
INTRODUCTION
Triple-negative breast cancer (TNBC) accounts for approximately 15%–20% of newly diagnosed breast cancer worldwide and occurs more prevalently in Hispanics, American and African women [1–3]. In fact, TNBC is a class of highly heterogeneous tumors incorporating various molecular and clinic pathologic features and clinical outcomes. Approximately 80% of TNBC overlaps with the basal-like breast cancer phenotype that is classified by gene expression profiling, although TNBC and basal-like breast cancer are never synonymous [4]. In general, TNBC is more destructive, with higher rates of relapse compared to other types of breast cancer, and shows frequently metastasis to the visceral and central nervous system [5]. In the absence of obvious targets, which could benefit pharmaceutical development, TNBC cases are mainly treated by tumor excision, radiation therapy and chemotherapy using cytotoxic agents. A lot of well-established target therapies that have been approved for the treatment of other cancers are found to be beneficial in the case of TNBC, including agents that target Poly [ADP ribose] polymerase 1 (PARP1) [6], androgen receptor (AR) [7], vascular endothelial growth factor receptor (VEGFR) and epidermal growth factor receptor (EGFR) under certain circumstances [8].
Although multiple targeted agents and monoclonal antibodies are under investigation for TNBCs, the majority of them have failed to achieve a satisfying therapeutic outcome. The lack of biomarkers to stratify TNBC and identify sensitive patients who are most likely to respond to different targeted therapy remains a major reason for these disappointing results. Here, we have reviewed different studies which classify TNBC into subtypes, and investigated representative therapeutic strategies for these subtypes.
FURTHER SUBTYPING OF TNBC
Vast advances have been made to classify TNBC into more molecular subtypes, which have enormous potential for personalized medicine and guidance for clinical trial. The pioneer studies were conducted by Brian et al. (Figure 1), who reported six TNBC subtypes based on gene expression profiling (GEP), i.e., basal-like 1 (BL1), basal-like 2 (BL2), an immunomodulatory (IM), a mesenchymal (M), a mesenchymal stem-like (MSL), and a luminal androgen receptor (LAR) subtype [9]. More importantly, they identified representative TNBC cell lines of these subtypes to show that analysis of distinct gene expression profiles can inform therapy selection. BL-TNBC is characterized by DNA-repair deficiency, and the relevant cell models responded to cisplatin treatment. The M and MSL subtypes have higher expression of genes involved in epithelial to mesenchymal transition (EMT) and activation of receptor tyrosine kinase pathways, and representative cell lines preferentially responded to PI3K/mTOR inhibitors and ABL/SRC inhibitors. The LAR subtype is characterized by androgen receptor signaling and LAR cell lines were uniquely sensitive to AR antagonists.
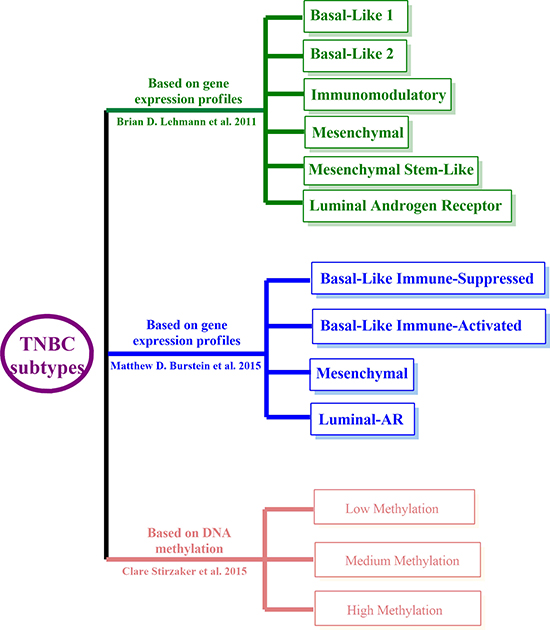
Figure 1: TNBC classifications. Brian’s (Green), Matthew’s (Blue), and Clare’s methylation subtyping (Red).
More recently, Matthew et al. revisited the grouping of TNBC by analyzing the RNA and DNA profile of 198 TNBC tumors, and classed these TNBCs into four, rather than six subtypes (Figure 1), i.e., Luminal-AR (LAR), Mesenchymal (MES), Basal-Like Immune-Suppressed (BLIS), Basal-Like Immune-Activated (BLIA) [10]. They also identified putative subtype-specific targets: androgen receptor and the cell surface mucin MUC1 for the LAR subtype; PDGF receptor A and c-Kit for the MES subtype; VTCN1, an immune suppressing molecule for the BLIS subtype; and STAT signal transduction molecules and cytokines for the BLIA subtype. These studies imply a promising future for personalized therapy in TNBC based on molecular subtype, and further studies for subtype-specific therapy are rationally warranted.
The development of a tumor is a multistep process resulting from the accumulation of genetic alterations, which involve not only changes in the DNA sequence, but also epigenetic alterations such as DNA methylation and histone modification. Recently, emerging epigenetic analyses, especially for the DNA methylation, have contributed to the further stratification of TNBC. Clare et al. tested whether DNA methylation signatures can classify TNBC, and whether they could be used to predict specific clinical outcomes [11]. A total of 25 TNBC patient tumor samples were analyzed by whole-genome methylation capture sequencing assay, and 865 differentially methylated regions (DMRs) were identified. Among them, they found 36 DMRs were specific to TNBC by comparing these with 542 samples from the TCGA breast cancer cohort. Specifically, they utilized DMRs to stratify TNBC patients into three distinct methylation clusters, which were strongly associated with overall survival (Figure 1). Using a similar strategy, 17 DMRs were identified from the TCGA breast cancer cohort, which were also associated with survival in TNBC samples.
Vast efforts have also been made to stratify breast cancer utilizing the DNA methylation patterns [12]. In the global methylation profile, TNBC (the basal-like variety in their study) was the least frequently methylated, and another study also showed nine epigenetic biomarker genes were hypo-methylated in the basal-like and claudin-low breast cancers (normally TNBC) [13].
These works indicate that DNA methylation signatures could extend our ability to classify breast cancer, which will not only improve diagnosis and prognosis of breast cancer but also help to develop new therapeutic targets, which is specific to different subgroups of TNBC.
BASAL-LIKE TNBC
BL-TNBC is the predominant molecular subgroup of TNBC. In Brian’s study, two basal-like subtypes were identified. The GEP of BL1 is enriched for cell cycle check point and DNA damage response genes, and BL2 is enriched for genes involved in growth factor pathways. Particularly, TNBC cell lines from the BL1 and BL2 groups are sensitive to cisplatin treatment. Matthew identified two basal-like subtypes (BLIA and BLIS), which have down-regulation (BLIS) or up-regulation (BLIA) in immune cell-regulating pathways and cytokine pathways. Hence, immune-based strategies may be useful treatments for BLIS tumors which will be further discussed in the “immune associated TNBC” part, and BLIA tumors are sensitive to STAT inhibitors, cytokine or cytokine receptor antibodies.
Platinum salt drug based chemotherapy
As reported by Brian, BL1 is enriched for tumors that harbor a deficiency in (homologous recombination) HR repair, which is largely caused by mutations or epigenetic changes in the BRCA1/2; and the BL2 subgroup, on the other hand, is uniquely enriched in growth factor signaling pathways (EGF, NGF and MET pathways). Thus, targeting DNA-repair deficiency by DNA damage agents appears to be a promising treatment for BL-TNBC (more likely to be effective for BL1). Indeed, good response rates to platinum-based chemotherapy have been associated with low BRCA1-mRNA expression and high BRCA1 methylation [14, 15]. Platinum-based chemotherapy appears to significantly increase the pathological complete response (pCR) rate in TNBC patients, which is relatively higher in patients with a family history of BRCA-mutation than the rest of the population [16]. Platinum salts have been increasingly tested for TNBC in combination with various other chemotherapy drugs e.g. gemcitabine [17, 18], which masquerades as cytidine and inhibits DNA synthesis. A phase III trial involving 236 patients demonstrated that cisplatin plus gemcitabine is an alternative or even the preferred first-line chemotherapy strategy for patients with metastatic triple-negative breast cancer [19].
Inhibition of poly (ADP ribose) polymerase 1
Besides platinum salt-based agents, this subtype of TNBC is also believed to respond to PARP inhibitors, which cause synthetic lethal effects with HR-repair deficiency (Figure 2). PARP1 is involved in the process of responding to single-strand DNA damage, and maintains genomic integrity via base excision repair [20]. Double-strand DNA damages are normally repaired through HR, which requires normal functions of the tumor suppressor proteins BRCA1/2 [21]. Furthermore, it has been estimated that up to three quarters of BRCA1-associated tumors are BLBCs [22], and many TNBCs are frequently found to have defects in BRCA-related HR [23, 24]. Thus, it has provided a strong rationale for the use of PARP inhibitors for the treatment of TNBC with HR deficiency, and devoid of side-effects on the remaining normal cells. The American Society of Clinical Oncology (ASCO, 2017) state that Olaparib (PARP inhibitor) slows the growth of BRCA-related metastatic Breast Cancer. Findings suggest that such PARP inhibitors could play a key role in Breast Cancer treatment. Indeed, clinical trials showed that Olaparib significantly improved the overall survival in phase I/II clinical trials [25, 26], and Iniparib, another PARP inhibitor, is assessed in phase III clinical trials in advanced TNBC. However, these trials failed to meet the primary study end points (mainly failed in PFS and overall survival), which is thought to be due to a lack of powerful selection of BRCA1 mutated TNBC [27]. Thus, further studies are needed to better understand and target the resistance to PARP1 inhibitors. More recently, a phase III trial (the patients involved were HER2-negative metastatic breast cancer cases with a germline BRCA mutation) reported that the median progression-free survival was significantly longer in Olaparib monotherapy group than in the standard chemotherapy group, and the risk of disease progression or death was lower than for standard chemotherapy [28].
