. Adiponectin and prostate cancer mortality: to be or not to be skinny? Clin Chem. 2010; 56:1–3.
91. Weber M, Iliopoulos C, Moebius P, Buttner-Herold M, Amann K, Ries J, Preidl R, Neukam FW, Wehrhan F. Prognostic significance of macrophage polarization in early stage oral squamous cell carcinomas. Oral Oncol. 2016; 52:75–84.
92. Dadi S, Chhangawala S, Whitlock BM, Franklin RA, Luo CT, Oh SA, Toure A, Pritykin Y, Huse M, Leslie CS, Li MO. Cancer Immunosurveillance by Tissue-Resident Innate Lymphoid Cells and Innate-like T Cells. Cell. 2016; 164:365–377.
93. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998; 392:245–252.
94. Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, Pulendran B, Palucka K. Immunobiology of dendritic cells. Annu Rev Immunol. 2000; 18:767–811.
95. Matheoud D, Perie L, Hoeffel G, Vimeux L, Parent I, Maranon C, Bourdoncle P, Renia L, Prevost-Blondel A, Lucas B, Feuillet V, Hosmalin A. Cross-presentation by dendritic cells from live cells induces protective immune responses in vivo. Blood. 2010; 115:4412–4420.
96. Duman RS, Nakagawa S, Malberg J. Regulation of adult neurogenesis by antidepressant treatment. Neuropsychopharmacology. 2001; 25:836–844.
97. Dunn GP, Old LJ, Schreiber RD. The Three Es of Cancer Immunoediting. Annual Review of Immunology. 2004; 22:329–360.
98. Zitvogel L, Tesniere A, Kroemer G. Cancer despite immunosurveillance: immunoselection and immunosubversion. Nat Rev Immunol. 2006; 6:715–727.
99. Sun J, Ming GL, Song H. Epigenetic regulation of neurogenesis in the adult mammalian brain. Eur J Neurosci. 33:1087–1093.
100. Yagyuu T, Hatakeyama K, Imada M, Kurihara M, Matsusue Y, Yamamoto K, Obayashi C, Kirita T. Programmed death ligand 1 (PD-L1) expression and tumor microenvironment: Implications for patients with oral precancerous lesions. Oral Oncol. 2017; 68:36–43.
101. Teng MW, Galon J, Fridman WH, Smyth MJ. From mice to humans: developments in cancer immunoediting. J Clin Invest. 2015; 125:3338–3346.
102. Lyford-Pike S, Peng S, Young GD, Taube JM, Westra WH, Akpeng B, Bruno TC, Richmon JD, Wang H, Bishop JA, Chen L, Drake CG, Topalian SL, et al. Evidence for a role of the PD-1:PD-L1 pathway in immune resistance of HPV-associated head and neck squamous cell carcinoma. Cancer Res. 2013; 73:1733–1741.
103. Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, Carbone DP, Gabrilovich DI. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol. 2001; 166:678–689.
104. Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer. 2013; 13:739–752.
105. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012; 12:253–268.
106. Mazzoni A, Bronte V, Visintin A, Spitzer JH, Apolloni E, Serafini P, Zanovello P, Segal DM. Myeloid suppressor lines inhibit T cell responses by an NO-dependent mechanism. J Immunol. 2002; 168:689–695.
107. Gabrilovich DI, Velders MP, Sotomayor EM, Kast WM. Mechanism of immune dysfunction in cancer mediated by immature Gr-1+ myeloid cells. J Immunol. 2001; 166:5398–5406.
108. Sinha P, Clements VK, Ostrand-Rosenberg S. Reduction of myeloid-derived suppressor cells and induction of M1 macrophages facilitate the rejection of established metastatic disease. J Immunol. 2005; 174:636–645.
109. Liu C, Yu S, Kappes J, Wang J, Grizzle WE, Zinn KR, Zhang HG. Expansion of spleen myeloid suppressor cells represses NK cell cytotoxicity in tumor-bearing host. Blood. 2007; 109:4336–4342.
110. Bunt SK, Yang L, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res. 2007; 67:10019–10026.
111. Bunt SK, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Inflammation Induces Myeloid-Derived Suppressor Cells that Facilitate Tumor Progression. J. Immunol. 2005; 176:284–290.
