
INTRODUCTION
In these last decades, many researches focused on the possible role of mesenchymal stem cells (MSCs) to promote tumor progression by interacting with tumor cells and other stroma cells in the complex network of microenvironment [1]. The multiple properties of these cells such as self-renewal, plasticity to differentiate into several cell types and ability to modulate immune response as well as strong tropism to tumors make them crucial players in the development of metastatic phenotype.
Once the MSCs come into contact with the tumor microenvironment (TME) they are “educated” to evolve and differentiate in tumor-associated MSCs (TA-MSCs) and cancer associated fibroblasts (CAFs) [2–4]. Both these cells cooperate to support all hallmarks of cancer including sustaining proliferative signaling, evading growth suppressors, resisting cell death, enabling replicative immortality, inducing angiogenesis, and activating invasion and metastasis [5] (Figure 1).
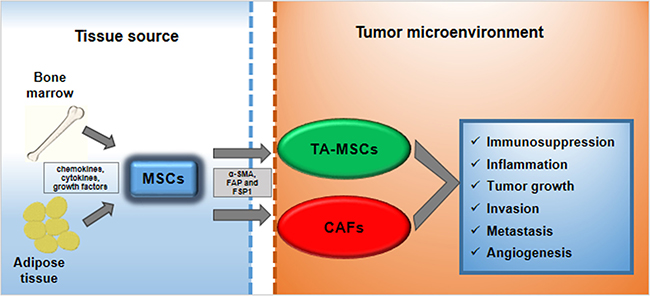
Figure 1: MSCs trans-differentiation into tumor microenvironment. MSCs deriving from bone marrow or from adipose tissue can be recruited to tumor microenvironment in response to multiple signals: chemokines, cytokines and growth factors. Here, MSCs can trans-differentiate into tumor-associated MSCs (TA-MSCs) and cancer-associated fibroblasts (CAFs). They are characterized by specific markers such as α-smooth muscle actin (α-SMA), fibroblast activating protein (FAP) and fibroblast-specific protein 1 (FSP1). TA-MSCs and CAFs promote metastatic phenotype acting on: immunosuppression, inflammation, tumor growth, invasion, metastasis and angiogenesis.
Many signals are involved in the cross-talk between TA-MSCs and other component of tumor microenvironment. Several chemokines, cytokines, growth factors and others are produced by tumor cells to recruit MSCs from bone marrow and adipose tissue. In turn, TA-MSCs release the same molecules to repress immune surveillance, to induce epithelial-mesenchymal transition (EMT) program and to promote tumor cell migration and invasion. Recently, on this topic we reported that bone-marrow MSCs (BM-MSCs) derived from patients induced the metastatic phenotype of osteosarcoma and hepatocellular carcinoma through stromal derived factor 1 (SDF-1)-C-X-C-Chemokine receptor type 4 (CXCR4) axis and also through the aquaporin 1 (AQP1) membrane channel [6, 7]. Furthermore, it has been observed that MSCs can contribute to prepare the pre-metastatic sites by inducing a favorable microenvironment for the colonization of circulating tumor cells [1].
Although many studies reported the pro-tumorigenic activity of MSCs many others showed their tumor suppressive properties [8].
Here, we mainly focused on the progress made to elucidate the key mechanisms in which MSCs are involved to promote a pro-metastatic phenotype.
Nowadays, there are few reports regarding the possibility to target MSCs to hamper cancer progression. However, recently Ramos et al. (2017) reported that the inhibition of Histone deacetylases 8 in MSCs derived from myeloproliferative neoplasms selectively decreases their hematopoietic-supporting ability [9].
TROPISM OF MESENCHYMAL STEM CELLS TOWARDS TUMORS
Currently, there has been a heightened focus on the homing abilities of MSCs into tumors and their role in promoting tumor progression [10, 11] but the specific mechanisms behind this are not yet well elucidated. MSCs that show a strong tropism to cancer, are derived from bone marrow [12, 13], adipose tissue [14] and also the umbilical cord [15].
Nakamizo et al. (2005) isolated human MSCs (hMSCs) from the bone marrow and followed their fate labeled them using fluorescent protein. Then, they injected labeled-MSCs into the opposite hemisphere of mouse brain of a orthotopic glioma model and after 14 days observed that the fluorescent hMSCs were exclusively within the brain tumors [13].
Moreover, fluorescent-labeled MSCs were detected in metastatic breast tumors in mice after their systemic administration and monocyte chemotactic protein-1 (MCP-1) produced by tumor cells was involved in their recruitment [16].
Interestingly, it has been reported that human adipose-derived MSCs (AD-MSCs) transduced with a retroviral vector encoding full-length human tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) specifically localized into xenografts and mediated tumor cell apoptosis without significant apparent toxicities to normal tissues [14].
Furthermore, Hu et al. (2011) observed that human umbilical blood mononuclear cell (UBMC)-derived mesenchymal stem cells (UBMC-MSCs) transfected with recombinant pIRES2-IL-21-enhancement green fluorescent protein delayed tumor growth and prolonged survival in ovarian-cancer-bearing mice [15].
Tumors are considered the “wounds that never heal” and due to this are in a constant inflammatory status [17]. Indeed, cancer cells and tumor associated stroma cells produce various inflammatory molecules such as cytokines, chemokines and growth factors that are involved in MSC recruitment [18–20]. Moreover, it has been reported that MSCs respond to these signals by expressing high levels of specific receptors, adhesion molecules, cell surface markers and toll-like receptors [21].
At present, although much research is being carried out in this field, it has not yet been cleared which factor could play the most important role in this mechanism, it would seem that more factors are involved in the mediation of MSC tropism to a specific tumor site. In TME there is a chemotactic gradient consisting of different chemokines: CC-chemokine ligand 2 and 5 (CCL2, CCL5), CXC – chemokine ligand 12 and 16 (CXCL12, CXCL16), which are able to recruit MSCs [1]. Recently, it has been reported that the chemokine CCL2 plays a crucial role in the ionizing radiation induced tropism of MSCs to gliomas [22]. Furthermore, it has been shown that CCL2 and CCL5 produced by macrophages induce MSC recruitment in tumor sites [23]. Interestingly, a study evaluated the role of SDF-1a/CXCL12 in the migration of MSCs in response to tumor cells elucidating that Jak/STAT, MEK/ERK as well as NFkB pathways are activated downstream of SDF-1 [24]. Jung et al. (2013) provided evidence of an important cross-talk between MSCs and tumor cells in promoting a metastatic phenotype of prostate tumors. MSCs were both recruited as well as transformed in CAFs by CXCL16 produced by tumor cells. In turn, MSC-like CAFs secreted CXCL12 that binding to CXCR4 on tumor cells induced an EMT, which ultimately promoted metastasis to secondary tumor sites [25].
Many growth factors such as, platelet derived growth factor (PDGF), vascular endothelial growth factor (VEGF), insulin-like growth factor 1 (IGF-1), transforming growth factor-β (TGF-β) and basic fibroblast growth factor (bFGF) have been found to effectively mediate MSC homing to TME [21, 26, 27].
Doucette et al. (2011) showed that syngeneic MSCs are capable of homing to endogenous gliomas in immunocompetent mice. This model of high-grade glioma was induced by overexpression of PDGF-BB, that previously has been well characterized to be a critical mediator of MSC tropism to tumor sites [28–30].
Recent studies suggest that TGF-β1 is a crucial player in inducing MSC migration towards prostatic carcinoma cells (PC3 DU145) as well as to tumor stroma components [31]. In addition, Beckermann et al. (2008) showed that MSCs migrated towards growth factors produced by pancreatic tumors, such as PDGF, EGF, VEGF and that specific inhibitors Glivec, Erbitux and Avastin hampered their recruitment [32]. Furthermore, it has been demonstrated that bFGF and downstream Erk/Smad3 signaling pathway are involved in BM-MSC tropism to 4T1 breast cancer cells by using a specific neutralizing antibody [33].
In breast cancer, hypoxic TME, through activation of hypoxia-inducible factors (HIFs), is able to regulate the complex bidirectional MSC-tumor cell interaction. It has been demonstrated that hypoxia-induced expression of Placental growth factor (PGF) and CXCL16 in breast cancer cells is required for MSC recruitment and their pro-metastatic activity [33–36]. Likewise, breast cancer cells under hypoxic conditions (1.5% O2) were able to secrete high levels of interleukin-6 (IL-6), which served to activate and attract MSCs through Stat3 and MAPK signaling pathways [34].
MicroRNA’s (miRNAs) have been shown to modulate MSC migration towards breast cancer. Indeed, miR-126/miR-126(*) suppressed the sequential recruitment of MSCs and inflammatory monocytes into the tumor stroma by directly inhibiting SDF-1α expression by cancer cells. [37].
A critical event of MSCs homing to cancer is to execute trans-migration through endothelial cells of the vessel. It has been observed that similarly to hematopoietic cells, MSCs express high levels of E-selectin, whereas lack the expression of platelet endothelial cell adhesion molecule-1 (PECAM-1), L-selectin and β2 integrins [38]. On the other hand, intravital microscopy demonstrated the capacity of MSCs to roll and adhere to post-capillary venules in vivo in a mouse model through a P-selectin and vascular cell adhesion molecule-1 (VCAM-1)/ very late antigen-4 (VLA-4) dependent manner [39].
At the present, many studies have been performed and are still underway to clarify the mechanisms underlying MSC tumor tropism and to evidence responsible factors which induce their recruitment in different tumor sites (Table 1).