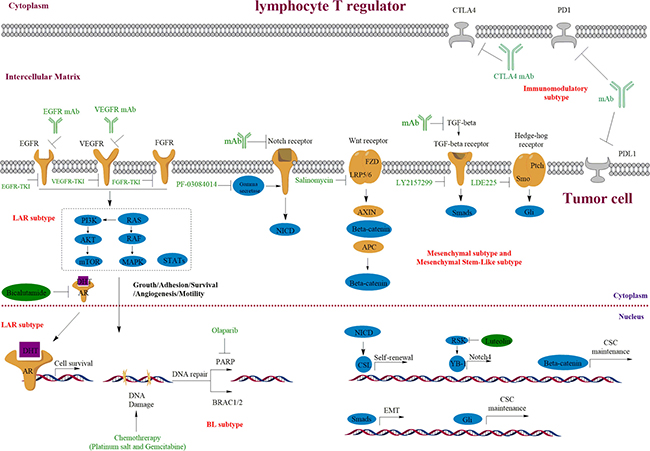
Figure 2: Targeting the growth factor receptors and PARP in TNBC and the important roles of Notch, Wnt/β-catenin, Hedge-hog and TGF-β signaling pathways in TNBC. Overexpression or mutations of the EGFR, VEGFR, AR and FGFR are common in TNBC, which result in the deregulation of downstream signaling. Receptor specific-monoclonal antibody (mAb) and TKIs are used to block ligand-receptor interaction or kinase activity, which further turnoff their downstream signaling. The BL2 subtype of TNBC could be especially sensitive to these growth signaling inhibition. BRCA1/2 mutations or decreased expression are frequently involved in TNBC initiation and development, which also causes HR deficiency and hypersensitive to PARP inhibition (BL1 subtype). Mesenchymal-like subgroup of TNBC is enriched for genes involved in CSCs regulation and EMT, and corresponding tumors could be sensitive to mAb and inhibitors in these pathways.
In 2006, De Soto evaluated the sensitivity of multiple cell lines (non-cancerous mouse embryonic stem cells and hamster cells; human and mouse breast tumor cells) with BRCA1 or BRCA2 deficiency to three PARP1 inhibitors (NU1025, 3-aminobenzamide, and AG14361) [29]. They showed that AG14361 has high selectivity to kill BRCA1-knockout embryonic stem cells. Whereas to human and mouse breast tumor cells tested, the PARP1 inhibitors were either ineffective or eliminate these cells irrespective of BRCA1 status. During BRCA1 mutation and carcinogenesis, the cells may go through two distinct phases according to their sensitivity to PARP1 inhibitors. In the initial stage of BRCA1 mutation, cells are generally naïve and sensitive before they acquire multiple genetic mutations and eventually develop into cancer cells, which are resistant to PARP1 inhibition. In the same year, Hochegger et al. reported that Ku-70 or Ligase IV mutation is responsible for the resistance to double-strand breaks inducing drugs in chicken DT40 cells with a heterozygous PARP1 mutation [30].
Currently, other PARP inhibitors (Veliparib and Talazoparib) are still under investigation in the cases of BRCA mutation-associated breast cancer (ClinicalTrials.gov NCT01945775 and NCT02163694), and further analyses are necessary to determine if a specific subset of TNBC patients can benefit from use of the PARP inhibitors. Olaparib was approved by Food and Drug Administration for the treatment of BRCA1-deficient ovarian cancer in 2014 [31].
MESENCHYMAL ASSOCIATED TNBC
Mesenchymal and mesenchymal stem like TNBCs generally indicate the subgroups of TNBCs, which are enriched for genes involved in EMT and the biological regulation of CSCs (Figure 2). TNBC cell lines from the mesenchymal subgroup (e.g. MDA-MB-231) are highly resistant to multiple cytotoxic agents and possess stem cell phenotypes.
Human breast cancer cells with CD44+/CD24−/EpCAM+ expression markers and/or possessing ALDH1 enzyme activity are recognized as CSCs and are responsible for maintaining tumor growth. Unlike other cells within the tumor bulk, these cells are commonly insensitive to current chemotherapies [32]. Over-expression of multidrug resistance proteins [33] and anti-apoptotic proteins [34] are responsible for the resistance to some cytotoxic agents. It’s widely recognized that the CSC population can be enriched after chemotherapy through various mechanisms. In this regard, we recently showed that PI3K/AKT signaling plays a significant role in cisplatin-resistance of CSCs, which is dramatically enriched after treatment [35].
Although TNBCs have a high complete response rate to neoadjuvant therapy, a substantial proportion of patients eventually develop chemo resistance and relapse after treatment. These characteristics are believed to be due to the existence of CSCs, thus targeting abnormal signal pathways to eliminate CSCs might be a promising strategy to manage TNBC (Figure 2). The TGF-β, Notch, Wnt/β-catenin signaling pathways and tyrosine-kinase receptors [9, 36, 37] that regulate EMT and CSCs function are abnormally regulated in TNBC. Thus, we reviewed these pathways plus their regulations, which not only be targeted in the mesenchymal-like TNBCs but also in other TNBCs and serve as potential therapeutic targets.
Notch signaling pathway
Notch signaling actively regulates normal mammary stem cell self-renewal and differentiation, which is essential for ensuring normal mammary gland development [38, 39]. It is also widely recognized that abnormal expression of Notch pathway members is also involved in breast tumor development. Gallahan et al. showed that the mutagenic insertion of mouse mammary tumor virus (MMTV) generates a truncated and constitutively active form of the Notch1/4 receptor, leading to the formation of mammary tumors in mice [40]. Dysregulation of Notch signaling pathway leads to abnormal self-renewal and transformation of stem cells/progenitors, which undergo aberrant differentiation processes and result in carcinogenesis [41, 42]. Rustighi and coworkers found that activities of Notch1/4 are essential for maintaining the ‘stemness’ character of both normal stem cells and breast cancer stem cells (BCSCs), and that Notch1/4 activity is strongly correlated with self-renewal and chemo resistance of BCSCs [43]. Harrison and coworkers isolated BCSCs from breast cancer cell lines and primary breast cancer samples by sorting cells resistant to anoikis or cells containing markers of ESA+/CD44+/CD24low, and they found that Notch4 is specifically overexpressed in these BCSCs [44]. Furthermore, pharmacological or genetic inhibition of Notch4 signaling markedly reduces BCSC proliferation and self-renewal in vitro and tumor formation in vivo. In 2013, Reipas demonstrated that p90 ribosomal S6 kinase (RSK) is an activator of Notch4 signal in TNBC by phosphorylation of Y-box binding protein-1 (YB-1), which is an oncogenic transcription factor binding to the Notch4 promoter [45]. Thus in vitro kinase assays and molecular docking were employed to screen for RSK inhibitors from off-patent drugs, and a lead candidate luteolin was found. As anticipated, luteolin showed a high ability to block the Notch4 signaling and suppress proliferation of TNBC, especially BCSC-enriched populations. Similarly, Notch1 also plays a key role in the regulation of BCSC. In vivo studies showed that the Notch1 signal is responsible for the chemo resistance in TNBC cells after treatment of docetaxel. Furthermore, docetaxel-treated cells are enriched for CSCs and became more tumorigenic when replanted into xenograft models [46, 47]. In contrast, γ secretase inhibitor could reverse the chemo resistance of these cells and diminish the CSCs pool in combination with docetaxel. In a TNBC patient-derived xenograft model, Notch1 monoclonal antibodies exhibited synthetically antitumor efficacy combined with docetaxel via inhibition of CSCs [48].
Wnt/β-catenin signaling pathway
The Wnt receptor frizzled-7 and LRP5/6 have been found to be up-regulated in many TNBCs and are associated with poor prognosis [49, 50]. Lack of β-catenin around the membrane or abundant accumulation of β-catenin in the nucleus is regarded as a surrogate marker of Wnt signaling pathway activation, which is found to be enriched in TNBC and basal-like breast cancer [51]. Compelling evidence indicates that Wnt10B may be a valuable therapeutic target for TNBC, and a transgenic murine model with a Wnt10B-driven tumor is shown to have characteristics of human TNBC [52].
The Wnt/β-catenin signaling is believed to be implicated in the control of various stem cells even from distinct tissues and may act as a niche factor to maintain the self-renewing of stem cells [53–55]. There are reports demonstrating that IHC staining of nuclear β-catenin was overlapped with CD44+/CD24low staining [56]. Recently, through high throughput screening, Gupta and colleagues identified salinomycin as a selective inhibitor of CSCs through inhibition of Wnt/β-catenin signaling pathway and degradation of LRP6 [57]. The reduction of β-catenin significantly inhibits tumorigenic ability of TNBC cell lines both in vitro and in vivo, and the stemness of cancer cells is also reduced [58]. In addition, Johnson has developed a mouse model of breast cancer that mimics TNBC by eliminating of Retinoblastoma (Rb), p53, and BRCA1. These tumors have demonstrated a stem phenotype that can be suppressed by Wnt pathway inhibitors [59].
Hedgehog signaling pathway
Hh signaling plays important roles in embryonic mammary gland induction and ductal morphogenesis, which is demonstrated by the synergetic regulation of Ptch1 and Gli-2 in mediating epithelial-stromal interactions [60, 61]. Disruption of either Ptch1 or Gli-2 leads to defects in ductal morphogenesis, which suggests a role for abnormal Hh signaling in breast cancer formation. Meanwhile, it has been reported that Hh signaling components were over-expressed in both BCSCs and human normal mammary stem/progenitor cells [62].