112. Jordan RC, Daley T. Oral squamous cell carcinoma: new insights. J Can Dent Assoc. 1997; 63:517–518, 521–515.
113. Saranath D, Bhoite LT, Deo MG. Molecular lesions in human oral cancer: the Indian scene. Eur J Cancer B Oral Oncol. 1993; 29B:107–112.
114. Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002; 3:349–363.
115. De Wever O, Demetter P, Mareel M, Bracke M. Stromal myofibroblasts are drivers of invasive cancer growth. Int J Cancer. 2008; 123:2229–2238.
116. Chaudhary M, Gadbail AR, Vidhale G, Mankar Gadbail MP, Gondivkar SM, Gawande M, Patil S. Comparison of myofibroblasts expression in oral squamous cell carcinoma, verrucous carcinoma, high risk epithelial dysplasia, low risk epithelial dysplasia and normal oral mucosa. Head Neck Pathol. 2012; 6:305–313.
117. Kapse SC, Rathod N, Baad R, Mandlik J, Sharma AS, Bommanavar S. Quantitative assessment of myofibroblast in severe dysplasia, microinvasion and oral squamous cell carcinoma: an immunohistochemical study. J Contemp Dent Pract. 2013; 14:34–38.
118. Zidar N, Gale N, Kambic V, Fischinger J. Proliferation of myofibroblasts in the stroma of epithelial hyperplastic lesions and squamous carcinoma of the larynx. Oncology. 2002; 62:381–385.
119. Angadi PV, Kale AD, Hallikerimath S. Evaluation of myofibroblasts in oral submucous fibrosis: correlation with disease severity. J Oral Pathol Med. 2011; 40:208–213.
120. Vered M, Allon I, Buchner A, Dayan D. Stromal myofibroblasts accompany modifications in the epithelial phenotype of tongue dysplastic and malignant lesions. Cancer Microenviron. 2009; 2:49–57.
121. Etemad-Moghadam S, Khalili M, Tirgary F, Alaeddini M. Evaluation of myofibroblasts in oral epithelial dysplasia and squamous cell carcinoma. J Oral Pathol Med. 2009; 38:639–643.
122. Weaver VM, Gilbert P. Watch thy neighbor: cancer is a communal affair. J Cell Sci. 2004; 117(Pt 8):1287–1290.
123. Lohr M, Schmidt C, Ringel J, Kluth M, Muller P, Nizze H, Jesnowski R. Transforming growth factor-beta1 induces desmoplasia in an experimental model of human pancreatic carcinoma. Cancer Res. 2001; 61:550–555.
124. Aoyagi Y, Oda T, Kinoshita T, Nakahashi C, Hasebe T, Ohkohchi N, Ochiai A. Overexpression of TGF-beta by infiltrated granulocytes correlates with the expression of collagen mRNA in pancreatic cancer. Br J Cancer. 2004; 91:1316–1326.
125. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006; 6:392–401.
126. Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007; 67:10123–10128.
127. Zhang L, Chen Y, Li G, Chen M, Huang W, Liu Y, Li Y. TGF-beta1/FGF-2 signaling mediates the 15-HETE-induced differentiation of adventitial fibroblasts into myofibroblasts. Lipids Health Dis. 2016; 15:2.
128. Nayak S, Goel MM, Makker A, Bhatia V, Chandra S, Kumar S, Agarwal SP. Fibroblast Growth Factor (FGF-2) and Its Receptors FGFR-2 and FGFR-3 May Be Putative Biomarkers of Malignant Transformation of Potentially Malignant Oral Lesions into Oral Squamous Cell Carcinoma. PLoS One. 2015; 10:e0138801.
129. De Wever O, Mareel M. Role of tissue stroma in cancer cell invasion. J Pathol. 2003; 200:429–447.
130. Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006; 7:131–142.
131. Nawshad A, Lagamba D, Polad A, Hay ED. Transforming growth factor-beta signaling during epithelial-mesenchymal transformation: implications for embryogenesis and tumor metastasis. Cells Tissues Organs. 2005; 179:11–23.
132. Orr B, Riddick AC, Stewart GD, Anderson RA, Franco OE, Hayward SW, Thomson AA. Identification of stromally expressed molecules in the prostate by tag-profiling of cancer-associated fibroblasts, normal fibroblasts and fetal prostate. Oncogene. 2012; 31:1130–1142.
133. Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP, Chaudhry SI, Harrington K, Williamson P, Moeendarbary E, Charras G, Sahai E. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol. 2013; 15:637–646.
