
INTRODUCTION
Cytokines profiles are now well-defined in the pathophysiology of multiple diseases mainly by influencing the growth, proliferation, differentiation of a variety of cell types [1]. Theses cytokines signaling pathways are triggered as a consequence of enhanced interaction of cytokines with their specific cell surface receptors, which leads to the activation of intracellular molecules, such as Janus kinases (JAKs) and signal transducers and activators of transcription (STATs) [2, 3]. Studies have reported that macrophages are important immune cells and function as the direct target of some cytokines, which might contribute to the pathogenesis of many diseases [4, 5]. Monocyte-macrophage lineage has long been recognized as heterogeneous cell type that could undergo a continuum from M1 to M2 activation states depending on the microenvironment signals [6, 7]. The M1/M2 subpopulation represents the dynamic shift between inflammatory and reparative macrophages, which significantly contribute to either beneficial or detrimental effects in different diseases [8, 9]. As expected, these distinct and opposing subsets are often exposed to the various cytokines stimuli, for example interleukins (ILs), interferons (IFNs) and growth factors [10–12].
In response to pathogens or injury, cytokines and intracellular signaling pathways may promote the M1/M2 macrophage polarization in cancer and other disorders [13–15]. Suppressors of cytokine signaling (SOCS) proteins act as inducible negative feedback regulators of cytokine signaling by a generic mechanism of targeting associated proteins for degradation [16, 17]. Profound new discoveries have showed that the disparate SOCS family molecules may serve as the molecular switch that controls immune activation/suppression and M1/M2 macrophage polarization [18, 19]. Herein, this review provides an overview of the emerging roles and mechanisms of SOCS proteins in cytokines-induced M1/M2 macrophage polarization. Taken together, an updated understanding of the fate of SOCS-directed macrophage polarization and function could guide the development of novel therapeutic targets for various diseases.
THE CLASSIFICATION, STRUCTURE, AND ACTION MECHANISM OF SOCS FAMILY OF PROTEINS
At present, the mammalian SOCS family of proteins is believed to consist of eight members: SOCS1-7 and the alternatively named cytokine-inducible Src homology 2 (SH2)-containing protein (CIS) [20, 21]. The SOCS/CIS family is a group of intracellular proteins, several of which downregulate cytokines signaling following cytokines engagement of their specific receptors complex through a negative feedback loop [22].
Lines of evidence suggest that SOCS/CIS family negatively regulates the cytokines signaling is largely attributing to their domain structure characteristics [23]. Each SOCS protein shares a common modular organization that generally contains three differentially conserved domains: an amino (N)-terminal region of varied length and amino acid sequence (low conserved), a central SH2-domain (conserved) and a carboxy (C)-terminal 40 amino acid module, e.g. SOCS box motif (highly conserved), which has been well-described in previously published literatures [24, 25]. Particularly, the SOCS3 and CIS-SH2 domains exist a 35-residue (PEST) (proline-, glutamicacid-, serine- and threonine-rich)-motif that inserted between the α β helix and the BG loop, which can affect the SOCS3 turnover and stability [26]. Another feature of the SOCS family is the extended SH2 sequence called (ESS) that is important for the interaction with phosphotyrosine residues on the target protein [27]. There exists an amino acid sequence homology in pairs between all SOCS family members: CIS and SOCS2, SOCS1 and SOCS3, SOCS4 and SOCS5, and SOCS6 and SOCS7, which have marked pair-wise homology across the entire protein sequence [28, 29]. However, several important distinctions among different SOCS members are also unambiguous in view of firstly based on a short(CIS, SOCS1-3) or long, N-terminal region(SOCS4-7) and secondly, it is the former group, which are most clearly induced in response to cytokine signaling and act in a classical negative-feedback loop [30] (Figure 1).
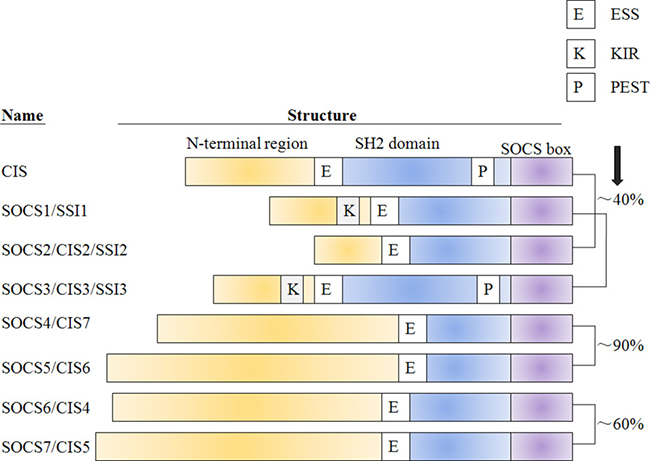
Figure 1: The classification and domain structure of the SOCS protein family. All of the eight SOCS-family members have a central SH2 domain,an amino-terminal domain of variable length and a 40-amino-acid motif at the carboxy terminus that is known as the SOCS box. Although there is sequence homology between all family members-particularly in the SOCS box and SH2 domain-CIS and SOCS2, SOCS1 and SOCS3, SOCS4 and SOCS5, and SOCS6 and SOCS7 have marked pair-wise homology across the entire protein sequence, as indicated. In SOCS1 and SOCS3, a kinase-inhibitory region (K) adjacent to the SH2 domain that is required for high-affinity binding to Jaks and the inhibition of kinase activity has also been defined. The alternative nomenclature for each SOCS protein is given in parentheses. CIS, cytokine-induced SH2 protein; JAB, Janus kinase(Jak)-binding protein; NAP4, Nck, Ash and phospholipase-C binding protein; SH2, SRC-homology 2; SOCS, suppressor of cytokine signalling; SSI, Stat-induced Stat inhibitor; Stat, signal transducer and activator of transcription.
There are mainly two kinds of action modes that are necessary for the proper regulatory functions of CIS-SOCS-family proteins. The first approach of all SOCS proteins to degrade the targets is dependent on the ubiquitin pathway [31]. The SOCS box interacts with elongin B and C, cullin-5 and RING-box-2 (RBX2) to recruit E2 ubiquitin transferase and E3 ubiquitin ligases complex. This could lead to the polyubiquitylation of bound signaling proteins, and their consequent proteasomal degradation, resulting in the termination of signaling [32, 33]. Second, both SOCS1 and SOCS3 can inhibit the catalytic-activity function of JAK tyrosine kinase by acting as a pseudosubstrate and competing with substrates through their SH2 domain and kinase inhibitory region (KIR), which locates N-terminal domain [34, 35] (Figure 1). Meanwhile, CIS and SOCS2 have been thought to act by competing with STAT proteins for binding to phosphorylated tyrosine residues within the receptor cytoplasmic domains [36]. In contrast, the functions and mechanisms of SOCS4-7 will be discussed later in this review (Figure 2).
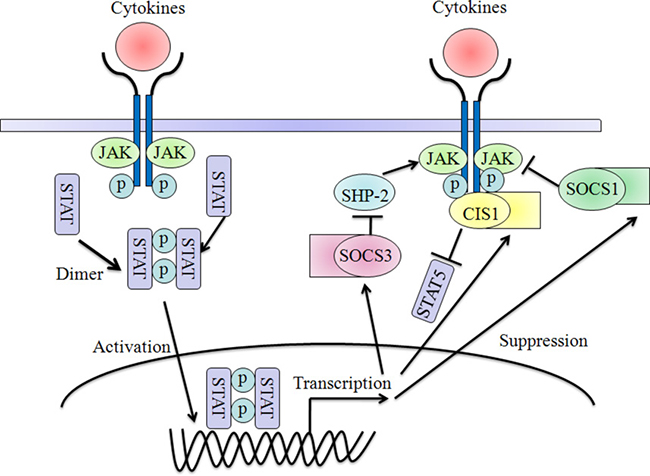
Figure 2: Negative-feedback loop regulation of cytokines signaling by SOCS proteins. Cytokine stimulation activates the Jak-Stat pathway, leading to the induction of CIS, SOCS1 and SOCS3. CIS, SOCS1 and SOCS3 could inhibit signaling by different mechanisms: SOCS1 binds to the JAKs and inhibits catalytic activity; SOCS3 binds to JAK-proximal sites on cytokine receptors and inhibits JAK activity; and CIS blocks the binding of Stats to cytokine receptors. Both SOCS1 and SOCS3 contain a kinase inhibitory region (KIR) for the suppression of Jak tyrosine kinase activity.