Table 1: Factors involved in mesenchymal stem cell tropism to tumor microenvironment
Factor |
Tumor Type |
Reference |
|
|---|---|---|---|
Cytokines |
TFN-a |
Glioma |
[151] |
IFN-g |
Glioma |
[151] |
|
IL-1b |
Melanoma Breast Cancer |
[152] |
|
IL-6 |
Breast Cancer |
||
IL-8 |
Lung Cancer Breast Cancer |
[154] |
|
Growth Factors |
TGF-b |
Glioma Prostate Cancer |
|
PGF |
Breast Cancer |
[35] |
|
PDGF |
Glioma Renal Cancer Breast Cancer |
||
HGF |
Gastric Cancer Lung Cancer |
[157] |
|
Chemokines |
SDF-1/CXCR4 |
Glioma Breast Cancer Skin Cancer |
|
CCL2 CCL5 |
Glioma |
||
CXCL (GRO-a) |
Breast Cancer |
||
MCP-1 |
Breast Cancer |
[16] |
|
Other Factors |
HIF-1 |
Glioma Breast Cancer |
[35] |
MMP |
Glioma |
[158] |
|
VCAM |
Glioma |
[159] |
|
LL-37 |
Ovarian Cancer |
[161] |
MESENCHYMAL STEM CELLS MODULATE TUMOR IMMUNE RESPONSE
MSCs are well known to be important modulators of inflammatory and immune responses affecting both the adaptive as well as innate immune systems [40]. In 1998, McIntosh KR et al. reported, for the first time, the use of MSCs for prevention and immune responses in transplantation [41]. A few years after, haploidentical MSCs were transplanted to treat severe acute graft-versus-host disease [42].
In TME cancer cells produce many inflammatory factors that are able to recruit non only MSCs but also many immune cells. A described above, MSCs into tumor sites are modified in TA-MSCs that contribute to generate malignant phenotype also excercising an immunosuppressive funcion [43, 44]. Han et al. (2011) reported that, when B16 melanoma cells were co-injected with MSCs pre-incubated with interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α) in syngeneic mice, xenografts were developed faster than those obtained from B16 cells alone whereas tumor incidence was increased in allogeneic recipients [45]. These immunosuppressive effects were due to an increase of inducible nitric oxide synthase (iNOS) expression in MSCs [45]. Similarly, MSCs pre-treated with IL-1α effectively promoted the growth of prostate cancer cells in vivo through TGF-β upregulation [46].
In addition, It has been shown that MSCs isolated from spontaneous lymphomas in mice (L-MSCs) were more effective in recruiting monocytes/macrophages and in promoting tumor growth than BM-MSCs and their activity was mediated via C-C-Chemokine receptor type 2 (CCR2). Importantly, when BM-MSCs were TNFα-pretreated they mimicked L-MSCs in their chemokine production profile and in their ability to promote tumorigenesis not only of lymphoma but also melanoma, and breast carcinoma [47]. Recently, Yu et al. (2016) showed that TNFα-activated MSCs expressed CXCR2 ligands (CXCL1, CXCL 2 and CXCL5) and through them efficiently recruited CXCR2+ neutrophils into breast cancer microenvironment.
These neutrophils directly enhanced tumor lung metastasis, inducing tumor cells to express pro-metastatic genes [48]. In addition, in breast cancer cells indoleamine 2,3-dioxygenase (IDO)-expressing humanized MSCs (MSC-IDO) were capable of suppressing T-lymphocyte proliferation in vitro as well as reducing tumor-infiltrating CD8+ T cells and B cells in vivo , similar effects were also observed in melanoma and lymphoma tumor models [49].
Conversely, in breast cancer MSCs conferred immune protection through TGF-β1-mediated generation of forkhead box P3 (FoxP3)+ Tregs (regulatory t cells), that in turn suppressed tumor cell cytolysis by CD8+ T cells and natural killer (NK) cells [50]. In triple negative breast cancer, the cross-talk between tumor cells and MSCs, caused the production of macrophage colony-stimulating factor 1 (CSF1) that recruited in the TME, tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs) [51]. Macrophages and MSCs may engage in a bidirectional interaction where M2 or M2-like macrophages determinate an increase of MSCs growth and motility [52]. In turn, MSCs can induce macrophages to acquire an anti-inflammatory phenotype with immunosuppressive abilities and pro-tumor functions [53, 54]. Recently, Yamada et al. (2016) reported that mouse BM-MSCs in vivo caused the increase of melanoma growth and M2 macrophage polarization through milk fat globule EGP factor 8 protein (MFG-E8) [55]. BM-MSCs obtained from patients with follicular lymphoma showed a different gene expression profile respect to MSCs obtained from healthy donors (HD-MSCs). These cells were able to recruit and polarize monocytes more efficiently than HD-MSCs thus sustaining malignant B-cell growth. Conversely, when MSCs were transfected to overexpress an NAD-dependent deacetylase sirtuin 1 (MSCs-Sirt1), they inhibited the growth of breast and prostate carcinomas by recruiting NK cells and macrophages [56]. Interestingly, MSCs associated in pancreatic carcinoma microenvironment had an increased tumor-promoting potential in respect to MSCs obtained from normal pancreas. This effect was mediated by their ability to induce macrophage polarization [57]. Chiassone et al (2016) showed that MSCs were able to induce the polarization of macrophages toward a novel M2-like phenotype (MMSC) that in turn could inhibit NK cells activation and could cause the expansion of Tregs cells [58]. In addition, the engagement of tool-like receptor (TLR) reverted MMSC toward a M1 phenotype with pro-inflammatory and immunostimulatory activities [58] thus becoming detrimental for tumor progression. Conversely, it has been reported that MSCs derived from bone marrow of patients with low/intermediate risk leukemia at diagnosis enhanced the NK cell antitumor cytolytic activity and their pro-inflammatory cytokine production [59].
TRANS-DIFFERENTIATION OF TUMOR-ASSOCIATED MESENCHYMAL STEM CELLS INTO CANCER ASSOCIATED FIBROBLASTS
When MSCs arrive into TME they can differentiate not only in TA-MSCs but also in CAFs. Among stromal cells that constitute TME, CAFs are known to play a crucial role in promoting tumor progression [60]. They are involved in all tumor events preceding the metastatic spread such as of EMT, neo-angiogenesis, immune surveillance, tumor cell migration and invasion [60]. CAFs were found in different forms of cancer and their high heterogeneity probably was due to different sources: fibroblasts, smooth muscle cells, endothelial cells and epithelial cells [61]. Recently, it has been reported that important CAF precursors are MSCs. These cells for their high plastic abilities, when stimulated directly or indirectly by factors produced from tumor cells or others stroma cells can trans-differentiate into a CAF-like phenotype [1]. For the first time, Mishra et al. (2008) observed that the effect of prolonged exposure of MSCs to factors produced by a human breast cancer cell line MDA-MB-231 caused up-regulation of 53 CAF-associated genes and an higher expression of α smooth muscle actin (α-SMA), vimentin, fibroblast surface protein (FSP) and SDF-1 [2]. An important evidence that CAF may derive from MSCs was shown by injecting green fluorescent protein (GFP)-labelled BM-MSCs in a mouse model of inflammation-dependent interleukin 1β (IL 1β) gastric cancer. About 20% of CAFs were found to be GFP-positive, indicating that GFP-BM-MSCs were their pre-cursors [4]. Importantly, in 2009 the first report was published showing that in female patients with gastric cancer and rectal adenoma, which had received bone marrow transplants from male donors, were identified several Y-chromosome positive CAFs [62].
In the last years, many studies investigated the key role played by TGF-β in the mechanism underlying MSC differentiation to CAFs. Recently, an interesting paper reported that in prostate cancer, MSCs are both recruited as well as induced to differentiate into CAFs in response to TGF-β produced by tumor cells. In addition, respect to normal MSCs, CAF-like MSCs performed vascular mimicry and recruited monocytes, which were polarized to M2 macrophages within the prostate cancer (PCa) environment [31]. Previously, Shangguan et al. (2012) observed that, when human BM-MSCs were transduced with a lentiviral vector encoding bone morphogenetic protein and activin membrane-bound inhibitor (BAMBI, a decoy TGF-β receptor), TGF-β/Smad signaling was significantly inhibited. Consequently, CAF markers were down-regulated in human BM-MSCs treated with TGF-β1 or tumor-conditioned medium or co-cultured with cancer cells [63]. Moreover, it has been reported that an endoplasmic reticulum (ER) chaperone GRP78, overexpressed in a variety of tumors, was able to induce BM-MSCs differentiation into CAFs through activating TGF-β/Smad signalling pathway. The importance of TGF-β involvement in MSC transformation was confirmed by another study where a TGF-β type I receptor kinase inhibitor, SB431542, caused a significant decrease of CAF markers expression [64].
It has also been observed that BM-MSCs were able to migrate towards 4T1 breast cancer cells and there trans-differentiate into CAFs in response to bFGF signaling pathway [33].
Several CAFs markers were identified in trans-differentiated MSCs, the main ones are α-SMA, the fibroblast activation protein (FAP) and FSP, but also thrombospondin-1, tenascin-C, desmin-1, and VEGF-AA can be involved [5, 65, 66]. α-SMA has been known to play a pivotal role in the embryonic stem cell-derived cardiomyocyte differentiation. On the other hand, expression of α-SMA in the stroma increases fibroblasts contractile ability and contributes to alterations in the cytoskeletal organization [61, 66, 67]. CAFs may alter the extracellular matrix (ECM) through the production of proteases such as FAP. High levels of this enzyme are expressed in over 90% of human epithelial carcinomas including breast, lung, and ovarian cancers. Conversely, normal healthy adult tissues have almost no detectable FAP expression [68]. Generally, MSC-like CAFs that are positive for FSP and FAP originate from MSCs which derive from the bone marrow, whereas it has been observed that adipose-derived MSC mainly differentiate into vascular and fibrovascular stromal cells. [65]. Compared to normal fibroblasts and myofibroblasts, CAFs are perpetually activated, and cannot revert back to their original phenotype nor undergo apoptosis [61], confirming their dramatic role in tumor progression.