134. Fukumura D, Xavier R, Sugiura T, Chen Y, Park EC, Lu N, Selig M, Nielsen G, Taksir T, Jain RK, Seed B. Tumor induction of VEGF promoter activity in stromal cells. Cell. 1998; 94:715–725.
135. Gandolfo M, Keszler A, Lanfranchi H, Itoiz ME. Increased subepithelial vascularization and VEGF expression reveal potentially malignant changes in human oral mucosa lesions. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2011; 111:486–493.
136. Blanpain C. Stem cells: Skin regeneration and repair. Nature. 2010; 464:686–687.
137. Alonso L, Fuchs E. Stem cells of the skin epithelium. Proc Natl Acad Sci U S A. 2003; 100:11830–11835.
138. Aida J, Izumiyama-Shimomura N, Nakamura K, Ishikawa N, Poon SS, Kammori M, Sawabe M, Arai T, Matsuura M, Fujiwara M, Kishimoto H, Takubo K. Basal cells have longest telomeres measured by tissue Q-FISH method in lingual epithelium. Exp Gerontol. 2008; 43:833–839.
139. Janes SM, Lowell S, Hutter C. Epidermal stem cells. J Pathol. 2002; 197:479–491.
140. Phinney DG, Prockop DJ. Concise review: mesenchymal stem/multipotent stromal cells: the state of transdifferentiation and modes of tissue repair--current views. Stem Cells. 2007; 25:2896–2902.
141. Beltrami AP, Cesselli D, Bergamin N, Marcon P, Rigo S, Puppato E, D'Aurizio F, Verardo R, Piazza S, Pignatelli A, Poz A, Baccarani U, Damiani D, et al. Multipotent cells can be generated in vitro from several adult human organs (heart, liver, and bone marrow). Blood. 2007; 110:3438–3446.
142. Zhang Z, Song J, Han Y, Mu D, Su S, Ji X, Liu H. Impairment of mesenchymal stem cells derived from oral leukoplakia. Int J Clin Exp Pathol. 2015; 8:10026–10037.
143. Tamamura R, Nagatsuka H, Siar CH, Katase N, Naito I, Sado Y, Nagai N. Comparative analysis of basal lamina type IV collagen alpha chains, matrix metalloproteinases-2 and -9 expressions in oral dysplasia and invasive carcinoma. Acta Histochem. 2013; 115:113–119.
144. Fan HX, Li HX, Chen D, Gao ZX, Zheng JH. Changes in the expression of MMP2, MMP9, and ColIV in stromal cells in oral squamous tongue cell carcinoma: relationships and prognostic implications. J Exp Clin Cancer Res. 2012; 31:90.
145. Le Bars P, Piloquet P, Daniel A, Giumelli B. Immunohistochemical localization of type IV collagen and laminin (alpha1) in denture stomatitis. J Oral Pathol Med. 2001; 30:98–103.
146. Galateau-Salle FB, Luna RE, Horiba K, Sheppard MN, Hayashi T, Fleming MV, Colby TV, Bennett W, Harris CC, Stetler-Stevenson WG, Liotta L, Ferrans VJ, Travis WD. Matrix metalloproteinases and tissue inhibitors of metalloproteinases in bronchial squamous preinvasive lesions. Hum Pathol. 2000; 31:296–305.
147. Gilbert L, He X, Farmer P, Boden S, Kozlowski M, Rubin J, Nanes MS. Inhibition of osteoblast differentiation by tumor necrosis factor-alpha. Endocrinology. 2000; 141:3956–3964.
148. Brailo V, Vucicevic-Boras V, Cekic-Arambasin A, Alajbeg IZ, Milenovic A, Lukac J. The significance of salivary interleukin 6 and tumor necrosis factor alpha in patients with oral leukoplakia. Oral Oncol. 2006; 42:370–373.
149. Wenghoefer M, Pantelis A, Najafi T, Deschner J, Allam JP, Novak N, Reich R, Martini M, Berge S, Fischer HP, Jepsen S, Winter J. Gene expression of oncogenes, antimicrobial peptides, and cytokines in the development of oral leukoplakia. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2010; 110:351–356.
150. Zhang Z, Han Y, Song J, Luo R, Jin X, Mu D, Su S, Ji X, Ren YF, Liu H. Interferon-gamma regulates the function of mesenchymal stem cells from oral lichen planus via indoleamine 2,3-dioxygenase activity. J Oral Pathol Med. 2015; 44:15–27.
