
Introduction
Prostate cancer is the third leading cause of cancer death in developed countries and the most common cancer in men [1]. The androgen receptor (AR) plays a key role in both normal prostate biology and prostate cancer progression and, as a result, targeting of AR signalling is a major therapeutic strategy for advanced prostate cancer [2]. While initial response rates to androgen deprivation therapy (ADT) are high, therapeutic options for castration resistant disease are limited. Notably, castration-resistant prostate cancer (CRPC) remains dependent on AR signalling, but becomes insensitive to ADT through a variety of mechanisms [3-8].
During recent years, the observation that cancer cells frequently display an altered metabolism has gained increasing attention, to the point where metabolic deregulation is now considered one of the emerging hallmarks of cancer, and strong evidence suggests that metabolic alterations also play an important role in prostate cancer [9, 10]. Intriguingly, recent studies have unveiled a high degree of crosstalk between AR signalling and metabolic pathways in prostate cancer cells. The AR has been shown to regulate prostate cancer metabolism by driving the expression of an extensive network of metabolic genes, such as fatty acid synthase and alpha-methylacyl-CoA racemase, resulting in stimulation of both aerobic glycolysis and anabolic pathways [11-17]. Another gene whose expression is driven by the AR is Calcium/Calmodulin-Dependent Protein Kinase Kinase 2 (CAMKK2), one of the upstream kinases of the metabolic master regulator AMPK. AMPK is a highly conserved heterotrimeric serine/threonine kinase consisting of a catalytic alpha and regulatory beta and gamma subunits [18, 19]. It acts as an energy sensor that is activated in response to numerous stress factors that decrease cellular ATP:AMP ratio, such as hypoxia or glucose deprivation, and facilitates the restoration of cellular energy balance by activating catabolic pathways and inhibiting anabolic pathways. In prostate cancer cells, AR-driven up-regulation of CAMKK2 has been shown to result in increased AMPK activity upon androgen stimulation [11, 20]. This mechanism has been proposed to drive prostate cancer cell growth through multiple mechanisms, including increased glycolysis and mitochondrial biogenesis [11, 21].
Despite a large number of studies investigating AMPK function, its role in prostate cancer remains controversial. Several lines of evidence suggest that AMPK has tumour suppressor properties in the prostate. Knock-out of its upstream kinase LKB1 results in prostatic intraepithelial neoplasia (PIN) in mouse models [22]. Moreover, retrospective studies suggest that metformin, a commonly used anti-diabetic drug that is thought to partially act through activation of AMPK, may decrease the risk of developing various types of cancer, including prostate [23-25]. In line with this, pharmacologic activation of AMPK has been reported to decrease growth and viability of several prostate cancer cell lines in vitro and to inhibit lipid synthesis induced by the synthetic androgen R1881 [26]. Multiple mechanisms may contribute to these effects of AMPK, including inhibition of the mTOR pathway, up-regulation of p53 and p21 [27] and induction of apoptosis through generation of reactive oxygen species as well as through cross-talk with TNF signalling [28, 29]. In addition to its anti-proliferative effects, AMPK may also enhance the sensitivity of cancer cells to therapeutic treatments. For example, metformin has been shown to sensitise cancer cells to chemotherapy while exerting protective effects on normal epithelial cells [30]. Similarly, AMPK activation can sensitise cancer cells to ionizing radiation [31] and to treatment with the multi-tyrosine kinase inhibitor dasatinib [32]. Finally, the growth inhibitory effects of adiponectin and a number of natural compounds on prostate cancer cells have been at least partially attributed to their ability to activate AMPK [33-35]. However, other studies suggest that AMPK could have tumour-promoting functions in the prostate and that AMPK activation is higher in prostate cancer than in normal tissue [21, 36]. Indeed, it has been proposed that AMPK may actually contribute to resistance to anti-cancer therapy in some settings [37-39].
Intriguingly, metformin has been reported to not only reduce the risk of cancer, but of other age-related diseases, opening up possibilities for novel applications of this well-established drug [40, 41]. Taken together, these findings have sparked considerable interest both in the development of novel drugs targeting AMPK and related pathways in a clinical setting, and in a potential use of existing AMPK activators such as metformin in the treatment of cancer and other diseases.
In light of the contradictory findings on the role of AMPK in cancer, we sought to improve our understanding of this signalling pathway in prostate cancer cells by characterising the transcriptional output of AMPK activation. Genome-wide expression profiling revealed that AMPK signalling resulted in the repression of genes that are commonly overexpressed in prostate cancer. Intriguingly, the AR was identified as a potential downstream mediator of AMPK signalling. We then went on to show that activation of AMPK decreased AR activity and nuclear localisation, suggesting the presence of a negative feedback loop between AMPK and AR in prostate cancer cells. Our study thus provides a novel mechanism of cross-talk between these two major metabolic drivers in prostate cancer cells.
Results
Transcriptional impact of AMPK activation in prostate cancer cells
We used genome-wide expression profiling to determine differentially expressed genes in the LNCaP cell line upon AMPK activation. To take account of potential off-target effects of AMPK activating drugs, we used two structurally unrelated, commonly used AMPK activators, 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) and metformin [42, 43], and compared their transcriptional effects in prostate cancer cells. After 8 h, we identified 489 differentially expressed genes following AICAR treatment, but only 41 after treatment with metformin (FDR 0.01; Table 1). In contrast, 711 and 3864 genes were differentially expressed after 24 h of treatment with AICAR or metformin, respectively. We compared up- and down-regulated genes between treatments irrespective of time point and found a highly significant overlap between the two AMPK activators (Figure 1A). Approximately 65% of AICAR- and 90% of metformin-regulated genes were not shared between treatments. As differences in the gene expression profiles of these two drugs could potentially represent AMPK-independent effects, we focussed our downstream analysis on the 362 genes that were concordantly regulated by both drugs and were thus more likely to be AMPK-regulated (Figure 1B). In line with the established role of AMPK as a metabolic master regulator, biological processes enriched among these 362 genes were predominantly metabolism-related (Figure 1C). Using Connectivity Map 02 (http://www.broadinstitute.org/cmap/;[44]) to compare our AMPK signature to a collection of gene-expression signatures associated with 1,309 bioactive small molecules we found that our gene set closely resembled signatures of three compounds known to inhibit the PI3K/Akt/mTOR pathway (thioridazine, wortmannin and LY-294002; Supplementary Table 1). In accordance with this, inhibition of mTOR is one of the best-described functions of AMPK [45-47]. Our set of 362 putative AMPK target genes was thus consistent with the established biological function of AMPK.
Table 1: Differentially expressed genes in prostate cancer cells following AMPK activation. LNCaP cells were treated with AICAR or metformin for 8 h and 24 h. Gene expression was analysed using Illumina Humanv4 BeadChip arrays. Numbers of differentially expressed genes (DEG’s) identified using a globally applied FDR-corrected p-value cut-off of 0.01 are shown.