Recently, some reports have suggested that the irreversible activation of fibroblasts might be driven by epigenetic alterations [69, 70]. Albrengues et al (2015) identified the DNA methyltransferase (DNMT) family epigenetic modifiers as regulators of the pro-invasive CAF activity that affects Janus kinase 1 (JAK1)–signal transducer and activator of transcription 3 (STAT3) activation [71]. Dual inhibition of DNMT and JAK activity restored the non-invasive phenotype of CAFs. Conversely, a global gene hypometylation was observed in CAFs isolated from human gastric cancers [72]. Moreover, Shakya et al found globally decreased 5-methyl-cytosine (5-mC), along with increased amounts of 5-hydroxymethyl-cytosine (5-HmC) in CAFs, in progression from pancreatic intraepithelial neoplasia to pancreatic ductal adenocarcinoma [73]. Among these epigenic changes the post-trascriptional control involving miRNA statutes plays an important role [69]. For example, it has been observed that upregulation of miRNA-21 in CAFs is associated with high proliferation in breast cancer [74, 75], poor-disease-free survival in colorectal carcinoma [76] and invasion of esophageal squamous cell carcinoma [77].
Notably, CAFs are able also to migrate with epithelial cancer cells through endothelial cell layers, thus contributing to establishment of new pre-metastatic niches [78–80].
MESENCHYMAL STEM CELLS PROMOTE CANCER CELL EPITHELIAL–MESENCHYMAL TRANSITION AND INVASION
In these last years, research studies on mechanisms underlying tumor progression and acquisition of pre-metastatic phenotype have demonstrated well that MSCs actively participate in inducing oncogenic EMT (Figure 2) [81, 82].
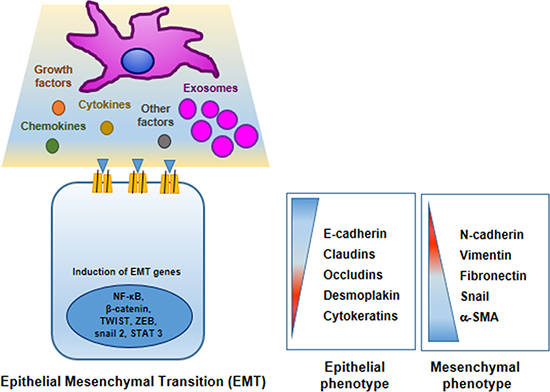
Figure 2: Mesenchymal stem cells promote cancer cell epithelial–mesenchymal transition (EMT). MSCs in TME produce many factors that induce or repress in tumor cells different genes encoding proteins involved in EMT program. In tumor cells epithelial markers are down-regulated: E-cadherin, claudins, occludins, desmoplakin and cytokeratins; whereas mesenchymal markers are up-regulated: N-cadherin, vimentin, fibronectin, snail and and smooth muscle actin.
In particular, in TME where a complex crosstalk between tumor cells and stromal cells takes place, the epithelial tumor cells undergo profound morphological and functional changes trans-differentiating in a mesenchymal-like phenotype with a higher metastatic potential [83]. Generally, EMT represents a physiological developmental process by which the epithelial cells in particular conditions (normal embryogenesis, tissue repair and in tumors), upon extracellular cues, undergo profound morphogenetic changes, to become cells with phenotype and morphology similar to stem cells [81, 84]. EMT of tumor cells is characterized by a sequence of molecular events: 1) down-regulation of E-cadherin, 2) secretion of enzymes (i.e. matrix metalloproteinases), 3) up regulation of mesenchymal markers (i.e vimentin, N-cadherin and fibronectin) [85, 86].
Many EMT inducible factors have been demonstrated to be produced or secreted by MSCs such as cytokines (IL1, IL6) [87], chemokines (CCL5 , CXCL1, CXCL5, CXCL7 and CXCL8) [88, 89], growth factors (TGF-β, FGF, Hepatocyte growth factor HGF and epidermal growth factor, EGF) [90, 91] as well as hypoxia inducible factors and reactive oxygen species. Interestengly, in breast cancer a part from MSCs, other stromal cells such as adipocytes are able to promote a more aggressive phenotype by producing CCL5 and IGF-1 [92, 93].
These molecules acting in a paracrine manner are able to orchestrate EMT program. It has been reported the importance of expression and functional activation of a group of EMT-inducing transcription factors (Twist, Snail, Slug, zinc finger E-box-binding homeobox 1 (ZEB1) and ZEB2 [81, 94]. The activation of these transcription factors causes the down-regulation of genes responsible to encode epithelial junction proteins determining a disassembly of adherens junctions, desmosomes, and tight junctions [95]. Conversely, they can up-regulate expression of mesenchymal genes encoding N-cadherin, fibronectin and vimentin [96]. The trigger of these transcription factors is sufficient to induce EMT in tumor epithelial cells as observed in different invasive carcinomas [97]. On this regard, recently we reported that CXCL12/CXCR4 axis is involved in migration and invasion of osteosarcoma and hepatocellular carcinoma cell lines through EMT activation [6].
The pivotal role of EMT program in cancer progression, has been investigated in several pre-clinical cancer models such as breast [98], ovarian [99], colon [100], and esophageal carcinomas [101].
Most recently, it has been reported that EMT program is also under miRNAs control that may promote mesenchymal or inhibit epithelial gene expression. miRNA-9 which is up-regulated in breast cancer cells, directly targets the E-cadherin-encoding messenger RNA, leading to an increase in cell motility and invasiveness [102]. Conversely, miRNA 200b was able to revert EMT in prostate carcinoma animal model increasing pan-epithelial marker such as E-cadherin, cytokeratins 8 and 1 and down-regulating mesenchymal markers, fibronectin and vimentin [103]. Furthermore, miRNA-21 promoted the acquisition of luminal markers and EMT in prostate cells suppressing B-cell translocation gene 2 (BTG2) expression [104].
In conclusion, EMT process can be drawn as very interesting and complex phenomena that offers a way to move cancer cells from primary tumor to the confined tissues and blood vessels thus facilitating metastatization process [85]. In addition, it has been well demonstrated that tumor-like mesenchymal stem cell cells can also undergo a reverse phenotype switching to again become phenotypically epithelial cells via mesenchymal-to-epithelial transition (MET) [105]. This process is associated with formation of metastatic niches [97].
A particular pre-requisite for tumor invasion is represented by modifications of ECM involving the degradation of its components by enzymes as the metalloproteinases 2 and 9 (MMP-2 and MMP-9) [106–108]. It has been reported that MSCs increased MMPs expression in lung and pancreas carcinoma activating EMT program [109, 110]. In particular, it seem plausible that MMPs could be directly involved in EMT program during cancer progression by different mechanisms: (a) the increase of MMPs in the tumor microenvironment can prompt EMT in epithelial cells, (b) cancer cells that use EMT programming produce extra MMPs and (c) epithelial cells can be subjected to EMT through additional MMP [111]. MSCs can also act on ECM modifying its organization by production of molecules as collagen [112]. Furthermore, Kaplan et al. (2005) investigating the role of MSCs to contribute to pre-metastatic sites, observed that bone marrow-derived hematopoietic progenitor cells that expressed vascular endothelial growth factor receptor 1 (VEGFR1; also known as Flt1) homed to tumor-specific pre-metastatic sites and formed cellular clusters before the arrival of tumor cells [113].
MESENCHYMAL STEM CELLS AS VEHICLES FOR CELL-BASED CANCER THERAPY
MSCs are being investigated as cellular vehicles for the delivery of anti-cancer agents on the basis of their pronounced tropism and integration capacity into tumor microenvironment as well as for their immunomodulatory abilities [114] (Table 2). Indeed, MSCs can be infused into HLA-non-identical recipients because they do not activate the host immune response and escape immunological rejection [115]. They may be genetically engineered to express anti-proliferative, anti-angiogenic, pro-apoptotic factors as well as cytokines and suicide genes. Recently, Marini et al. reported the therapeutic activity of MSC stably transfected to express a TNF-Related Apoptosis-Inducing Ligand (TRAIL)-EGFR specific against Colo205 xenograft tumor model [116]. Similarly, when MSC expressing TRAIL were injected in pre-established Ewing’s Sarcoma mouse model they caused significant tumor apoptosis and showed anti-angiogenic function respect to control group [117]. It has been observed that MSCs engineered to produce and deliver scFv-Ftd-tBid, a novel γ-SM-targetd immuno-proapoptotic molecule, inhibited prostate cancer growth both in vitro and in vivo [118]. Furthermore, MSCs combined with an adenovirus vector to deliver NK4 caused a decrease of growth and migration of high metastatic liver carcinoma cells as well as neo-angiogenesis [119]. Many other anti-angiogenic approaches have been used to impair tumor progression using MSCs as cargo. These cells have been engineered to produce anti-angiogenic factors in different tumor models such as soluble VEGF receptor-1 (sFLT-1) in Lewis lung cancer [120], endostatin in colorectal carcinoma [121], throspondin-1 (TSP-1) in glioblastoma [122] and pigment epithelium-derived factor (PEDF) in prostate carinoma [123]. Interestingly, MSCs can be transfected to express different cytokines to elicit an immune response and/or to inhibit tumor progression. Several findings reported the ability of IFN-β gene modified MSCs to reduce tumor growth in hepatocellular carcinoma [124], glioblastoma [125] and pancreatic tumors [126]. In addition, it has been observed that the secretion of IL-12 from engineered MSCs in tumor microenvironment of different cancer models such as glioma, Ewing sarcoma, renal cell carcinoma and breast cancer, causes reduction of tumor growth [126–130]. Another interesting therapeutic approach use MSCs loaded with suicide genes such as the herpes simplex virus-thimidine kinase (HSV-TK) and cytosine deaminase (CD) to hamper tumor progression. HSV-TK converts non-toxic ganciclovir (GCV) into phosphorylated toxic compound (GCV-ppp) whereas CD modifides low-toxic substrat 5-fluorocytosine into a 5-fluorouracil potent anticancer agent, in both cases MSCs delivery efficiently these genes in tumor sites. A significant decrease in tumor growth and a subsequent increase in survival were observed when mice bearing highly aggressive GBM were treated with MSC co-expressing S-TRAIL and HSV-TK [131], while sequential combination gene therapy using MSC/dTRAIL-TK achieved long-term remission of metastatic renal cell carcinoma without noticeable toxicity [132]. Importantly, it has been reported a single-arm phase I/II study which assessed the safety and efficacy of HSV-TK genetically modified autologous MSCs as delivery vehicles for a cell-based gene therapy, for advanced recurrent, metastatic gastrointestinal and hepatopancreatobiliary adenocarcinoma [133]. Furtheremore, CD-MSCs showed high therapeutical efficacy to treat animal models of osteosarcoma [134], glioma [135], melanoma [136] and prostate carcinoma [137].