151. Kannan R, Bijur GN, Mallery SR, Beck FM, Sabourin CL, Jewell SD, Schuller DE, Stoner GD. Transforming growth factor-alpha overexpression in proliferative verrucous leukoplakia and oral squamous cell carcinoma: an immunohistochemical study. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996; 82:69–74.
152. Tang L, Li N, Xie H, Jin Y. Characterization of mesenchymal stem cells from human normal and hyperplastic gingiva. J Cell Physiol. 2011; 226:832–842.
153. Luz-Crawford P, Kurte M, Bravo-Alegria J, Contreras R, Nova-Lamperti E, Tejedor G, Noel D, Jorgensen C, Figueroa F, Djouad F, Carrion F. Mesenchymal stem cells generate a CD4+CD25+Foxp3+ regulatory T cell population during the differentiation process of Th1 and Th17 cells. Stem Cell Res Ther. 2013; 4:65.
154. Kamal R, Dahiya P, Goyal N, Kumar M, Sharma N, Saini H. Mast cells and oral pathologies: A Review. J Nat Sci Biol Med. 2015; 6:35.
155. Telagi N, Ahmed Mujib BR, Kulkarni P, Naik R. The master switch: Comparative study of mast cell in oral epithelial dysplasia, oral submucous fibrosis and oral squamous cells carcinoma and their association with inflammation and angiogenesis. J Oral Maxillofac Pathol. 2015; 19:25.
156. Ribatti D, Crivellato E. Mast cells, angiogenesis, and tumour growth. Biochim Biophys Acta. 2012; 1822:2–8.
157. Conti P, Castellani ML, Kempuraj D, Salini V, Vecchiet J, Tetè S, Mastrangelo F, Perrella A, De Lutiis MA, Tagen M, Theoharides TC. Role of Mast Cells in Tumor Growth. Ann Clin Lab Sci. 2007; 37:315–322.
158. Sivapathasundharam B, Sharma B, Sriram G, Saraswathi TR. Immunohistochemical evaluation of mast cells and angiogenesis in oral squamous cell carcinoma. Indian J Dent Res. 2010; 21:260.
159. Michailidou EZ, Markopoulos AK, Antoniades DZ. VEGF expression from human dysplastic or malignant oral epithelium may be related to mast cell density and the subsequent angiogenetic phenomena. Int J Oral Max Surg. 2012; 41:1467–1473.
160. Newton SS, Thome J, Wallace TL, Shirayama Y, Schlesinger L, Sakai N, Chen J, Neve R, Nestler EJ, Duman RS. Inhibition of cAMP response element-binding protein or dynorphin in the nucleus accumbens produces an antidepressant-like effect. J Neurosci. 2002; 22:10883–10890.
161. Sathyakumar M, Sriram G, Saraswathi TR, Sivapathasundharam B. Immunohistochemical evaluation of mast cells and vascular endothelial proliferation in oral precancerous lesion-leukoplakia. J Oral Maxillofac Pathol. 2012; 16:343.
162. Ramsridhar S. Immunohistochemical Evaluation of Mast Cells in Leukoplakia and Oral Squamous Cell Carcinoma. J Clin and Diagn Res. 2016; 0:ZC100–103.
163. Ge S, Sailor KA, Ming GL, Song H. Synaptic integration and plasticity of new neurons in the adult hippocampus. J Physiol. 2008; 586:3759–3765.
164. Kinra M, Ramalingam K, Sarkar S, Rehman F, Girish KL. Comparison of mast cell count and mast cell density in normal mucosa, oral leukoplakia, oral lichen planus, oral submucous fibrosis and oral squamous cell carcinoma – a study on 50cases. J Pharm Sci Inn. 2012;1;4–11.
165. Pujari R, Vidya N. Mast cell density in oral submucous fibrosis: a possible role in pathogenesis. Int J Health Sci (Qassim). 2013; 7:23–29.
166. Nakagawa S, Kim JE, Lee R, Chen J, Fujioka T, Malberg J, Tsuji S, Duman RS. Localization of phosphorylated cAMP response element-binding protein in immature neurons of adult hippocampus. J Neurosci. 2002; 22:9868–9876.
167. Teh MT, Yadav A, Desai RS, Bhuta BA, Singh JS, Mehta R, Nehete AP. Altered Immunohistochemical Expression of Mast Cell Tryptase and Chymase in the Pathogenesis of Oral Submucous Fibrosis and Malignant Transformation of the Overlying Epithelium. PLoS ONE. 2014; 9:e98719.