Time point |
AICAR |
Metformin |
8 h |
489 |
41 |
24 h |
711 |
3864 |
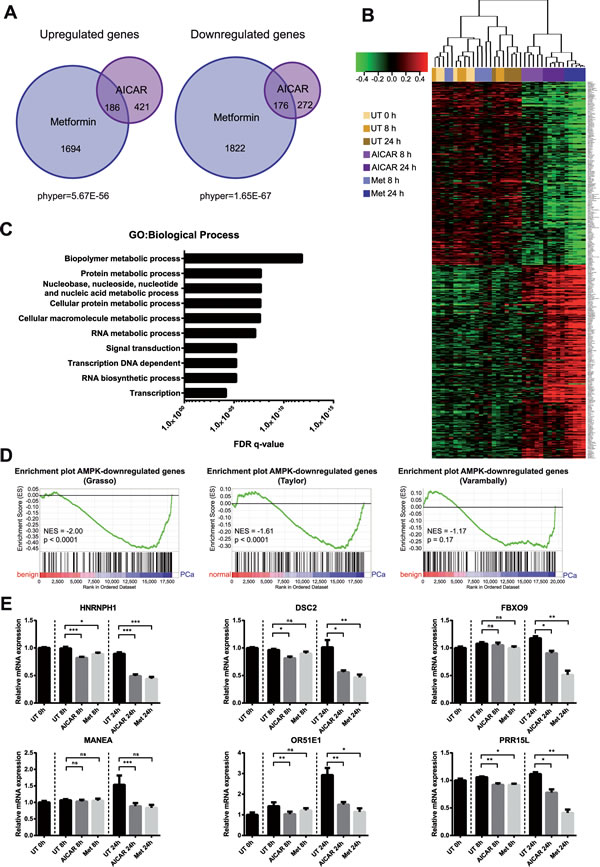
Figure 1: Genome-wide expression profiling reveals the transcriptional impact of AMPK activation in prostate cancer cells. A: LNCaP cells were treated with AICAR or metformin for 8 h and 24 h. Gene expression was analysed using Illumina Humanv4 BeadChip arrays. Differentially expressed genes (DEG’s) were identified using a globally applied FDR-corrected p-value cut-off of 0.01 and compared between treatments. The overlap between gene lists is depicted as an area-proportional Venn diagram. Hypergeometric test was performed to test significance of overlaps between treatments. B: Heatmap showing expression of 362 putative AMPK-regulated genes. Samples were clustered according to Pearson correlation. C: Biological processes enriched among 362 putative AMPK target genes according to gene ontology terms. D: Enrichment plots of AMPK-repressed genes in published prostate cancer datasets. Genes in the Grasso, Taylor and Varambally datasets were ranked according to their expression in benign/normal prostate tissue compared to primary prostate cancer. Gene set enrichment analysis (GSEA) was used to determine enrichment of AMPK-repressed genes within these phenotypes. NES: normalised enrichment score. E: qRT-PCR validation of selected AMPK-repressed genes that are overexpressed in primary prostate cancer. n=6.
To investigate whether AMPK-regulated genes might have a role in prostate cancer, we used gene set enrichment analysis (GSEA) [48, 49] to compare our set of AMPK target genes to three published expression datasets comparing benign/normal prostate tissue with primary and metastatic prostate cancer (Grasso, Taylor and Varambally datasets) [50-52]. Strikingly, genes that were down-regulated upon AMPK activation were significantly enriched among genes over-expressed in primary prostate cancer, compared to normal/benign tissue, in both the Grasso and Taylor datasets (NES=-2.00 and NES=-1.61, respectively). A similar trend was observed in the Varambally dataset (NES=-1.17), although it did not reach statistical significance. AMPK-induced genes did not show significant enrichment in any of the three datasets.
We then selected six genes (HNRNPH1, DSC2, FBXO9, MANEA, OR51E1, PRR15L) for validation by qRT-PCR that were over-expressed in prostate cancer across all datasets and showed high fold changes in our microarray (Supplementary Figure 1B). To our knowledge, none of these genes have previously been shown to be AMPK regulated. Of note, one of the selected genes, OR51E1, has been proposed as a prostate cancer biomarker [53-55] and formed part of the core enrichment in all three GSEA analyses (Supplementary Figure 1C). Reduced expression of all six genes upon AICAR or metformin treatment was confirmed by qRT-PCR (Figure 1E). Together, these results showed that activation of AMPK resulted in reduced expression of genes that are associated with primary prostate cancer.
The AR is a candidate transcription factor acting downstream of AMPK in prostate cancer cells
To identify pathways mediating the transcript-level effect of AMPK activation in prostate cancer cells, we used Ingenuity Pathway Analysis (IPA) to predict potential transcriptional regulators of the AICAR- and metformin-regulated genes in our dataset (Figure 2A). Comparing the fifteen most significantly enriched transcriptional regulators for each treatment, we found four that were shared between both treatments: AR, p53, glucocorticoid receptor (NR3C1) and MYC. Regulation of p53 [56] and NR3C1 [57] by AMPK has been previously demonstrated, confirming that our approach was able to identify established AMPK-regulated transcriptional pathways in prostate cancer cells. After AICAR treatment, the AR was the most significantly enriched transcriptional regulator, and NKX3.1, a well-established AR regulated gene [58, 59], was ranked third. The enrichment after treatment with metformin was not as striking, as the AR was only ranked tenth in this condition. However, we still found a highly significant overlap between metformin- and androgen receptor regulated genes (Figure 2A).
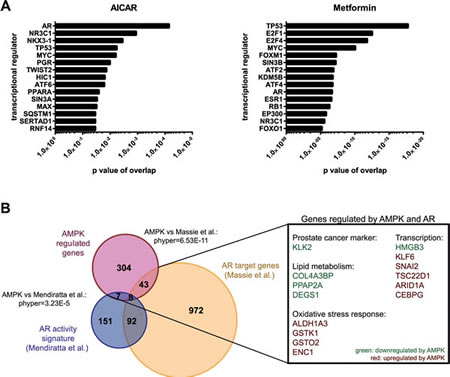
Figure 2: The androgen receptor is a potential transcription factor downstream of AMPK. A: Ingenuity Pathway analysis was used to predict significantly enriched transcriptional regulators among differentially expressed genes following 24 h of AICAR or metformin treatment. B: AMPK target genes were compared to two published datasets describing AR target genes [11, 60]. The overlap between gene lists is depicted as an area-proportional Venn diagram. Hypergeometric test was performed to test significance of overlaps between treatments. Examples of genes regulated by AMPK and by AR in at least one dataset are shown.
This finding, together with the central role of AR in prostate cancer development and progression, prompted us to further investigate the potential link between AMPK signalling and AR activity. We thus compared our subset of putative AMPK target genes to previously published data on AR target genes [11] as well as a published AR activity signature [60] and confirmed significant overlap between AMPK- and AR-regulated genes (Figure 2B). Specifically, 16 % of AMPK regulated genes also showed evidence of being AR-regulated in at least one of the two datasets. Of note, among the genes repressed by AMPK was KLK2, a prostate cancer marker and a well-established AR target gene that belongs to the same gene family (kallikrein-related peptidases) as prostate specific antigen (PSA / KLK3). Additionally, a number of genes involved in lipid metabolism (DEGS1, PPAP2A, COL4A3BP) that were induced by AR were also down-regulated by AMPK, consistent with the previously-established roles of both of these proteins in the regulation of lipid metabolism. Taken together, these findings led us to focus on the AR as a potential downstream effector of AMPK signalling.
