
Introduction
Myeloablative conditioning regimens comprising the bi-functional alkylating agent busulfan (BU) were introduced in the late 1970s as an alternative to total body irradiation [1]. Since then BU has been extensively used, especially in combination with cyclophosphamide in patients undergoing hematopoietic stem cell transplantation (HSCT) [2]. Studies in children and adults demonstrate reduced toxicity and increased efficacy, when the BU area under the curve concentration (AUC) is within optimal target range [3, 4]. Hence, dose adjustment guided by therapeutic drug monitoring is performed to prevent treatment-related toxicities. In a recent study comparing the performance of 12 pediatric dosing guidelines, the therapeutic target window can be reached in approximately 51% to 74% in pediatric cases and 45% to 64% in infants [5]. Nevertheless, the authors and others also caution about emerging evidence that the therapeutic AUC range of 3.7 - 6.2 mg.h/L per dose (equivalent ~ steady state concentration, Css, of 615 - 1031.3 ng/mL) should not be universally applied as the optimal AUC may depend on the indication for transplant or other patient-related factors [5-8]. Recently, a narrower optimal i.v. BU cumulative AUC of 78-101 mg.h/L (equivalent to a Css of ~ 830-1050 ng/mL) has been suggested to improve outcomes irrespective of disease condition, based on the analysis of 674 pediatric patients [4]. This proposed narrower therapeutic window constitutes a need to identify the underlying factors responsible for the inter-individual variability of BU clearance (CL), particularly at first dose before BU adjustment.
BU is eliminated via conjugation with glutathione catalyzed by glutathione S-transferase enzymes (GST) especially by the Glutathione S Transferase Alpha1 (GSTA1) isoform followed, to a lesser extent, by the Glutathione S Transferase Mu 1 (GSTM1) and Glutathione S Transferase Pi 1 (GSTP1) [9]. Factors that could affect the metabolism or elimination of BU are the availability of glutathione, efflux of conjugates by transporter proteins Unmarked set by huezodia and the different activity of GSTs [2, 10]. The GSTs are highly polymorphic; the promoter region of GSTA1 contains polymorphic variants that influence enzyme function [3, 11, 12]. A null variant is encountered for GSTM1, whereby the entire gene is deleted in a considerable proportion of different populations, resulting in the complete absence of the corresponding enzyme activity [13]. GSTP1 contains the 313A>G polymorphism leading to an Ile-to-Val substitution that has shown to decrease enzyme activity [14]. Our team and other groups have investigated genetic variants in GSTs for their association with BU exposure and/or clinical outcomes [15-33], summarized in Supplementary Table 1. Most studies demonstrated an association between BU pharmacokinetics (PK) and GSTA1-69 C>T, [15-28] which delineates haplotypes *A and *B. Nevertheless, functional assessment of GSTA1 sub-haplotypes and more detailed insight into their relationship with clinical outcomes in a larger patient population is still lacking. In this report, we analyzed promoter activity of each GSTA1 haplotype subgroup and have extended our previous analysis of pediatric patients from a single center [20] to a larger multicenter cohort to validate the association of GST genes, particularly GSTA1 haplotype combinations as diplotypes, with BU exposure and clinical outcomes of HSCT.
Results
Functional characterization of the GSTA1 polymorphisms
To explore how GSTA1 function is related to each GSTA1 haplotype (Figure 1a), we estimated promoter activity by luciferase gene reporter using six haplotype constructs that were transiently transfected in human hepatoma (HEPG2) cells. Results are illustrated in Figure 1b, where a significant increase of luciferase activity was observed when *A1 was mutated at position -631 forming *A2 and at position -1142 forming*A3 haplotype (p < 0.001). In contrast, the promoter activity was significantly decreased in the case of all *B haplotypes that are conjunctly delineated from haplotype *A by changes at three positions in full linkage disequilibrium (-52, -69, -567). The lowest activity among *B haplotypes was observed for *B1b (defined by position -513, p = 0.00001) that equalled the activity of the promoterless plasmid.
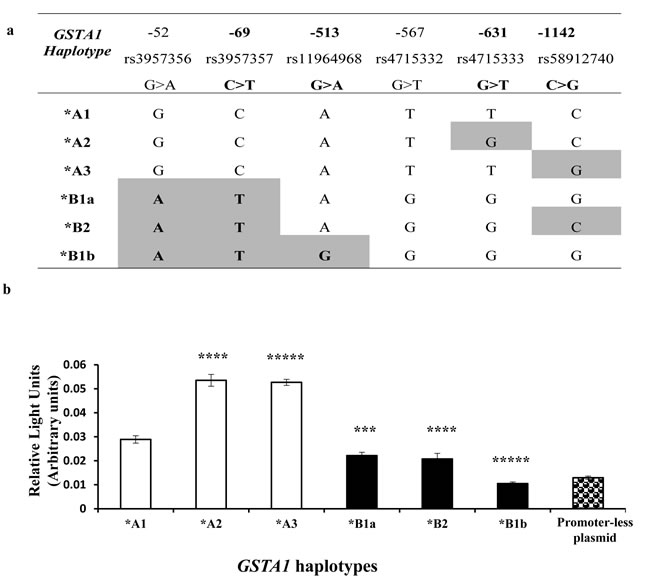
Figure 1: GSTA1 Haplotype and Reporter Gene assay of GSTA1 promoter. A. Haplotypes investigated with luciferase reporter assay. rs (reference SNP ID) numbers correspond to each SNP included for site directed mutagenesis. SNPs used for genotyping and for inferring sub-haplotypes in patients are highlighted in bold. B. Luciferase activities of the proximal promoters of GSTA1 variants (GSTA1*A1, GSTA1*A2, GSTA1*A3, GSTA1*B1a, GSTA1*B2, GSTA1*B1b) in transient transfection in HepG2 cells. Error bars represent the standard deviations. Pairwise comparisons by analysis of variance (ANOVA) between data for the GSTA1*A1 vs. any other haplotype, after Bonferroni correction *** = p < 0.001; ****= p <0.0001, ***** = p < 0.000001.
Pharmacogenetics vs pharmacokinetics and dose requirement
Based on the functional effect of each haplotype, predicted activity of each diplotype and the relationship with CL (ml.min/kg), four major functional GSTA1 groups were revealed Table 1; I (in 9.4% cases), defined by two copies of rapid metabolizing alleles, mostly represented by *A2*A2 individuals, group IV (14.5%) represented by two copies of slow metabolizing alleles (defined in all cases but one by*B1a*B1a diplotype) and by the presence of one copy of very slow metabolizing *B1b allele. Group II (28.2%) and III (47.8%) had intermediate to normal metabolizing capacity and were defined by the presence of *A2 and *A1, respectively. Linear effect was observed, whereby group I demonstrated highest and group IV lowest CL (p = 0.009, Figure 2a) with even more evident correlation seen in girls (p ≤ 0.0005, Figure 2b).