168. Sabarinath B, Sriram G, Saraswathi TR, Sivapathasundharam B. Immunohistochemical evaluation of mast cells and vascular endothelial proliferation in oral submucous fibrosis. Indian J Dent Res. 2011; 22:116–121.
169. Jontell M, Hansson HA, Nygren H. Mast cells in oral lichen planus. J Oral Pathol. 1986; 15:273–5.
170. Zhao ZZ, Savage NW, Sugerman PB, Walsh LJ. Mast cell/T cell interactions in oral lichen planus. J Oral Pathol Med. 2002; 31:189–195.
171. Mane DR, Bhat K, Kale AD, Hallikerimath S. Immunoexpression of tenascin as a predictor of the malignancy potential of oral leukoplakia associated with a tobacco habit. Biotechnic & Histochemistry. 2015; 90:544–551.
172. Bajracharya D, Shrestha B, Kamath A, Menon A, Radhakrishnan R. Immunohistochemical Correlation of Matrix Metalloproteinase-2 and Tissue Inhibitors of Metalloproteinase-2 in Tobacco Associated Epithelial Dysplasia. Dis Markers. 2014; 2014:1–9.
173. de Carvalho Fraga CA, Farias LC, de Oliveira MV, Domingos PL, Pereira CS, Silva TF, Roy A, Gomez RS, de Paula AM, Guimaraes AL. Increased VEGFR2 and MMP9 protein levels are associated with epithelial dysplasia grading. Pathol Res Pract. 2014; 210:959–964.
174. Rhodus NL, Ho V, Miller CS, Myers S, Ondrey F. NF-kappaB dependent cytokine levels in saliva of patients with oral preneoplastic lesions and oral squamous cell carcinoma. Cancer Detect Prev. 2005; 29:42–45.
175. Piva MR, DE Souza LB, Martins-Filho PR, Nonaka CF, DE Santana Santos T, DE Souza Andrade ES, Piva D. Role of inflammation in oral carcinogenesis (Part II): CD8, FOXP3, TNF-alpha, TGF-beta and NF-kappaB expression. Oncol Lett. 2013; 5:1909–1914.
176. Chang KP, Kao HK, Wu CC, Fang KH, Chang YL, Huang YC, Liu SC, Cheng MH. Pretreatment interleukin-6 serum levels are associated with patient survival for oral cavity squamous cell carcinoma. Otolaryngol Head Neck Surg. 2013; 148:786–791.
177. Chen Z, Malhotra PS, Thomas GR, Ondrey FG, Duffey DC, Smith CW, Enamorado I, Yeh NT, Kroog GS, Rudy S, McCullagh L, Mousa S, Quezado M, et al. Expression of proinflammatory and proangiogenic cytokines in patients with head and neck cancer. Clin Cancer Res. 1999; 5:1369–1379.
178. van der Waal I. Potentially malignant disorders of the oral and oropharyngeal mucosa; present concepts of management. Oral Oncol. 2010; 46:423–425.
179. Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, Gutkovich-Pyest E, Urieli-Shoval S, Galun E, Ben-Neriah Y. NF-kB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004; 431:461–6.
180. Aggarwal BB. Nuclear factor-kappaB: the enemy within. Cancer Cell. 2004; 6:203–208.
181. Shishodia S, Aggarwal BB. Nuclear factor-kappaB activation mediates cellular transformation, proliferation, invasion angiogenesis and metastasis of cancer. Cancer Treat Res. 2004; 119:139–173.
182. Ye X, Zhang J, Lu R, Zhou G. Signal regulatory protein alpha associated with the progression of oral leukoplakia and oral squamous cell carcinoma regulates phenotype switch of macrophages. Oncotarget. 2016; 7:81305–81321. https://doi.org/10.18632/oncotarget.12874.
183. Cohen RF, Contrino J, Spiro JD, Mann EA, Chen LL, Kreutzer DL. Interleukin-8 expression by head and neck squamous cell carcinoma. Arch Otolaryngol Head Neck Surg. 1995; 121:202–209.
184. Naba A, Clauser KR, Hoersch S, Liu H, Carr SA, Hynes RO. The Matrisome: In Silico Definition and In Vivo Characterization by Proteomics of Normal and Tumor Extracellular Matrices. Mol Cell Proteomics. 2012; 11:M111.014647.