Table 2: Mesenchymal stem cell as cargo for anti-cancer agents
Agent |
Tumor Type |
Reference |
|
|---|---|---|---|
Suicide genes |
HSV-TK |
Glioma |
[131] |
Renal Cell Carcinoma |
[132] |
||
Gastrointestinal and Hepatopancreatobiliary adenocarcinoma |
[133] |
||
CD |
Osteosarcoma |
[134] |
|
Glioma |
[135] |
||
Melanoma |
[136] |
||
Prostate carcinoma |
[137] |
||
Anti-angiogenic factors |
Soluble VEGF Receptor-1 |
Lewis lung cancer |
[120] |
Endostatin |
Colorectal carcinoma |
[121] |
|
Throspondin-1 |
Glioblastoma |
[122] |
|
Interferon-b |
Hepatocellular carcinoma |
[124] |
|
Glioblastoma |
[125] |
||
Pancreatic cancer |
[126] |
||
Glioma |
[127] |
||
Cytokines |
Interleukin-12 |
Ewing sarcoma |
[128] |
Renal cell carcinoma |
[129] |
||
Breast cancer |
[130] |
||
Interleukin-18 |
Glioma |
[162] |
|
Interleukin-21 |
Ovarian cancer |
[121] |
|
Interleukin-15 |
Pancreatic cancer |
[163] |
HSV-TK: herpes simplex virus-thimidine kinase; CD: cytosine deaminase.
ANTI-TUMORIGENIC ACTIVITY OF MESENCHYMAL STEM CELLS
MSCs derived from different sources and acting in different tumor contexts can have a pro- or an anti-tumorigenic behavior. Probably, these opposite effects of MSCs can depend on the experimental setting in cell and in the animal models. It seems that among the MSCs which that originate from umbilical cord /cord blood (UC-MSCs) show more anti-cancer properties. Many studies report that UC-MSCs not only suppress tumor progression, but also promote drug sensitivity of different blood cancer cells including Jurkat leukemia cells [138], K562 erythromyeloblastoid leukemia cells [139], Burkitt’s limphoma cells [140] and multiple myeloma cells [141]. In addition, UC-MSCs exert a suppressive effect on tumor growth of several solid tumor cell lines: breast cancer [142], liver cancer [143], prostate and bladder cancer [144]. Similarly, adipose tissue MSCs (AT-MSCs) inhibit proliferation and induce apoptosis of hepatc cancer cells [145], breast cancer cells [146] as well as prostate cancer cells [147] and melanoma [148]. BM-MSCs mainly show pro-tumorigenic effects even if in some cases it has observed a their anti-proliferative activity on tumor growth [149, 150]. This peculiar aspect of MSCs to repress progression of specific tumors in particular conditions through production of endogenous factors suggest the possibility to use naïve MSCs not only as drug cargo but also themselves as anti-cancer agents.
CONCLUSIONS AND PERSPECTIVES
In the present review, we highlight the multiple activities of TA-MSCs in the TME with particular emphasis on their ability to stimulate cancer immunosuppression, to induce EMT program, to trans-differentiate into CAFs and finally to promote pro-metastatic phenotype (Figure 3).
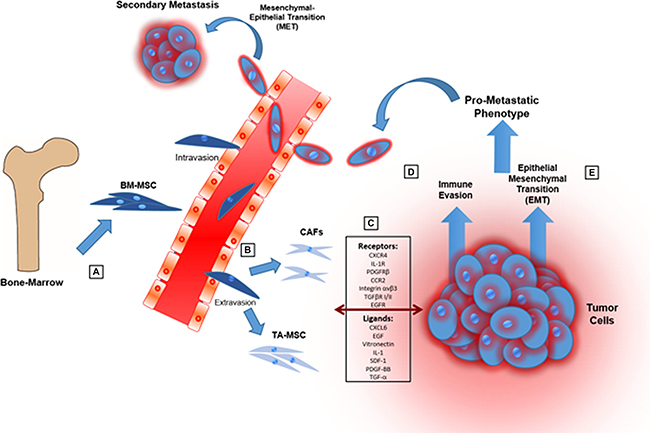
Figure 3: Cartoon showing the main steps through which MSCs promote tumor progression and a pro-metastatic phenotype. (A) MSCs are recruited from bone-marrow and adipose tissue in response of mediators produced by cancer and stroma cells in TME. (B) MSCs into TME can be “educated” to evolve in tumor-associated MSCs (TA-MSCs) and differentiate in cancer-associated fibroblasts (CAFs). (C) MSCs can engage in bidirectional communication with tumor cells through different signals. (D) MSCs can promote immunosuppression in TME modulating both innate and adaptive immune systems. (E) MSCs induce epithelial-mesenchymal transition causing a more aggressive phenotype of tumor cells.
Many mechanisms underlying MSCs role in TME have been elucidated but still further studies are needed to understand completely their involvement in tumor progression.
These investigations could provide information for targeting TA-MSCs with therapeutic approaches and eventually for using them as vehicles to deliver specifically in TME anti-cancer agents.
ACKNOWLEDGMENTS
B.H. is supported by INCIPIT international PhD program cofounded by Marie Skłodowska - Curie Actions.
CONFLICTS OF INTEREST
The authors have no conflicts of interests to declare.
REFERENCES
1. Shi Y, Du L, Lin L, Wang Y. Tumor-associated mesenchymal stem/stromal cells: emerging therapeutic targets. Nat Rev Drug Discov. 2017; 16:35–52.
2. Mishra PJ, Mishra PJ, Humeniuk R, Medina DJ, Alexe G, Mesirov JP, Ganesan S, Glod JW, Banerjee D. Carcinoma associated fibroblast like differentiation of human mesenchymal stem cells. Cancer Res. 2008; 68:4331–4339.
3. Spaeth EL, Dembinski JL, Sasser AK, Watson K, Klopp A, Hall B, Andreeff M, Marini F. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS One. 2009; 4:e4992.
4. Quante M, Tu SP, Tomita H, Gonda T, Wang SS, Takashi S, Baik GH, Shibata W, Diprete B, Betz KS, Friedman R, Varro A, Tycko B, Wang TC. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell. 2011; 19:257–272.
5. Barcellos-de-Souza P, Gori V, Bambi F, Chiarugi P. Tumor microenvironment: bone marrow-mesenchymal stem cells as key players. Biochim Biophysica Acta. 2013; 1836:321–335.
6. Fontanella R, Pelagalli A, Nardelli A, D'Alterio C, Ieranò C, Cerchia L, Lucarelli E, Scala S, Zannetti A. A novel antagonist of CXCR4 prevents bone marrow-derived mesenchymal stem cell-mediated osteosarcoma and hepatocellular carcinoma cell migration and invasion. Cancer Lett. 2016; 1:100–107.
7. Pelagalli A, Nardelli A, Fontanella R, Zannetti A. Inhibition of aqp1 hampers osteosarcoma and hepatocellular carcinoma progression mediated by bone marrow-derived mesenchymal stem cells. Int J Mol Sci. 2016; 17:1102.
8. Klopp AH, Gupta A, Spaeth E, Andreeff M, Marini F. Concise review: Dissecting a discrepancy in the literature: do mesenchymal stem cells support or suppress tumor growth? Stem Cells. 2011; 29:11–19.
9. Ramos TL, Sánchez-Abarca LI, Redondo A, Hernández-Hernández Á, Almeida AM, Puig N, Rodriguez C, Ortega R, Preciado S, Rico A, Muntión S, Porras JRG, Dei Cañizo C, Sánchez-Guijo F. HDAC8 overexpression in mesenchymal stromal cells from JAK2+ myeloproliferative neoplasms: a new therapeutic target? Oncotarget. 2017; 8:28187–28202. http://doi.org/10.18632/oncotarget.15969.
10. Li Z, Fan D, Xiong D. Mesenchymal stem cells as delivery vectors for anti-tumor therapy. Stem Cell Invest. 2015; 2:6.
11. Ullah I, Subbarao RB, Rho GJ. Human mesenchymal stem cells – current trends and future prospects. Biosci Rep. 2015; 35:e00191.
12. Hung SC, Deng WP, Yang WK, Liu RS, Lee CC, Su TC, Lin RJ, Yang DM, Chang CW, Chen WH, Wei HJ, Gelovani JG. Mesenchymal stem cell targeting of microscopic tumors and tumor stromal development monitored by noninvasive in vivo positron emission tomography imaging. Clin Cancer Res. 2005; 11:7749–7756.
13. Nakamizo A, Marini F, Amano T, Khan A, Studeny M, Gumin J, Chen J, Hentschel S, Vecil G, Dembinski J, Andreeff M, Lang FF. Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Res. 2005; 65:3307–3318.
14. Grisendi G, Bussolari R, Cafarelli L, Petak I, Rasini V, Veronesi E, De Santis G, Spani C, Tagliazzuochi M, Barti-Juhasz H, Scarabelli L, Bambi F, Frassoldati A, et al. Adipose-derived mesenchymal stem cells as stable source of tumor necrosis factor-related apoptosis-inducing ligand delivery for cancer therapy. Cancer Res. 2010; 70:3718–3729.