Table 1: GSTA1 diplotype frequencies in the study population and proposed functional groups
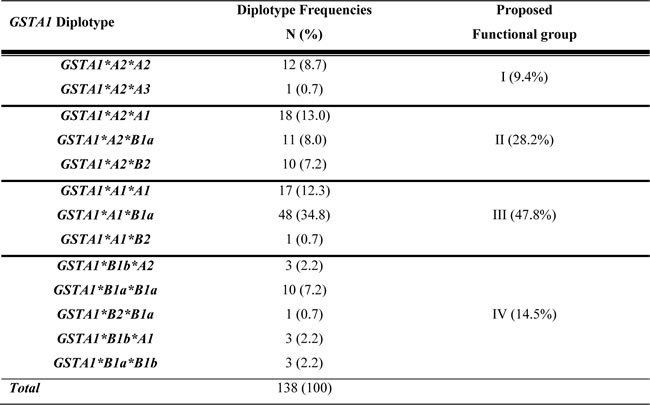
Abbreviations: GSTA1: glutathione S transferase alpha1. The proposed functional groups arise from in vitro reporter-gene assays and in vivo PK data performed in this study. Group I: considered as rapid metabolizers; Group II: considered as intermediate metabolizers; Group III: considered as normal metabolizers and Group IV: considered as slow metabolizers.
Due to the difference in dose adjustment across participating centers, the ratio of adjusted to initial dose and cumulative AUC obtained in a single center (University Hospital Center Sainte Justine, Montreal) was compared among GSTA1 diplotype groups. Patients in group I had on average a higher dose requirement compared to the other patients, whereas group IV cases had on average very little change from the initial dose (p = 0.03, Figure 2c). Cumulative AUC was also significantly associated in an additive manner with four diplotype groups with the highest exposure seen in the group IV (p = 0.01, Figure 2d).
Table 2: GSTM1 and GSTP1 genotypes and minor allele frequencies in the study population
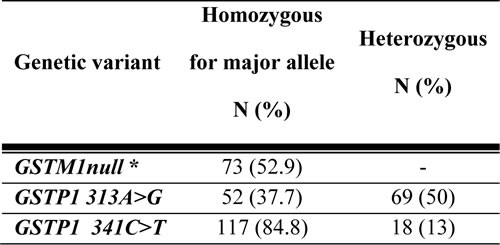
Abbreviations: GSTM1: glutathione S transferase Mu1; GSTM1 null- homozygous individuals for deletion; GSTP1: glutathione S transferase Pi1. *Distinction cannot be made between GSTM1 non-null heterozygous and homozygous individuals by the method used in the study, therefore observed frequencies could not be provided. GSTM1 null individuals are considered as homozygous for major allele.
Genotype frequencies for GSTM1 and GSTP1 variants are summarized in Table 2. When all patients were analyzed there was no significant association between PK and GSTP1 or GSTM1 genotypes (Table 3). But BU CL was associated with GSTM1 genotypes in children above 4 years of age (Figure 2e). GSTA1 diplotypes correlated with different ethnicities with group I being more frequent in other populations than in Caucasians (p < 0.0005). Among non-genetic factors, CL adjusted for weight correlated significantly with age (p < 0.0005). Age (p < 0.0005), gender (p = 0.09) and GSTA1 diplotypes (p = 0.01) were retained in the final multivariate linear model that explained 28% of overall variability in BU CL (Table 3). The diplotype contribution was relatively minor (5%) when all diplotypes and all patients are included in the analysis. However, when all girls (n = 64) or patients with diplotype groups I and IV representing the extreme of CL distribution (n = 33) were considered, diplotype contribution increased to 19% and 23%, respectively, and the model explained 46% and 42% of variability in CL.
Table 3: Relationship between GST genotypes and the clearance in univariate and multivariate linear regression
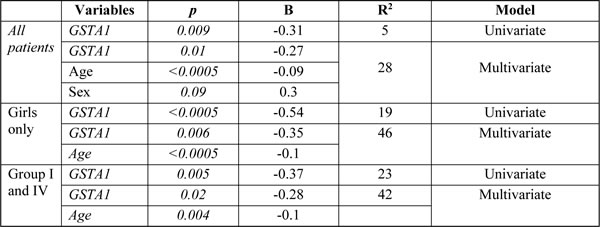
Co-variables include age (continuous), sex (dichotomized), conditioning regimen (busulfan-cyclophophamide vs others) and diagnosis (malignant versus non-malignant); Co-variables with p<0.1 were retained in final model. When all patients and girls only were analyzed, GSTA1 included all 4 diplotype groups; B, unstandardized coefficient, R2, % of variability explained by the genotype or the model (in multivariate analysis).
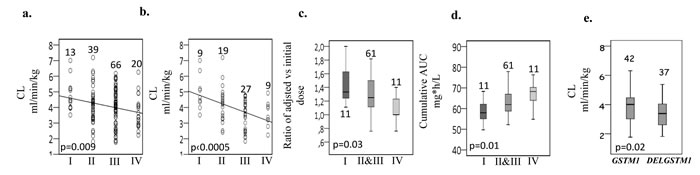
Figure 2: Pharmacokinetic parameters of BU and dose requirement in relation to GSTA1 functional diplotype groups and GSTM1 genotypes. A. Busulfan first dose clearance (CL, in ml/min/kg) against GSTA1 diplotypes B. Busulfan first dose CL in females only against GSTA1 genotypes. C. Dose requirement (ratio of adjusted to initial dose) against GSTA1 diplotypes. D. Cumulative AUC (mg.h/L) against GSTA1 diplotypes. CHU Sainte-Justine patients only were included for analysis presented in C and D. Diplotype groups II and III were combined into a single group in C and D. E. Busulfan first dose clearance in children above 4 yrs of age against GSTM1 genotypes. DELGSTM = Deleted GSTM1 gene. Number of patients and p values are depicted on the plots.
Pharmacogenetics vs HSCT-related toxicities
Analyses between HSCT-related toxicities and the four GSTA1 diplotype groups revealed a strong association with Sinusoidal Obstruction Syndrome (SOS) (p < 0.0005; Figure 3a), whereby Group IV carriers had seven-fold higher risk of SOS (HR = 7.1; 95% CI: 2.5-20.4) compared to patients with other GSTA1 diplotypes. Likewise, group IV carriers were also associated with the highest risk of acute Graft versus Host Disease (aGvHD), grades 1-4 (p < 0.0005, Figure 3b) and with Treatment Related Toxicity (TRT: combining SOS, hemorrhagic cystitis, lung toxicity and aGvHD grades 1-4, p < 0.0005, Figure 3c). The association between GSTA1 diplotypes and TRT was also maintained if aGvHD grades 2-4 were included in the analysis rather than all grades of aGvHD (p = 0.03, Figure 3d). Individuals with group IV who received BU-cyclophosphamide conditioning regimen had also lowest overall survival (OS) compared to group I (p = 0.02, Figure 3e). This was likely due to the high TRT rates, since when the cumulative incidence of competing events analysis was performed, with TRT, relapse and death as competing events, only the association with TRT remained significant (p < 0.0005, Figure 3f).