Activation of AMPK inhibits the transcriptional activity of AR
To determine if AMPK activation regulates androgen receptor-driven transcription, we employed an androgen-responsive luciferase reporter system (MMTV-Luc). Treatment of transiently transfected LNCaP cells with the synthetic androgen R1881 resulted in an approximately 800-fold increase in luciferase activity compared to vehicle control, an induction that could be almost completely abrogated by the anti-androgen bicalutamide. Importantly, co-treatment with either AICAR or metformin resulted in a significant decrease in luciferase induction compared to R1881 alone, suggesting that activation of AMPK does indeed modulate AR activity (Figure 3A). We obtained similar results in the castrate-resistant LNCaP derived cell line C4-2, suggesting that the negative regulation of AR by AMPK is maintained in this model of castration-resistant prostate cancer. To further confirm these findings, we created a second androgen-responsive luciferase reporter containing an androgen-responsive element derived from the promoter of CAMKK2 and introduced it in a lentiviral vector in order to generate stable AR-reporter cell lines. In LNCaP cells stably transduced with the CAMKK2 reporter, AICAR and metformin significantly decreased R1881-induced luciferase activity, confirming the inhibitory effect of AMPK activation on AR activity (Figure 3B).
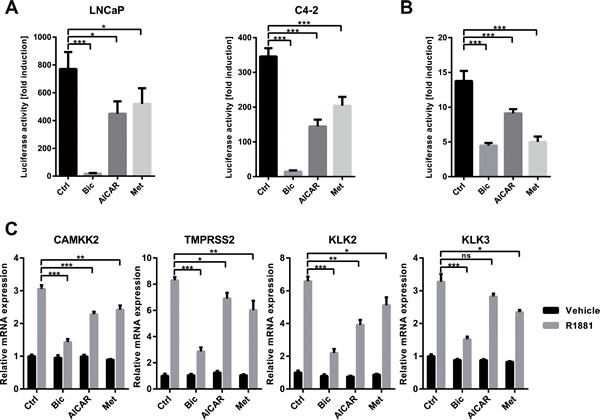
Figure 3: Activation of AMPK inhibits the transcriptional activity of AR. A: An androgen-responsive dual luciferase reporter assay was used to assess effects of AMPK activation on AR activity. LNCaP and C4-2 cells were transfected with MMTV-Luc and pRLTK and treated with R1881 and bicalutamide, AICAR or metformin for 24 h. Fold induction of luciferase activity was calculated with respect to a control without R1881. n=3. B: LNCaP cells were stably transduced with an androgen-responsive luciferase reporter derived from the CAMKK2 promoter. Cells were treated with R1881 and bicalutamide, AICAR or metformin for 24 h. Fold induction of luciferase activity was calculated with respect to a control without R1881. n=5. C: To assess effects of AMPK activation on induction of endogenous AR-target genes by R1881, C4-2 cells were grown in androgen-free medium for three days and stimulated with R1881 and bicalutamide, AICAR or metformin for 12 h. mRNA expression was assessed by qRT-PCR; values are depicted relative to vehicle control. n=4 for CAMKK2 and KLK2, n=3 for KLK3 and TMPRSS2.
We then tested the effect of AMPK activation on the androgen-mediated induction of endogenous AR target genes (Figure 3C). In C4-2 cells, treatment with AICAR or metformin significantly attenuated induction of established AR target genes CAMKK2, KLK2, and TMPRSS2 by R1881 (Figure 3C). Induction of KLK3 (PSA) was also attenuated, but only reached significance with metformin. Comparable results were obtained in LNCaP cells (Supplementary Figure 2). In summary, these findings demonstrate that activation of AMPK negatively regulates AR transcriptional activity.
Silencing of AMPK catalytic subunits stimulates the transcriptional activity of AR
To further demonstrate that the effects of AICAR and metformin on AR activity are mediated through AMPK, we made use of two different combinations of siRNAs directed against the catalytic AMPKα subunits to achieve good levels of knock-down for both isoforms, AMPKα1 (PRKAA1) and AMPKα2 (PRKAA2) (Figures 4A and B). Silencing of AMPKα resulted in an increased activation of AR by R1881 in both LNCaP and C4-2 cells (Figure 4C) and an increased androgen-induced expression of the AR target genes CAMKK2, KLK2, KLK3 and TMPRSS2 (Figure 4D). These data ruled out that results observed from pharmacological activation of AMPK were off-target effects and further supported the finding that AMPK is a negative regulator of AR.
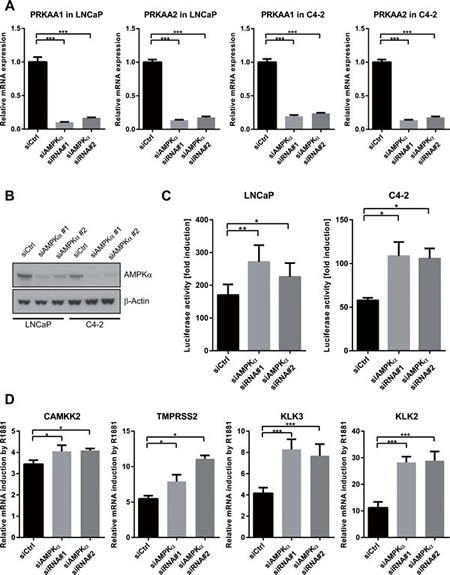
Figure 4: Silencing of AMPK increases the transcriptional activity of AR. A: LNCaP and C4-2 cells were transfected with two different combinations of siRNAs directed against the AMPKα isoforms (PRKAA1 and PRKAA2). Successful knock-down was validated using qRT-PCR three days after transfection. n=4. B: LNCaP and C4-2 cells were transfected with two different combinations of siRNAs directed against both AMPKα isoforms. Successful knock-down was validated by Western Blot four days after transfection. C: An androgen-responsive dual luciferase reporter assay was used to assess effects of AMPK knock-down on AR activity. LNCaP or C4-2 cells were co-transfected with MMTV-Luc, pRL-TK and siRNAs for 24 h and treated with R1881 for 40 h. Fold induction of luciferase activity was calculated with respect to vehicle control for each siRNA. n=5 for LNCaP cells, n=4 for C4-2 cells. D: To assess effects of AMPK knock-down on induction of endogenous AR-target genes by R1881, LNCaP cells were transfected with siRNAs and grown in androgen-free medium for three days, then stimulated with R1881 for 12 h. mRNA expression was assessed by qRT-PCR; values are depicted relative to vehicle control for each siRNA. n=4, except for TMPRSS2: n=3.
Activation of AMPK reduces nuclear localisation of AR
Two previous studies had reported that metformin treatment of prostate cancer cells down-regulates the expression of AR, though neither of them addressed potential effects on the expression of androgen-regulated genes [61, 62]. To test whether this mechanism could explain our finding that AMPK activation inhibits AR activity, we investigated the dynamics of AR inhibition by AICAR and metformin in more detail. Notably, in previous studies, reduction of AR expression was observed after relatively long metformin treatments (96 h) when used at comparable concentrations to our own study, whereas we observed decreased induction of AR-regulated genes already at 12 h (Figure 3C). We performed a time course of the expression of CAMKK2 and another established AR target gene, NKX3.1, which was chosen due to its rapid response to androgen, after co-treatment with R1881 and AICAR or metformin (Figure 5A). In both cases, the inhibitory effects of AICAR and metformin on the expression of these genes became apparent as soon as appreciable R1881-induced induction was observed (four and eight hours for NKX3.1 and CAMKK2, respectively). AR mRNA was slightly decreased by all treatments, consistent with previous reports that androgens reduce AR mRNA levels [63, 64], but no further decrease was observed following AICAR or metformin treatment. More importantly, there was no appreciable decrease in AR protein level with AICAR or metformin treatment during this time frame (Figure 5A and B). Taken together, these results demonstrate that regulation of AR expression levels cannot account for the rapid inhibitory effect of AMPK activation on AR activity that we observe.