In fact, many genes of Hh pathways are known oncogenes, including Gli1/2, Sonic Hedge-hog (Shh) and Smo, and Ptch1 can also be classified as a tumor suppressor. The Ptch1 locus is one of the most commonly detected changes among the tumor suppressor genes, which occur in about 19% of human breast cancers and in up to 33% of breast cancer cell lines [63]. Interestingly, the Gli1 expression is inversely associated with ER expression and Smo/Gli1 expression is significantly higher in the TNBC than non-TNBC forms [64]. Mukherjee reported that Ptch1 and Smo are expressed at low level in the normal tissue, and Smo is over-expressed in nearly 70% of ductal carcinomas and in about 30% of metastasis breast cancer tissues [65].
The role of Hh signaling pathway in the self-renewal of mammary stem cells is well established. Nevertheless, the regulation role of this pathway in BCSCs has yet to be determined [42]. Liu has shown that Hh pathway activation alters the expression of Bmi-1, and further alters tumorigenic potential of BCSCs [62]. Hh pathway members are highly expressed in CD44+CD24-/low populations, and inhibition of Hh signaling by cyclopamine or Gli1 siRNA suppresses the proliferation and maintenance of these cells [66].
TGF-β signaling pathway
Tumor invasion is often associated with EMT, as disseminated cancer cells seem to require the capacities of the stem cells to form secondary tumors. Thus, the EMT process might also confer the self-renewal capability to disseminated cancer cells. In 2008, Sendurai and colleagues reported a direct association between EMT and stem cell properties [67]. They showed that exposure to TGF-β1 dramatically enhanced mammary stem cells proportion and increased their ability to form mammospheres, and the gene expression profile of EMT resembled that of breast cancer stem cells (e.g. N-cadherin, Slug, E-cadherin, and Snail). Furthermore, more differentiated neoplastic cells that undergo EMT process could generate the cancer stem-like cells, as demonstrated by their increased ability to form mammospheres and tumors in mouse hosts [67]. In 2011, Michaelhas reported the generation of CSCs by inducing EMT, which is induced via exposure of tumor cells to cytokines (TGF-β/TNFα) [68]. These induced CSC-like cells were equipped with increased self-renewing capacity, more aggressive tumorigenicity and increased resistance to chemo agents. More recently, Deng et al. reported that CD24 expression is regulated by TGF-βR1 signaling, which contributed to chemo resistance in TNBCs [69].
The vital role of TGF-β signaling in EMT induction and subsequent acquisition of stemness has suggested this pathway as a new therapeutic approach to target BCSCs. Indeed, inhibition of TGF-βRI combined with paclitaxel significantly reduced CSC frequency and disease progression in an in vivo model [70]. Li has shown that caffeic acid attenuated the CSCs-like properties of CSCs derived from TNBC cell lines by inhibition of TGF-β/Smad2 signaling pathway both in vitro and in vivo [71].
IMMUNE ASSOCIATED TNBC
The immunomodulatory TNBC identified by Brian and basal-like immune activated TNBC identified by Matthew are enriched in gene ontologies of immune cell signaling, cytokine signaling, antigen processing-presentation, and core immune signal transduction pathways [9, 10]. It is not surprising that the immune associated TNBC subtype is associated with enhanced levels of immune cell infiltration and resulted in a good clinical outcome, and tumor-infiltrating lymphocytes seem predictive of neoadjuvant chemotherapy response [72–75]. The immune checkpoint is an elaborate machine that prevents the excessive activation of T-cells under normal conditions, and immune checkpoint blockade-based cancer therapies are currently being increasingly evaluated in the clinical studies. Cytotoxic-T-lymphocyte-antigen-4 (CTLA-4) is a cell surface receptor of lymphocyte T regulators and suppresses T-cell activation. Blockage of CTLA-4 via anti-CTLA-4- mAbs (e.g. ipilimumab and tremelimumab) is predicted to enhance T-cell activity against tumor cells and is under clinical trial for breast cancers (NCT02892734 and NCT02563925) [76]. The PD-L1/PD-1 pathway is a potent mechanism by which tumor cells evade host immune surveillance, and anti-PD-1 and anti-PD-L1-mAbs could also enhance T-cell immune response towards tumor cells (clinical trials in breast cancer include NCT02838823 and NCT02129556) [77, 78]. Recently, there has been great interest in investigating immune checkpoint inhibition in combination with novel agents. In 2017, ASCO demonstrated the potency of combined HDAC and PD-1 inhibition in TNBC. However, there is no currently active trial to assess their efficacy in the various stratifications of TNBC.
LUMINAL ANDROGEN RECEPTOR (LAR) SUBTYPE
The LAR subtype identified by Matthew and Brian is characterized by overexpression of AR and hyper-activation of this pathway [9, 10]. Compared to the rest of the TNBC subtypes, the LAR subtype seems especially resistant to various chemotherapies both in vitro and in vivo, as judged by retrospective analyses of data derived from clinical trials [79, 80]. While the LAR subtype of TNBC showed high response rate to anti-androgens in preclinical studies and clinical trials.
Inhibition of androgen receptor
AR belongs to a kind of transcription factors that are activated by the binding of either androgenic hormones, dihydrotestosterone, or testosterone in the cytoplasm and then translocate into the nucleus [81]. The AR and AR targeted genes were over-expressed in one-third of TNBCs detected via immunohistochemistry [82]. In the in vitro cell culture model, the LAR subtype was shown to depend on AR signaling, as elimination of AR greatly decreased cell viability and tumor growth [9]. Furthermore, the anti-androgen bicalutamide and orteronel are currently under phase II study in AR+, ER−/PR− metastatic breast cancer patients [83, 84]. Together, these studies provide a strong rationale for further investigation of this therapeutic strategy in TNBC. Meanwhile, some LAR TNBC cell lines were found to harbor an activating mutation in Phosphatidylinositol-4, 5-Bisphosphate 3-Kinase, Catalytic Subunit Alpha (PIK3CA), which illuminates the mechanism of resistance to bicalutamide via a compensatory activation of PI3K signaling. In addition, synergistic activity between bicalutamide and PI3K inhibitors was observed in preclinical studies, and providing a rationale for further clinical trials [9]. More recently, Xiaoxiang et al. (2016) reported that PARP1 and AR expression were positively correlated with each other, and BRCA1 expression is negatively correlated with these. They also demonstrated that inhibition of PARP1 and AR had a strong synergy effect in AR positive TNBCs [85].
Recently, there has been an increasing interest in the study of AR negative TNBC, which mainly falls into the basal-like molecular subtype. This so called quadruple negative breast cancer (QNBC) [86–88] lacks the defined targetable pathways mentioned above, and continuous effort has been made to look for targets that are dysregulated in QNBC. In 2017, the ASCO annual meeting showed an investigation of AR expression in TNBC patients. They found that AR expression was positive in 30% of TNBC, and AR negative is significantly associated with the younger age group, higher grade, and higher tumor stage.
Phosphatidylinositol-4, 5-bisphosphate 3-kinase, catalytic subunit alpha
In the last decades, various high throughput analyses in TNBC have discovered a spectrum of gene alteration. Among them, TP53 is the most frequently mutated locus (62%), followed by PIK3CA mutation (10%) [89]. At the same time, the PIK3CA activating mutations seem to be enriched in the LAR molecular subtype of TNBC [9], which sensitize this subtype of TNBC to combined AR and PI3K inhibition [90]. When targeted clinically, patients with PIK3CA mutations showed a favorable response to PI3K signaling inhibition [91]. Besides activating mutations of PIK3CA, alterations in INPP4B (function as an antagonist of PI3K/AKT signaling) and PTEN were also found to frequently occur in TNBC [92], making the PI3K/AKT/mTOR pathway an attractive intervention point for a large fraction of TNBC cases.
OVERLAPPING THERAPEUTIC TARGETS
Tremendous efforts have been made in the identification and characterization of therapeutic targets for TNBC in the past years. Some of these targets have been entered in difference phases of clinic trials (Table 1). If successful, these studies should greatly benefit the long-term survival of cancer patients.
Table 1: Widely studied therapeutic targets that under investigations in the clinical trial for TNBC
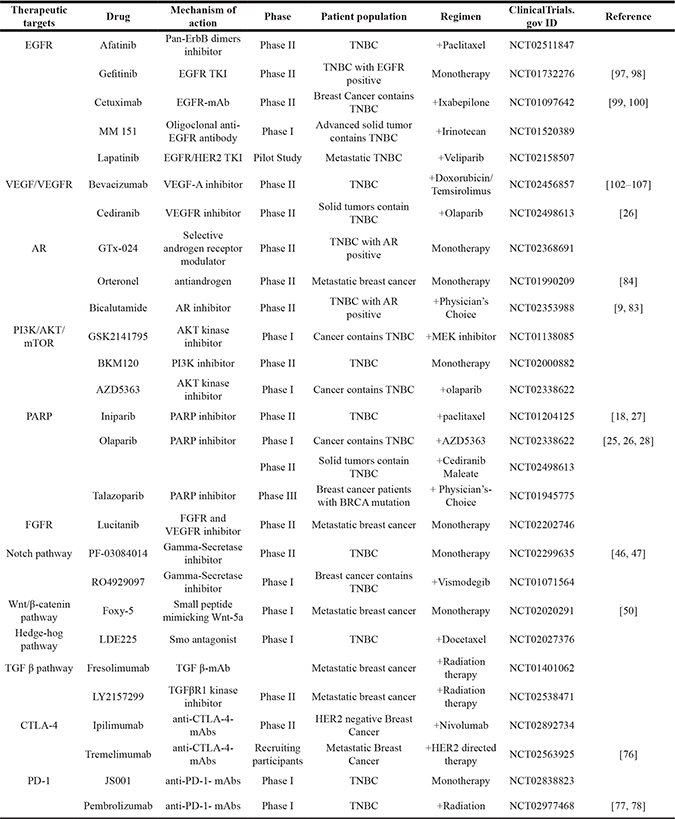
Drugs for each target are listed with its mechanism of action, and patients’ condition and treatment regimen are listed below.