Table 4: Relationship between GST genotypes and other variables with clinical outcomes in univariate and multivariate logistic regression
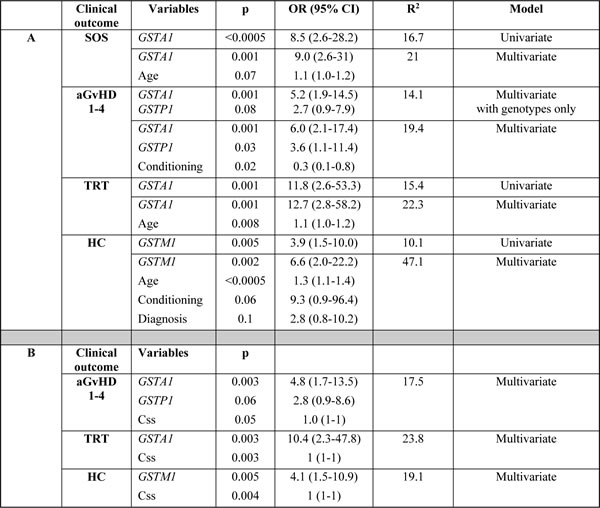
In A: co-variables include age, sex, diagnosis and conditioning regimen; in B: co-variable is Busulfan exposure represented as Busulfan Steady state concentration (Css). Co-variables with p<0.1 were retained in final model. Dichotomized variables were GSTP1 (GG vs others), GSTA1 (group IV vs others), GSTM1 (non-null vs null), conditioning regimen (busulfan-cyclophosphamide vs others) and diagnosis (malignant versus non-malignant) whereas Css and age were continuous variables, OR, odds ratio, CI, confidence interval, R2, % of variability explained by the genotype or the model (in multivariate analysis).
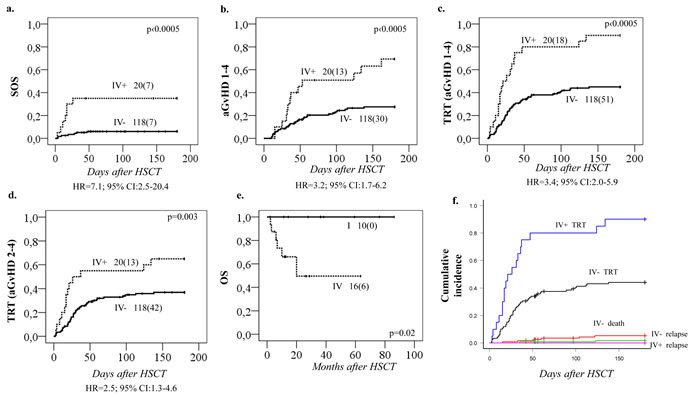
Figure 3: Incidence of SOS, aGvHD and TRT in relation to GSTA1 functional diplotype groups. Cumulative incidences of A. sinusoidal obstruction syndrome (SOS), B. acute graft versus host disease (aGvHD) 1-4, C. treatment related toxicity (TRT) including aGvHD 1-4 and D. TRT including aGvHD 2-4. Results plotted for diplotype group IV (IV+) versus groups I, II & III (IV-). E. Overall survival (OS) in relation to GSTA1 extreme diplotype status (group I vs. group IV), in patients who received busulfan-cyclophosphamide conditioning regimen. Total number of patients represented by each curve with number of patients with indicated toxicities in parenthesis, and p value are depicted on each plot; group IV associated hazard ratios are depicted below each plot. F. Association of TRT with diplotype group IV in a competing events risk analysis. IV+ and IV- indicates the presence of this GSTA1 diplotype group. Competing events for TRT incidence were: death and relapse. p values for the difference in cumulative incidence of TRT, death, and relapse, between haplotype groups (IV vs others) is 0.000003, 0.3 and 0.5, respectively.
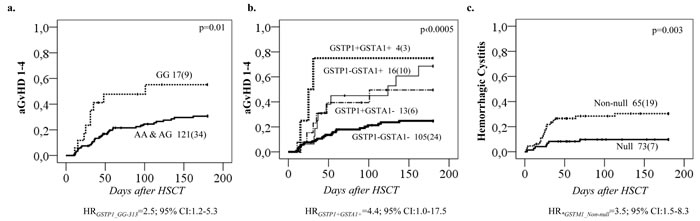
Figure 4: Complications of HSCT in relation to GSTP1 genotypes and GSTA1 diplotypes. A. Acute GvHD 1-4 incidence according to GSTP1 c.313A>G genotypes; and B. Acute GvHD 1-4 incidence according to combinatory GSTA1-GSTP1 status. A plus sign represents the risk genotypes, which is presence of GSTP1*GG and/or GSTA1 diplotype group IV. C. Hemorrhagic Cystitis (HC) incidences in relation to GSTM1 Null and Non-null genotype.
Regarding the remaining GST genes, GSTP1 (GG 313) was associated with a higher probability of aGvHD 1-4 (p = 0.01, Figure 4a). This effect was independent of GSTA1 haplotype and each genotype (GSTP1 GG 313 and GSTA1 group IV) independently and combined contributed to aGvHD development with highest risk seen in individuals with both risk genotypes (p < 0.0005, Figure 4b). Incidence of hemorrhagic cystitis was higher in GSTM1 non-null individuals compared to patients with GSTM1 deletion (p = 0.003, Figure 4c). Css after first dose categorized according to historical target correlated with event free survival (EFS), OS and TRT (p < 0.0005) (Figure 5a). Css above 900 ng/mL was associated with TRT irrespective of diplotype groups (Figure 6a), whereas high risk of TRT for Css below 900 ng/mL was evident only for group IV carriers (Figure 6b). In multiple logistic regression models the best predictors of SOS and TRT were age and GSTA1 diplotypes (Table 4a), explaining 21-22% of variability of which 15-17% was attributed to GSTA1. For aGvHD, the final model included conditioning regimen, GSTA1 and GSTP1 that explained 19% of the variability of which 14% was accounted for by the two GSTs (Table 4a). Multivariate modelling for hemorrhagic cystitis included GSTM1, diagnosis, conditioning regimen and age, explaining 47% of variability of which 10% was attributed to GSTM1(Table 4a). BU exposure was significantly associated with aGvHD, TRT and hemorrhagic cystitis and was further analyzed together with genotypes/diplotypes in a multivariate model in which both variables remained significant predictors of respective outcomes (GSTA1, p = 0.003, GSTM1, p = 0.005, Css p ≤ 0.05, (Table 4b).