15. Hu W, Wang J, He X, Zhang H, Yu F, Jiang L, Chen D, Chen J, Dou J. Human umbilical blood mononuclear cell-derived mesenchymal stem cells serve as interleukin-21 gene delivery vehicles for epithelial ovarian cancer therapy in nude mice. Biotechnol Appl Biochem. 2011; 58:397–404.
16. Dwyer RM, Potter-Beirne SM, Harrington KA, Lowery AJ, Hennessy E, Murphy JM, Barry FP, O’Brien T, Kerin MJ. Monocyte chemotactic protein-1 secreted by primary breast tumors stimulates migration of mesenchymal stem cells. Clin Cancer Res. 2007; 13:5020–5027.
17. Dvorak HF, Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986; 315:1650–1659.
18. Chavey C, Bibeau F, Gourgou-Bourgade S, Burlinchon S, Boissière F, Laune D, Roques S, Lazennec G. Oestrogen-receptor negative breast cancers exhibit a high cytokine content. Breast Cancer Res. 2007; 9:R15.
19. Lazennec G, Richmond A, Chemokines and chemokine receptors: new insights into cancer-related inflammation, Trends Mol Med. 2010; 16:133–144.
20. Boissière-Michot F, Lazennec G, Frugier H, Jarlier M, Roca L, Duffour, Du Paty E, Laune D, Blanchard, Le Pessot F, Sabourin JC, Bibeau F. Characterization of an adaptive immune response in microsatellite-instable colorectal cancer. Oncoimmunology. 2014; 3:e29256.
21. Spaeth E, Klopp A, Dembinski J, Andreeff M, Marini F, Inflammation and tumor microenvironments: defining the migratory itinerary of mesenchymal stem cells. Gene Ther. 2008; 15:730–738.
22. Thomas JG, Parker Kerrigan BC, Hossain A, Gumin J, Shinojima N, Nwajei F, Ezhilarasan R, Love P, Sulman EP, Lang FF. Ionizing radiation augments glioma tropism of mesenchymal stem cells. J Neurosurg. 2017; 31:1–9.
23. Anton K, Banerjee D, Glod J. Macrophage-associated mesenchymal stem cells assume an activated, migratory, pro-inflammatory phenotype with increased IL-6 and CXCL10 secretion. PLoS One. 2012; 7:35036.
24. Gao H, Priebe W, Glod J, Banerjee D. Activation of signal transducers and activators of transcription 3 and focal adhesion kinase by stromal cell-derived factor 1 is required for migration of human mesenchymal stem cells in response to tumor cell-conditioned medium. Stem Cells. 2009; 27:857–65.
25. Jung Y, Kim JK, Shiozawa Y, Wang J, Mishra A, Joseph J, Berry JE, McGee S, Lee E, Dun H, Wang J, Jin T, Zhang H, et al. Recruitment of mesenchymal stem cells into tumours promotes metastasis. Nat Commun. 2013; 4:1795.
26. Saklaida I, Terai S, Yamamoto N, Aoyama K, Ishikawa T, Nishina H, Okita K. Transplantation of bone marrow cells reduces CCl4-induced liver fibrosis in mice. Hepatology. 2004; 40:1304–1311.
27. Wang L, Wang X, Wang L, Chiu JD, van de Ven G, Gaarde WA, DeLeve LD, Hepatic vascular endothelial growth factor regulates recruitment of rat liver sinusoidal endothelial cell progenitor cells. Gastroenterology. 2012; 143:1555–1563.
28. Cheng P, Gao ZQ, Liu YH, Xue YX. Platelet-derived growth factor BB promotes the migration of bone marrow-derived mesenchymal stem cells towards C6 glioma and up-regulates the expression of intracellular adhesion molecule-1. Neurosci Lett. 2009; 451:52–56.
29. N Hata, N Shinojima, J Gumin, R Yong, F Marini, M Andreeff, Lang FF. Platelet-derived growth factor BB mediates the tropism of human mesenchymal stem cells for malignant gliomas. Neurosurgery. 2010; 66:144–156.
30. Doucette T, Rao G, Yang Y, Gumin J, Shinojima N, Bekele BN, Qiao W, Zhang W, Lang FF. Mesenchymal stem cells display tumor-specific tropism in an RCAS/Ntv-a glioma model. Neoplasia. 2011; 13:716–725.
31. Barcellos-de-Souza P, Comito G, Pons-Segura C, Taddei ML, Gori V, Becherucci V, Bambi F, Margheri F, Laurenzana A, Del Rosso M, Chiarugi P. Mesenchymal Stem Cells are Recruited and Activated into Carcinoma-Associated Fibroblasts by Prostate Cancer Microenvironment-Derived TGF-β1. Stem Cells. 2016; 34:2536–2547.
32. Beckermann BM, Kallifatidis G, Groth A, Frommhold D, Apel A, Mattern J, Salnikov AV, Moldenhauer G, Wagner W, Diehlmann A, Saffrich R, Schubert M, Ho AD, et al. VEGF expression by mesenchymal stem cells contributes to angiogenesis in pancreatic carcinoma. Br J Cancer. 2008; 99:622–631.
33. Yang X, Hao J, Mao Y, Cao R, Liu XH, Ding XL, Wang XD, Chen D, Wu XZ. BFGF promotes migration and induces cancer-associated fibroblast differentiation of mouse bone mesenchymal stem cells to promote tumor growth. Stem Cell Dev. 2016; 25:1629–1629.
34. Rattigan Y, Hsu JM, Mishra PJ, Glod J, Banerjee D. Interleukin 6 mediated recruitment of mesenchymal stem cells to the hypoxic tumor milieu. Exp Cell Res. 2010; 316:3417–3424.
35. Chaturvedi P, Gilkes DM, Wong CC; Kshitiz, Luo W, Zhang H, Wei H, Takano N, Schito L, Levchenko A, Semenza GL. Hypoxia-inducible factor–dependent breast cancer–mesenchymal stem cell bidirectional signaling promotes metastasis. J Clin Invest. 2013; 123:189–205.
36. Chaturvedi P, Gilkes DM, Takano N, Semenza GL. Hypoxia-inducible factor-dependent signaling between triple-negative breast cancer cells and mesenchymal stem cells promotes macrophage recruitment. Proc Natl Acad Sci U S A. 2014; 111:2120–2129.
37. Zhang Y, Yang P, Sun T, Li D, Xu X, Rui Y, Li C, Ching M, Ibrahim T, Mercantali L, Amadori D, Lu X, Xie D, Li QJ, Wang XF. miR-126 and miR126* represses recruitment of mesenchymal stem cells and inflammatory monocytes to inhibit breast cancer metastasis. Nat Cell Biol. 2013; 15:284–294.
38. Ciuculescu F, Giesen M, Deak E, Lang V, Seifried E, Henschler R. Variability in chemokine-induced adhesion of human mesenchymal stromal cells, Cytotherapy. 2011; 13:1172–1179.
39. Rüster B, Göttig S, Ludwig RJ, Bistrian R, Müller S, Seifried E, Gille J, Henschler R. Mesenchymal stem cells display coordinated rolling and adhesion behavior on endothelial cells. Blood. 2006; 108:3938–3944.
40. Ma S, Xie N, Li W, Yuan B, Shi Y, Wang Y. Immunobiology of mesenchymal stem cells. Cell Death Differ. 2014; 21:216–225.
41. McIntosh KR, Mosca JD, Klyushnenkova EN. Mesenchymal stem cells for prevention and treatment of immune responses in transplantation. WO Patent 1999047163 A2 1998. Available from: https://www.google.com/patents/WO1999047163A2?cl=3Den.
42. Le Blanc K, Rasmusson I, Sundberg B, Götherström C, Hassan M, Uzunel M, Ringdén O. Treatment of severe acute graft-versus-host disease with third party haploidentical mesenchymal stem cells. Lancet. 2004; 363:1439–1441.
43. Sun Z, Wang S, Zhao RC. The roles of mesenchymal stem cells in tumor inflammatory microenvironment. J Hematol Oncol. 2014; 7:14.
44. Trivanović D, Krstić J, Djordjević IO, Mojsilović S, Santibanez JF, Bugarski D, Jauković A. The roles of mesenchymal stromal/stem cells in tumor microenvironment associated with inflammation. Mediators Inflamm. 2016; ID 7314016.
45. Han Z, Tian Z, Lv G, Zhang L, Jiang G, Sun K, Wang C, Bu X, li R, Shi Y, Wu M, Wei L. Immunosuppressive effect of bone marrow-derived mesenchymal stem cells in inflammatory microenvironment favours the growth of B16 melanoma cells. J Cell Mol Med. 2011; 15:2343–2352.
46. Cheng J, Li L, Liu Y, Wang Z, Zhu X, Bai X, Interleukin-1α induces immunosuppression by mesenchymal stem cells promoting the growth of prostate cancer cells. Mol Med Rep. 2012; 6:955–960.
47. Ren G, Zhao X, Wang Y, Zhang X, Chen X, Xu C, et al. CCR2-dependent recruitment of macrophages by tumor-educated mesenchymal stromal cells promotes tumor development and is mimicked by TNF alpha, Cell Stem Cell. 2012; 11:812–824.
48. Yu PF, Huang Y, Han YY, Lin LY, Sun WH, Rabson AB, Wang Y, Shi YF. TNFα-activated mesenchymal stromal cells promote breast cancer metastasis by recruiting CXCR2+ neutrophils. Oncogene. 2016; 36:482–490.
49. Ling W, Zhang J, Yuan Z, Ren G, Zhang L, Chen X, Rabson AB, Roberts AI, Wang Y, Shi Y. Mesenchymal stem cells use IDO to regulate immunity in tumor microenvironment. Cancer Res. 2014; 74:1576–1587.
50. Patel SA, Meyer JR, Greco SJ, Corcoran KE, Bryan M, Rameshwar P. Mesenchymal stem cells protect breast cancer cells through regulatory T cells: role of mesenchymal stem cell-derived TGF-beta. J Immunol. 2010; 184:5885–5894.