Epidermal growth factor receptor (EGFR)
EGFR was first identified as an important therapeutic target in lung cancer, and it is also a negative prognostic factor for TNBC [93]. It was reported that approximately half of TNBC cases exhibit EGFR expression, and EGFR signaling amplification is common in this aggressive form of breast cancer [94, 95]. Teng et. al. investigated a cohort of 653 TNBC patients and showed that up to 11% of the patients had activating mutation in EGFR locus and were sensitive to EGFR-tyrosine kinase inhibitor (TKI) [96]. However, many studies of clinical trials so far have been disappointing. Gefitinib, a small-molecule against EGFR, was shown to have minimal activity in a phase II trial for treatment of metastatic breast cancer [97] and have modest activity in combination with standard chemotherapy [98]. Some clinical trials investigated EGFR monoclonal antibody in combination with platinum agents for the treatment of TNBC, but failed to achieve improved outcomes [99, 100]. In a phase II trial of cetuximab in combination with carboplatin among TNBC patients, this combination produced responses only in fewer than 20% of patients [99]. In these trials, although the majority of patients harbor EGFR pathway activation, cetuximab failed to block the expression of EGFR pathway in most cases, suggesting that alternative mechanisms were involved. In 2013 Yi et al. reported that the effect of EGFR kinase inhibitors in TNBC is attenuated by activation of PI3K/AKT pathway, and combined inhibition of EGFR and PI3K is warranted [101]. More importantly, in some randomized clinical trials, lack of biomarkers to stratify the heterogeneous tumor accurately and identify sensitive patients who are most likely to respond to EGFR blocking remains a major reason for these disappointing results.
Vascular endothelial growth factor receptor and angiogenesis (VEGFR)
Like EGFR, the VEGFR has also been studied as a therapeutic target for patients with TNBC. Bevacizumab, a humanized monoclonal antibody of VEGFA, has been evaluated in several phase III trials for the treatment of metastatic breast cancer. Two of these phase III trials investigated the curative effect of bevacizumab in a pooled subset of 621 patients [102, 103]. Although bevacizumab has showed improvements in progression-free survival (PFS) of patients, there is no overall survival benefit in the TNBC subgroup [104, 105]. Several phase III trials demonstrated that bevacizumab was ineffective in unselected TNBC patients, including the latest results of phase III Beatrice trial (failure in PFS) [106, 107]. Meanwhile, another newer antiangiogenic agent ramucirumab (IMC-1121B, ImClone) was under investigation. In combined therapy, ramucirumab and docetaxel is currently under phase III trial designed for breast cancer patients with negative HER2 expression (Clinical trials ID: NCT00703326).
Similarly, VEGFR TKIs were also widely assessed in patients with intractable breast cancer. There is little preclinical data that indicates the value of targeting the VEGF pathway in TNBC. The initial evidence came from a phase II trial of a VEGFR TKI (sunitinib), and the overall response to monotherapy was significantly higher in TNBC than in heavily pretreated patients [108]. These results led to the recruitment of a phase III trial via a combination of sunitinib to capcecitabine, but no improvement in PFS of TNBC patients was observed [109].
Fibroblast growth factor receptors (FGFRs)
The aberrant signaling transduced by fibroblast growth factor receptors (FGFRs) is believed to be involved in the pathogenesis of multiple cancer types. However, compared to other receptor tyrosine kinases, targeting the FGFR signaling as a therapeutic strategy for cancer patients has lagged. FGFR1 and FGFR2 were amplified in about 9% and 4% of TNBC, respectively [2, 110]. Genome-wide association studies also identify FGFR2 as one of the major novel susceptibility loci for breast cancer formation [111, 112]. Preclinical study demonstrated that breast cancer cell lines with FGFR amplification were sensitive to FGFR inhibitor PD173074 [110]. Moreover, dovitinib, a pan-FGFR TKI, was found to have an anticancer effect on breast cancer in preclinical studies [113]. The novel anticancer drug NVP-BGJ398 is a selective FGFR inhibitor, and oncogenic FGFR1 amplification serves as a biomarker for cancer cells and predicts sensitivity to NVP-BGJ398 administration [114].
OTHER THERAPEUTIC TARGETS FOR TNBC
Beyond these intensively studied targets above, vast efforts have been made to find new therapeutic targets (Table 2) [115, 116]. Wael et al. showed geminin, which is over-expressed in TNBC, is activated by tyrosine kinase c-Abl [117]. Inhibition of c-Abl by imatinib/nilotonib caused tumor recession. Chinois’s study (2009) demonstrated that the HSP90 inhibitor PU-H71 is a potential chemotherapy in a xenograft model, and induced complete responses in triple-negative breast cancer [118, 119]. To identify new therapeutic targets in TNBC, Toker performed a short hairpin RNA screening for protein kinases (about 26 kinases) commonly dysregulated in breast cancer [120]. They identified AKT3 as a gene preferentially required for the growth of TNBCs, and inhibition of AKT3 significantly abolished cell growth in three-dimensional spheroid culture model. Semenza et. al. reported that hypoxia-inducible factors (HIFs) signaling is essential for maintenance of BCSCs, and HIFs inhibitors combine with cytotoxic chemotherapy showed favorable synergy [121].
Table 2: Some other therapeutic targets under investigations in the clinical trial for TNBC
Therapeutic targets |
Drug |
Mechanism of action |
Phase |
Patient population |
Regimen |
Reference |
|
|---|---|---|---|---|---|---|---|
Tyrosine kinase |
SU011248 |
Inhibitor of PDGFR/VEGFR/KIT/FLT3 |
Phase II |
TNBC |
Monotherapy |
NCT00246571 |
|
MET |
Tivantinib |
MET inhibitor |
Phase II |
Metastatic breast cancer |
Monotherapy |
NCT01575522 |
|
XL184 |
MET and VEGFR2 inhibitor |
Phase II |
TNBC |
Monotherapy |
NCT01738438 |
||
Src tyrosine kinase |
Dasatinib |
Src/c-kit/PDGFR inhibitor |
Phase II |
TNBC |
Monotherapy |
NCT00817531 |
|
HSP90 |
AT13387 |
HSP90 inhibitor |
Phase I |
TNBC |
+paclitaxel |
NCT02474173 |
|
Ganetespib |
HSP90 inhibitor |
Phase II |
Breast cancer contains TNBC |
Monotherapy |
NCT01677455 |
[119] |
|
STAT3 pathway |
Ruxolitinib |
JAK1/2 inhibitor |
Phase I |
Recurrent breast cancer |
Monotherapy |
NCT02041429 |
|
TP53/WEE1 pathway |
MK-1775 |
WEE1 inhibitor |
Phase I |
Advanced solid tumors |
Carboplatin/Paclitaxel |
NCT02341456 |
|
Nuclear export comptlex |
Selinexor |
CRM1 inhibitors |
Phase II |
TNBC |
Monotherapy |
NCT02402764 |
Drugs for each target are listed with its mechanism of action, and patients’ condition and treatment regimen are listed below.
CONCLUSION AND FUTURE ASPECTS
We have discussed the potential therapeutic targets and related signaling pathways for TNBC based on its intrinsic subtypes. Among these known subtypes, the Basal-like TNBC (especially those harboring mutations or dysfunctions in BRCA1) is quite outstanding, which is believed to be sensitive to platinum compounds and PARP inhibitors. Besides the Basal-like subtype, the LAR TNBC is also quite notable because it is reliably identified by both Brian and Matthew. Despite lacking ERs and PRs, the LAR subtype is enriched in hormonally regulated pathways, which imply a targeted therapy for AR signaling. Indeed, bicalutamide showed clinical benefit in ER/PR-negative and AR-positive breast cancers [83], and enzalutamide is under trial for metastatic AR-positive breast cancers [122].
Managing intractable breast cancer via the development of precision medicine has drawn quite a degree of scientific interest, and this concept also holds great promise to TNBC [123, 124]. The alleged scientific and logistical challenges are the main obstacle that hinders clinical implementation of precision medicine. Future studies should be directed towards identifying novel driver mutations by bioinformatics combined with assessment of pathway activation, and to employ more precise medicines for targeting key driver mutations using more reliable and efficient approaches [125]. In this regard, a recent study by Bruna et al. (2016) developed and studied breast-cancer-patient-derived xenografts (PDTXs) and PDTX-derived cells cultures (PDTCs). The comparison between PDTXs and PDTCs revealed that both models share a similar feature in that they both preserve the intra-tumor heterogeneity of the original tumors and can be used for drug screening. Their study further demonstrated that drugs identified using PDTCs were also highly effective for the treatment of PDTXs. This leads to a promising application that PDTCs may serve as a robust platform for pre-clinical pharmacogenomics studies [126].