Table 5: Participating centers, demographic and transplantation characteristics
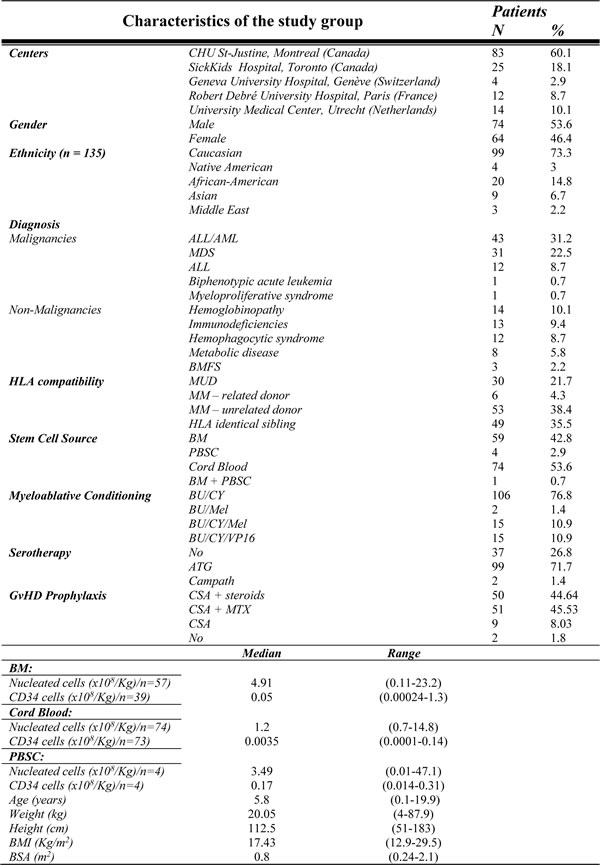
Abbreviations ALL: acute lymphoblastic leukemia; AML: acute myeloid leukemia; ATG: anti-thymocyte globulin; BM: bone marrow; BMFS: bone marrow failure syndrome; BMI: Body mass index; BSA: body surface area; BU: busulfan; Campath: Alemtuzumab; CY: cyclophosphamide; CSA: cyclosporine; Flu: fludarabine; MDS: myelodysplastic syndrome; Mel: melphalan; MM: mismatch; MRD: matched related donor; MUD: matched unrelated donor; MTX: methotrexate; CSA: cyclosporine; SD: sibling donor; GvHD: graft-versus-host-disease; PBSC: peripheral blood stem cells; VP16:etoposide
Table 6: Administration of BU and Pharmacokinetic parameter estimation per Center (n=138)
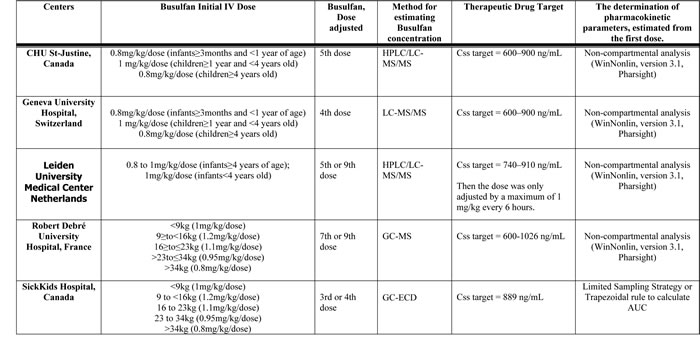
Evaluation of Pharmacokinetic parameter estimations: A cross calibration study was conducted by our group with the collaboration of Pierre Fabre Laboratories between all centers participating in the study to validate analytical method used to determine the PK parameters from the first dose (data available upon request). Only centers with the measured BU concentrations falling within ± 20% of the therapeutic drug target concentrations were included for analysis of PK parameters. Abbreviations: AUC: Area under the curve; Css: Steady state concentration; GC-MS: Gas Chromatography Mass Spectrometry; GC-ECD: Gas Chromatography with Electron Capture Detector; HPLC: High Performance Liquid Chromatography; LC-MS/MS:Liquid chromatography–mass spectrometry
Table 7: Busulfan pharmacokinetic parameters observed in the study subjects after administration of the first dose.
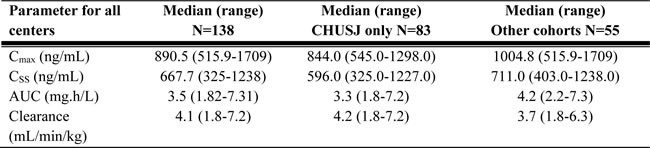
Cmax: maximum plasma concentration; Cmin: Minimum plasma concentration; Css: Steady state plasma concentration; AUC: Area under the curve; CHUSJ: Center Hospitalier universitaire Sainte-Justine
Table 8: Clinical outcomes observed in the study subjects
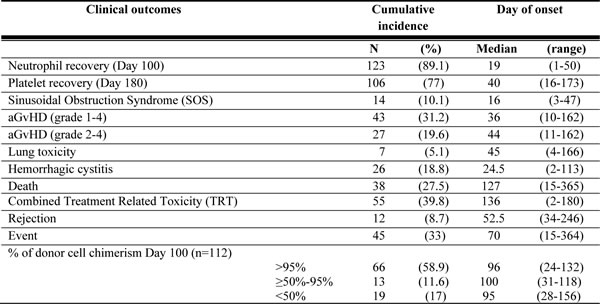
Abbreviation: aGvHD - acute Graft versus Host Disease
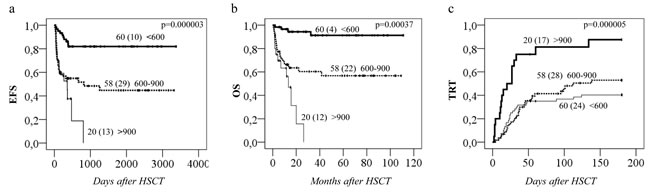
Figure 5: Busulfan plasma exposure and clinical outcomes of HSCT. Incidences of event-free survival (EFS), overall survival (OS), and treatment related toxicity (TRT) plotted against 3 groups based on first dose steady state concentration (Css) i.e. <600, 600-900 and > 900 ng/mL in all patients (n = 138). Total number of patients in each group (number of patients with events) are depicted on all plots. P values are shown on the plots.
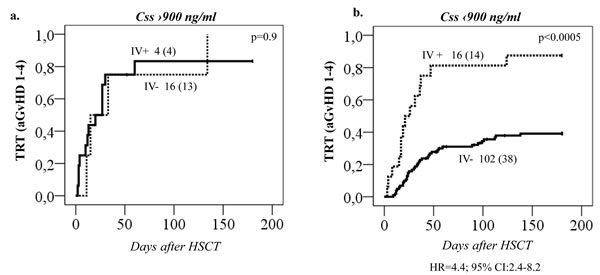
Figure 6: Treatment related toxicity (TRT) in relation to both 1st dose Css and GSTA1 groups. TRT (all cases combined) plotted against A. Busulfan Css below 900 ng/mL or B. Css above 900 ng/mL, dependent on whether patients are in GSTA1 diplotype group I, II, III (IV-) or IV (IV+). Total number of patients in each group with number of patients with TRT in brackets is depicted on all plots. Hazard ratio for group IV is depicted for plot (A) only.
Discussion
This study is conducted in a multicenter pediatric HSCT cohort and provides the first evidence for the association of functional GSTA1 diplotype groups with BU PK and clinical outcomes. Several studies including our own (summarized in Supplementary Table 1), performed in childhood and adult patients who received either iv or oral BU, have previously demonstrated an association between first dose PK and GSTA1 gene [3, 15-20, 22-26, 28, 33]. These studies, however, rarely included clinical outcome and none (except the one conducted by our group) included GSTA1 sub-haplotypes. There are only two studies [19, 26] that are comparable to the present report since they were conducted in childhood patients diagnosed mainly with malignancies who received i.v. BU- cyclophosphamide combination. Still, genetic association with clinical outcomes were not investigated in both of these studies and were also limited by the sample numbers. Overall, positive associations between GSTA1 gene and PK are characterized by higher BU exposure in *B individuals and lower in *A carriers, which is functionally driven by a -52 G>A promoter SNP (in complete linkage disequilibrium with -69 C>T and -567 A>T) delineating *A and*B haplotypes [34]. GSTA1 gene has an additional three SNPs within its promoter further diversifying haplotypes into subgroups that can potentially contribute to GSTA1 functionality. Using a gene reporter assay, we defined for the first time the promoter activity of each subgroup showing that *A1 could be classified as a normal metabolizer, *A2 and *A3 as rapid metabolizers, whereas slow metabolizing capacity was assigned to *B1a and *B2 and very slow to *B1b. Four major diplotype groups that reflected well GSTA1-PK relationship were defined. Group I (rapid metabolizers) was mostly represented by *A2*A2 diplotype and group IV (slow metabolizers) by *B1a*B1a diplotype and the presence of *B1b haplotype. Group I correlated with highest and group IV, with lowest CL. The relationship observed with group I is in accordance with our previous study [20, 33] which reported higher BU CL in*A2 *A2 individuals. More evident association of diplotype groups with PK was seen in girls, also in accordance with previous observation [20], which could be due to the higher level of GSTA1 in females compared to males particularly in liver cytosol [35, 36], or to more prominent induction and inhibition of GSTA1 [37].