CYTOKINES SIGNALING AND MACROPHAGE POLARIZATION
Within differential cytokines microenvironment, macrophage could undergo polarization toward M1 or M2 phenotype. Though the mechanisms of cytokines signaling controlling macrophage polarization remain not fully clear, we discuss some recently important findings regarding the roles of cytokines signaling in the regulation of macrophage polarization.
INTERLEUKINS (ILS)
Interleukins (ILs) are a wide range of peptidic substances released by a variety of cell types in response to various inflammatory processes [37]. They are known as soluble factors and thought to have a critical role in the modulation of many immune pathways [38, 39]. Remarkably, it has now been approved that monocytes and macrophages M1/M2 phenotype formation required after the ILs stimulation.
It has been widely accepted that IL-4 or/and IL-13 can stimulate the macrophage into the M2 subpopulation [10, 40]. IL-4Rα binding of IL-4 activates JAK1 and JAK3, which lead to the activation and translocation of STAT6 into nucleus where it binds to the promoter region of target M2 genes [41, 42]. IL-4-provoked M2 functions are largely mediated by the activation of IL-4/STAT6 signaling pathway and enhanced Arg-1 production [43, 44]. Furthermore, IL-13 receptor alpha 1 (IL-13Rα1) specifically heterodimerizes with the IL-4Rα chain to form a type II heteroreceptor, which is able to phosphorylate STAT6 efficiently and controls the differentiation and function of M2 macrophages [45, 46].Thus, utilizing an IL-4/STAT6-dependent mechanism to shift macrophage polarization to the M2/anti-inflammatory phenotype might ameliorate inflammatory and autoimmune diseases in clinical treatment. IL-5 activity inhibition may lead to the substantial reduction of IL-13-triggered M2 responses, which were associated with increased production of the cytokine IFN-γ [47]. Conversely, tumor associated macrophages (TAMs) from the STAT6-null mice showed a functional M1 phenotype upon activation as characterized by i.e. NO production, which firmly indicated that JAK1, 3/STAT6 signaling pathway are essential for the M2 activation [48, 49]. IL-4 in combination with IL-13 has been widely used as the inducers in vitro of skewing monocytes/macrophages into the M2 polarization. However, IL-4 and IL-13 signaling in monocytes/macrophages might differentially regulate the expression of several inflammatory genes starting from IL-4/IL-13 cytokine receptors to ultimately control Jak/Stat-mediated signaling pathways [50]. IL-13 utilizes both IL-4Rα/Jak2/Stat3 and IL-13Rα1/Tyk2/Stat1/Stat6 signaling pathways, while IL-4 can use only the IL-4Rα/Jak1/Stat3/Stat6 cascade to promotethe expression of some critical inflammatory genes [51]. These conclusions provide novel insights into the mechanisms and functions of alternatively activated monocytes/macrophages stimulated by IL-4 and IL-13, which have important implications for the potential treatment of multiple inflammatory diseases.
In addition, though IL-6 is commonly recognized as a pro-inflammatory mediator and is associated with the pathogenesis of many inflammatory processes, the pleiotrophic nature of IL-6 are also controversial [52, 53]. IL-6 is considered to assign an unexpected homeostatic role to limit inflammation and as an important inducer in promoting M2 polarization, which is mainly dependent on the upregulation of the IL-4Rα [54]. IL-6 could induce the expression of the receptor for IL-4 and augment the response to IL-4 in macrophages, but IL-6ra∆myel mice are resistant to IL-4-mediated M2 polarization and exhibit enhanced susceptibility to lipopolysaccharide (LPS)-induced endotoxemia [55]. Besides, the anti-inflammatory cytokine IL-10 promotes M2 polarization through the induction of p50 NF-κB homodimer, c-Maf, and STAT3 activities [56]. On the contrary, the M1 subtype is a phenotype characterized by the marked increase in cytokines(IL-1β, IL-12) and decrease in cytokines(IL-4, 10 and 13) [57, 58]. IL-12p40 and L-12p35 bind to IL-12Rβ1 and β2, respectively, which results in transphosphorylation of associated JAKs (JAK2 and TYK2) and then leads the activation and translocation of STAT4 into nucleus where they bind to STAT binding sites in the IFN-γ promoter of targeting M1 genes [59]. In conclusion, different STATs seem to be pivotal factors and relevant to the in M1/ M2 balance (Figure 3).
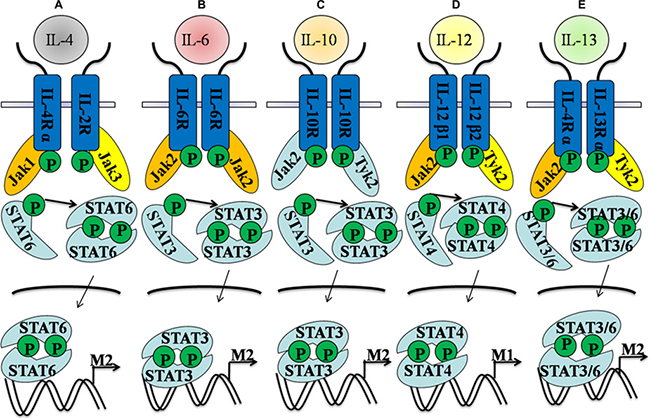
Figure 3: Different cytokines stimuli direct the effects of M1- and M2-like polarization. Here, we summarize key signaling mediators and receptors in common and distinct pathways. (A) IL-4-stimulated Stat6 activation is mediated by Jak1, which is required for M2-like polarization. (B) IL-6-treated macrophage polarization is induced by the activation of Jak2/Stat3 signaling pathway. (C) Jak2 and Tyk2 are required for Stat3 activation in IL-10-dependent M2-like polarization. (D) Jak2 controls Stat4 activation in IL-12-treated monocytes, which could induce M1-like polarization. (E) Jak2 and Tyk2 are the upstream regulators of Stat3 and Stat6 activation in IL-13-stimulated monocytes, which eventually induce the M2-like polarization.
OTHER CYTOKINES
Chemokines are a superfamily of small proteins with a crucial role in the polarized immune responses [60]. Particularly, chemokines and their receptors are able to trigger the differentiation and activation of mononuclear phagocytes by influencing the expression of functionally relevant and polarization-associated genes [61]. The crosstalk between chemokines and other cytokines has been reported to play critical roles in macrophage polarization. For example, CCL2 and IL-6 were found to contribute to the survival of CD11b+ myeloid monocytes recruited to the tumor microenvironment and skew the phenotype toward tumor-promoting CD14+/CD206+ M2-type macrophages [62]. CCL2-CCR2 axis was found to down-regulate the extent of pro-inflammatory M1 polarization by influencing the polarization-associated genes, including TNF-α, IL-6, granulocyte macrophage colony-stimulating factor (GM-CSF) and macrophage colony-stimulating factor (M-CSF) [63, 64]. GM-CSF and M-CSF are commonly recognized as critical factors controlling the M1 and M2 polarization, respectively [65]. GM-CSF favors the M1 phenotype through the activation of Jak2-STAT5 signalling [66], but M-CSF-dependent M2 phenotype is largely skewed by the activation of STAT3 [67]. Moreover, activin A preferentially released by M1 (GM-CSF) macrophages, and activin A-initiated Smad2 and prolyl hydroxylase PHD3 signaling both skew macrophage polarization toward the acquisition of a proinflammatory phenotype [68, 69]. Investigation of GM-CSF knockout alveolar macrophages (AMs) revealed intrinsic overexpression of IFN-γ, a potent inducer of the M1 phenotype, which as a causative factor for activin A, iNOS, CCL5, and IL-6 upregulation [70]. Examination of M2 markers in GM-CSF knockout mice are also simultaneously elevated, suggesting a unique mix of M1-M2 macrophage phenotypes in GM-CSF knockout mice. Besides, TGF-β is believed to control the M2-like polarization in part through the activation of Jak-STAT signaling [71]. Apparently, the activation of various cytokines signaling has been shown to act as the critical modulator of macrophage polarization based on the aforementioned researches. Thus, the molecules that could switch M1/M2 polarization derived from the cytokines signaling might provide a basis for macrophage-centered therapeutic strategies.