51. Freytes DO, Kang JW, Marcos-Campos I, Vunjak-Novakovic G. Macrophages modulate the viability and growth of human mesenchymal stem cells. J Cell Biochem. 2012; 114:220–229.
52. Kim J, Hematti P. Mesenchymal stem cell-educated macrophages: a novel type of alternatively activated macrophages. Exp Hematol. 2009; 37:1445–1453.
53. Zhang J, Lu Y, Pienta KJ. Multiple roles of chemokine (C-C motif) ligand 2 in promoting prostate cancer growth. J Natl Cancer Inst. 2010; 102:522–528.
54. Mantovani A. MSCs, macrophages, and cancer: a dangerous ménage-à-trois. Cell Stem Cell. 2012; 11:730–732.
55. Yamada K, Uchiyama A, Uehara A, Perera B, Ogino S, Yokoyama Y, Takeucho Y, Udey MC, Ishikawa O, Motegi S. Mfg-e8 drives melanoma growth by stimulating mesenchymal stromal cell-induced angiogenesis and m2 polarization of tumor-associated macrophages. Cancer Res. 2016; 76:4283–4292.
56. Yu Y, Liu Y, Zong C, Yu Q, Yang X, Liang L, Ye F, Nong L, Jia Y, Lu Y, Han Z. Mesenchymal stem cells with Sirt1 overexpression suppress breast tumor growth via chemokine-dependent natural killer cells recruitment. Sci Rep. 2016; 26:6:35998.
57. Mathew E, Brannon AL, Del Vecchio AC, Garcia PE, Penny MK, Kane KT, Vinta A, Buckanovich RJ, di Magliano MP. Mesenchymal stem cells promote pancreatic tumor growth by inducing alternative polarization of macrophages. Neoplasia. 2016; 18:142–151.
58. Chiassone L, Conte R, Spaggiari GM, Serra M, Romei C, Bellora F, Becchetti F, Andaloro A, Moretta L, Bottino C. Mesenchymal stromal cells induce peculiar alternatively activated macrophages capable of dampening both innate and adaptive immune responses. Stem Cells. 2016; 34:1909–21.
59. Entrena A, Varas A, Vázquez M, Melen GJ, Fernández-Sevilla LM, García-Castro J, Ramírez M, Zapata AG, Vicente Á. Mesenchymal stem cells derived from low risk acute lymphoblastic leukemia patients promote NK cell antitumor activity. Cancer Lett. 2015; 363:156–165.
60. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006; 6:392–401.
61. Xing F, Saidou J, Watabe K. Cancer associated fibroblasts (CAFs) in tumor microenvironment. Front Biosci. 2010; 15:166–179.
62. Worthley DL, Ruszkiewicz A, Davies R, Moore S, Nivison-Smith I, Bik To L, Browett P, Western R, Durrant S, So J, Young GP, Mullighan CG, Bardy PG, et al. Human gastrointestinal neoplasia-associated myofibroblasts can develop from bone marrow-derived cells following allogeneic stem cell transplantation. Stem Cells. 2009; 27:1463–1468.
63. Shangguan L, Ti X, Krause U, Hai B, Zhao Y, Yang Z, Liu F. Inhibition of TGF-β/Smad signaling by BAMBI blocks differentiation of human mesenchymal stem cells to carcinoma-associated fibroblasts and abolishes their protumor effects. Stem Cells. 2012; 30:2810–2819.
64. Peng Y, Li Z, Li Z. GRP78 secreted by tumor cells stimulates differentiation of bone marrow mesenchymal stem cells to cancer-associated fibroblasts. Biochem Biophys Res Commun. 2013; 440:558–563.
65. Guan J, Chen J. Mesenchymal stem cells in the tumor microenvironment. Biomed Rep. 2013; 1:517–521.
66. Rhee KJ, Lee JI, Eom YW. Mesenchymal stem cell-mediated effects of tumor support or suppression. Int J Mol Sci. 2015; 16:30015–30033.
67. Rønnov-Jessen L, Petersen OW. A function for filamentous alpha-smooth muscle actin: retardation of motility in fibroblasts. J Cell Biol. 1996; 134:67–80.
68. Brennen WN, Rosen DM, Wang H, Isaacs JT, Denmeade SR, Targeting carcinoma- associated fibroblasts within the tumor stroma with a fibroblast activation protein- activated prodrug. J Natl Cancer Inst. 2012; 104:1320–1334.
69. Du H, Che G. Genetic alterations and epigenetic alterations of cancer associated fibroblasts. Oncol Lett. 2017; 13:3–12.
70. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016; 16:582–598.
71. Albrengues J, Bertero T, Grasset E, Bonan S, Maiel M, Bourget I, Philippe C, Herraiz Serrano C, Benamar S, Croce O, Sanz-Moreno V, Meneguzzi G, Feral CC, Cristofari G, Gaggioli C. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat Commun. 2015; 15:10204.
72. Jiang L, Gonda TA, Gamble MV, Salas M, Seshan V, Tu S, Twaddell WS, Hegyi P, Lazar G, Steele I, Varro A, Wang TC, Tycko B. Global hypomethylation of genomic DNA in cancerassociated myofibroblasts. Cancer Res. 2008; 68:9900–9908.
73. Shakya R, Gonda T, Quante M, Salas M, Kim S, Brooks J, Hirsch S, Davies J, Cullo A, Olive K, Wang TC, Szabolcs M, Tycko B, Ludwig T. Hypomethylating therapy in an aggressive stroma-rich model of pancreatic carcinoma. Cancer Res. 2013; 73:885–96.
74. Rask L, Balslev E, Jørgensen S, Eriksen J, Flyger H, Møller S, Høgdall E, Litman T, Nielsen BS. High expression of miR-21 in tumor stroma correlates with increased cancer cell proliferation in human breast cancer. APMIS. 2011; 119:663–673.
75. Pathamanathan N, Balleine RL. Ki67 and proliferation in breast cancer. J Clin Pathol 2013; 66:512–516.
76. Yamamichi N, Shimomura R, Inada K, Sakurai K, Haraguchi T, Ozaki Y, Fujita S, Mizutani T, Furukawa C, Fujishiro M, Ichinose M, Shiogama K, Tsutsumi Y, et al. Locked nucleic acid in situ hybridization analysis of miR-21 expression during colorectal cancer development. Clin Cancer Res. 2009; 15:4009–4016.
77. Nouraee N, Roosbroeck K, Vasei M, Semnani S, Samaei NM, Naghshvar F, Omidi AA, Calin GA, Mowla SJ: Expression, tissue distribution and function of miR-21 in esophageal squamous cell carcinoma. PLoS One. 2013; 8:73009.
78. Choi YP, Lee JH, Gao MQ, Kim BG, Kang S, Kim SH, Cho NH. Cancer-associated fibroblast promote transmigration through endothelial brain cells in three-dimensional in vitro models. Int J Cancer. 2014; 135:2024–2033.
79. Luo H, Tu G, Liu Z, Liu M. Cancer-associated fibroblasts: a multifaceted driver of breast cancer progression, Cancer Lett. 2015; 361:155–163.
80. Cammarota F, Laukkanen MO. Mesenchymal stem/stromal cells in stromal evolution and cancer progression. Stem Cells Int. 2016; 4824573.
81. Thiery JP. Epithelial-mesenchymal transitions in tumor progression. Nat Rev Cancer. 2002; 2:442–454.
82. Cannito S, Novo E, di Bonzo LV, Busletta C, Colombatto S, Parola M. Epithelial-mesenchymal transition: From molecular mechanisms, redox regulation to implications in human health and disease. Antioxid Redox Signal. 2010; 12:1383–1430.
83. Liu J, Shen JX, Hu JL, Dou XW, Zhang GJ. Role of epithelial-mesenchymal transition in invasion and metastasis of breast cancers. OA Cancer. 2013; 1:16.
84. Grunert S, Jechlinger M, Beug H. Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nat Rev Mol Cell Biol. 2003; 4:657–665.
85. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009; 119:1420–1428.
86. Smith BN, Bhowmick NA. Role of EMT in metastasis and therapy resistance. J Clin Med. 2016; 5:17.
87. Sasser AK, Sullivan NJ, Studebaker AW, Hendey LF, Axel AE, Hall BM. Interleukin-6 is a potent growth factor for ER-{alpha}-positive human breast cancer. FASEB J. 2007; 21:3763–3770.
88. Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, Richardson AL, Polyak K, Tubo R, Weinberg RA. Mesenchymal stem cells within tumor stroma promote breast cancer metastasis. Nature. 2007; 449:557–563.
89. Gwendal L, Paula YL. Recent discoveries concerning the tumor - mesenchymal stem cell interactions. Biochim Biophys Acta. 2016; 1866:290–299.
90. McAndrews KM, McGrail DJ, Ravikumar N, Dawson MR. Mesenchymal stem cells induce directional migration of invasive breast cancer cells through TGF beta, Sci Rep. 2015; 5:16941.
91. Berger L, Shamai Y, Skorecki KL, Tzukerman M. Tumor specific recruitment and reprogramming of mesenchymal stem cells in tumorigenesis. Stem Cells. 2016; 34:1011–1026.
92. D'Esposito V, Liguoro D, Ambrosio MR, Collina F, Cantile M, Spinelli R, Raciti GA, Miele C, Valentino R, Campiglia P, De Laurentiis M, Di Bonito M, Botti G, et al. Adipose microenvironment promotes triple negative breast cancer cell invasiveness and dissemination by producing CCL5. Oncotarget. 2016; 7:24495–509. http://doi.org/10.18632/oncotarget.8336.
93. D'Esposito V, Passaretti F, Hammarstedt A, Liguoro D, Terracciano D, Molea G, Canta L, Miele C, Smith U, Beguinot F, Formisano P. Adipocyte-released insulin-like growth factor-1 is regulated by glucose and fatty acids and controls breast cancer cell growth in vitro. Diabetologia. 2012; 55:2811–2822.