Association of GSTA1 with PK was also reflected in GSTA1 association with dose requirements and with cumulative AUC, as seen by the analysis performed in CHU Sainte Justine patients. Indeed, on average there was no dose reduction in group IV, whereas in group I, in spite of the dose increase in most of these patients, cumulative AUC stayed the lowest. For group IV higher cumulative AUC’s may suggest that 1) Initial doses were too high for some patients; 2) Subsequent dose adjustments in these patients were not sufficient and thus dose reduction needs to be greater; 3) Toxic damage may have already been inflicted after first doses, 4) Possibility of a significant reduction in CL in group IV upon time. This emphasizes the need to adjust first dose according to genotype rather than adjusting only after therapeutic drug monitoring. Our results are in accordance with a study looking at oral BU dose requirement based on GSTA1 haplotypes [18] in which the first two oral doses were kept constant and later doses were adjusted to target a Css of 900 ng/mL (AUC of 5.4 mg.h/L), but despite dose adjustments, the average BU Css of the 3 dosing intervals remained significantly higher among GSTA1*B carriers [18].
We did not observe an association of GSTM1 with PK when all patients were analyzed. However, GSTM1’s involvement in BU PK could be quantitatively and qualitatively different in infants and toddlers (0-4 years old) compared to children and adolescents (4-18 year olds) as a consequence of developmental changes in gene expression as reported in ontogeny data [38]. This shows the limitation of the model with CL in L/h/kg that does not capture well the data for all patients over a varied age range. In future studies a more physiological related PK model should be used [39, 40]. Indeed, similar to our previous finding [20] patients above 4 years of age with GSTM1 null genotype had lower CL (Figure 2e). Bremer and colleagues [18] reported that GSTA1*B*B individuals who are also GSTM1 null tend to have higher Css after oral BU and patients with a combined GSTA1*B*B, GSTM1 null and GSTT1 null presented the highest Css levels of all genotype groups. We did not notice the combined effect of GSTA1 diplotypes and GSTM1 null genotype on the PK. Oral BU used in the study of Bremer et al., [18] usually resulting in larger inter-individual variability, might have also contributed to this difference.
Attention should be given to the association of GSTA1 diplotypes with clinical outcomes, given the paucity of such data in the literature [15-18, 20, 25, 27, 29, 31, 32], summarized in Supplementary Table 1. Interestingly, despite the fast BU metabolizing capacity and concentrations falling below the target range individuals with rapid metabolizing capacity (GSTA1 group I) showed a protective role in terms of OS, likely related to lower probability of TRT. This was most evident in individuals who received BU-cyclophosphamide conditioning regimen. A similar association was seen in a study performed in a Chinese population, where the GSTA1*A*A carriers showed significantly lower AUC than the GSTA1*A*B group with minimal toxicity when BU was administered once daily, highlighting the protective role of GSTA1*A*A [15]. This is also in accordance with our previous report [20] in which patients with two copies of haplotype *A2 had better EFS. On the other hand, individuals in group IV with slow metabolizing capacity demonstrated lower OS. These reductions seem to be related to higher frequency of TRT reflected by more frequent SOS and aGvHD suggesting that these patients are at a higher risk of TRT and might benefit from adjustment of the target AUC and initial dose reduction.
The association of GSTA1 gene with clinical outcomes might be related to GSTA1-PK relationship or might reflect additional involvement of GSTA1 beyond BU metabolism. When both GSTA1 diplotype groups and BU exposure were entered in a multivariate model, GSTA1 diplotypes were the only predictors of SOS, whereas aGvHD and combined TRT were predicted by both BU exposure and GSTA1 diplotypes independently. This was also reflected by the fact that Css above 900 ng/mL was associated with TRT irrespective of diplotype groups, whereas high risk of TRT for Css below 900 ng/mL was evident only for group IV carriers. The patients were recruited during a large time span, which could have influenced some of the associations observed, however, neither prophylactic measures nor TRT incidences differed significantly over time. GSTA1 also participates in the metabolism of cyclophosphamide [41] possibly explaining modulation of associations of clinical outcomes in BU-cyclophosphamide conditioning regimen. Interestingly in lupus nephritis patients, GSTA1*B genotypes have higher exposure to activated cyclophosphamide metabolites [42]. Therefore, these vulnerable patients might benefit from administration of cyclophosphamide before BU or alternative conditioning regimens such as BU/Fludarabine. Non-catalytic functions of the GST enzymes could be considered as well, for example GSTM1 and GSTP1 have been linked to inhibition of the mitogen-activated protein (MAP) kinase pathway [43] modulating apoptotic signalling. Additionally, glutathione S-transferase family has the potential to behave as minor histocompatibility antigens (miHA). miHA disparities have been attributed to GvHD in the HLA-matched transplantation setting [44] potentially explaining independent effect of GSTA1 and GSTP1 genotypes in aGvHD susceptibility.
The association observed with GSTM1 and hemorrhagic cystitis could derive from the interaction between busulfan and cyclophosphamide affecting the clearance of cyclophosphamide metabolites such as acrolein, which can be damaging to the kidney and bladder epithelium [45]. GSTM1 non-null carriers might deplete the glutathione pool, limiting conjugation of these metabolites by other specific GSTs such as GSTP1. The additional role of GSTM1 in determining clinical outcomes of HSCT, which is distinct from its role in BU metabolism is also a possible explanation for the higher incidences of early mortality observed in the study by Bremer et al. for GSTM1 non null individuals receiving BU-cyclophosphamide regimen [18].
Conclusion
We provide evidence for an association of GSTA1 diplotypes with BU PK and clinical outcomes of HSCT supported by functional studies. GSTA1 diplotypes can explain in some models ~20% of the variability seen in BU CL and can contribute to HSCT -related complications acting within and beyond BU metabolism. Prior genotyping may be helpful in deciding on BU first dose, thus optimizing therapeutic drug monitoring and decreasing TRT, which is particularly important for GSTA1 group IV carriers. It might also help to define each patient risk to toxicity and introduce the possibility of individualised prophylaxis. Other GST polymorphisms seem also to contribute to HSCT-related toxicities and may include additional mechanisms. Further studies are needed to define prospectively how to adjust dose according to the genotype, including non-genetic factors and different BU administration schedules.
Materials and Methods
Patients
The study includes 138 pediatric patients who underwent allogeneic HSCT with i.v. BU as part of a myeloablative conditioning regimen from five different centers in Europe and Canada (Geneva, Leiden, Montreal, Paris, Toronto), recruited between May 2000 and April 2013. The Institutional Review Board at each center approved the study and all patients and/or parents provided informed consent. Details of inclusion criteria are available at Clinicaltrials.gov site (NCT01257854). Patients’ characteristics are provided in Table 5.