An important step in the activation of AR is its translocation to the nucleus upon ligand binding [65]. We tested whether AMPK activity affected the localisation of the AR using nuclear-cytoplasmic fractionation of cellular lysates (Figure 5C and D). As expected, we observed increased nuclear localisation of the AR upon androgen treatment, a process which was reversed by bicalutamide. Treatment with AICAR or metformin reduced nuclear levels of AR. This suggests that AMPK might exert its inhibitory effect on AR activity by reducing its nuclear localisation following ligand binding, rather than by down-regulating its expression.
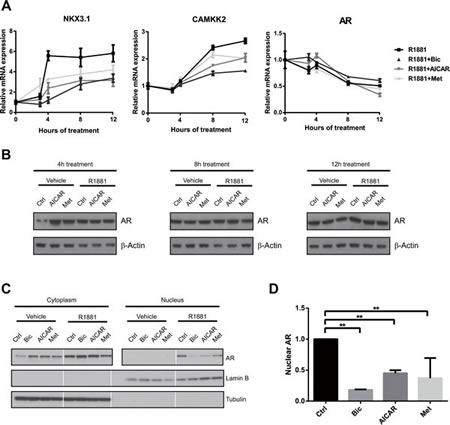
Figure 5: Activation of AMPK reduces nuclear localisation, but not expression of AR. A: To assess the dynamics of AR inhibition following AMPK activation, LNCaP cells were grown in androgen-free medium for three days, then stimulated with R1881 and bicalutamide, AICAR or metformin for 3, 4, 8 and 12 h. Expression of NKX3.1 , CAMKK2 and AR was quantified by qRT-PCR. n=3. B: To evaluate effects of AICAR and metformin on AR protein level, LNCaP cells were grown in androgen-free medium for three days, then stimulated with R1881 and bicalutamide, AICAR or metformin for 4, 8 and 12 h. AR protein levels were assessed by Western Blot. C: To assess effects of AICAR and metformin on localisation of the AR, C4-2 cells were grown in androgen-free medium and stimulated with drugs as indicated for 12 h. Cytoplasmic and nuclear fractions were separated, and AR protein levels were assessed by Western Blot. Lamin B and tubulin were used as loading controls and to confirm successful fractionation. White lines separate non-contiguous bands run on the same gel. D: Nuclear AR levels after androgen stimulation were quantified using Western Blots from three independent experiments and normalised to the Lamin B signal.
Discussion
While a large body of evidence confirms that metabolic alterations play an important role in prostate cancer, the role of AMPK in this context has remained controversial. A number of signalling pathways through which AMPK could both promote and suppress prostate cancer development and progression have been described. Using an unbiased approach, we have now identified the AR, the major driver of prostate cancer, as a downstream mediator of AMPK signalling in prostate cancer cells, adding an important new dimension to the role of AMPK in this cancer type.
To our knowledge, only one previous study has investigated the transcriptional impact of AMPK signalling in a prostate cancer context [27]. In that case, a stably transfected cell line expressing dominant-negative AMPK was used, and AMPK inhibition was found to result in differential expression of several tumour-relevant genes. Our study now extends these findings by identifying a number of additional putative AMPK targets, and by demonstrating that genes repressed by AMPK activation are overexpressed in prostate cancer specimens. Our data thus provides a resource for the field that could be used as a basis for future studies on the role of AMPK in prostate cancer.
Our data also adds further weight to a number of studies that have demonstrated off-target effects of AMPK activators, particularly metformin. While it has previously been proposed that anti-tumour effects of metformin may at least partially be mediated through AMPK-independent pathways [66, 67], our finding that approximately 90% of metformin-regulated genes are not differentially expressed after AICAR treatment demonstrates that caution must be exercised in assigning any observed effects of metformin to activation of AMPK without further validation. We believe the observed differences in dynamics of gene expression changes as well as in the extent of off-target effects between the two drugs may be due to their different mechanisms of action in activating AMPK. While AICAR is converted to AICAR 5′-Monophosphate (ZMP) within the cell, which activates AMPK by acting as an AMP mimetic [68], metformin inhibits complex I of the electron transport chain, prompting a decrease in cellular ATP/AMP ratio which then results in activation of AMPK [69]. The recent development of novel, potentially more specific activators of AMPK [70] will likely be an important step towards a better characterisation of AMPK function.
Nevertheless, we believe that our strategy of investigating gene expression changes that are shared between AICAR and metformin, two structurally unrelated AMPK activators, has yielded relevant insights into AMPK function. We have shown that biological processes enriched among our AMPK target gene signature are representative of the described function of AMPK, that our AMPK target gene signature resembles PI3K/Akt/mTor inhibition, and that predicted transcription factors shared between AICAR and metformin include known AMPK targets. All of this evidence suggests that our data is indeed representative of AMPK function.
Using this approach we were able to identify and validate the AR as a novel downstream mediator of AMPK signalling in prostate cancer cells. The AR drives both proliferation and anabolic pathways in prostate cancer cells [11]. In contrast, upon its activation, AMPK down-regulates anabolic pathways, stimulates catabolic pathways and reduces proliferation [9]. Notably, AR has previously been shown to regulate AMPK activity by inducing the expression of CAMKK2 [11, 20]. Therefore, our results suggest the presence of a negative feedback loop whereby the AR increases the activity of AMPK, which in turn feeds back to reduce AR activity (Figure 6). Thus, one could speculate that AMPK functions as a safety mechanism to prevent overshooting of AR activity and resulting stimulation of anabolism and proliferation under low-energy conditions.
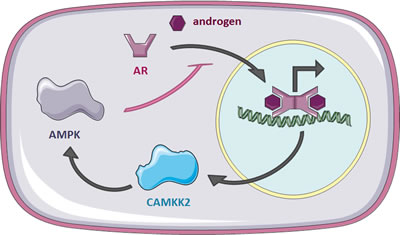
Figure 6: A negative feedback loop between AR and AMPK in prostate cancer cells. AR translocates to the nucleus upon ligand binding to activate transcription of its target genes, including CAMKK2. CAMKK2 phosphorylates AMPK, resulting in increased AMPK activity following androgen stimulation. Activated AMPK, in turn, reduces nuclear localisation of the AR, thereby attenuating its activity.
The exact mechanism by which AMPK is able to interfere with nuclear localisation of the AR will require further investigation. Notably, the activity of other nuclear receptors, including progesterone receptor, peroxisome proliferator-activated receptors and glucocorticoid receptor, has previously been shown to be regulated by AMPK [57, 71, 72], suggesting potential mechanisms of transcriptional regulation by AMPK that need to be explored further. First, AMPK has been shown to phosphorylate the transcriptional co-activator p300 at its serine 89 residue, thereby blocking its interaction with a number of nuclear receptors. It is thus conceivable that activation of AMPK could also disrupt interaction of p300 with AR [71]. As acetylation of the AR by p300 is thought to promote its nuclear localisation [73, 74], this mechanism could explain the decreased nuclear AR levels following AMPK activation that we observe.