SOCS PROTEINS FAMILY AND MACROPHAGE POLARIZATION
SOCS1
SOCS1 is one of the best-studied SOCS proteins and is initially reported as a molecule induced by STATs [34, 72].The expression of SOCS1 has now been confirmed to be induced by diverse cytokines, including insulin and LPS [73]. It is a well-known negative regulator of JAKs-STATs by accessing the activation loop of JAKs with its KIR domain and these effects are associated with the attenuated signaling of LPS, IFN-γ and IL-4 in many diseases [74–76].
SOCS1 has been reported to exert a significant role in the regulation of macrophage polarization and function [77, 78]. Macrophage phenotypes alterations have been shown to be influenced by the epigenetic mechanism by which SOCS1 plays as a capacitor for M1/M2 polarization. Upregulation of DNA methyltransferases1 (DNMT1) appears to be linked with the SOCS1hypermethylation, which lead to the activation of JAK2/STAT3 pathway and the releasement of LPS-induced pro-inflammatory cytokines(TNF-α, IL-6) in macrophages [79, 80]. This result indicated that loss of SOCS1 expression might direct the pro-inflammatory M1 effect of macrophage by activating the JAK/STAT pathway. In addition, microRNAs (miRs) were also shown to play pivotal roles in the SOCS1 function. MicroRNA-155 (miR155) as a pro-inflammatory regulator could enhance macrophage inflammatory responses by targeting and degrading SOCS-1 [81, 82]. RNA virus infection induces miR-155 expression in macrophages via TLR/MyD88-independent and the inducible miR-155 feedback positively regulates host antiviral innate immune response by promoting type I IFN signaling via targeting SOCS1 [83, 84]. In human macrophages, miR-155 directly targets IL13Rα1 and reduces the levels of IL13Rα1 protein, leading to diminished activation of STAT6. MiR-155 affects the IL-13-dependent regulation of several genes (SOCS1, DC-SIGN, CCL18, CD23, and SERPINE) involved in the establishment of an M2/pro-Th2 phenotype in macrophages [85]. Further studies showed that miR-155 might promote the inflammation in atherosclerosis (AS) via the SOCS1-STAT3-PDCD4 axis, which increased the IL-6 and TNF-α expression in macrophages [86]. Thus we could find that miR-155-mediated SOCS1 downregulation is extremely associated with M1 polarization. As expected, Ma et al suggested that resveratrol exerts anti-inflammatory effects due to the upregulation of SOCS1 in macrophages, which is a potential target of miR-155 [82]. In contrast, inhibiting the STAT1-miR-155-SOCS1 signaling axis might enhance the development of tumor-promoting M2 macrophages in colon cancer [87]. These findings thus reveal a novel role of miR-155-SOCS1 pathway in the balance between pro-inflammatory M1 macrophages and anti-inflammatory M2 macrophages during different kinds of diseases [86]. Additionally, upregulation of miR-142-5p and downregulation of miR-130a-3p in macrophages play a pivotal role in the profibrogenic M2 effect of tissue fibrogenesis partially through targeting the SOCS1-STAT6 signaling [88]. SOCS1 is a key regulator of M1/M2 functions that coordinates with various cellular signaling pathways underlies the pathogenesis of many diseases, which could be developed as a useful therapy target (Figure 4).
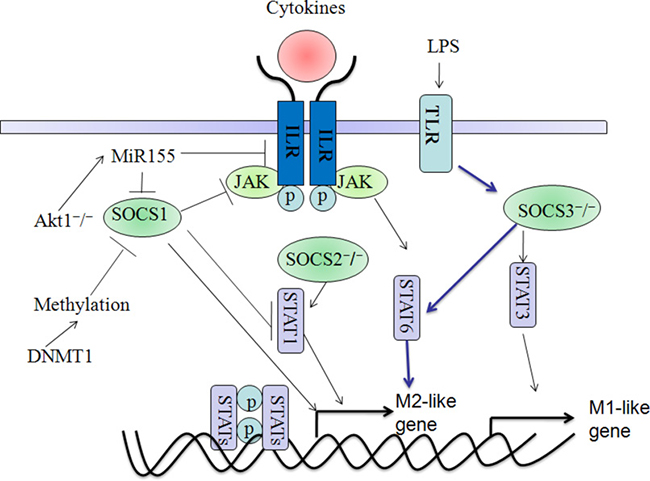
Figure 4: Functions and molecular mechanisms of SOCS1, SOCS2 and SOCS3 are implicated in directing macrophage polarization. Firstly, SOCS1 suppresses Stat1 and stimulates the M2-type polarization which could be influenced by different molecular mechanisms. Particularly, Akt1 ablation might influence MiR-155 expression and promote the M1-like polarization by targeting and inhibiting SOCS1. Moreover, DNMT1-mediated the methylation of SOCS1 could influence the M2-like macrophage. Secondly, SOCS2 knock out might stimulate M2-like polarization via the activation of Stat1. Thirdly, SOCS3 knock out could stimulate the M2- and M1-like polarization via the activation of Stat6 and Stat3, respectively. However, there seems to be still elusive about the SOCS proteins in the regulation of macrophage polarization and the real situation is much more complicated.
SOCS3
SOCS3 protein is commonly known to serve as a negative regulator of STAT3, which is the key physiological regulator in immune homeostasis and diseases pathogenesis [89–91]. SOCS3 has been shown to be induced by a wide variety of the cytokines and growth factors, such as IL-1β, 2, 4 and M-CSF [92, 93]. Unlike its closest homologue, SOCS-3 molecules differ greatly in their mechanism of function of SOCS-1. SOCS1 can inhibit activation of JAK by directly binding to JAK, whereas SOCS-3 sufficiently inhibits the action of JAK only in the presence of receptors, such as gp130 [94]. SOCS3 is a repressor of cytokine signaling which could inhibit the inflammatory genes expression in macrophages [95–97]. Recently, Sun et al investigated the anti-inflammatory effect of L. paracasei on the inhibition of TNF-α, IL-1β and IL-6 expressions by PBMC and THP-1 cell line owing to the expression of negative regulators of the NF-κB signaling pathway, including the SOCS1, SOCS3 [97]. Recombinant human IL-37 (rhIL-37) has both preventive and therapeutic effects in gouty arthritis by enhancing the activation of SOCS3 to trigger multiple intracellular switches to block inflammation [98]. Of note, several reports are available for the roles of SOCS3 in modulating macrophages M1/M2 polarization governed by the transcriptional and post-transcriptional mechanisms [99–101]. For example, SOCS3-deficient macrophages exhibit heightened STAT3 activation and are polarized toward the M1 phenotype [102]. Intriguingly, M2 macrophage (expressing upregulated Arg-1 and SOCS3) could be switched from M1 through the apolipoprotein E (APOE) signaling via very-low-density lipoprotein receptor (VLDL-R) or APOE receptor-2 (APOER2) [103]. These results collectively imply that SOCS3 is involved in repressing the M1 pro-inflammatory phenotype, thereby ameliorating inflammatory responses in macrophages [104]. SOCS3 is highly and preferentially expressed in hapten-induced contact hypersensitivity (CHS), which promotes the M2 polarization and participates the attenuation of CHS by suppressing MMP12 production [105]. Reprogramming macrophages to M2c subtype effectively suppressed the inflammation and fibroproliferarion in acute lung injury (ALI) partially mediated by activating the JAK1/STAT3/SOCS3 signaling pathway due to the production of IL-10 [106]. Collectively, we concluded that STAT3 and SOCS3 signaling are extremely required for the pro- and anti-inflammatory effects of macrophages, respectively.