94. De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013; 13:97–110.
95. Qin Y, Capaldo C, Gumbiner BM, Macara IG. The mammalian Scribble polarity protein regulates epithelial cell adhesion and migration through E-cadherin. J Cell Biol. 2005; 171:1061–1071.
96. Porta-de-la-Riva M, Stanisavljevic J, Curto J, Franci C, Dıaz VM, Garcıa de Herreros A, Baulida J. TFCP2c/LSF/LBP-1c is required for Snail1-induced fibronectin gene expression. Biochem J. 2011; 435:563–568.
97. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008; 133:704–715.
98. Trimboli AJ, Fukino K, Bruin A, Wei G, Shen L, Tanner SM, Creasap N, Rosol TJ, Robinson ML, Eng C, Ostrowshi MC, Leone G. Direct evidence for epithelial-mesenchymal transitions in breast cancer. Cancer Res. 2008; 68:937–945.
99. Vergara D, Merlot B, Lucot JP, Collinet P, Vinatier D, Fournier I, Salzet M. Epithelial-mesenchymal transition in ovarian cancer. Cancer Lett. 2009; 291:59–66.
100. Brabletz T, Hlubek F, Spaderna S, Schmalhofer O, Hiendlmeyer E, Jung A, Kirchner T. Invasion and metastasis in colorectal cancer: epithelial-mesenchymal transition, mesenchymal-epithelial transition, stem cells and beta-catenin. Cells Tissues Organs. 2005; 179:56–65.
101. Usami Y, Satake S, Nakayama F, Matsumoto M, Ohnuma K, Komori T, Semba S, Ito A, Yokozaki H. Snail-associated epithelial-mesenchymal transition promotes oesophageal squamous cell carcinoma motility and progression. J Pathol. 2008; 215:330–339.
102. Ma L, Young J, Prabhala H, Pan E, Mestdagh P, Muth D, Teruya-Feldstein J, Reinhardt F, Onder TT, Valastyan S, Westermann F, Speleman F, Vandesompele J, et al. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol. 2010; 12:247–256.
103. Williams LV, Veliceasa D, Vinokour E, Volpert OV. miR-200b inhibits prostate cancer EMT, growth and metastasis. PLoS one. 2013; 8:83991.
104. Coppola V, Musumeci M, Patrizii M, Cannistraci A, Addario A, Maugeri-Saccà M, Biffoni M, Francescangeli F, Cordenonsi M, Piccolo S, Memeo L, Pagliuca A, Muto G, et al. BTG2 loss and miR-21 upregulation contribute to prostate cell transformation by inducing luminal markers expression and epithelial−mesenchymal transition. Oncogene. 2013; 32:1843–1853.
105. Yao D, Dai C, Peng S. Mechanism of the mesenchymal–epithelial transition and its relationship with metastatic tumor formation. Mol Cancer Res. 2011; 9:1608–1620.
106. Deryugina EI, Quigley JP. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006; 25:9–34.
107. Ries C, Egea V, Karow M, Kolb H, Jochum M, Neth P, MMP-2, MT1-MMP, and TIMP-2 are essential for the invasive capacity of human mesenchymal stem cells: differential regulation by inflammatory cytokines. Blood. 2007; 109:4055–4063.
108. Calvo F, Ege N, Garcia AG, Hooper S, Jenkins RP, Chaudhry SI, Harrington K, Williamson P, Moeendabary E, Charras G, Sehai E. Mechanotransduction and YAP dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol. 2013; 15:637–646.
109. Jones LE, Humphreys MJ, Campbell F, Neoptolemos JP, Boyd MT. Comprehensive analysis of matrix metalloproteinase and tissue inhibitor expression in pancreatic cancer: increased expression of matrix metalloproteinase-7 predicts poor survival. Clin Cancer Res. 2004; 10:2832–2845.
110. Liu D, Nakano J, Ishikawa S, Yokomise H, Ueno M, Kadota K, Urushihara M, Huang CL. Overexpression of matrix metalloproteinase-7 (MMP-7) correlates with tumor proliferation, and a poor prognosis in non-small cell lung cancer, Lung cancer. 2007; 58:384–391.
111. Son H, Moon A. Epithelial-mesenchymal transition and cell invasion. Toxicol Res. 2010; 26:245–252.
112. Gilkes DM, Semenza GL, Wirtz D. Hypoxia and the extracellular matrix: drivers of tumor metastasis, Nat Rev Cancer. 2014; 14:430–439.
113. Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincen L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA, Zhu Z, Hicklin D, Wu Y, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005; 438:820–827.
114. Hagenhoff A, Bruns CJ, Zhao Y, von Lüttichau I, Niess H, Spitzweg C, Nelson PJ. Harnessing mesenchymal stem cell homing as an anticancer therapy. Expert Opin Biol Ther. 2016; 16:1079–92.
115. Uchibori R, Tsukahara T, Ohmine K, Ozawa K. Cancer gene therapy using mesenchymal stem cells. Int J Hematol. 2014; 99:377–382.
116. Marini I, Siegemund M, Hutt M, Kontermann RE, Pfizenmaier K. Antitumor Activity of a Mesenchymal Stem Cell Line Stably Secreting a Tumor-Targeted TNF-Related Apoptosis-Inducing Ligand Fusion Protein. Front Immunol. 2017; 8:536.
117. Grisendi G, Spano C, D'souza N, Rasini V, Veronesi E, Prapa M, Petrachi T, Piccinno S, Rossignoli F, Burns JS, Fiorcari S, Granchi D, Baldini N, et al. Mesenchymal progenitors expressing TRAIL induce apoptosis in sarcomas. Stem Cells. 2015; 33:859–869.
118. Yan F, Li X, Li N, Zhang R, Wang Q, Ru Y, Hao X, Ni J, Wang H, Wu G. Immunoproapoptotic molecule scFv-Fdt-tBid modified mesenchymal stem cells for prostate cancer dual-targeted therapy. Cancer Lett. 2017; 402:32–42.
119. Cai C, Hou L, Zhang J, Zhao D, Wang Z, Hu H, He J, Guan W, Ma Y. The Inhibitory Effect of Mesenchymal Stem Cells with rAd-NK4 on Liver Cancer. Appl Biochem Biotechnol. 2017. [Epub ahead of print].
120. Hu M, JL Yang, H Teng, YQ Jia, R Wang, XW Zhang, Y Wu, Y Luo, Chen XC, Zhang R, Tian L, Zhao X, Wei YQ. Anti-angiogenesis therapy based on the bone marrow-derived stromal cells genetically engineered to express sFlt-1 in mouse tumor model. BMC Cancer . 2008; 8:306.
121. Zhang D, Zheng L, Shi H, Chen X, Wan Y, Zhang H, Li M, Lu L, Luo S, Yin T, Lin H, He S, Luo Y, Yang L. Suppression of peritoneal tumorigenesis by placenta-derived mesenchymal stem cells expressing endostatin on colorectal cancer. Int J Med Sci. 2014; 11:870–879.
122. Choi SH, Tamura K, Khajuria RK, Bhere D, Nesterenko I, Lawler J, Shah K. Antiangiogenic variant of TSP-1 targets tumor cells in glioblastomas. Mol Ther. 2015; 23:235–43.
123. Zolochevska O, Yu G, Gimble JM, Figueiredo ML. Pigment epithelial-derived factor and melanoma differentiation associated gene-7 cytokine gene therapies delivered by adipose-derived stromal/mesenchymal stem cells are effective in reducing prostate cancer cell growth. Stem Cells Dev. 2012; 21:1112–23.
124. Xie C, Xie DY, Lin BL, Zhang GL, Wang PP, Peng L, Gao ZL. Interferon-b gene-modified human bone marrow mesenchymal stem cells attenuate hepatocellular carcinoma through inhibiting AKT/FOXO3a pathway. Br J Cancer. 2013; 109:1198–1205.
125. Nakamizo A, Marini F, Amano T, Khan A, Studeny M, Gumin J, Chen J, Hentschel S, Vecil G, Dembinski J, Andreeff M, Lang FF. Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Res. 2005; 65:3307–3318.
126. Kidd S, Caldwell L, Dietrich M, Samudio I, Spaeth EL, Watson K, Shi Y, Abbruzzese J, Konopleva M, Andreeff M, Marini FC. Mesenchymal stromal cells alone or expressing interferon-beta suppress pancreatic tumors in vivo, an effect countered by anti-inflammatory treatment. Cytotherapy. 2010; 12:615–625.
127. Ryu CH, Park SH, Park SA, Kim SM, Lim JY, Jeong CH, Yoon WS, Oh WI, Sung YC, Jeun SS. Gene therapy of intracranial glioma using interleukin 12-secreting human umbilical cord blood-derived mesenchymal stem cells. Hum Gene Ther. 2011; 22:733–743.
128. Duan X, Guan H, Cao Y, Kleinerman ES. Murine bone marrow-derived mesenchymal stem cells as vehicles for interleukin-12 gene delivery into Ewing sarcoma tumors. Cancer. 2009; 115:13–22.
129. Gao P, Ding Q, Wu Z, Jiang H, Fang Z. Therapeutic potential of human mesenchymal stem cells producing IL-12 in a mouse xenograft model of renal cell carcinoma. Cancer Lett. 2010; 290:157–166.
130. Eliopoulos N, Francois M, Boivin MN, Martineau D, Galipeau J. Neo-organoid of marrow mesenchymal stromal cells secreting interleukin-12 for breast cancer therapy. Cancer Res. 2008; 68:4810–4818.
131. Martinez-Quintanilla J, Bhere D, Heidari P, He D, Mahmood U, Shah K. Therapeutic efficacy and fate of bimodal engineered stem cells in malignant brain tumors. Stem Cells. 2013; 31:1706–1714.
132. Kim SW, Kim SJ, Park SH, Yang HG, Kang MC, Choi YW, Kim SM, Jeun SS, Sung YC. Complete regression of metastatic renal cell carcinoma by multiple injections of engineeredmesenchymal stemcells expressing dodecameric TRAIL and HSV-TK. Clin Cancer Res. 2013; 19:415–427.