Second, AMPK has been shown to stimulate the activity of p38 and JNK [57, 75-77]. These two kinases have been reported to phosphorylate AR on its serine 650 residue, promoting its nuclear export [78]. Finally, investigating links between AMPK and AR based on published data using MetaCore reveals a number of additional pathways through which AMPK may affect AR activity, including p53, PARP-1 and β-catenin signalling, hinting at the potential complexity of this regulatory system (Supplementary Figure 3). Further research will be required to unravel this intricate network of signalling pathways in order to determine the exact mechanisms of action by which AMPK interferes with AR localisation.
Interestingly, it has recently been reported that metformin can inhibit membrane-initiated androgen signalling [79], and two previous publications have shown that metformin treatment reduces AR expression levels over the course of several days [61, 62]. However, both of these effects were demonstrated to be either partially or entirely independent of AMPK. Our study thus provides the first evidence that classical AR signalling is regulated by AMPK, and suggests a mechanism involving decreased nuclear localisation of the AR. Notably, this acute inhibitory effect on AR occurs within several hours of treatment of cells with AICAR or metformin, lending further support to the notion that it represents a direct consequence of AMPK activation in the cell, rather than more indirect effects observed after long-term treatment. Considering our finding that the vast majority of the widespread metformin-induced transcriptional changes in prostate cancer cells cannot be replicated by AICAR treatment and are thus possibly independent of AMPK, it is not surprising that metformin would also affect the AR via pathways that do not involve AMPK.
In conclusion, we propose that AMPK and AR regulate each other via a negative feedback loop. A full understanding of how these two major metabolic regulators influence each other should provide important insights for the design of successful therapeutic strategies targeting prostate cancer cell metabolism.
Methods
Cell culture conditions and drug treatments
Cell lines were maintained in RPMI 1640 (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Hyclone / Thermo Scientific, Waltham, MA, USA) and passaged twice per week. Androgen treatments were carried out in phenol-red free RPMI 1640 (Invitrogen) supplemented with 10% charcoal/dextrane-treated FBS (Hyclone). AICAR (Tocris Bioscience, Bristol, UK) and metformin (Sigma-Aldrich, St Louis, USA) were used at final concentrations of 0.5 mM and 2 mM, respectively. R1881 and bicalutamide (Sigma-Aldrich, St Louis, USA) were used at concentrations of 1 nM and 10 μM, respectively.
Genome-wide expression profiling
LNCaP cells grown in RPMI with 10% FBS were treated with AICAR or metformin in five independent biological replicates. RNA was extracted after 0 h, 8 h and 24 h of treatment using RNeasy Plus Mini Kit (Qiagen, Hilden, Germany). cDNA was generated, hybridised to Illumina HumanHT-12 v4 BeadChip arrays and scanned using standard Illumina protocols. Data was analysed using R (R Development Core Team, 2010) and Bioconductor [80]. Spatial artefacts were removed using BASH [81] and HULK algorithms from the beadarray package [82]. Data was log2 transformed and quantile normalised, and differentially expressed genes were identified with a global false discovery rate of 0.01. Expression data will be deposited in Gene Expression Omnibus (GEO). Significance of gene list overlaps was tested in R using the phyper function. Heatmaps were generated using a Cancer Research UK Cambridge Institute Bioinformatics Core Facility Galaxy tool. Enrichment of gene ontology terms was tested using the Molecular Signature Database v4.0 (Broad Institute, http://www.broadinstitute.org/gsea/index.jsp). To test enrichment of gene sets of interest within publicly available prostate cancer datasets, genes were ranked from most underexpressed to most overexpressed in primary cancer compared to normal/benign tissue according to t statistic, and gene set enrichment analysis was performed using the GSEA software (Broad Institute, http://www.broadinstitute.org/gsea/index.jsp). Enriched transcription factors were identified using Ingenuity Pathway Analysis (http://www.ingenuity.com/products/ipa). Due to the extremely high number of significantly differentially expressed genes after 24 h metformin treatment, only genes with a log fold change of ± 0.65 or above were used for this analysis.
Quantitative reverse transcription PCR
RNA from cells was isolated using the RNeasy Plus Mini Kit according to the manufacturer’s instructions. cDNA was synthesized using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA) following the manufacturer’s instructions. qRT-PCR reactions were performed on an ABI PRISM 7900 HT Sequence Detection System. Relative gene expression was calculated according to the ∆∆Ct method; ACTB and SDH were used as housekeeping genes. Details of primers used are given in Supplementary Table 2.
Luciferase reporter assay
Cells were seeded in 48-well plates and transfected with luciferase reporter constructs MMTV-Luc and pRL-TK one day after seeding using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. To evaluate the effects of pharmacological AMPK activators, the medium was replaced with phenol-red free RPMI supplemented with charcoal-stripped serum, R1881 or vehicle and AMPK activators 6 h post-transfection. After a further 24 h, dual luciferase reporter assay was carried out using the Dual Luciferase Reporter Assay System (Promega, Madison, WI, USA) according to the manufacturer’s instructions. To determine the effects of gene silencing, the constructs were co-transfected with siRNAs and the medium was replaced with phenol-red free RPMI supplemented with charcoal-stripped FBS 6 h after transfection. R1881 or vehicle was added 24 h post-transfection to allow knock-down of target genes before stimulation. Dual luciferase reporter assay was performed 40 h after stimulation with R1881.
Generation of an androgen-responsive luciferase reporter cell line
The pGL4.16 vector from Promega was cut with XhoI and BglII, and a polylinker including ClaI, AsclI and NheI was inserted. The Luc2CP gene was retrieved by cutting the resulting vector with ClaI, XbaI and EagI HF and cloned into a pCSC-SP-PW-CMV-LacZ vector restricted with ClaI and XbaI. To add a selection marker, the PGK-Puromycin resistance cassette was obtained from a pSICOR-PGK-Puro plasmid by cutting it with BamHI and ScaI and ligated into the pCSC-Luc2CP, previously cut with BamHI and PmeI. The CAMKK2 androgen-receptor binding sequence was cloned through PCR from genomic DNA from LNCaP cells (forward primer: ATGGCGCGCCGCTAGCGAATGCA TGCGGCAGTGTTCCAAT; reverse primer GGCCAGATCTGCTAGCTAAAGA AGGAAGGGAGGTGGCTGA) and inserted by recombination into the pCSC-Luc2CP-Puro vector cut with NheI.
For generation of lentiviral particles, HEK293TLA cells were transfected with the pCSC-CAMKK2-Luc2CP-Puro vector and packaging, envelope and reverse expression plasmids using the calcium phosphate method. Media was changed after 24 h and fresh media applied. After a further 24 h viral supernatant was filtered (45 μm pore size, Millipore, Billerica, MA) and mixed with fresh media. LNCaP cells were infected with lentiviral particles and expanded as a stable cell line (LNCaP-CAMKK2-Luc).
Transient transfection of siRNAs
siRNAs against AMPK (AMPKα1: J-005027-06, J-005027-07; AMPKα2: J-005361-06, J-005361-07) were purchased from Dharmacon and transfected using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s reverse transfection protocol. To achieve knock-down of both AMPKα subunits, we combined two siRNAs, one directed against each subunit. Two different combinations of siRNA were used to account for potential off-target effects. siAllStars (Qiagen) was used as a non-targeting control. siRNAs were transfected at a final concentration of 20 nM.