There is also increasing evidence suggests that SOCS3 is involved in suppressing the M2 polarization and reciprocally promoting the M1 polarization. For example, macrophages lacking the SOCS3 gene or carrying a mutation of the SOCS3-binding site in gp130, the production of TNF and IL-12 is suppressed by the anti-inflammatory response originated from IL-6 signaling [107]. The carbohydrate kinase-like protein (CARKL) is rapidly downregulated in vitro and in vivo upon LPS stimulation in both mice and humans and the CARKL-dependent metabolic reprogramming is required for proper M2 polarization, which is associated with enhanced STAT3 phosphorylation and decreased SOCS3 expression, without influencing SOCS1 levels [108]. Rat bone marrow-derived macrophages (BMDM) in incubation with IFN-γ and LPS suppressed SOCS1 while uniquely polarizing macrophages to SOCS3 expressing macrophages, which proposing that SOCS3 is essential for development of M1 macrophages. In contrast, knockdown of SOCS3 by siRNA had enhanced STAT3 activity; induction of macrophage mannose receptor, Arg-1 and SOCS1 [109]. Myeloid-restricted SOCS3 deletion (Socs3Lyz2cre) resulted in resistance to LPS-induced endotoxic shock and observed striking bias toward M2 macrophages in Socs3Lyz2cre mice, enhanced regulatory T (Treg) cell recruitment by Socs3Lyz2cre cells coincided with enhanced interleukin-4 (IL-4) plus IL-13-induced STAT6 phosphorylation in Socs3Lyz2cre macrophages. [110] Taken together, these observations strongly suggest that phenotypic and functional heterogeneity of SOCS3-expressing macrophages within different diseases might be dependent on the specific local microenvironments. Finally, understanding how the M1/M2 polarization occurs in vivo, which is essential for designing pharmacologic and genetic approaches that employed to treat various diseases (Figure 4).
Other SOCS proteins
The function as well as the mechanism of SOCS-2 has been reported to be involved in the SHP2-binding site of activated growth hormone (GH) receptors, and it attenuates GH signaling by inhibiting the activation of JAK2 and STAT5b axis [111]. SOCS2 is considered as essential controller of macrophage activation and function. Genetic studies using transgenic approaches have shown that SOCS2 over-expressing transgenic (SOCS2Tg) mice showed functional improvement of anti-inflammatory response with the increased numbers of CD11b+CD206+ M2 macrophages than wildtype littermates following mild or moderately severe traumatic brain injury (TBI) [112]. As expected, M1 population was enriched in SOCS2(-/-) mice and the altered polarization coincided with enhanced IFN-γ-induced STAT1 activation in SOCS2(-/-) macrophages [110]. Therefore, SCOS2 is known as the M2 marker and the mRNA levels are downregulated with increasing disease severity in isolated lesions of atherosclerotic disease [113].
Previous studies have supported that miRNAs are functionally involved in macrophage polarization and function by the translational repression mechanism [114]. Up-regulation of let-7b is characteristic of prostatic TAMs (Tumor-associated macrophages), which targeting of the SOCS4 3’ untranslated region and inhibition of SOCS4 promoted phosphorylation of STAT3 and STAT6 [115]. Signal activation of Jak2/STAT3/STAT6 pathway could exert an impact on the M2 polarization [116]. Given the aforementioned observations, the detailed analysis of the mechanisms of SOCS4 underlying macrophages polarization merits consideration. In addition, SOCS5 was particularly suppressed in the M1-IFNγ status confirmed by the RNA-Seq Data with the genome-wide analysis of the expression of transcription factors (TFs) in SOCS families [117]. Of note, SOCS5 recently has been shown to contribute to M1 polarization by binding the IL-4Rα and blocking STAT6 phosphorylation [118, 119]. The role of SOCS6 and SOCS7 in M1/M2 polarization is still obscure. For example, miR-199a-3p accumulation was associated with the P53 induction in renal fibrosis and SOCS7 was identified as a target gene of miR-199a-3p. Silencing of SOCS7 promoted STAT3 activation and the infiltration of macrophage was suppressed in p53-KO mice [120]. These lesser-studied SOCS, such as SOCS6 and SOCS7 may play a yet undefined role in macrophage polarization and is needed to be further investigated [121].
CONCLUDING REMARKS
Altogether, M1/M2 subtypes might appear to be indispensable for fine-tuning the host responses to the various pathogens in different diseases. An understanding of how to program the macrophage polarization process is a keystone of deciphering homeostasis and disease pathogenesis. heThese observations that SOCS proteins participate in directing the dynamics of macrophage polarization have drawn attention to SOCS-dependent macrophage functions as a potential therapeutic target. Collectively, more in-depth analysis of how SOCS members operate the M1/M2 polarization is vital for developing novel strategies in the antiviral, antibacterial and antitumor responses.
Abbreviations
SOCS, suppressors of cytokine signaling; JAK, Janus kinases; ILs, interleukins; STAT, signal transducers and activators of transcription; IFNs, interferons; CIS, Src homology 2 (SH2)-containing protein; TAMs, tumor associated macrophages; RBX2, RING-box-2; KIR, kinase inhibitory region; TAMs, tumor associated macrophages; MAO-A, monoamine oxidase A; LPS, lipopolysaccharide; GM-CSF, granulocyte macrophage colony-stimulating factor; DNMT1, DNA methyltransferases1; TFs, transcription factors; M-CSF, macrophage colony-stimulating factor; PHD3, prolyl hydroxylase; AMs, alveolar macrophages; PDGF, platelet-derived growth factor; MOG, myelin oligodendrocyte glycoprotein; EAE, experimental autoimmune encephalomyelitis; APOE, apolipoprotein E; VLDL-R, very-low-density lipoprotein receptor; CARKL, carbohydrate kinase-like protein; GH, growth hormone; iNOS, inducible nitric oxide synthase; Arg-1, arginine-1.
ACKNOWLEDGMENTS
All the authors contributed to the concept, design, and writing of this paper. DXZ and JJL contributed to the concept and design of the figures and tables. All authors read, reviewed and approved the final manuscript. This work was supported by grants from Provincial Natural Science Foundation of Anhui (1608085MH178); Provincial Natural Science Research Project of Department of Education of Anhui (NO: KJ2014A262).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. McInnes IB, Buckley CD, Isaacs JD. Cytokines in rheumatoid arthritis-shaping the immunological landscape. Nat Rev Rheumatol. 2016; 12:63–68.
2. Elshaer D, Begun J. The role of barrier function, autophagy, and cytokines in maintaining intestinal homeostasis. Semin Cell Dev Biol. 2017; 61:51–59.
3. Villarino AV, Kanno Y, O’Shea JJ. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat Immunol. 2017; 18:374–384.
4. McLaughlin T, Ackerman SE, Shen L, Engleman E. Role of innate and adaptive immunity in obesity-associated metabolic disease. J Clin Invest. 2017; 127:5–13.