Future studies should also be directed at identifying more subtypes of TNBC based on their specific features of DNA, RNA, protein, and epigenetics. Given the intrinsic molecular and clinical heterogeneity of TNBC, it is highly possible to further classify it into more different subtypes. Targeted therapies developed against these specific subtypes should greatly benefit patient care and treatment, and effectively extend the health life of these patients.
ACKNOWLEDGMENTS AND FUNDING
We thank for the University of Macau. This work was supported, in part, by the Chair Professor Grant (CPG2017-00026-FHS), Startup Research Grant (SRG2015-00045-FHS) of University of Macau and FDCT grants (065/2015/A2 and 094/2015/A3) to C.X.D.
CONFLICTS OF INTEREST
None.
REFERENCES
1. Chacon RD, Costanzo MV. Triple-negative breast cancer. Breast Cancer Res. 2010; 12:S3.
2. Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012; 490:61–70.
3. Reis-Filho JS, Lakhani SR. Breast cancer special types: why bother? J Pathol. 2008; 216:394–398.
4. Weigelt B, Baehner FL, Reis-Filho JS. The contribution of gene expression profiling to breast cancer classification, prognostication and prediction: a retrospective of the last decade. J Pathol. 2010; 220:263–280.
5. Smid M, Wang Y, Zhang Y, Sieuwerts AM, Yu J, Klijn JG, Foekens JA, Martens JW. Subtypes of breast cancer show preferential site of relapse. Cancer Res. 2008; 68:3108–3114.
6. Chuang HC, Kapuriya N, Kulp SK, Chen CS, Shapiro CL. Differential anti-proliferative activities of poly(ADP-ribose) polymerase (PARP) inhibitors in triple-negative breast cancer cells. Breast Cancer Res Treat. 2012; 134:649–659.
7. He J, Peng R, Yuan Z, Wang S, Peng J, Lin G, Jiang X, Qin T. Prognostic value of androgen receptor expression in operable triple-negative breast cancer: a retrospective analysis based on a tissue microarray. Med Oncol. 2012; 29:406–410.
8. Liu D, He J, Yuan Z, Wang S, Peng R, Shi Y, Teng X, Qin T. EGFR expression correlates with decreased disease-free survival in triple-negative breast cancer: a retrospective analysis based on a tissue microarray. Med Oncol. 2012; 29:401–405.
9. Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011; 121:2750–2767.
10. Burstein MD, Tsimelzon A, Poage GM, Covington KR, Contreras A, Fuqua SA, Savage MI, Osborne CK, Hilsenbeck SG, Chang JC, Mills GB, Lau CC, Brown PH. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin Cancer Res. 2015; 21:1688–1698.
11. Stirzaker C, Zotenko E, Song JZ, Qu W, Nair SS, Locke WJ, Stone A, Armstong NJ, Robinson MD, Dobrovic A, Avery-Kiejda KA, Peters KM, French JD, et al. Methylome sequencing in triple-negative breast cancer reveals distinct methylation clusters with prognostic value. Nat Commun. 2015; 6:5899.
12. Szyf M. DNA methylation signatures for breast cancer classification and prognosis. Genome Med. 2012; 4:26.
13. Roll JD, Rivenbark AG, Sandhu R, Parker JS, Jones WD, Carey LA, Livasy CA, Coleman WB. Dysregulation of the epigenome in triple-negative breast cancers: basal-like and claudin-low breast cancers express aberrant DNA hypermethylation. Exp Mol Pathol. 2013; 95:276–287.
14. Silver DP, Richardson AL, Eklund AC, Wang ZC, Szallasi Z, Li Q, Juul N, Leong CO, Calogrias D, Buraimoh A, Fatima A, Gelman RS, Ryan PD, et al. Efficacy of neoadjuvant Cisplatin in triple-negative breast cancer. J Clin Oncol. 2010; 28:1145–1153.
15. Sikov WM, Berry DA, Perou CM, Singh B, Cirrincione CT, Tolaney SM, Kuzma CS, Pluard TJ, Somlo G, Port ER, Golshan M, Bellon JR, Collyar D, et al. Impact of the addition of carboplatin and/or bevacizumab to neoadjuvant once-per-week paclitaxel followed by dose-dense doxorubicin and cyclophosphamide on pathologic complete response rates in stage II to III triple-negative breast cancer: CALGB 40603 (Alliance). J Clin Oncol. 2015; 33:13–21.
16. von Minckwitz G, Schneeweiss A, Loibl S, Salat C, Denkert C, Rezai M, Blohmer JU, Jackisch C, Paepke S, Gerber B, Zahm DM, Kummel S, Eidtmann H, et al. Neoadjuvant carboplatin in patients with triple-negative and HER2-positive early breast cancer (GeparSixto; GBG 66): a randomised phase 2 trial. Lancet Oncol. 2014; 15:747–756.
17. Maisano R, Zavettieri M, Azzarello D, Raffaele M, Maisano M, Bottari M, Nardi M. Carboplatin and gemcitabine combination in metastatic triple-negative anthracycline- and taxane-pretreated breast cancer patients: a phase II study. J Chemother. 2011; 23:40–43.
18. O’Shaughnessy J, Schwartzberg L, Danso MA, Miller KD, Rugo HS, Neubauer M, Robert N, Hellerstedt B, Saleh M, Richards P, Specht JM, Yardley DA, Carlson RW, et al. Phase III study of iniparib plus gemcitabine and carboplatin versus gemcitabine and carboplatin in patients with metastatic triple-negative breast cancer. J Clin Oncol. 2014; 32:3840–3847.
19. Hu XC, Zhang J, Xu BH, Cai L, Ragaz J, Wang ZH, Wang BY, Teng YE, Tong ZS, Pan YY, Yin YM, Wu CP, Jiang ZF, et al. Cisplatin plus gemcitabine versus paclitaxel plus gemcitabine as first-line therapy for metastatic triple-negative breast cancer (CBCSG006): a randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. 2015; 16:436–446.
20. Dantzer F, de La Rubia G, Menissier-De Murcia J, Hostomsky Z, de Murcia G, Schreiber V. Base excision repair is impaired in mammalian cells lacking Poly(ADP-ribose) polymerase-1. Biochemistry. 2000; 39:7559–7569.
21. McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, Giavara S, O’Connor MJ, Tutt AN, Zdzienicka MZ, Smith GC, Ashworth A. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006; 66:8109–8115.
22. Waddell N, Arnold J, Cocciardi S, da Silva L, Marsh A, Riley J, Johnstone CN, Orloff M, Assie G, Eng C, Reid L, Keith P, Yan M, et al. Subtypes of familial breast tumours revealed by expression and copy number profiling. Breast Cancer Res Treat. 2010; 123:661–677.
23. Tommiska J, Bartkova J, Heinonen M, Hautala L, Kilpivaara O, Eerola H, Aittomaki K, Hofstetter B, Lukas J, von Smitten K, Blomqvist C, Ristimaki A, Heikkila P, et al. The DNA damage signalling kinase ATM is aberrantly reduced or lost in BRCA1/BRCA2-deficient and ER/PR/ERBB2-triple-negative breast cancer. Oncogene. 2008; 27:2501–2506.
24. Yamashita N, Tokunaga E, Kitao H, Hitchins M, Inoue Y, Tanaka K, Hisamatsu Y, Taketani K, Akiyoshi S, Okada S, Oda Y, Saeki H, Oki E, et al. Epigenetic Inactivation of BRCA1 Through Promoter Hypermethylation and Its Clinical Importance in Triple-Negative Breast Cancer. Clin Breast Cancer. 2015; 15:498–504.
25. Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, Friedlander M, Arun B, Loman N, Schmutzler RK, Wardley A, Mitchell G, Earl H, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010; 376:235–244.
26. Liu JF, Tolaney SM, Birrer M, Fleming GF, Buss MK, Dahlberg SE, Lee H, Whalen C, Tyburski K, Winer E, Ivy P, Matulonis UA. A Phase 1 trial of the poly(ADP-ribose) polymerase inhibitor olaparib (AZD2281) in combination with the anti-angiogenic cediranib (AZD2171) in recurrent epithelial ovarian or triple-negative breast cancer. Eur J Cancer. 2013; 49:2972–2978.
27. O’Shaughnessy LSSJ, Danso MA, Rugo HS, Miller K, Yardley DA, Carlson RW, Finn RS, Charpentier E, Freese M, Gupta S, Blackwood-Chirchir A, Winer EP. A randomized phase III study of iniparib (BSI-201) in combination with gemcitabine/carboplatin (G/C) in metastatic triple-negative breast cancer (TNBC). J Clin Oncol. 2011; 29(suppl):1007.
28. Robson M, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, Delaloge S, Li W, Tung N, Armstrong A, Wu W, Goessl C, Runswick S, Conte P. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N Engl J Med. 2017; 377:523–533.
29. De Soto JA, Wang X, Tominaga Y, Wang RH, Cao L, Qiao W, Li C, Xu X, Skoumbourdis AP, Prindiville SA, Thomas CJ, Deng CX. The inhibition and treatment of breast cancer with poly (ADP-ribose) polymerase (PARP-1) inhibitors. Int J Biol Sci. 2006; 2:179–185.
30. Hochegger H, Dejsuphong D, Fukushima T, Morrison C, Sonoda E, Schreiber V, Zhao GY, Saberi A, Masutani M, Adachi N, Koyama H, de Murcia G, Takeda S. Parp-1 protects homologous recombination from interference by Ku and Ligase IV in vertebrate cells. EMBO J. 2006; 25:1305–1314.