Western blot analysis
Cells were lysed in ice-cold M-PER buffer (Pierce / Thermo Scientific) containing protease and phosphatase inhibitor cocktails and harvested by scraping. Lysates were cleared by centrifugation at maximum speed and 4°C for 10 min in a benchtop centrifuge. Protein content was quantified using a Direct Detect Spectrometer (Millipore). Samples for Western Blot were prepared by addition of 5x Laemmli buffer and boiled for 5 min. Equal amounts of protein were loaded onto 4-12% Criterion precast gels (Bio-Rad Laboratories, Hercules, CA, USA) and separated by SDS-PAGE. Proteins were transferred to nitrocellulose membranes using the iBlotTM Dry Blotting System (Invitrogen), and transfer was assessed by staining with Ponceau S (Sigma). Membranes were blocked in 5% milk (Marvel) or BSA (Sigma) in Tris-buffered saline with 0.1% Tween-20 and incubated with primary antibodies and HPRC-conjugated secondary antibodies (Dako). Detection was carried out using Western Lightning ECL Pro (Perkin Elmer). Films were scanned and quantification of intensity of bands was carried out using ImageJ. pACC Ser79(#3661), pAMPK Thr172 (40H9) and AMPKα (#2532) antibodies were from Cell Signalling Technology; β-actin (AC-40) antibody was from Sigma; lamin B (M-20) antibody was from Santa Cruz Biotechnology; β-tubulin (D66) antibody was from Abcam; AR (441) antibody was from Dako.
Nuclear-cytoplasmic fractionation
To obtain nuclear and cytoplasmic fractions, cell pellets were resuspended in Buffer A (10 mM Hepes pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol) supplemented with 0.1% Triton X-100 and incubated on ice for 10 min. Nuclei were pelleted by centrifugation at 4°C and 1,300 G for 4 min. The supernatants were cleared by centrifugation to obtain cytoplasmic fractions. Nuclei were washed three times in Buffer A without Triton X-100, resuspended in Buffer B (3 mM EDTA, 0.2 mM EGTA) and disrupted by sonication. Nuclear fractions were cleared by centrifugation.
Statistical analysis
Statistical analysis was carried out using GraphPad Prism 6. To test significant differences between groups, one-way or randomized blocks ANOVA was performed depending on the experimental design. If significance at the level of p=0.05 was reached, multiple comparisons of means were performed using Holm-Sidak test. Unless otherwise stated, all figures show means of biological replicates and error bars indicate SEM. Significance levels are indicated as follows: * p < 0.05; ** p < 0.01; *** p < 0.001.
Acknowledgement
We thank the core facilities at the Cambridge Research Institute led by James Hadfield (Genomics) and Matt Eldridge (Bioinformatics). Charlie Massie, Mohammad Asim, Helen Ross-Adams, Hayley Whitaker, Hélène Bon, Karan Wadhwa, Nelma Pértega-Gomes and Steve Hawkins provided valuable discussion and assistance.
References
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E and Forman D. Global cancer statistics. CA Cancer J Clin. 2011; 61(2):69-90.
2. Denmeade SR and Isaacs JT. A history of prostate cancer treatment. Nat Rev Cancer. 2002; 2(5):389-396.
3. Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG and Sawyers CL. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004; 10(1):33-39.
4. Koivisto P, Kononen J, Palmberg C, Tammela T, Hyytinen E, Isola J, Trapman J, Cleutjens K, Noordzij A, Visakorpi T and Kallioniemi OP. Androgen receptor gene amplification: a possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Res. 1997; 57(2):314-319.
5. Yuan X, Li T, Wang H, Zhang T, Barua M, Borgesi RA, Bubley GJ, Lu ML and Balk SP. Androgen receptor remains critical for cell-cycle progression in androgen-independent CWR22 prostate cancer cells. Am J Pathol. 2006; 169(2):682-696.
6. Taplin ME, Bubley GJ, Ko YJ, Small EJ, Upton M, Rajeshkumar B and Balk SP. Selection for androgen receptor mutations in prostate cancers treated with androgen antagonist. Cancer Res. 1999; 59(11):2511-2515.
7. Zhao XY, Malloy PJ, Krishnan AV, Swami S, Navone NM, Peehl DM and Feldman D. Glucocorticoids can promote androgen-independent growth of prostate cancer cells through a mutated androgen receptor. Nat Med. 2000; 6(6):703-706.
8. Gregory CW, He B, Johnson RT, Ford OH, Mohler JL, French FS and Wilson EM. A mechanism for androgen receptor-mediated prostate cancer recurrence after androgen deprivation therapy. Cancer Res. 2001; 61(11):4315-4319.
9. Flavin R, Zadra G and Loda M. Metabolic alterations and targeted therapies in prostate cancer. J Pathol. 2010; 223(2):283-294.
10. Hanahan D and Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144(5):646-674.
11. Massie CE, Lynch A, Ramos-Montoya A, Boren J, Stark R, Fazli L, Warren A, Scott H, Madhu B, Sharma N, Bon H, Zecchini V, Smith DM, Denicola GM, Mathews N, Osborne M, et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011; 30(13):2719-2733.
12. Swinnen JV, Esquenet M, Goossens K, Heyns W and Verhoeven G. Androgens stimulate fatty acid synthase in the human prostate cancer cell line LNCaP. Cancer Res. 1997; 57(6):1086-1090.
13. Rossi S, Graner E, Febbo P, Weinstein L, Bhattacharya N, Onody T, Bubley G, Balk S and Loda M. Fatty acid synthase expression defines distinct molecular signatures in prostate cancer. Mol Cancer Res. 2003; 1(10):707-715.
14. Shurbaji MS, Kalbfleisch JH and Thurmond TS. Immunohistochemical detection of a fatty acid synthase (OA-519) as a predictor of progression of prostate cancer. Hum Pathol. 1996; 27(9):917-921.
15. Shah US, Dhir R, Gollin SM, Chandran UR, Lewis D, Acquafondata M and Pflug BR. Fatty acid synthase gene overexpression and copy number gain in prostate adenocarcinoma. Hum Pathol. 2006; 37(4):401-409.
16. Rubin MA, Zhou M, Dhanasekaran SM, Varambally S, Barrette TR, Sanda MG, Pienta KJ, Ghosh D and Chinnaiyan AM. alpha-Methylacyl coenzyme A racemase as a tissue biomarker for prostate cancer. JAMA. 2002; 287(13):1662-1670.
17. Mukherji M, Schofield CJ, Wierzbicki AS, Jansen GA, Wanders RJ and Lloyd MD. The chemical biology of branched-chain lipid metabolism. Prog Lipid Res. 2003; 42(5):359-376.
18. Davies SP, Hawley SA, Woods A, Carling D, Haystead TA and Hardie DG. Purification of the AMP-activated protein kinase on ATP-gamma-sepharose and analysis of its subunit structure. Eur J Biochem. 1994; 223(2):351-357.
19. Woods A, Cheung PC, Smith FC, Davison MD, Scott J, Beri RK and Carling D. Characterization of AMP-activated protein kinase beta and gamma subunits. Assembly of the heterotrimeric complex in vitro. J Biol Chem. 1996; 271(17):10282-10290.
20. Frigo DE, Howe MK, Wittmann BM, Brunner AM, Cushman I, Wang Q, Brown M, Means AR and McDonnell DP. CaM kinase kinase beta-mediated activation of the growth regulatory kinase AMPK is required for androgen-dependent migration of prostate cancer cells. Cancer Res. 2011; 71(2):528-537.