5. Abraham C, Dulai PS, Vermeire S, Sandborn WJ. Lessons Learned From Trials Targeting Cytokine Pathways in Patients With Inflammatory Bowel Diseases. Gastroenterology. 2017; 152:374–388.e4.
6. Barrett JP, Henry RJ, Villapol S, Stoica BA, Kumar A, Burns MP, Faden AI, Loane DJ. NOX2 deficiency alters macrophage phenotype through an IL-10/STAT3 dependent mechanism: implications for traumatic brain injury. J Neuroinflammation. 2017; 14:65.
7. Zhou D, Huang C, Lin Z, Zhan S, Kong L, Fang C, Li J. Macrophage polarization and function with emphasis on the evolving roles of coordinated regulation of cellular signaling pathways. Cell Signal. 2014; 26:192–197.
8. Shan B, Wang X, Wu Y, Xu C, Xia Z, Dai J, Shao M, Zhao F, He S, Yang L, Zhang M, Nan F, Li J, et al. The metabolic ER stress sensor IRE1alpha suppresses alternative activation of macrophages and impairs energy expenditure in obesity. Nature immunology. 2017; 18:519–529.
9. Schuch K, Wanko B, Ambroz K, Castelo-Rosa A, Moreno-Viedma V, Grun NG, Leitner L, Staffler G, Zeyda M, Stulnig TM. Osteopontin affects macrophage polarization promoting endocytic but not inflammatory properties. Obesity. 2016; 24:1489–1498.
10. Chen Y, Liu W, Wang Y, Zhang L, Wei J, Zhang X, He F, Zhang L. Casein Kinase 2 Interacting Protein-1 regulates M1 and M2 inflammatory macrophage polarization. Cell Signal. 2017; 33:107–121.
11. Yamane K, Leung KP. Rabbit M1 and M2 macrophages can be induced by human recombinant GM-CSF and M-CSF. FEBS open bio. 2016; 6:945–953.
12. Li M, Piao L, Chen CP, Wu X, Yeh CC, Masch R, Chang CC, Huang SJ. Modulation of Decidual Macrophage Polarization by Macrophage Colony-Stimulating Factor Derived from First-Trimester Decidual Cells: Implication in Preeclampsia. Am J Pathol. 2016; 186:1258–1266.
13. Petruzzi MN, Cherubini K, Salum FG, de Figueiredo MA. Role of tumour-associated macrophages in oral squamous cells carcinoma progression: an update on current knowledge. Diagn Pathol. 2017; 12:32.
14. Taniguchi K, Karin M. IL-6 and related cytokines as the critical lynchpins between inflammation and cancer. Semin Immunol. 2014; 26:54–74.
15. Hirahara K, Schwartz D, Gadina M, Kanno Y, O’Shea JJ. Targeting cytokine signaling in autoimmunity: back to the future and beyond. Curr Opin Immunol. 2016; 43:89–97.
16. McCormick SM, Gowda N, Fang JX, Heller NM. Suppressor of Cytokine Signaling (SOCS)1 Regulates Interleukin-4 (IL-4)-activated Insulin Receptor Substrate (IRS)-2 Tyrosine Phosphorylation in Monocytes and Macrophages via the Proteasome. J Biol Chem. 2016; 291:20574–20587.
17. Luckey MA, Kimura MY, Waickman AT, Feigenbaum L, Singer A, Park JH. The transcription factor ThPOK suppresses Runx3 and imposes CD4(+) lineage fate by inducing the SOCS suppressors of cytokine signaling. Nat Immunol. 2014; 15:638–645.
18. Arnold CE, Whyte CS, Gordon P, Barker RN, Rees AJ, Wilson HM. A critical role for suppressor of cytokine signalling 3 in promoting M1 macrophage activation and function in vitro and in vivo. Immunology. 2014; 141:96–110.
19. Iwahara N, Hisahara S, Kawamata J, Matsumura A, Yokokawa K, Saito T, Fujikura M, Manabe T, Suzuki H, Matsushita T, Suzuki S, Shimohama S. Role of Suppressor of Cytokine Signaling 3 (SOCS3) in Altering Activated Microglia Phenotype in APPswe/PS1dE9 Mice. J Alzheimers Dis. 2017; 55:1235–1247.
20. Mishra KK, Gupta S, Banerjee K. SOCS3 induces neurite differentiation and promotes neuronal cell survival. IUBMB life. 2016; 68:468–476.
21. Linossi EM, Nicholson SE. Kinase inhibition, competitive binding and proteasomal degradation: resolving the molecular function of the suppressor of cytokine signaling (SOCS) proteins. Immunol Rev. 2015; 266:123–133.
22. Wang S, Song X, Zhang Z, Li H, L K, Yin B, He J, Li C. Shrimp with knockdown of LvSOCS2, a negative feedback loop regulator of JAK/STAT pathway in Litopenaeus vannamei, exhibit enhanced resistance against WSSV. Dev Comp Immunol. 2016; 65:289–298.
23. Kim WS, Kim MJ, Kim DO, Byun JE, Huy H, Song HY, Park YJ, Kim TD, Yoon SR, Choi EJ, Jung H, Choi I. Suppressor of Cytokine Signaling 2 Negatively Regulates NK Cell Differentiation by Inhibiting JAK2 Activity. Sci Rep. 2017; 7:46153.
24. Naudin C, Chevalier C, Roche S. The role of small adaptor proteins in the control of oncogenic signalingr driven by tyrosine kinases in human cancer. Oncotarget. 2016; 7:11033–11055. doi: 10.18632/oncotarget.6929.
25. Beaurivage C, Champagne A, Tobelaim WS, Pomerleau V, Menendez A, Saucier C. SOCS1 in cancer: An oncogene and a tumor suppressor. Cytokine. 2016; 82:87–94.
26. Delconte RB, Kolesnik TB, Dagley LF, Rautela J, Shi W, Putz EM, Stannard K, Zhang JG, Teh C, Firth M, Ushiki T, Andoniou CE, Degli-Esposti MA, et al. CIS is a potent checkpoint in NK cell-mediated tumor immunity. Nat Immunol. 2016; 17:816–824.
27. Feng Y, Sanders AJ, Morgan LD, Harding KG, Jiang WG. Potential roles of suppressor of cytokine signaling in wound healing. Regen Med. 2016; 11:193–209.
28. Shin SH, Lee JY, Lee TH, Park SH, Yahng SA, Yoon JH, Lee SE, Cho BS, Lee DG, Kim YJ, Lee S, Min CK, Cho SG, et al. SOCS1 and SOCS3 are expressed in mononuclear cells in human cytomegalovirus viremia after allogeneic hematopoietic stem cell transplantation. Blood Res. 2015; 50:40–45.
29. Maehr T, Vecino JL, Wadsworth S, Wang T, Secombes CJ. Four CISH paralogues are present in rainbow trout Oncorhynchus mykiss: differential expression and modulation during immune responses and development. Mol Immunol. 2014; 62:186–198.
30. Croker BA, Kiu H, Nicholson SE. SOCS regulation of the JAK/STAT signalling pathway. Semin Cell Dev Biol. 2008; 19:414–422.
31. Peng HY, Cheng YC, Hsu YM, Wu GH, Kuo CC, Liou JP, Chang JY, Jin SL, Shiah SG. MPT0B098, a Microtubule Inhibitor, Suppresses JAK2/STAT3 Signaling Pathway through Modulation of SOCS3 Stability in Oral Squamous Cell Carcinoma. PloS One. 2016; 11:e0158440.
32. Alexander WS, Hilton DJ. The role of suppressors of cytokine signaling (SOCS) proteins in regulation of the immune response. Annu Rev Immunol. 2004; 22:503–529.
33. Linossi EM, Babon JJ, Hilton DJ, Nicholson SE. Suppression of cytokine signaling: the SOCS perspective. Cytokine Growth Factor Rev. 2013; 24:241–248.