Sampling, genotyping, administration of BU and PK estimation
GST genotyping was performed according to the previously described procedures [33, 46]. BU was administered every 6h as a 2 h infusion for a total of 16 doses. The first dose was either age or weight-based and dose adjustment (based on the first dose PK parameter estimates) was performed on the 3rd; 4th; 5th; or 9th dose onwards as detailed on Table 6 which summarizes the dosing schedule followed by each center. The median BU PK parameters observed in the study subjects after administration of the first dose are summarized in Table 7.
Clinical outcomes (Table 8) were defined as per the standard guidelines of the European Society for Blood and Marrow Transplantation and Center for International Blood and Marrow Transplant Research as detailed in previous reports [6, 20]. TRT was defined as the occurrence of first toxicity, either SOS, aGvHD, lung toxicity or HC. An event was defined as graft rejection, relapse or death from any cause.
Reporter-gene assay
Plasmid constructs were prepared by a gene assembly service (GeneScript, Piscataway, USA). DNA region from -1275 to +126 relative to the translational start codon was cloned into pGl4.10 (Promega, Maddison, USA). HepG2 cells were co-transfected with the pGL4.10 GSTA1 constructs (with site specific mutations underlying GSTA1 haplotypes) and the pRL-SV40 vector (Promega) that codes for Renilla luciferase for transfection control and normalisation. Difference in promoter activity between haplotypes was assessed by t-test or ANOVA.
Statistical analyses
BU clearance (mL/min/kg), ratio of adjusted to initial dose and cumulative AUCs were compared across genotypes or diplotype groups using non-parametric tests or linear regression. Cumulative incidences of OS and EFS and of adverse events were estimated in relation to the genotype/ diplotype groups, using Kaplan-Meier framework and log-rank test. Univariate Cox regression was used to estimate hazard ratios. The analyses were also performed by cumulative incidence of competing events and the difference among groups estimated by Gray’s test [47]. The relationship of GST with CL and clinical outcomes was additionally explored through stratified and multivariate analysis. Stratified analyses were performed according to age, gender, diagnosis and conditioning regimen. Multivariate analysis included co-variables that were either significantly associated with outcome studied, correlated with genotypes/diplotypes or modulated genotype-phenotype associations: age, gender, diagnosis, conditioning regimen, ethnicity and BU exposure. The allele and genotype frequencies, and Hardy-Weinberg equilibrium were analysed using Haploview [48]; haplotypes were resolved using PHASE [49]. Statistical analyses were performed using IBM® SPSS® statistics (version 19, SPSS Inc, NY) and EZR (Version 1.31) [50].
Abbreviations
HSCT: Hematopoietic stem cell transplantation; GST: glutathione S transferase; GSTA1: glutathione S transferase Alpha1; GSTP1: glutathione S transferase Pi 1; GSTM1: glutathione S transferase Mu 1; GSTT: glutathione S transferase Theta; BU: Busulfan; AUC: Area under the curve; Css: Stead state concentration; PK: pharmacokinetics; TRT: Treatment Related Toxicity; SOS: Sinusoidal Obstruction Syndrome; aGvHD: acute Graft versus Host Disease; HepG2: Human hepatoma; OS: Overall survival; EFS: Events Free Survival.
Author contributions
M.A, P.HDC, CRS.U, T.N, MA.R, V.M, L.L, Y.T performed experiments, M.A, P.HDC, CRS.U, MA.R and M.K performed the analysis, M.A and MK designed the research, M.A, P.HDC, CRS.U and M.K drafted the article; all authors contributed to the interpretation of data and revised the manuscript critically.
Acknowledgments
This study was performed under the supervision of the Swiss National Science Foundation and CANSEARCH foundation. We warmly thank the patients and their parents for consenting to participate in this study. We also thank R. Lo Piccolo, S. Mezziani, M-F.Vachon, and M. Cortier for the help in this study as well as N. Von Der Weid and the Swiss Pediatric Oncology Group for being the sponsors of this study.
Conflicts of interest
H.B has acted as a consultant for Jazz Pharmaceuticals and obtained an education grant from them. H.B also acted as a consultant for Seattle Genetics. The authors declare that they have no other financial relationship(s) to disclose.
Funding
This study was supported by grants from the Swiss National Science Foundation (Grant number153389), CANSEARCH Foundation, the Geneva Cancer League, the Dr. Henri Dubois-Ferrière Dinu Lipatti Foundation, and Foundation of Charles-Bruneau Cancer Center.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
Supplementary Data
Supplementary Data available in Supplementary Files.
References
1. Santos GW, Tutschka PJ, Brookmeyer R, Saral R, Beschorner WE, Bias WB, Braine HG, Burns WH, Elfenbein GJ, Kaizer H. Marrow transplantation for acute nonlymphocytic leukemia after treatment with busulfan and cyclophosphamide. The New England Journal of Medicine. 1983; 309: 1347-53. doi: 10.1056/NEJM198312013092202.
2. Hassan M, Andersson BS. Role of pharmacogenetics in busulfan/cyclophosphamide conditioning therapy prior to hematopoietic stem cell transplantation. Pharmacogenomics. 2013; 14: 75-87. doi: 10.2217/pgs.12.185.
3. Huezo-Diaz P, Uppugunduri CR, Tyagi AK, Krajinovic M, Ansari M. Pharmacogenetic aspects of drug metabolizing enzymes in busulfan based conditioning prior to allogenic hematopoietic stem cell transplantation in children. Current Drug Metabolism. 2014; 15: 251-64.
4. Bartelink IH, Lalmohamed A, Van Reij EML, Dvorak CC, Savic RM, Zwaveling J, Bredius RGM, Egberts ACG, Bierings M, Kletzel M, Shaw PJ, Nath CE, Hempel G, et al. A New Harmonized Approach to Associate Busulfan Exposure with Survival and Toxicity after Hematopoietic Cell Transplantation in Children and Young Adults: a Multicenter Retrospective Cohort Analysis. The Lancet Haematology. 2016; 3: e526-e36.
5. Zao JH, Schechter T, Liu WJ, Gerges S, Gassas A, Egeler RM, Grunebaum E, Dupuis LL. Performance of Busulfan Dosing Guidelines for Pediatric Hematopoietic Stem Cell Transplant Conditioning. Biology of Blood and Marrow Transplantation. 2015; 21: 1471-8. doi: 10.1016/j.bbmt.2015.05.006.
6. Ansari M, Theoret Y, Rezgui MA, Peters C, Mezziani S, Desjean C, Vachon MF, Champagne MA, Duval M, Krajinovic M, Bittencourt H, Pediatric Disease Working Parties of the European B, Marrow Transplant G. Association between busulfan exposure and outcome in children receiving intravenous busulfan before hematopoietic stem cell transplantation. Therapeutic Drug Monitoring. 2014; 36: 93-9. doi: 10.1097/FTD.0b013e3182a04fc7.
7. Baker KS, Bostrom B, DeFor T, Ramsay NK, Woods WG, Blazar BR. Busulfan pharmacokinetics do not predict relapse in acute myeloid leukemia. Bone Marrow Transplantation. 2000; 26: 607-14. doi: 10.1038/sj.bmt.1702590.