21. Tennakoon JB, Shi Y, Han JJ, Tsouko E, White MA, Burns AR, Zhang A, Xia X, Ilkayeva OR, Xin L, Ittmann MM, Rick FG, Schally AV and Frigo DE. Androgens regulate prostate cancer cell growth via an AMPK-PGC-1alpha-mediated metabolic switch. Oncogene. 2013.
22. Pearson HB, McCarthy A, Collins CM, Ashworth A and Clarke AR. Lkb1 deficiency causes prostate neoplasia in the mouse. Cancer Res. 2008; 68(7):2223-2232.
23. Wright JL and Stanford JL. Metformin use and prostate cancer in Caucasian men: results from a population-based case-control study. Cancer Causes Control. 2009; 20(9):1617-1622.
24. Currie CJ, Poole CD, Jenkins-Jones S, Gale EA, Johnson JA and Morgan CL. Mortality after incident cancer in people with and without type 2 diabetes: impact of metformin on survival. Diabetes Care. 2012; 35(2):299-304.
25. Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR and Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005; 330(7503):1304-1305.
26. Xiang X, Saha AK, Wen R, Ruderman NB and Luo Z. AMP-activated protein kinase activators can inhibit the growth of prostate cancer cells by multiple mechanisms. Biochem Biophys Res Commun. 2004; 321(1):161-167.
27. Zhou J, Huang W, Tao R, Ibaragi S, Lan F, Ido Y, Wu X, Alekseyev YO, Lenburg ME, Hu GF and Luo Z. Inactivation of AMPK alters gene expression and promotes growth of prostate cancer cells. Oncogene. 2009; 28(18):1993-2002.
28. Sauer H, Engel S, Milosevic N, Sharifpanah F and Wartenberg M. Activation of AMP-kinase by AICAR induces apoptosis of DU-145 prostate cancer cells through generation of reactive oxygen species and activation of c-Jun N-terminal kinase. Int J Oncol. 2012; 40(2):501-508.
29. Zhou J, Yang Z, Tsuji T, Gong J, Xie J, Chen C, Li W, Amar S and Luo Z. LITAF and TNFSF15, two downstream targets of AMPK, exert inhibitory effects on tumor growth. Oncogene. 2011; 30(16):1892-1900.
30. Apontes P, Leontieva OV, Demidenko ZN, Li F and Blagosklonny MV. Exploring long-term protection of normal human fibroblasts and epithelial cells from chemotherapy in cell culture. Oncotarget. 2011; 2(3):222-233.
31. Sanli T, Rashid A, Liu C, Harding S, Bristow RG, Cutz JC, Singh G, Wright J and Tsakiridis T. Ionizing radiation activates AMP-activated kinase (AMPK): a target for radiosensitization of human cancer cells. Int J Radiat Oncol Biol Phys. 2010; 78(1):221-229.
32. Lin YC, Wu MH, Wei TT, Huang WC, Huang LY, Lin YT and Chen CC. Metformin sensitizes anticancer effect of dasatinib in head and neck squamous cell carcinoma cells through AMPK-dependent ER stress. Oncotarget. 2014; 5(1):298-308.
33. Zakikhani M, Dowling RJ, Sonenberg N and Pollak MN. The effects of adiponectin and metformin on prostate and colon neoplasia involve activation of AMP-activated protein kinase. Cancer Prev Res (Phila). 2008; 1(5):369-375.
34. Lin VC, Tsai YC, Lin JN, Fan LL, Pan MH, Ho CT, Wu JY and Way TD. Activation of AMPK by Pterostilbene Suppresses Lipogenesis and Cell-Cycle Progression in p53 Positive and Negative Human Prostate Cancer Cells. J Agric Food Chem. 2012; 60(25):6399-6407.
35. Lee CR, Chun JN, Kim SY, Park S, Kim SH, Park EJ, Kim IS, Cho NH, Kim IG, So I, Kim TW and Jeon JH. Cyclosporin A suppresses prostate cancer cell growth through CaMKKbeta/AMPK-mediated inhibition of mTORC1 signaling. Biochem Pharmacol. 2012.
36. Park HU, Suy S, Danner M, Dailey V, Zhang Y, Li H, Hyduke DR, Collins BT, Gagnon G, Kallakury B, Kumar D, Brown ML, Fornace A, Dritschilo A and Collins SP. AMP-activated protein kinase promotes human prostate cancer cell growth and survival. Mol Cancer Ther. 2009; 8(4):733-741.
37. Chhipa RR, Wu Y and Ip C. AMPK-mediated autophagy is a survival mechanism in androgen-dependent prostate cancer cells subjected to androgen deprivation and hypoxia. Cell Signal. 2011; 23(9):1466-1472.
38. Jung SN, Park IJ, Kim MJ, Kang I, Choe W, Kim SS and Ha J. Down-regulation of AMP-activated protein kinase sensitizes DU145 carcinoma to Fas-induced apoptosis via c-FLIP degradation. Exp Cell Res. 2009; 315(14):2433-2441.
39. Martinez Marignac VL, Smith S, Toban N, Bazile M and Aloyz R. Resistance to Dasatinib in primary chronic lymphocytic leukemia lymphocytes involves AMPK-mediated energetic re-programming. Oncotarget. 2013; 4(12):2550-2566.
40. Anisimov VN. Metformin and rapamycin are master-keys for understanding the relationship between cell senescent, aging and cancer. Aging (Albany NY). 2013; 5(5):337-338.
41. Blagosklonny MV. Selective anti-cancer agents as anti-aging drugs. Cancer Biol Ther. 2013; 14(12):1092-1097.
42. Zhou G, Sebhat IK and Zhang BB. AMPK activators--potential therapeutics for metabolic and other diseases. Acta Physiol (Oxf). 2009; 196(1):175-190.
43. Fogarty S and Hardie DG. Development of protein kinase activators: AMPK as a target in metabolic disorders and cancer. Biochim Biophys Acta. 2010; 1804(3):581-591.
44. Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, Reich M, Hieronymus H, Wei G, Armstrong SA, Haggarty SJ, Clemons PA, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006; 313(5795):1929-1935.
45. Inoki K, Zhu T and Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003; 115(5):577-590.
46. Corradetti MN, Inoki K, Bardeesy N, DePinho RA and Guan KL. Regulation of the TSC pathway by LKB1: evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev. 2004; 18(13):1533-1538.
47. Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE and Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008; 30(2):214-226.
48. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES and Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005; 102(43):15545-15550.
49. Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003; 34(3):267-273.
50. Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, Asangani IA, Ateeq B, Chun SY, Siddiqui J, Sam L, Anstett M, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012; 487(7406):239-243.
51. Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, Antipin Y, Mitsiades N, Landers T, Dolgalev I, Major JE, Wilson M, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010; 18(1):11-22.
52. Varambally S, Yu J, Laxman B, Rhodes DR, Mehra R, Tomlins SA, Shah RB, Chandran U, Monzon FA, Becich MJ, Wei JT, Pienta KJ, Ghosh D, Rubin MA and Chinnaiyan AM. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell. 2005; 8(5):393-406.
53. Wang J, Weng J, Cai Y, Penland R, Liu M and Ittmann M. The prostate-specific G-protein coupled receptors PSGR and PSGR2 are prostate cancer biomarkers that are complementary to alpha-methylacyl-CoA racemase. Prostate. 2006; 66(8):847-857.
54. Weng J, Wang J, Hu X, Wang F, Ittmann M and Liu M. PSGR2, a novel G-protein coupled receptor, is overexpressed in human prostate cancer. Int J Cancer. 2006; 118(6):1471-1480.