31. Kim G, Ison G, McKee AE, Zhang H, Tang S, Gwise T, Sridhara R, Lee E, Tzou A, Philip R, Chiu HJ, Ricks TK, Palmby T, et al. FDA Approval Summary: Olaparib Monotherapy in Patients with Deleterious Germline BRCA-Mutated Advanced Ovarian Cancer Treated with Three or More Lines of Chemotherapy. Clin Cancer Res. 2015; 21:4257–4261.
32. Liu S, Wicha MS. Targeting breast cancer stem cells. J Clin Oncol. 2010; 28:4006–4012.
33. Engelmann K, Shen H, Finn OJ. MCF7 side population cells with characteristics of cancer stem/progenitor cells express the tumor antigen MUC1. Cancer Res. 2008; 68:2419–2426.
34. Wang S, Yang D, Lippman ME. Targeting Bcl-2 and Bcl-XL with nonpeptidic small-molecule antagonists. Semin Oncol. 2003; 30:133–142.
35. Vassilopoulos A, Xiao C, Chisholm C, Chen W, Xu X, Lahusen TJ, Bewley C, Deng CX. Synergistic therapeutic effect of cisplatin and phosphatidylinositol 3-kinase (PI3K) inhibitors in cancer growth and metastasis of Brca1 mutant tumors. J Biol Chem. 2014; 289:24202–24214.
36. Ueno NT, Zhang D. Targeting EGFR in Triple Negative Breast Cancer. J Cancer. 2011; 2:324–328.
37. Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002; 2:442–454.
38. Dontu G, Jackson KW, McNicholas E, Kawamura MJ, Abdallah WM, Wicha MS. Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Res. 2004; 6:R605–615.
39. Clarke RB, Spence K, Anderson E, Howell A, Okano H, Potten CS. A putative human breast stem cell population is enriched for steroid receptor-positive cells. Dev Biol. 2005; 277:443–456.
40. Gallahan D, Callahan R. Mammary tumorigenesis in feral mice: identification of a new int locus in mouse mammary tumor virus (Czech II)-induced mammary tumors. J Virol. 1987; 61:66–74.
41. Pece S, Serresi M, Santolini E, Capra M, Hulleman E, Galimberti V, Zurrida S, Maisonneuve P, Viale G, Di Fiore PP. Loss of negative regulation by Numb over Notch is relevant to human breast carcinogenesis. J Cell Biol. 2004; 167:215–221.
42. Liu S, Dontu G, Wicha MS. Mammary stem cells, self-renewal pathways, and carcinogenesis. Breast Cancer Res. 2005; 7:86–95.
43. Rustighi A, Zannini A, Tiberi L, Sommaggio R, Piazza S, Sorrentino G, Nuzzo S, Tuscano A, Eterno V, Benvenuti F, Santarpia L, Aifantis I, Rosato A, et al. Prolyl-isomerase Pin1 controls normal and cancer stem cells of the breast. EMBO Mol Med. 2014; 6:99–119.
44. Harrison H, Farnie G, Howell SJ, Rock RE, Stylianou S, Brennan KR, Bundred NJ, Clarke RB. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res. 2010; 70:709–718.
45. Reipas KM, Law JH, Couto N, Islam S, Li Y, Li H, Cherkasov A, Jung K, Cheema AS, Jones SJ, Hassell JA, Dunn SE. Luteolin is a novel p90 ribosomal S6 kinase (RSK) inhibitor that suppresses Notch4 signaling by blocking the activation of Y-box binding protein-1 (YB-1). Oncotarget. 2013; 4:329–345. http://doi.org/10.18632/oncotarget.834.
46. Zhang CC, Yan Z, Zong Q, Fang DD, Painter C, Zhang Q, Chen E, Lira ME, John-Baptiste A, Christensen JG. Synergistic effect of the gamma-secretase inhibitor PF-03084014 and docetaxel in breast cancer models. Stem Cells Transl Med. 2013; 2:233–242.
47. Zhang CC, Pavlicek A, Zhang Q, Lira ME, Painter CL, Yan Z, Zheng X, Lee NV, Ozeck M, Qiu M, Zong Q, Lappin PB, Wong A, et al. Biomarker and pharmacologic evaluation of the gamma-secretase inhibitor PF-03084014 in breast cancer models. Clin Cancer Res. 2012; 18:5008–5019.
48. Qiu M, Peng Q, Jiang I, Carroll C, Han G, Rymer I, Lippincott J, Zachwieja J, Gajiwala K, Kraynov E, Thibault S, Stone D, Gao Y, et al. Specific inhibition of Notch1 signaling enhances the antitumor efficacy of chemotherapy in triple negative breast cancer through reduction of cancer stem cells. Cancer Lett. 2013; 328:261–270.
49. Yang L, Wu X, Wang Y, Zhang K, Wu J, Yuan YC, Deng X, Chen L, Kim CC, Lau S, Somlo G, Yen Y. FZD7 has a critical role in cell proliferation in triple negative breast cancer. Oncogene. 2011; 30:4437–4446.
50. Safholm A, Tuomela J, Rosenkvist J, Dejmek J, Harkonen P, Andersson T. The Wnt-5a-derived hexapeptide Foxy-5 inhibits breast cancer metastasis in vivo by targeting cell motility. Clin Cancer Res. 2008; 14:6556–6563.
51. Geyer FC, Lacroix-Triki M, Savage K, Arnedos M, Lambros MB, MacKay A, Natrajan R, Reis-Filho JS. beta-Catenin pathway activation in breast cancer is associated with triple-negative phenotype but not with CTNNB1 mutation. Mod Pathol. 2011; 24:209–231.
52. Wend P, Runke S, Wend K, Anchondo B, Yesayan M, Jardon M, Hardie N, Loddenkemper C, Ulasov I, Lesniak MS, Wolsky R, Bentolila LA, Grant SG, et al. WNT10B/beta-catenin signalling induces HMGA2 and proliferation in metastatic triple-negative breast cancer. EMBO Mol Med. 2013; 5:264–279.
53. Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005; 434:843–850.
54. Nusse R. Wnt signaling and stem cell control. Cell Res. 2008; 18:523–527.
55. Takebe N, Harris PJ, Warren RQ, Ivy SP. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol. 2011; 8:97–106.
56. Zhao Z, Lu P, Zhang H, Xu H, Gao N, Li M, Liu C. Nestin positively regulates the Wnt/beta-catenin pathway and the proliferation, survival and invasiveness of breast cancer stem cells. Breast Cancer Res. 2014; 16:408.
57. Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, Lander ES. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009; 138:645–659.
58. Xu J, Prosperi JR, Choudhury N, Olopade OI, Goss KH. beta-Catenin is required for the tumorigenic behavior of triple-negative breast cancer cells. PLoS One. 2015; 10:e0117097.
59. Johnson JP, Kumar P, Koulnis M, Patel M, Simin K. Crucial and novel cancer drivers in a mouse model of triple-negative breast cancer. Cancer Genomics Proteomics. 2014; 11:115–126.
60. Lewis MT, Veltmaat JM. Next stop, the twilight zone: hedgehog network regulation of mammary gland development. J Mammary Gland Biol Neoplasia. 2004; 9:165–181.
61. Werb Z, Sympson CJ, Alexander CM, Thomasset N, Lund LR, MacAuley A, Ashkenas J, Bissell MJ. Extracellular matrix remodeling and the regulation of epithelial-stromal interactions during differentiation and involution. Kidney Int Suppl. 1996; 54:S68–74.
62. Liu S, Dontu G, Mantle ID, Patel S, Ahn NS, Jackson KW, Suri P, Wicha MS. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006; 66:6063–6071.
63. Buller AJ, Chatzinikolas K, Giannopoulos N, Nair J, Kelleher SH, Raj SD, Roy K, Naylor G. Digit preference in Goldmann applanation tonometry: the hedgehog effect. Am J Ophthalmol. 2005; 140:527–529.
64. Tao Y, Mao J, Zhang Q, Li L. Overexpression of Hedgehog signaling molecules and its involvement in triple-negative breast cancer. Oncol Lett. 2011; 2:995–1001.
65. Mukherjee S, Frolova N, Sadlonova A, Novak Z, Steg A, Page GP, Welch DR, Lobo-Ruppert SM, Ruppert JM, Johnson MR, Frost AR. Hedgehog signaling and response to cyclopamine differ in epithelial and stromal cells in benign breast and breast cancer. Cancer Biol Ther. 2006; 5:674–683.
66. Tanaka H, Nakamura M, Kameda C, Kubo M, Sato N, Kuroki S, Tanaka M, Katano M. The Hedgehog signaling pathway plays an essential role in maintaining the CD44+CD24-/low subpopulation and the side population of breast cancer cells. Anticancer Res. 2009; 29:2147–2157.
67. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008; 133:704–715.
68. Asiedu MK, Ingle JN, Behrens MD, Radisky DC, Knutson KL. TGFbeta/TNF(alpha)-mediated epithelial-mesenchymal transition generates breast cancer stem cells with a claudin-low phenotype. Cancer Res. 2011; 71:4707–4719.
69. Deng X, Apple S, Zhao H, Song J, Lee M, Luo W, Wu X, Chung D, Pietras RJ, Chang HR. CD24 Expression and differential resistance to chemotherapy in triple-negative breast cancer. Oncotarget. 2017; 8:38294–38308. http://doi.org/10.18632/oncotarget.16203.
70. Bhola NE, Balko JM, Dugger TC, Kuba MG, Sanchez V, Sanders M, Stanford J, Cook RS, Arteaga CL. TGF-beta inhibition enhances chemotherapy action against triple-negative breast cancer. J Clin Invest. 2013; 123:1348–1358.