34. Maruoka M, Kedashiro S, Ueda Y, Mizutani K, Takai Y. Nectin-4 Co-stimulates the Prolactin Receptor by Interacting with SOCS1 and Inhibiting Its Activity on the JAK2-STAT5a Signaling Pathway. J Biol Chem. 2017; 292:6895–6909.
35. Zhao HM, Xu R, Huang XY, Cheng SM, Huang MF, Yue HY, Wang X, Zou Y, Lu AP, Liu DY. Curcumin Suppressed Activation of Dendritic Cells via JAK/STAT/SOCS Signal in Mice with Experimental Colitis. Front Pharmacol. 2016; 7:455.
36. Letellier E, Haan S. SOCS2: physiological and pathological functions. Front Biosci. 2016; 8:189–204.
37. Lubberts E. The IL-23-IL-17 axis in inflammatory arthritis. Nat Rev Rheumatol. 2015; 11:562.
38. Geginat J, Larghi P, Paroni M, Nizzoli G, Penatti A, Pagani M, Gagliani N, Meroni P, Abrignani S, Flavell RA. The light and the dark sides of Interleukin-10 in immune-mediated diseases and cancer. Cytokine Growth Factor Rev. 2016; 30:87–93.
39. Cavalli G, Koenders M, Kalabokis V, Kim J, Tan AC, Garlanda C, Mantovani A, Dagna L, Joosten LA, Dinarello CA. Treating experimental arthritis with the innate immune inhibitor interleukin-37 reduces joint and systemic inflammation. Rheumatology. 2016; 55:2220–2229.
40. Awad F, Assrawi E, Jumeau C, Georgin-Lavialle S, Cobret L, Duquesnoy P, Piterboth W, Thomas L, Stankovic-Stojanovic K, Louvrier C, Giurgea I, Grateau G, Amselem S, et al. Impact of human monocyte and macrophage polarization on NLR expression and NLRP3 inflammasome activation. PloS One. 2017; 12:e0175336.
41. Saito Y, Azuma A, Matsuda K, Kamio K, Abe S, Gemma A. Pirfenidone exerts a suppressive effect on CCL18 expression in U937-derived macrophages partly by inhibiting STAT6 phosphorylation. Immunopharmacol Immunotoxicol. 2016; 1–8.
42. Lu L, McCurdy S, Huang S, Zhu X, Peplowska K, Tiirikainen M, Boisvert WA, Garmire LX. Time Series miRNA-mRNA integrated analysis reveals critical miRNAs and targets in macrophage polarization. Sci Rep. 2016; 6:37446.
43. Zhai T, Sun Y, Li H, Zhang J, Huo R, Li H, Shen B, Li N. Unique immunomodulatory effect of paeoniflorin on type I and II macrophages activities. J Pharmacol Sci. 2016; 130:143–150.
44. Mandal P, Pratt BT, Barnes M, McMullen MR, Nagy LE. Molecular mechanism for adiponectin-dependent M2 macrophage polarization: link between the metabolic and innate immune activity of full-length adiponectin. J Biol Chem. 2011; 286:13460–13469.
45. Kiguchi N, Sakaguchi H, Kadowaki Y, Saika F, Fukazawa Y, Matsuzaki S, Kishioka S. Peripheral administration of interleukin-13 reverses inflammatory macrophage and tactile allodynia in mice with partial sciatic nerve ligation. J Pharmacol Sci. 2017; 133:53–56.
46. Cao H, Zhang J, Liu H, Wan L, Zhang H, Huang Q, Xu E, Lai M. IL-13/STAT6 signaling plays a critical role in the epithelial-mesenchymal transition of colorectal cancer cells. Oncotarget. 2016; 7:61183–61198. doi: 10.18632/oncotarget.11282.
47. Reiman RM, Thompson RW, Feng CG, Hari D, Knight R, Cheever AW, Rosenberg HF, Wynn TA. Interleukin-5 (IL-5) augments the progression of liver fibrosis by regulating IL-13 activity. Infect Immun. 2006; 74:1471–1479.
48. Yan D, Wang HW, Bowman RL, Joyce JA. STAT3 and STAT6 Signaling Pathways Synergize to Promote Cathepsin Secretion from Macrophages via IRE1alpha Activation. Cell Rep. 2016; 16:2914–2927.
49. Bashir S, Sharma Y, Elahi A, Khan F. Macrophage polarization: the link between inflammation and related diseases. Inflamm Res. 2016; 65:1–11.
50. Bhattacharjee A, Shukla M, Yakubenko VP, Mulya A, Kundu S, Cathcart MK. IL-4 and IL-13 employ discrete signaling pathways for target gene expression in alternatively activated monocytes/macrophages. Free Radic Biol Med. 2013; 54:1–16.
51. Yakubenko VP, Hsi LC, Cathcart MK, Bhattacharjee A. From macrophage interleukin-13 receptor to foam cell formation: mechanisms for alphaMbeta2 integrin interference. J Biol Chem. 2013; 288:2778–2788.
52. Jung IH, Choi JH, Chung YY, Lim GL, Park YN, Park SW. Predominant Activation of JAK/STAT3 Pathway by Interleukin-6 Is Implicated in Hepatocarcinogenesis. Neoplasia. 2015; 17:586–597.
53. Hasegawa H, Mizoguchi I, Chiba Y, Ohashi M, Xu M, Yoshimoto T. Expanding Diversity in Molecular Structures and Functions of the IL-6/IL-12 Heterodimeric Cytokine Family. Front Immunol. 2016; 7:479.
54. Braune J, Weyer U, Hobusch C, Mauer J, Bruning JC, Bechmann I, Gericke M. IL-6 Regulates M2 Polarization and Local Proliferation of Adipose Tissue Macrophages in Obesity. J Immunol. 2017; 198:2927–2934.
55. Mauer J, Chaurasia B, Goldau J, Vogt MC, Ruud J, Nguyen KD, Theurich S, Hausen AC, Schmitz J, Bronneke HS, Estevez E, Allen TL, Mesaros A, et al. Signaling by IL-6 promotes alternative activation of macrophages to limit endotoxemia and obesity-associated resistance to insulin. Nat Immunol. 2014; 15:423–430.
56. Ambade A, Satishchandran A, Saha B, Gyongyosi B, Lowe P, Kodys K, Catalano D, Szabo G. Hepatocellular carcinoma is accelerated by NASH involving M2 macrophage polarization mediated by hif-1alphainduced IL-10. Oncoimmunology. 2016; 5:e1221557.
57. Wang Y, Sun L, Song Z, Wang D, Bao Y, Li Y. Maspin inhibits macrophage phagocytosis and enhances inflammatory cytokine production via activation of NF-kappaB signaling. Mol Immunol. 2017; 82:94–103.
58. Osada Y, Fujiyama T, Kamimura N, Kaji T, Nakae S, Sudo K, Ishiwata K, Kanazawa T. Dual genetic absence of STAT6 and IL-10 does not abrogate anti-hyperglycemic effects of Schistosoma mansoni in streptozotocin-treated diabetic mice. Exp Parasitol. 2017; 177:1–12.
59. Watford WT, Moriguchi M, Morinobu A, O’Shea JJ. The biology of IL-12: coordinating innate and adaptive immune responses. Cytokine Growth Factor Rev. 2003; 14:361–368.
60. Timoteo RP, da Silva MV, Miguel CB, Silva DA, Catarino JD, Rodrigues Junior V, Sales-Campos H, Freire Oliveira CJ. Th1/Th17-Related Cytokines and Chemokines and Their Implications in the Pathogenesis of Pemphigus Vulgaris. Mediators Inflamm. 2017; 2017:7151285.