133. Niess H, von Einem JC, Thomas MN, Michl M, Angele MK, Huss R, Günther C, Nelson PJ, Bruns CJ, Heinemann V. Treatment of advanced gastrointestinal tumors with genetically modified autologous mesenchymal stromal cells (TREAT-ME1): study protocol of a phase I/II clinical trial. BMC Cancer. 2015; 15:237.
134. NguyenThai QA, Sharma N, Luong H, Sodhi SS, Kim JH, Kim N, Oh SJ, Jeong DK. Targeted inhibition of osteosarcoma tumor growth by bone marrow-derived mesenchymal. J Gene Med. 2015; 17:87–99.
135. Kosaka H, Ichikawa T, Kurozumi K, Kambara H, Inoue S, Maruo T, Nakamura K, Hamada H, Date I. Therapeutic effect of suicide gene-transferred mesenchymal stem cells in a rat model of glioma. Cancer Gene Ther. 2012; 19:572–578.
136. Kucerova L, J Zmajkovic, Toro L, Skolekova S, Demkova L, Matuskova M. Tumor-driven molecular changes in human mesenchymal stromal cells. Cancer Microenviron. 2014; 8:1–14.
137. Abrate A, Buono R, Canu T, Esposito A, Del Maschio A, Luciano R ,Bettiga A, Colciago G, Guazzoni G, Benigni F, Hedlund P, Altaner C, Montorsi F, et al. Mesenchymal stem cells expressing therapeutic genes induce autochthonous prostate tumour regression. Eur J Cancer. 2014; 50:2478–2488.
138. Liang X, Hao L, Chen X, Zhang X, Kong P, Peng X, Gao L, Zhang C, Wang Q. Effects of bone marrow stromal cells and umbilical cord blood-derived stromal cells on daunorubicin-resistant residual Jurkat cells. Transplant Proc. 2010; 42:3767–72.
139. Fonseka M, Ramasamy R, Tan BC, Seow HF. Human umbilical cord blood-derived mesenchymal stem cells (hUCB-MSC) inhibit the proliferation of K562 (human erythromyeloblastoid leukaemic cell line). Cell Biol Int. 2012; 36:793–801.
140. Lin HD, Fong CY, Biswas A, Choolani M, Bongso A. Human Wharton's jelly stem cells, its conditioned medium and cell-free lysate inhibit the growth of human lymphoma cells. Stem Cell Rev. 2014; 10:573–86.
141. Ciavarella S, Caselli A, Tamma AV, Savonarola A, Loverro G, Paganelli R, Tucci M, Silvestris F. A peculiar molecular profile of umbilical cord-mesenchymal stromal cells drives their inhibitory effects on multiple myeloma cell growth and tumor progression. Stem Cells Dev. 2015; 24:1457–70.
142. Leng L, Wang Y, He N, Wang D, Zhao Q, Feng G, Su W, Xu Y, Han Z, Kong D, Cheng Z, Xiang R, Li Z. Molecular imaging for assessment of mesenchymal stem cells mediated breast cancer therapy. Biomaterials. 2014; 35:5162–70.
143. Yulyana Y, Ho IA, Sia KC, Newman JP, Toh XY, Endaya BB, Chan JK, Gnecchi M, Huynh H, Chung AY, Lim KH, Leong HS, Iyer NG, et al. Paracrine factors of human fetal MSCs inhibit liver cancer growth through reduced activation of IGF-1R/PI3K/Akt signaling. Mol Ther. 2015; 23:746–56.
144. Han I, Yun M, Kim EO, Kim B, Jung MH, Kim SH. Umbilical cord tissue-derived mesenchymal stem cells induce apoptosis in PC-3 prostate cancer cells through activation of JNK and downregulation of PI3K/AKT signaling. Stem Cell Res Ther. 2014; 5:54.
145. Zhao W, Ren G, Zhang L, Liu J, Kuang P, Tin Z, Wang X. Efficacy of mesenchymal stem cells derived from human adipose tissue in inhibition of hepatocellular carcinoma cells in vitro. Cancer Biother Radiopharm. 2012; 27:606–13.
146. Ryu H, Oh JE, Rhee KJ, Baik SK, Kim J, Kang SJ, Sohn JH, Choi E, Shin HC, Kim YM, Kim HS, Bae KS, Eom YW. Adipose tissue-derived mesenchymal stem cells cultured at high density express IFN-β and suppress the growth of MCF-7 human breast cancer cells. Cancer Lett. 2014; 352:220–7.
147. Takahara K, Ii M, Inamoto T, Komura K, Ibuki N, Minami K, Uehara H, Hirano H, Nomi H, Kiyama S, Asahi M, Azuma H. Adipose-derived stromal cells inhibit prostate cancer cell proliferation inducing apoptosis. Biochem Biophys Res Commun. 2014; 446:1102–7.
148. Ahn JO, Coh YR, Lee HW, Shin IS, Kang SK, Youn HY. Human adipose tissue-derived mesenchymal stem cells inhibit melanoma growth in vitro and in vivo. Anticancer Res. 2015; 35:159–68.
149. Böhrnsen F, Fricke M, Sander C, Leha A, Schliephake H, Kramer FJ. Interactions of human MSC with head and neck squamous cell carcinoma cell line PCI-13 reduce markers of epithelia-mesenchymal transition. Clin Oral Investig. 2015; 19:1121–8.
150. Secchiero P, Zorzet S, Tripodo C, Corallini F, Melloni E, Caruso L, Bosco R, Ingrao S, Zavan B, Zauli G¬. Human bone marrow mesenchymal stem cells display anti-cancer activity in SCID mice bearing disseminated non-Hodgkin's lymphoma xenografts. PLoS One. 2010; 5:11140.
151. Egea V, von Baumgarten L, Schichor C, Berninger B, Popp T, Neth P, Goldbrunner R, Kienast Y, Winkler F, Jochum M, Ries C. TNF-α respecifies human mesenchymal stem cells to a neural fate and promotes migration toward experimental glioma. Cell Death Differ. 2011; 18:853–863.
152. Uchibori R, Tsukahara T, Mizuguchi H, Saga Y, Urabe M, Mizukami H, Kume A, Ozawa K. NF-κB activity regulates mesenchymal stem cell accumulation at tumor sites, Cancer Res. 2013; 73:364–372.
153. Liu S, Ginestier C, Ou SJ, Clouthier SG, Patel SH, Monville F, Korkaya H, Heath A, Dutcher J, Kleer CG, Jung Y, Dontu G, Taichman R et al. Breast cancer stem cells are regulated by mesenchymal stem cells through cytokine networks. Cancer Res. 2011; 71:614–624.
154. Lourenco S, Teixeira VH, Kalber T, Jose RJ, Floto RA, Janes SM, Macrophage migration inhibitory factor - cxcr4 is the dominant chemotactic axis in human mesenchymal stem cell recruitment to tumors, J Immunol. 2015; 194:3463–3474.
155. Shinojima N, Hossain A, Takezaki T, Fueyo J, Gumin J, Gao F, Nwajei F, Marini FC, Andreeff M, Kuratsu J, Lang FF. TGF-Β mediates homing of bone marrow-derived human mesenchymal stem cells to glioma stem cells. Cancer Res. 2014; 73:2333–2344.
156. Hsiao WC, Liao CH, Wu HC, Hsieh CL. Platelet derived growth factor AA mediated the tropism of human mesenchymal stem cells for renal cell carcinoma, Cancer Res. 2011; 71:410.
157. Berger L, Shamai Y, Skorecki KL, Tzukerman M, Tumor specific recruitment and reprogramming of mesenchymal stem cells in tumorigenesis, Stem Cells. 2016; 34:1011–1026.
158. Ho IAW, Chan KYW, Ng WH, Guo CM, Hui KM, Cheang P, Lam PY. Matrix metalloproteinase 1 is necessary for the migration of human bone marrow-derived mesenchymal stem cells toward human glioma. Stem Cells. 2009; 27:1366–1375.
159. Hu Y, Cheng P, Xue YX, Liu YH. Glioma cells promote the expression of vascular cell adhesion molecule-1 on bone marrow-derived mesenchymal stem cells: a possible mechanism for their tropism toward gliomas. J Mol Neurosci. 2012; 4:127–135.
160. Senst C, Nazari-Shafti T, Kruger S, Höner K, Zu Bentrup H, Dupin CL, Chaffin AE, Srivastav SK, Wörner PM, Abdel-Mageed AB, Alt EU, Izadpanah R. Prospective dual role of mesenchymal stem cells in breast tumor microenvironment. Breast Cancer Res Treat. 2013; 137:69–79.
161. Coffelt SB, Marini FC, Watson K, Zwezdaryk KJ, Dembinski JL, La Marca HL,Tomchuck SL, Honer zu Bentrup K, Danka ES, Henkle SL, Scandurro AB. The pro-inflammatory peptide LL-37 promotes ovarian tumor progression through recruitment of multipotent mesenchymal stromal cells. Proc Natl Acad Sci U S A. 2009; 106:3806–11.
162. Xu G, Jiang XD, Xu Y, Zhang J, Huang FH, Chen ZZ, Zhou DX, Shang JH, Zou YX, Cai YQ, Kou SB, Chen YZ, Xu RX, Zeng YJ. Adenoviral-mediated interleukin-18 expression in mesenchymal stem cells effectively suppresses the growth of glioma in rats. Cell Biol Int. 2009; 33:466–74.
163. Jing W, Chen Y, Lu L, Hu X1, Shao C, Zhang Y, Zhou X, Zhou Y, Wu L, Liu R, Fan K, Jin G. Human umbilical cord blood-derived mesenchymal stem cells producing IL15 eradicate established pancreatic tumor in syngeneic mice. Mol Cancer Ther. 2014; 13:2127–37.