185. Gomes CC, Fonseca-Silva T, Galvao CF, Friedman E, De Marco L, Gomez RS. Inter- and intra-lesional molecular heterogeneity of oral leukoplakia. Oral Oncol. 2015; 51:178–181.
186. Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012; 12:265–277.
187. Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer. 2012; 12:237–251.
188. Sharma P, Wagner K, Wolchok JD, Allison JP. Novel cancer immunotherapy agents with survival benefit: recent successes and next steps. Nat Rev Cancer. 2011; 11:805–812.
189. Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol. 2012; 12:269–281.
190. Roopashree MR, Gondhalekar RV, Shashikanth MC, George J, Thippeswamy SH, Shukla A. Pathogenesis of oral lichen planus--a review. J Oral Pathol Med. 2010; 39:729–734.
191. Jin X, Lu S, Xing X, Wang L, Mu D, He M, Huang H, Zeng X, Chen Q. Thalidomide: features and potential significance in oral precancerous conditions and oral cancer. J Oral Pathol Med. 2013; 42:355–362.
192. Wu Y, Zhou G, Zeng H, Xiong CR, Lin M, Zhou HM. A randomized double-blind, positive-control trial of topical thalidomide in erosive oral lichen planus. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2010; 110:188–195.
193. Kini R, Nagaratna DV, Saha A, Praveen BN. Therapeutic Management of Oral Lichen Planus: A Review for the Clinicians. World Journal of Dentistry. 2011; 2:249–253.
194. Arun P, Brown MS, Ehsanian R, Chen Z, Van Waes C. Nuclear NF-kappaB p65 phosphorylation at serine 276 by protein kinase A contributes to the malignant phenotype of head and neck cancer. Clin Cancer Res. 2009; 15:5974–5984.
195. Kia SJ, Shirazian S, Mansourian A, Khodadadi Fard L, Ashnagar S. Comparative Efficacy of Topical Curcumin and Triamcinolone for Oral Lichen Planus: A Randomized, Controlled Clinical Trial. J Dent (Tehran). 2015; 12: 789–796.
196. Coussens LM, Zitvogel L, Palucka AK. Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science. 2013; 339:286–291.
197. Yoke PC, Tin GB, Kim MJ, Rajaseharan A, Ahmed S, Thongprasom K, Chaimusik M, Suresh S, Machin D, Bee WH, Seldrup J, Asian Lichen Planus Study G. A randomized controlled trial to compare steroid with cyclosporine for the topical treatment of oral lichen planus. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2006; 102:47–55.
198. Dalirsani Z, Taghavi Zenouz A, Mehdipour M, Alavi F, Javadzadeh Y. Comparison of the effect of combination of triamcinolone acetonide and vitamin a mouthwash with triamcinolone mouthwash alone on oral lichen planus. J Dent Res Dent Clin Dent Prospects. 2010; 4:21–24.
199. Xia J, Li C, Hong Y, Yang L, Huang Y, Cheng B. Short-term clinical evaluation of intralesional triamcinolone acetonide injection for ulcerative oral lichen planus. J Oral Pathol Med. 2006; 35:327–331.
200. Motta AC, Domaneschi C, Komesu MC, Souza Cda S, Aoki V, Migliari DA. Double-blind, crossover, placebo-controlled clinical trial with clobetasol propionate in desquamative gingivitis. Braz Dent J. 2009; 20:231–236.
201. Carbone M, Conrotto D, Carrozzo M, Broccoletti R, Gandolfo S, Scully C. Topical corticosteroids in association with miconazole and chlorhexidine in the long-term management of atrophic-erosive oral lichen planus: a placebo-controlled and comparative study between clobetasol and fluocinonide. Oral Dis. 1999; 5:44–49.
202. Carbone M, Goss E, Carrozzo M, Castellano S, Conrotto D, Broccoletti R, Gandolfo S. Systemic and topical corticosteroid treatment of oral lichen planus: a comparative study with long-term follow-up. J Oral Pathol Med. 2003; 32:323–329.
203. Hegarty AM, Hodgson TA, Lewsey JD, Porter SR. Fluticasone propionate spray and betamethasone sodium phosphate mouthrinse: A randomized crossover study for the treatment of symptomatic oral lichen planus. J Am Acad Dermatol. 2002; 47:271–279.