61. Zhong TY, Arancibia S, Born R, Tampe R, Villar J, Del Campo M, Manubens A, Becker MI. Hemocyanins Stimulate Innate Immunity by Inducing Different Temporal Patterns of Proinflammatory Cytokine Expression in Macrophages. J Immunol. 2016; 196:4650–4662.
62. Roca H, Varsos ZS, Sud S, Craig MJ, Ying C, Pienta KJ. CCL2 and interleukin-6 promote survival of human CD11b+ peripheral blood mononuclear cells and induce M2-type macrophage polarization. The Journal of biological chemistry. 2009; 284:34342–34354.
63. Sierra-Filardi E, Nieto C, Dominguez-Soto A, Barroso R, Sanchez-Mateos P, Puig-Kroger A, Lopez-Bravo M, Joven J, Ardavin C, Rodriguez-Fernandez JL, Sanchez-Torres C, Mellado M, Corbi AL. CCL2 shapes macrophage polarization by GM-CSF and M-CSF: identification of CCL2/CCR2-dependent gene expression profile. J Immunol. 2014; 192:3858–3867.
64. Fritz JM, Tennis MA, Orlicky DJ, Lin H, Ju C, Redente EF, Choo KS, Staab TA, Bouchard RJ, Merrick DT, Malkinson AM, Dwyer-Nield LD. Depletion of tumor-associated macrophages slows the growth of chemically induced mouse lung adenocarcinomas. Front Immunol. 2014; 5:587.
65. Roussel M, Ferrell PB Jr, Greenplate AR, Lhomme F, Le Gallou S, Diggins KE, Johnson DB, Irish JM. Mass cytometry deep phenotyping of human mononuclear phagocytes and myeloid-derived suppressor cells from human blood and bone marrow. J Leukoc Biol. 2017.
66. Byrd D, Shepherd N, Lan J, Hu N, Amet T, Yang K, Desai M, Yu Q. Primary human macrophages serve as vehicles for vaccinia virus replication and dissemination. J Virol. 2014; 88:6819–31.
67. Domínguez-Soto A, Sierra-Filardi E, Puig-Kröger A, Pérez-Maceda B, Gómez-Aguado F, Corcuera MT, Sánchez-Mateos P, Corbí AL. Dendritic Cell-Specific ICAM-3–Grabbing Nonintegrin Expression on M2-Polarized and Tumor-Associated Macrophages Is Macrophage-CSF Dependent and Enhanced by Tumor-Derived IL-6 and IL-10. J Immunol. 2011; 186:2192–2200.
68. Sierra-Filardi E, Puig-Kroger A, Blanco FJ, Nieto C, Bragado R, Palomero MI, Bernabeu C, Vega MA, Corbi AL. Activin A skews macrophage polarization by promoting a proinflammatory phenotype and inhibiting the acquisition of anti-inflammatory macrophage markers. Blood. 2011; 117:5092–5101.
69. Escribese MM, Sierra-Filardi E, Nieto C, Samaniego R, Sanchez-Torres C, Matsuyama T, Calderon-Gomez E, Vega MA, Salas A, Sanchez-Mateos P, Corbi AL. The prolyl hydroxylase PHD3 identifies proinflammatory macrophages and its expression is regulated by activin A. J Immunol. 2012; 189:1946–1954.
70. Dalrymple H, Barna BP, Malur A, Malur AG, Kavuru MS, Thomassen MJ. Alveolar macrophages of GM-CSF knockout mice exhibit mixed M1 and M2 phenotypes. BMC Immunol. 2013; 14:41.
71. Liu Y, Holdbrooks AT, De Sarno P, Rowse AL, Yanagisawa LL, McFarland BC, Harrington LE, Raman C, Sabbaj S, Benveniste EN, Qin H. Therapeutic efficacy of suppressing the Jak/STAT pathway in multiple models of experimental autoimmune encephalomyelitis. J Immunol. 2014; 192:59–72.
72. Zahn M, Marienfeld R, Melzner I, Heinrich J, Renner B, Wegener S, Miessner A, Barth TF, Dorsch K, Bruderlein S, Moller P. A novel PTPN1 splice variant upregulates JAK/STAT activity in classical Hodgkin lymphoma cells. Blood. 2017; 129:1480–1490.
73. Christensen DP, Gysemans C, Lundh M, Dahllof MS, Noesgaard D, Schmidt SF, Mandrup S, Birkbak N, Workman CT, Piemonti L, Blaabjerg L, Monzani V, Fossati G, et al. Lysine deacetylase inhibition prevents diabetes by chromatin-independent immunoregulation and beta-cell protection. Proc Natl Acad Sci USA. 2014; 111:1055–1059.
74. Wu H, Ballantyne CM. Skeletal muscle inflammation and insulin resistance in obesity. The Journal of clinical investigation. 2017; 127:43–54.
75. Beldi-Ferchiou A, Skouri N, Ben Ali C, Safra I, Abdelkefi A, Ladeb S, Mrad K, Ben Othman T, Ben Ahmed M. Abnormal repression of SHP-1, SHP-2 and SOCS-1 transcription sustains the activation of the JAK/STAT3 pathway and the progression of the disease in multiple myeloma. PloS One. 2017; 12:e0174835.
76. Sachithanandan N, Graham KL, Galic S, Honeyman JE, Fynch SL, Hewitt KA, Steinberg GR, Kay TW. Macrophage deletion of SOCS1 increases sensitivity to LPS and palmitic acid and results in systemic inflammation and hepatic insulin resistance. Diabetes. 2011; 60:2023–2031.
77. Tang H, Liang YB, Chen ZB, Du LL, Zeng LJ, Wu JG, Yang W, Liang HP, Ma ZF. Soluble Egg Antigen Activates M2 Macrophages via the STAT6 and PI3K Pathways, and Schistosoma Japonicum Alternatively Activates Macrophage Polarization to Improve the Survival Rate of Septic Mice. J Cell Biochem. 2017. doi: 10.1002/jcb.26073.
78. Ma W, Wang Y, Lu S, Yan L, Hu F, Wang Z. Targeting androgen receptor with ASC-J9 attenuates cardiac injury and dysfunction in experimental autoimmune myocarditis by reducing M1-like macrophage. Biochem Biophys Res Commun. 2017; 485:746–752.
79. Cheng C, Huang C, Ma TT, Bian EB, He Y, Zhang L, Li J. SOCS1 hypermethylation mediated by DNMT1 is associated with lipopolysaccharide-induced inflammatory cytokines in macrophages. Toxicol Lett. 2014; 225:488–497.
80. Jiang M, Zhang WW, Liu P, Yu W, Liu T, Yu J. Dysregulation of SOCS-Mediated Negative Feedback of Cytokine Signaling in Carcinogenesis and Its Significance in Cancer Treatment. Front Immunol. 2017; 8:70.
81. Zhang Y, Mei H, Chang X, Chen F, Zhu Y, Han X. Adipocyte-derived microvesicles from obese mice induce M1 macrophage phenotype through secreted miR-155. J Mol Cell Biol. 2016; 8:505–517.
82. Ma C, Wang Y, Shen A, Cai W. Resveratrol upregulates SOCS1 production by lipopolysaccharide-stimulated RAW264.7 macrophages by inhibiting miR-155. Int J Mol Med. 2017; 39:231–237.
83. Wang P, Hou J, Lin L, Wang C, Liu X, Li D, Ma F, Wang Z, Cao X. Inducible microRNA-155 feedback promotes type I IFN signaling in antiviral innate immunity by targeting suppressor of cytokine signaling 1. J Immunol. 2010; 185:6226–6233.
84. Du F, Yu F, Wang Y, Hui Y, Carnevale K, Fu M, Lu H, Fan D. MicroRNA-155 deficiency results in decreased macrophage inflammation and attenuated atherogenesis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2014; 34:759–767.