71. Li Y, Jiang F, Chen L, Yang Y, Cao S, Ye Y, Wang X, Mu J, Li Z, Li L. Blockage of TGFbeta-SMAD2 by demethylation-activated miR-148a is involved in caffeic acid-induced inhibition of cancer stem cell-like properties in vitro and in vivo. FEBS Open Bio. 2015; 5:466–475.
72. Adams S, Gray RJ, Demaria S, Goldstein L, Perez EA, Shulman LN, Martino S, Wang M, Jones VE, Saphner TJ, Wolff AC, Wood WC, Davidson NE, et al. Prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancers from two phase III randomized adjuvant breast cancer trials: ECOG 2197 and ECOG 1199. J Clin Oncol. 2014; 32:2959–2966.
73. Desmedt C, Haibe-Kains B, Wirapati P, Buyse M, Larsimont D, Bontempi G, Delorenzi M, Piccart M, Sotiriou C. Biological processes associated with breast cancer clinical outcome depend on the molecular subtypes. Clin Cancer Res. 2008; 14:5158–5165.
74. Denkert C, Loibl S, Noske A, Roller M, Muller BM, Komor M, Budczies J, Darb-Esfahani S, Kronenwett R, Hanusch C, von Torne C, Weichert W, Engels K, et al. Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J Clin Oncol. 2010; 28:105–113.
75. Denkert C, von Minckwitz G, Brase JC, Sinn BV, Gade S, Kronenwett R, Pfitzner BM, Salat C, Loi S, Schmitt WD, Schem C, Fisch K, Darb-Esfahani S, et al. Tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy with or without carboplatin in human epidermal growth factor receptor 2-positive and triple-negative primary breast cancers. J Clin Oncol. 2015; 33:983–991.
76. Vonderheide RH, LoRusso PM, Khalil M, Gartner EM, Khaira D, Soulieres D, Dorazio P, Trosko JA, Ruter J, Mariani GL, Usari T, Domchek SM. Tremelimumab in combination with exemestane in patients with advanced breast cancer and treatment-associated modulation of inducible costimulator expression on patient T cells. Clin Cancer Res. 2010; 16:3485–3494.
77. Nanda R, Chow LQ, Dees EC, Berger R, Gupta S, Geva R, Pusztai L, Pathiraja K, Aktan G, Cheng JD, Karantza V, Buisseret L. Pembrolizumab in Patients With Advanced Triple-Negative Breast Cancer: Phase Ib KEYNOTE-012 Study. J Clin Oncol. 2016; 34:2460–2467.
78. Tolcher AW, Sznol M, Hu-Lieskovan S, Papadopoulos KP, Patnaik A, Rasco D, Di Gravio D, Huang B, Gambhire D, Chen Y, Thall A, Pathan N, Schmidt EV, et al. Phase Ib Study of Utomilumab (PF-05082566), a 4–1BB/CD137 Agonist, in Combination with Pembrolizumab (MK-3475) in Patients with Advanced Solid Tumors. Clin Cancer Res. 2017. [Epub ahead of print].
79. Loibl S, Muller BM, von Minckwitz G, Schwabe M, Roller M, Darb-Esfahani S, Ataseven B, du Bois A, Fissler-Eckhoff A, Gerber B, Kulmer U, Alles JU, Mehta K, et al. Androgen receptor expression in primary breast cancer and its predictive and prognostic value in patients treated with neoadjuvant chemotherapy. Breast Cancer Res Treat. 2011; 130:477–487.
80. Masuda H, Baggerly KA, Wang Y, Zhang Y, Gonzalez-Angulo AM, Meric-Bernstam F, Valero V, Lehmann BD, Pietenpol JA, Hortobagyi GN, Symmans WF, Ueno NT. Differential response to neoadjuvant chemotherapy among 7 triple-negative breast cancer molecular subtypes. Clin Cancer Res. 2013; 19:5533–5540.
81. Roy AK, Lavrovsky Y, Song CS, Chen S, Jung MH, Velu NK, Bi BY, Chatterjee B. Regulation of androgen action. Vitam Horm. 1999; 55:309–352.
82. Collins LC, Cole KS, Marotti JD, Hu R, Schnitt SJ, Tamimi RM. Androgen receptor expression in breast cancer in relation to molecular phenotype: results from the Nurses’ Health Study. Mod Pathol. 2011; 24:924–931.
83. Gucalp A, Tolaney S, Isakoff SJ, Ingle JN, Liu MC, Carey LA, Blackwell K, Rugo H, Nabell L, Forero A, Stearns V, Doane AS, Danso M, et al. Phase II trial of bicalutamide in patients with androgen receptor-positive, estrogen receptor-negative metastatic Breast Cancer. Clin Cancer Res. 2013; 19:5505–5512.
84. Hussain M, Corn PG, Michaelson MD, Hammers HJ, Alumkal JJ, Ryan CJ, Bruce JY, Moran S, Lee SY, Lin HM, George DJ, Prostate Cancer Clinical Trials Consortium a program of the Department of Defense Prostate Cancer Research Program and the Prostate Cancer Foundation. Phase II study of single-agent orteronel (TAK-700) in patients with nonmetastatic castration-resistant prostate cancer and rising prostate-specific antigen. Clin Cancer Res. 2014; 20:4218–4227.
85. Luo J, Jin J, Yang F, Sun Z, Zhang W, Shi Y, Xu J, Guan X. The Correlation Between PARP1 and BRCA1 in AR Positive Triple-negative Breast Cancer. Int J Biol Sci. 2016; 12:1500–1510.
86. Barton VN, D’Amato NC, Gordon MA, Christenson JL, Elias A, Richer JK. Androgen Receptor Biology in Triple Negative Breast Cancer: a Case for Classification as AR+ or Quadruple Negative Disease. Horm Cancer. 2015; 6:206–213.
87. Hon JD, Singh B, Sahin A, Du G, Wang J, Wang VY, Deng FM, Zhang DY, Monaco ME, Lee P. Breast cancer molecular subtypes: from TNBC to QNBC. Am J Cancer Res. 2016; 6:1864–1872.
88. Wu X, Li Y, Wang J, Wen X, Marcus MT, Daniels G, Zhang DY, Ye F, Wang LH, Du X, Adams S, Singh B, Zavadil J, et al. Long chain fatty Acyl-CoA synthetase 4 is a biomarker for and mediator of hormone resistance in human breast cancer. PLoS One. 2013; 8:e77060.
89. Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, Neve RM, Kuo WL, Davies M, Carey M, Hu Z, Guan Y, Sahin A, Symmans WF, Pusztai L, Nolden LK, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008; 68:6084–6091.
90. Lehmann BD, Bauer JA, Schafer JM, Pendleton CS, Tang L, Johnson KC, Chen X, Balko JM, Gomez H, Arteaga CL, Mills GB, Sanders ME, Pietenpol JA. PIK3CA mutations in androgen receptor-positive triple negative breast cancer confer sensitivity to the combination of PI3K and androgen receptor inhibitors. Breast Cancer Res. 2014; 16:406.
91. Janku F, Wheler JJ, Westin SN, Moulder SL, Naing A, Tsimberidou AM, Fu S, Falchook GS, Hong DS, Garrido-Laguna I, Luthra R, Lee JJ, Lu KH, et al. PI3K/AKT/mTOR inhibitors in patients with breast and gynecologic malignancies harboring PIK3CA mutations. J Clin Oncol. 2012; 30:777–782.
92. Marty B, Maire V, Gravier E, Rigaill G, Vincent-Salomon A, Kappler M, Lebigot I, Djelti F, Tourdes A, Gestraud P, Hupe P, Barillot E, Cruzalegui F, et al. Frequent PTEN genomic alterations and activated phosphatidylinositol 3-kinase pathway in basal-like breast cancer cells. Breast Cancer Res. 2008; 10:R101.
93. Tabach Y, Milyavsky M, Shats I, Brosh R, Zuk O, Yitzhaky A, Mantovani R, Domany E, Rotter V, Pilpel Y. The promoters of human cell cycle genes integrate signals from two tumor suppressive pathways during cellular transformation. Mol Syst Biol. 2005; 1:2005 0022.
94. Rakha EA, Elsheikh SE, Aleskandarany MA, Habashi HO, Green AR, Powe DG, El-Sayed ME, Benhasouna A, Brunet JS, Akslen LA, Evans AJ, Blamey R, Reis-Filho JS, et al. Triple-negative breast cancer: distinguishing between basal and nonbasal subtypes. Clin Cancer Res. 2009; 15:2302–2310.
95. Grob TJ, Heilenkotter U, Geist S, Paluchowski P, Wilke C, Jaenicke F, Quaas A, Wilczak W, Choschzick M, Sauter G, Lebeau A. Rare oncogenic mutations of predictive markers for targeted therapy in triple-negative breast cancer. Breast Cancer Res Treat. 2012; 134:561–567.
96. Teng YH, Tan WJ, Thike AA, Cheok PY, Tse GM, Wong NS, Yip GW, Bay BH, Tan PH. Mutations in the epidermal growth factor receptor (EGFR) gene in triple negative breast cancer: possible implications for targeted therapy. Breast Cancer Res. 2011; 13:R35.
97. Baselga J, Albanell J, Ruiz A, Lluch A, Gascon P, Guillem V, Gonzalez S, Sauleda S, Marimon I, Tabernero JM, Koehler MT, Rojo F. Phase II and tumor pharmacodynamic study of gefitinib in patients with advanced breast cancer. J Clin Oncol. 2005; 23:5323–5333.
98. Bernsdorf M, Ingvar C, Jorgensen L, Tuxen MK, Jakobsen EH, Saetersdal A, Kimper-Karl ML, Kroman N, Balslev E, Ejlertsen B. Effect of adding gefitinib to neoadjuvant chemotherapy in estrogen receptor negative early breast cancer in a randomized phase II trial. Breast Cancer Res Treat. 2011; 126:463–470.