55. Weigle B, Fuessel S, Ebner R, Temme A, Schmitz M, Schwind S, Kiessling A, Rieger MA, Meye A, Bachmann M, Wirth MP and Rieber EP. D-GPCR: a novel putative G protein-coupled receptor overexpressed in prostate cancer and prostate. Biochem Biophys Res Commun. 2004; 322(1):239-249.
56. Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ and Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005; 18(3):283-293.
57. Nader N, Ng SS, Lambrou GI, Pervanidou P, Wang Y, Chrousos GP and Kino T. AMPK regulates metabolic actions of glucocorticoids by phosphorylating the glucocorticoid receptor through p38 MAPK. Mol Endocrinol. 2010; 24(9):1748-1764.
58. He WW, Sciavolino PJ, Wing J, Augustus M, Hudson P, Meissner PS, Curtis RT, Shell BK, Bostwick DG, Tindall DJ, Gelmann EP, Abate-Shen C and Carter KC. A novel human prostate-specific, androgen-regulated homeobox gene (NKX3.1) that maps to 8p21, a region frequently deleted in prostate cancer. Genomics. 1997; 43(1):69-77.
59. Magee JA, Chang LW, Stormo GD and Milbrandt J. Direct, androgen receptor-mediated regulation of the FKBP5 gene via a distal enhancer element. Endocrinology. 2006; 147(1):590-598.
60. Mendiratta P, Mostaghel E, Guinney J, Tewari AK, Porrello A, Barry WT, Nelson PS and Febbo PG. Genomic strategy for targeting therapy in castration-resistant prostate cancer. J Clin Oncol. 2009; 27(12):2022-2029.
61. Shen M, Zhang Z, Ratnam M and Dou QP. The interplay of AMP-activated protein kinase and androgen receptor in prostate cancer cells. J Cell Physiol. 2013.
62. Demir U, Koehler A, Schneider R, Schweiger S and Klocker H. Metformin anti-tumor effect via disruption of the MID1 translational regulator complex and AR downregulation in prostate cancer cells. BMC Cancer. 2014; 14(1):52.
63. Quarmby VE, Yarbrough WG, Lubahn DB, French FS and Wilson EM. Autologous down-regulation of androgen receptor messenger ribonucleic acid. Mol Endocrinol. 1990; 4(1):22-28.
64. Krongrad A, Wilson CM, Wilson JD, Allman DR and McPhaul MJ. Androgen increases androgen receptor protein while decreasing receptor mRNA in LNCaP cells. Mol Cell Endocrinol. 1991; 76(1-3):79-88.
65. Georget V, Lobaccaro JM, Terouanne B, Mangeat P, Nicolas JC and Sultan C. Trafficking of the androgen receptor in living cells with fused green fluorescent protein-androgen receptor. Mol Cell Endocrinol. 1997; 129(1):17-26.
66. Ben Sahra I, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P, Tanti JF, Le Marchand-Brustel Y and Bost F. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene. 2008; 27(25):3576-3586.
67. Ben Sahra I, Regazzetti C, Robert G, Laurent K, Le Marchand-Brustel Y, Auberger P, Tanti JF, Giorgetti-Peraldi S and Bost F. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011; 71(13):4366-4372.
68. Sullivan JE, Brocklehurst KJ, Marley AE, Carey F, Carling D and Beri RK. Inhibition of lipolysis and lipogenesis in isolated rat adipocytes with AICAR, a cell-permeable activator of AMP-activated protein kinase. FEBS Lett. 1994; 353(1):33-36.
69. El-Mir MY, Nogueira V, Fontaine E, Averet N, Rigoulet M and Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. 2000; 275(1):223-228.
70. Zadra G, Photopoulos C, Tyekucheva S, Heidari P, Weng QP, Fedele G, Liu H, Scaglia N, Priolo C, Sicinska E, Mahmood U, Signoretti S, Birnberg N and Loda M. A novel direct activator of AMPK inhibits prostate cancer growth by blocking lipogenesis. EMBO Mol Med. 2014.
71. Yang W, Hong YH, Shen XQ, Frankowski C, Camp HS and Leff T. Regulation of transcription by AMP-activated protein kinase: phosphorylation of p300 blocks its interaction with nuclear receptors. J Biol Chem. 2001; 276(42):38341-38344.
72. Wu L, Huang XJ, Yang CH, Deng SS, Qian M, Zang Y and Li J. 5’-AMP-activated protein kinase (AMPK) regulates progesterone receptor transcriptional activity in breast cancer cells. Biochem Biophys Res Commun. 2011; 416(1-2):172-177.
73. Fu M, Wang C, Reutens AT, Wang J, Angeletti RH, Siconolfi-Baez L, Ogryzko V, Avantaggiati ML and Pestell RG. p300 and p300/cAMP-response element-binding protein-associated factor acetylate the androgen receptor at sites governing hormone-dependent transactivation. J Biol Chem. 2000; 275(27):20853-20860.
74. Shiota M, Yokomizo A, Masubuchi D, Tada Y, Inokuchi J, Eto M, Uchiumi T, Fujimoto N and Naito S. Tip60 promotes prostate cancer cell proliferation by translocation of androgen receptor into the nucleus. Prostate. 2010; 70(5):540-554.
75. Kefas BA, Cai Y, Ling Z, Heimberg H, Hue L, Pipeleers D and Van de Casteele M. AMP-activated protein kinase can induce apoptosis of insulin-producing MIN6 cells through stimulation of c-Jun-N-terminal kinase. J Mol Endocrinol. 2003; 30(2):151-161.
76. Lee YM, Uhm KO, Lee ES, Kwon J, Park SH and Kim HS. AM251 suppresses the viability of HepG2 cells through the AMPK (AMP-activated protein kinase)-JNK (c-Jun N-terminal kinase)-ATF3 (activating transcription factor 3) pathway. Biochem Biophys Res Commun. 2008; 370(4):641-645.
77. Yoon MJ, Lee GY, Chung JJ, Ahn YH, Hong SH and Kim JB. Adiponectin increases fatty acid oxidation in skeletal muscle cells by sequential activation of AMP-activated protein kinase, p38 mitogen-activated protein kinase, and peroxisome proliferator-activated receptor alpha. Diabetes. 2006; 55(9):2562-2570.
78. Gioeli D, Black BE, Gordon V, Spencer A, Kesler CT, Eblen ST, Paschal BM and Weber MJ. Stress kinase signaling regulates androgen receptor phosphorylation, transcription, and localization. Mol Endocrinol. 2006; 20(3):503-515.
79. Malaguarnera R, Sacco A, Morcavallo A, Squatrito S, Migliaccio A, Morrione A, Maggiolini M and Belfiore A. Metformin Inhibits Androgen-Induced Igf-Ir Upregulation in Prostate Cancer Cells by Disrupting Membrane Initiated Androgen Signaling. Endocrinology. 2014:en20131925.
80. Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004; 5(10):R80.
81. Cairns JM, Dunning MJ, Ritchie ME, Russell R and Lynch AG. BASH: a tool for managing BeadArray spatial artefacts. Bioinformatics. 2008; 24(24):2921-2922.
82. Dunning MJ, Smith ML, Ritchie ME and Tavare S. beadarray: R classes and methods for Illumina bead-based data. Bioinformatics. 2007; 23(16):2183-2184.