8. Maheshwari S, Kassim A, Yeh RF, Domm J, Calder C, Evans M, Manes B, Bruce K, Brown V, Ho R, Frangoul H, Yang E. Targeted Busulfan therapy with a steady-state concentration of 600-700 ng/mL in patients with sickle cell disease receiving HLA-identical sibling bone marrow transplant. Bone Marrow Transplantation. 2014; 49: 366-9. doi: 10.1038/bmt.2013.188.
9. Czerwinski M, Gibbs JP, Slattery JT. Busulfan conjugation by glutathione S-transferases alpha, mu, and pi. Drug Metabolism & Disposition. 1996; 24: 1015-9.
10. Ramsay EE, Dilda PJ. Glutathione S-conjugates as prodrugs to target drug-resistant tumors. Frontiers in Pharmacology. 2014; 5: 181. doi: 10.3389/fphar.2014.00181.
11. Coles BF, Morel F, Rauch C, Huber WW, Yang M, Teitel CH, Green B, Lang NP, Kadlubar FF. Effect of polymorphism in the human glutathione S-transferase A1 promoter on hepatic GSTA1 and GSTA2 expression. Pharmacogenetics. 2001; 11: 663-9.
12. Bredschneider M, Klein K, Murdter TE, Marx C, Eichelbaum M, Nussler AK, Neuhaus P, Zanger UM, Schwab M. Genetic polymorphisms of glutathione S-transferase A1, the major glutathione S-transferase in human liver: consequences for enzyme expression and busulfan conjugation. Clinical Pharmacology & Therapeutics. 2002; 71: 479-87. doi: 10.1067/mcp.2002.124518.
13. Board P, Coggan M, Johnston P, Ross V, Suzuki T, Webb G. Genetic heterogeneity of the human glutathione transferases: a complex of gene families. Pharmacology & Therapeutics. 1990; 48: 357-69.
14. Zimniak P, Nanduri B, Pikula S, Bandorowicz-Pikula J, Singhal SS, Srivastava SK, Awasthi S, Awasthi YC. Naturally occurring human glutathione S-transferase GSTP1-1 isoforms with isoleucine and valine in position 104 differ in enzymic properties. European Journal of Biochemistry. 1994; 224: 893-9.
15. Yin J, Xiao Y, Zheng H, Zhang YC. Once-daily i.v. BU-based conditioning regimen before allogeneic hematopoietic SCT: a study of influence of GST gene polymorphisms on BU pharmacokinetics and clinical outcomes in Chinese patients. Bone Marrow Transplantation. 2015; 50: 696-705. doi: 10.1038/bmt.2015.14.
16. Choi B, Kim MG, Han N, Kim T, Ji E, Park S, Kim IW, Oh JM. Population pharmacokinetics and pharmacodynamics of busulfan with GSTA1 polymorphisms in patients undergoing allogeneic hematopoietic stem cell transplantation. Pharmacogenomics. 2015; 16: 1585-94. doi: 10.2217/pgs.15.98.
17. Ansari M, Huezo-Diaz P, Rezgui MA, Marktel S, Duval M, Bittencourt H, Cappelli B, Krajinovic M. Influence of glutathione S-transferase gene polymorphisms on busulfan pharmacokinetics and outcome of hematopoietic stem-cell transplantation in thalassemia pediatric patients. Bone Marrow Transplantation. 2016; 51: 377-83. doi: 10.1038/bmt.2015.321.
18. Bremer S, Floisand Y, Brinch L, Gedde-Dahl T, Bergan S. Glutathione Transferase Gene Variants Influence Busulfan Pharmacokinetics and Outcome After Myeloablative Conditioning. Therapeutic Drug Monitoring. 2015; 37: 493-500. doi: 10.1097/FTD.0000000000000180.
19. ten Brink MH, van Bavel T, Swen JJ, van der Straaten T, Bredius RG, Lankester AC, Zwaveling J, Guchelaar HJ. Effect of genetic variants GSTA1 and CYP39A1 and age on busulfan clearance in pediatric patients undergoing hematopoietic stem cell transplantation. Pharmacogenomics. 2013; 14: 1683-90. doi: 10.2217/pgs.13.159.
20. Ansari M, Rezgui MA, Theoret Y, Uppugunduri CR, Mezziani S, Vachon MF, Desjean C, Rousseau J, Labuda M, Przybyla C, Duval M, Champagne M, Peters C, et al. Glutathione S-transferase gene variations influence BU pharmacokinetics and outcome of hematopoietic SCT in pediatric patients. Bone Marrow Transplantation. 2013; 48: 939-46. doi: 10.1038/bmt.2012.265.
21. ten Brink MH, Wessels JA, den Hartigh J, van der Straaten T, von dem Borne PA, Guchelaar HJ, Zwaveling J. Effect of genetic polymorphisms in genes encoding GST isoenzymes on BU pharmacokinetics in adult patients undergoing hematopoietic SCT. Bone Marrow Transplantation. 2012; 47: 190-5. doi: 10.1038/bmt.2011.55.
22. Kim SD, Lee JH, Hur EH, Lee JH, Kim DY, Lim SN, Choi Y, Lim HS, Bae KS, Noh GJ, Yun SC, Han SB, Lee KH. Influence of GST gene polymorphisms on the clearance of intravenous busulfan in adult patients undergoing hematopoietic cell transplantation. Biology of Blood and Marrow Transplantation. 2011; 17: 1222-30. doi: 10.1016/j.bbmt.2010.12.708.
23. Abbasi N, Vadnais B, Knutson JA, Blough DK, Kelly EJ, O’Donnell PV, Deeg HJ, Pawlikowski MA, Ho RJ, McCune JS. Pharmacogenetics of intravenous and oral busulfan in hematopoietic cell transplant recipients. The Journal of Clinical Pharmacology. 2011; 51: 1429-38. doi: 10.1177/0091270010382915.
24. Gaziev J, Nguyen L, Puozzo C, Mozzi AF, Casella M, Perrone Donnorso M, Gravina P, Sodani P, Marziali M, Isgro A, Simone MD, Andreani M, Formosa A, et al. Novel pharmacokinetic behavior of intravenous busulfan in children with thalassemia undergoing hematopoietic stem cell transplantation: a prospective evaluation of pharmacokinetic and pharmacodynamic profile with therapeutic drug monitoring. Blood. 2010; 115: 4597-604. doi: 10.1182/blood-2010-01-265405.
25. Elhasid R, Krivoy N, Rowe JM, Sprecher E, Adler L, Elkin H, Efrati E. Influence of glutathione S-transferase A1, P1, M1, T1 polymorphisms on oral busulfan pharmacokinetics in children with congenital hemoglobinopathies undergoing hematopoietic stem cell transplantation. Pediatric Blood & Cancer. 2010; 55: 1172-9. doi: 10.1002/pbc.22739.
26. Johnson L, Orchard PJ, Baker KS, Brundage R, Cao Q, Wang X, Langer E, Farag-El Maasah S, Ross JA, Remmel R, Jacobson PA. Glutathione S-transferase A1 genetic variants reduce busulfan clearance in children undergoing hematopoietic cell transplantation. The Journal of Clinical Pharmacology. 2008; 48: 1052-62. doi: 10.1177/0091270008321940.