99. Carey LA, Rugo HS, Marcom PK, Mayer EL, Esteva FJ, Ma CX, Liu MC, Storniolo AM, Rimawi MF, Forero-Torres A, Wolff AC, Hobday TJ, Ivanova A, et al. TBCRC 001: randomized phase II study of cetuximab in combination with carboplatin in stage IV triple-negative breast cancer. J Clin Oncol. 2012; 30:2615–2623.
100. Baselga J, Gomez P, Greil R, Braga S, Climent MA, Wardley AM, Kaufman B, Stemmer SM, Pego A, Chan A, Goeminne JC, Graas MP, Kennedy MJ, et al. Randomized phase II study of the anti-epidermal growth factor receptor monoclonal antibody cetuximab with cisplatin versus cisplatin alone in patients with metastatic triple-negative breast cancer. J Clin Oncol. 2013; 31:2586–2592.
101. Yi YW, Hong W, Kang HJ, Kim HJ, Zhao W, Wang A, Seong YS, Bae I. Inhibition of the PI3K/AKT pathway potentiates cytotoxicity of EGFR kinase inhibitors in triple-negative breast cancer cells. J Cell Mol Med. 2013; 17:648–656.
102. Miller K, Wang M, Gralow J, Dickler M, Cobleigh M, Perez EA, Shenkier T, Cella D, Davidson NE. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med. 2007; 357:2666–2676.
103. Robert NJ, Dieras V, Glaspy J, Brufsky AM, Bondarenko I, Lipatov ON, Perez EA, Yardley DA, Chan SY, Zhou X, Phan SC, O’Shaughnessy J. RIBBON-1: randomized, double-blind, placebo-controlled, phase III trial of chemotherapy with or without bevacizumab for first-line treatment of human epidermal growth factor receptor 2-negative, locally recurrent or metastatic breast cancer. J Clin Oncol. 2011; 29:1252–1260.
104. O’Shaughnessy J, Romieu G, Diéras V, et al. Meta-analysis of patients with triple-negative breast cancer (TNBC) from three randomized trials of first-line bevacizumab (BV) and chemotherapy treatment for metastatic breast cancer (MBC). Cancer Res. 2010; 70(24 suppl):abstract nr P6-12-03.
105. Carpenter D, Kesselheim AS, Joffe S. Reputation and precedent in the bevacizumab decision. N Engl J Med. 2011; 365:e3.
106. Cameron D, Brown J, Dent R, Jackisch C, Mackey J, Pivot X, Steger GG, Suter TM, Toi M, Parmar M, Laeufle R, Im YH, Romieu G, et al. Adjuvant bevacizumab-containing therapy in triple-negative breast cancer (BEATRICE): primary results of a randomised, phase 3 trial. Lancet Oncol. 2013; 14:933–942.
107. Bidard FC, Mathiot C, Degeorges A, Etienne-Grimaldi MC, Delva R, Pivot X, Veyret C, Bergougnoux L, de Cremoux P, Milano G, Pierga JY. Clinical value of circulating endothelial cells and circulating tumor cells in metastatic breast cancer patients treated first line with bevacizumab and chemotherapy. Ann Oncol. 2010; 21:1765–1771.
108. Burstein HJ, Elias AD, Rugo HS, Cobleigh MA, Wolff AC, Eisenberg PD, Lehman M, Adams BJ, Bello CL, DePrimo SE, Baum CM, Miller KD. Phase II study of sunitinib malate, an oral multitargeted tyrosine kinase inhibitor, in patients with metastatic breast cancer previously treated with an anthracycline and a taxane. J Clin Oncol. 2008; 26:1810–1816.
109. Crown JP, Dieras V, Staroslawska E, Yardley DA, Bachelot T, Davidson N, Wildiers H, Fasching PA, Capitain O, Ramos M, Greil R, Cognetti F, Fountzilas G, et al. Phase III trial of sunitinib in combination with capecitabine versus capecitabine monotherapy for the treatment of patients with pretreated metastatic breast cancer. J Clin Oncol. 2013; 31:2870–2878.
110. Turner N, Lambros MB, Horlings HM, Pearson A, Sharpe R, Natrajan R, Geyer FC, van Kouwenhove M, Kreike B, Mackay A, Ashworth A, van de Vijver MJ, Reis-Filho JS. Integrative molecular profiling of triple negative breast cancers identifies amplicon drivers and potential therapeutic targets. Oncogene. 2010; 29:2013–2023.
111. Douglas JF, Iwasawa H, Sun Z, Fedorov AV, Ishikado M, Saitoh T, Eisaki H, Bando H, Iwase T, Ino A, Arita M, Shimada K, Namatame H, et al. Superconductors: unusual oxygen isotope effects in cuprates? Nature. 2007; 446:E5.
112. Gaudet MM, Kuchenbaecker KB, Vijai J, Klein RJ, Kirchhoff T, McGuffog L, Barrowdale D, Dunning AM, Lee A, Dennis J, Healey S, Dicks E, Soucy P, et al. Identification of a BRCA2-specific modifier locus at 6p24 related to breast cancer risk. PLoS Genet. 2013; 9:e1003173.
113. Andre F, Bachelot T, Campone M, Dalenc F, Perez-Garcia JM, Hurvitz SA, Turner N, Rugo H, Smith JW, Deudon S, Shi M, Zhang Y, Kay A, et al. Targeting FGFR with dovitinib (TKI258): preclinical and clinical data in breast cancer. Clin Cancer Res. 2013; 19:3693–3702.
114. Guagnano V, Kauffmann A, Wohrle S, Stamm C, Ito M, Barys L, Pornon A, Yao Y, Li F, Zhang Y, Chen Z, Wilson CJ, Bordas V, et al. FGFR genetic alterations predict for sensitivity to NVP-BGJ398, a selective pan-FGFR inhibitor. Cancer Discov. 2012; 2:1118–1133.
115. Finn RS, Bengala C, Ibrahim N, Roche H, Sparano J, Strauss LC, Fairchild J, Sy O, Goldstein LJ. Dasatinib as a single agent in triple-negative breast cancer: results of an open-label phase 2 study. Clin Cancer Res. 2011; 17:6905–6913.
116. Finn RS, Dering J, Ginther C, Wilson CA, Glaspy P, Tchekmedyian N, Slamon DJ. Dasatinib, an orally active small molecule inhibitor of both the src and abl kinases, selectively inhibits growth of basal-type/”triple-negative” breast cancer cell lines growing in vitro. Breast Cancer Res Treat. 2007; 105:319–326.
117. Blanchard Z, Mullins N, Ellipeddi P, Lage JM, McKinney S, El-Etriby R, Zhang X, Isokpehi R, Hernandez B, Elshamy WM. Geminin overexpression promotes imatinib sensitive breast cancer: a novel treatment approach for aggressive breast cancers, including a subset of triple negative. PLoS One. 2014; 9:e95663.
118. Caldas-Lopes E, Cerchietti L, Ahn JH, Clement CC, Robles AI, Rodina A, Moulick K, Taldone T, Gozman A, Guo Y, Wu N, de Stanchina E, White J, et al. Hsp90 inhibitor PU-H71, a multimodal inhibitor of malignancy, induces complete responses in triple-negative breast cancer models. Proc Natl Acad Sci USA. 2009; 106:8368–8373.
119. Jhaveri K, Chandarlapaty S, Lake D, Gilewski T, Robson M, Goldfarb S, Drullinsky P, Sugarman S, Wasserheit-Leiblich C, Fasano J, Moynahan ME, D’Andrea G, Lim K, et al. A phase II open-label study of ganetespib, a novel heat shock protein 90 inhibitor for patients with metastatic breast cancer. Clin Breast Cancer. 2014; 14:154–160.
120. Chin YR, Yoshida T, Marusyk A, Beck AH, Polyak K, Toker A. Targeting Akt3 signaling in triple-negative breast cancer. Cancer Res. 2014; 74:964–973.
121. Samanta D, Gilkes DM, Chaturvedi P, Xiang L, Semenza GL. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc Natl Acad Sci USA. 2014; 111:E5429–5438.
122. Schwartzberg LS, Yardley DA, Elias AD, Patel M, LoRusso P, Burris HA, Gucalp A, Peterson AC, Blaney ME, Steinberg JL, Gibbons JA, Traina TA. A Phase I/Ib Study of Enzalutamide Alone and in Combination with Endocrine Therapies in Women with Advanced Breast Cancer. Clin Cancer Res. 2017; 23:4046–4054.
123. Wong AH, Deng CX. Precision Medicine for Personalized Cancer Therapy. Int J Biol Sci. 2015; 11:1410–1412.
124. Lehmann BD, Pietenpol JA. Identification and use of biomarkers in treatment strategies for triple-negative breast cancer subtypes. J Pathol. 2014; 232:142–150.
125. Arnedos M, Vicier C, Loi S, Lefebvre C, Michiels S, Bonnefoi H, Andre F. Precision medicine for metastatic breast cancer-limitations and solutions. Nat Rev Clin Oncol. 2015; 12:693–704.
126. Bruna A, Rueda OM, Greenwood W, Batra AS, Callari M, Batra RN, Pogrebniak K, Sandoval J, Cassidy JW, Tufegdzic-Vidakovic A, Sammut SJ, Jones L, Provenzano E, et al. A Biobank of Breast Cancer Explants with Preserved Intra-tumor Heterogeneity to Screen Anticancer Compounds. Cell. 2016; 167:260–274 e222.