204. Malhotra AK, Khaitan BK, Sethuraman G, Sharma VK. Betamethasone oral mini-pulse therapy compared with topical triamcinolone acetonide (0.1%) paste in oral lichen planus: A randomized comparative study. J Am Acad Dermatol. 2008; 58:596–602.
205. Liu C, Xie B, Yang Y, Lin D, Wang C, Lin M, Ge L, Zhou H. Efficacy of intralesional betamethasone for erosive oral lichen planus and evaluation of recurrence: a randomized, controlled trial. Oral Surg Oral Med Oral Pathol Oral Radiol. 2013; 116:584–590.
206. Buajeeb W, Pobrurksa C, Kraivaphan P. Efficacy of fluocinolone acetonide gel in the treatment of oral lichen planus. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000; 89:42–45.
207. Thongprasom K, Chaimusig M, Korkij W, Sererat T, Luangjarmekorn L, Rojwattanasirivej S. A randomized-controlled trial to compare topical cyclosporin with triamcinolone acetonide for the treatment of oral lichen planus. J Oral Pathol Med. 2007; 36:142–146.
208. Ungphaiboon S, Nittayananta W, Vuddhakul V, Maneenuan D, Kietthubthew S, Wongpoowarak W, Phadoongsombat N. Formulation and efficacy of triamcinolone acetonide mouthwash for treating oral lichen planus. Am J Health Syst Pharm. 2005; 62:485–491.
209. Carbone M, Arduino PG, Carrozzo M, Caiazzo G, Broccoletti R, Conrotto D, Bezzo C, Gandolfo S. Topical clobetasol in the treatment of atrophic-erosive oral lichen planus: a randomized controlled trial to compare two preparations with different concentrations. J Oral Pathol Med. 2009; 38:227–233.
210. Cilurzo F, Gennari CG, Selmin F, Epstein JB, Gaeta GM, Colella G, Minghetti P. A new mucoadhesive dosage form for the management of oral lichen planus: formulation study and clinical study. Eur J Pharm Biopharm. 2010; 76:437–442.
211. Lee YC, Shin SY, Kim SW, Eun YG. Intralesional injection versus mouth rinse of triamcinolone acetonide in oral lichen planus: a randomized controlled study. Otolaryngol Head Neck Surg. 2013; 148:443–449.
212. Sonthalia S, Singal A. Comparative efficacy of tacrolimus 0.1% ointment and clobetasol propionate 0.05% ointment in oral lichen planus: a randomized double-blind trial. Int J Dermatol. 2012; 51:1371–1378.
213. McCaughey C, Machan M, Bennett R, Zone JJ, Hull CM. Pimecrolimus 1% cream for oral erosive lichen planus: a 6-week randomized, double-blind, vehicle-controlled study with a 6-week open-label extension to assess efficacy and safety. J Eur Acad Dermatol Venereol. 2011; 25:1061–1067.
214. Wee JS, Shirlaw PJ, Challacombe SJ, Setterfield JF. Efficacy of mycophenolate mofetil in severe mucocutaneous lichen planus: a retrospective review of 10 patients. Br J Dermatol. 2012; 167:36–43.
215. Thongprasom K, Dhanuthai K. Steriods in the treatment of lichen planus: a review. J Oral Sci. 2008; 50:377–385.
216. Salazar-Sanchez N, Lopez-Jornet P, Camacho-Alonso F, Sanchez-Siles M. Efficacy of topical Aloe vera in patients with oral lichen planus: a randomized double-blind study. J Oral Pathol Med. 2010; 39:735–740.
217. Mansourian A, Momen-Heravi F, Saheb-Jamee M, Esfehani M, Khalilzadeh O, Momen-Beitollahi J. Comparison of aloe vera mouthwash with triamcinolone acetonide 0.1% on oral lichen planus: a randomized double-blinded clinical trial. Am J Med Sci. 2011; 342:447–451.
218. Reddy RL, Reddy RS, Ramesh T, Singh TR, Swapna LA, Laxmi NV. Randomized trial of aloe vera gel vs triamcinolone acetonide ointment in the treatment of oral lichen planus. Quintessence Int. 2012; 43:793–800.
219. Choonhakarn C, Busaracome P, Sripanidkulchai B, Sarakarn P. The efficacy of aloe vera gel in the treatment of oral lichen planus: a randomized controlled trial. Br J Dermatol. 2008; 158:573–577.