27. Kim I, Keam B, Lee KH, Kim JH, Oh SY, Ra EK, Yoon SS, Park SS, Kim CS, Park S, Hong YC, Kim BK. Glutathione S-transferase A1 polymorphisms and acute graft-vs.-host disease in HLA-matched sibling allogeneic hematopoietic stem cell transplantation. Clinical Transplantation. 2007; 21: 207-13. doi: 10.1111/j.1399-0012.2006.00624.x.
28. Kusama M, Kubota T, Matsukura Y, Matsuno K, Ogawa S, Kanda Y, Iga T. Influence of glutathione S-transferase A1 polymorphism on the pharmacokinetics of busulfan. Clinica Chimica Acta. 2006; 368: 93-8. doi: 10.1016/j.cca.2005.12.011.
29. Gaziev J, Isgro A, Mozzi AF, Petain A, Nguyen L, Ialongo C, Dinallo V, Sodani P, Marziali M, Andreani M, Testi M, Paciaroni K, Gallucci C, et al. New insights into the pharmacokinetics of intravenous busulfan in children with sickle cell anemia undergoing bone marrow transplantation. Pediatric Blood & Cancer. 2015; 62: 680-6. doi: 10.1002/pbc.25376.
30. Ten Brink MH, Swen JJ, Bohringer S, Wessels JA, van der Straaten T, Marijt EW, von dem Borne PA, Zwaveling J, Guchelaar HJ. Exploratory analysis of 1936 SNPs in ADME genes for association with busulfan clearance in adult hematopoietic stem cell recipients. Pharmacogenetics and Genomics. 2013; 23: 675-83. doi: 10.1097/FPC.0000000000000007.
31. Zwaveling J, Press RR, Bredius RG, van Derstraaten TR, den Hartigh J, Bartelink IH, Boelens JJ, Guchelaar HJ. Glutathione S-transferase polymorphisms are not associated with population pharmacokinetic parameters of busulfan in pediatric patients. Therapeutic Drug Monitoring. 2008; 30: 504-10. doi: 10.1097/FTD.0b013e3181817428.
32. Srivastava A, Poonkuzhali B, Shaji RV, George B, Mathews V, Chandy M, Krishnamoorthy R. Glutathione S-transferase M1 polymorphism: a risk factor for hepatic venoocclusive disease in bone marrow transplantation. Blood. 2004; 104: 1574-7. doi: 10.1182/blood-2003-11-3778.
33. Ansari M, Lauzon-Joset JF, Vachon MF, Duval M, Theoret Y, Champagne MA, Krajinovic M. Influence of GST gene polymorphisms on busulfan pharmacokinetics in children. Bone Marrow Transplantation. 2010; 45: 261-7. doi: 10.1038/bmt.2009.143.
34. Morel F, Rauch C, Coles B, Le Ferrec E, Guillouzo A. The human glutathione transferase alpha locus: genomic organization of the gene cluster and functional characterization of the genetic polymorphism in the hGSTA1 promoter. Pharmacogenetics. 2002; 12: 277-86.
35. Hoensch H, Morgenstern I, Petereit G, Siepmann M, Peters WH, Roelofs HM, Kirch W. Influence of clinical factors, diet, and drugs on the human upper gastrointestinal glutathione system. Gut. 2002; 50: 235-40.
36. Mulder TP, Court DA, Peters WH. Variability of glutathione S-transferase alpha in human liver and plasma. Clinical Chemistry. 1999; 45: 355-9.
37. Mitchell AE, Burns SA, Rudolf JL. Isozyme- and gender-specific induction of glutathione S-transferases by flavonoids. Archives of Toxicology. 2007; 81: 777-84. doi: 10.1007/s00204-007-0210-9.
38. Strange RC, Howie AF, Hume R, Matharoo B, Bell J, Hiley C, Jones P, Beckett GJ. The development expression of alpha-, mu- and pi-class glutathione S-transferases in human liver. Biochimica et Biophysica Acta. 1989; 993: 186-90.
39. Diestelhorst C, Boos J, McCune JS, Russell J, Kangarloo SB, Hempel G. Physiologically based pharmacokinetic modelling of Busulfan: a new approach to describe and predict the pharmacokinetics in adults. Cancer Chemotherapy and Pharmacology. 2013; 72: 991-1000. doi: 10.1007/s00280-013-2275-x.
40. Diestelhorst C, Boos J, McCune JS, Russell J, Kangarloo SB, Hempel G. Predictive performance of a physiologically based pharmacokinetic model of busulfan in children. Pediatric Hematology and Oncology. 2014; 31: 731-42. doi: 10.3109/08880018.2014.927945.
41. Ekhart C, Doodeman VD, Rodenhuis S, Smits PH, Beijnen JH, Huitema AD. Influence of polymorphisms of drug metabolizing enzymes (CYP2B6, CYP2C9, CYP2C19, CYP3A4, CYP3A5, GSTA1, GSTP1, ALDH1A1 and ALDH3A1) on the pharmacokinetics of cyclophosphamide and 4-hydroxycyclophosphamide. Pharmacogenetics and Genomics. 2008; 18: 515-23. doi: 10.1097/FPC.0b013e3282fc9766.
42. Wang HN, Zhu XY, Zhu Y, Xie QH, Lai LY, Zhao M, Chen YC, Xue J, Hao CM, Gu Y, Lin SY. The GSTA1 polymorphism and cyclophosphamide therapy outcomes in lupus nephritis patients. Clinical Immunology. 2015; 160: 342-8. doi: 10.1016/j.clim.2015.07.010.
43. Townsend DM, Tew KD. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene. 2003; 22: 7369-75. doi: 10.1038/sj.onc.1206940.
44. Martinez-Bravo MJ, Calderon-Cabrera C, Marquez-Malaver FJ, Rodriguez N, Guijarro M, Espigado I, Nunez-Roldan A, Perez-Simon JA, Aguilera I. Mismatch on glutathione S- transferase T1 increases the risk of graft-versus-host disease and mortality after allogeneic stem cell transplantation. Biology of Blood and Marrow Transplantation. 2014; 20: 1356- 62. doi: 10.1016/j.bbmt.2014.05.008.
45. Conklin DJ, Haberzettl P, Lesgards JF, Prough RA, Srivastava S, Bhatnagar A. Increased sensitivity of glutathione S-transferase P-null mice to cyclophosphamide-induced urinary bladder toxicity. Journal of Pharmacology and Experimental Therapeutics. 2009; 331: 456-69. doi: 10.1124/jpet.109.156513.
46. Zhong S, Wyllie AH, Barnes D, Wolf CR, Spurr NK. Relationship between the GSTM1 genetic polymorphism and susceptibility to bladder, breast and colon cancer. Carcinogenesis. 1993; 14: 1821-4.
47. Gray RJ. A Class of K-Sample Tests for Comparing the Cumulative Incidence of a Competing Risk. The Annals of Statistics. 1988; 16: 1141-54.
48. Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005; 21: 263-5. doi: 10.1093/bioinformatics/bth457.
49. Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. The American Journal of Human Genetics. 2001; 68: 978-89. doi: 10.1086/319501.
50. Kanda Y. Investigation of the freely available easy-to-use software ‘EZR’ for medical statistics. Bone Marrow Transplantation. 2013; 48: 452-8. doi: 10.1038/bmt.2012.244.