85. Martinez-Nunez RT, Louafi F, Sanchez-Elsner T. The interleukin 13 (IL-13) pathway in human macrophages is modulated by microRNA-155 via direct targeting of interleukin 13 receptor alpha1 (IL13Ralpha1). J Biol Chem. 2011; 286:1786–1794.
86. Ye J, Guo R, Shi Y, Qi F, Guo C, Yang L. miR-155 Regulated Inflammation Response by the SOCS1-STAT3-PDCD4 Axis in Atherogenesis. Mediators of inflammation. 2016; 2016:8060182.
87. Jiang X, Zhou T, Xiao Y, Yu J, Dou S, Chen G, Wang R, Xiao H, Hou C, Wang W, Shi Q, Feng J, Ma Y, et al. Tim-3 promotes tumor-promoting M2 macrophage polarization by binding to STAT1 and suppressing the STAT1-miR-155 signaling axis. Oncoimmunology. 2016; 5:e1211219.
88. Su S, Zhao Q, He C, Huang D, Liu J, Chen F, Chen J, Liao JY, Cui X, Zeng Y, Yao H, Su F, Liu Q, et al. miR-142-5p and miR-130a-3p are regulated by IL-4 and IL-13 and control profibrogenic macrophage program. Nat Commun. 2015; 6:8523.
89. McFarland BC, Marks MP, Rowse AL, Fehling SC, Gerigk M, Qin H, Benveniste EN. Loss of SOCS3 in myeloid cells prolongs survival in a syngeneic model of glioma. Oncotarget. 2016; 7:20621–20635. doi: 10.18632/oncotarget.7992.
90. Caldow MK, Ham DJ, Chee A, Trieu J, Naim T, Stapleton DI, Swiderski K, Lynch GS, Koopman R. Muscle-specific deletion of SOCS3 does not reduce the anabolic response to leucine in a mouse model of acute inflammation. Cytokine. 2017; 96:274–278.
91. Zhao J, Yu H, Liu Y, Gibson SA, Yan Z, Xu X, Gaggar A, Li PK, Li C, Wei S, Benveniste EN, Qin H. Protective effect of suppressing STAT3 activity in LPS-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2016; 311:L868-L880.
92. Gui T, He BS, Gan Q, Yang C. Enhanced SOCS3 in osteoarthiritis may limit both proliferation and inflammation. Biotech Histochem. 2017; 92:107–114.
93. Ushiki T, Huntington ND, Glaser SP, Kiu H, Georgiou A, Zhang JG, Metcalf D, Nicola NA, Roberts AW, Alexander WS. Rapid Inflammation in Mice Lacking Both SOCS1 and SOCS3 in Hematopoietic Cells. PloS One. 2016; 11:e0162111.
94. Huang G, Yuan M, Zhang J, Li J, Gong D, Li Y, Zhang J, Lin P, Huang L. IL-6 mediates differentiation disorder during spermatogenesis in obesity-associated inflammation by affecting the expression of Zfp637 through the SOCS3/STAT3 pathway. Sci Rep. 2016; 6:28012.
95. Xue F, Nie X, Shi J, Liu Q, Wang Z, Li X, Zhou J, Su J, Xue M, Chen WD, Wang YD. Quercetin Inhibits LPS-Induced Inflammation and ox-LDL-Induced Lipid Deposition. Front Pharmacol. 2017; 8:40.
96. Alston CI, Dix RD. Murine cytomegalovirus infection of mouse macrophages stimulates early expression of suppressor of cytokine signaling (SOCS)1 and SOCS3. PloS One. 2017; 12:e0171812.
97. Sun KY, Xu DH, Xie C, Plummer S, Tang J, Yang XF, Ji XH. Lactobacillus paracasei modulates LPS-induced inflammatory cytokine release by monocyte-macrophages via the up-regulation of negative regulators of NF-kappaB signaling in a TLR2-dependent manner. Cytokine. 2017; 92:1–11.
98. Liu L, Xue Y, Zhu Y, Xuan D, Yang X, Liang M, Wang J, Zhu X, Zhang J, Zou H. Interleukin 37 limits monosodium urate crystal-induced innate immune responses in human and murine models of gout. Arthritis Res Ther. 2016; 18:268.
99. Goodman WA, Omenetti S, Date D, Di Martino L, De Salvo C, Kim GD, Chowdhry S, Bamias G, Cominelli F, Pizarro TT, Mahabeleshwar GH. KLF6 contributes to myeloid cell plasticity in the pathogenesis of intestinal inflammation. Mucosal Immunol. 2016; 9:1250–1262.
100. Yildirim-Buharalioglu G, Bond M, Sala-Newby GB, Hindmarch CC, Newby AC. Regulation of Epigenetic Modifiers, Including KDM6B, by Interferon-gamma and Interleukin-4 in Human Macrophages. Front Immunol. 2017; 8:92.
101. Yu T, Zuo Y, Cai R, Huang X, Wu S, Zhang C, Chin YE, Li D, Zhang Z, Xia N, Wang Q, Shen H, Yao X, et al. SENP1 regulates IFN-gamma-STAT1 signaling through STAT3-SOCS3 negative feedback loop. J Mol Cell Biol. 2017; 9:144–153.
102. Qin H, Yeh WI, De Sarno P, Holdbrooks AT, Liu Y, Muldowney MT, Reynolds SL, Yanagisawa LL, Fox TH, Park K. Signal transducer and activator of transcription-3/suppressor of cytokine signaling-3 (STAT3/SOCS3) axis in myeloid cells regulates neuroinflammation. Proc Natl Acad Sci U S A. 2012; 109:5004–5009.
103. Baitsch D, Bock HH, Engel T, Telgmann R, Muller-Tidow C, Varga G, Bot M, Herz J, Robenek H, von Eckardstein A, Nofer JR. Apolipoprotein E induces antiinflammatory phenotype in macrophages. Arterioscler Thromb Vasc Biol. 2011; 31:1160–1168.
104. Qin H, Holdbrooks AT, Liu Y, Reynolds SL, Yanagisawa LL, Benveniste EN. SOCS3 deficiency promotes M1 macrophage polarization and inflammation. J Immunol. 2012; 189:3439–3448.
105. Suzuki K, Meguro K, Nakagomi D, Nakajima H. Roles of alternatively activated M2 macrophages in allergic contact dermatitis. Allergol Int. 2017.
106. Tang L, Zhang H, Wang C, Li H, Zhang Q, Bai J. M2A and M2C Macrophage Subsets Ameliorate Inflammation and Fibroproliferation in Acute Lung Injury Through Interleukin 10 Pathway. Shock. 2016.
107. Yasukawa H, Ohishi M, Mori H, Murakami M, Chinen T, Aki D, Hanada T, Takeda K, Akira S, Hoshijima M, Hirano T, Chien KR, Yoshimura A. IL-6 induces an anti-inflammatory response in the absence of SOCS3 in macrophages. Nat Immunol. 2003; 4:551–556.
108. Haschemi A, Kosma P, Gille L, Evans CR, Burant CF, Starkl P, Knapp B, Haas R, Schmid JA, Jandl C, Amir S, Lubec G, Park J, et al. The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab. 2012; 15:813–826.
109. Liu Y, Stewart KN, Bishop E, Marek CJ, Kluth DC, Rees AJ, Wilson HM. Unique expression of suppressor of cytokine signaling 3 is essential for classical macrophage activation in rodents in vitro and in vivo. J Immunol. 2008; 180:6270–6278.
110. Spence S, Fitzsimons A, Boyd CR, Kessler J, Fitzgerald D, Elliott J, Gabhann JN, Smith S, Sica A, Hams E, Saunders SP, Jefferies CA, Fallon PG, et al. Suppressors of cytokine signaling 2 and 3 diametrically control macrophage polarization. Immunity. 2013; 38:66–78.
111. Mense K, Meyerholz M, Gil Araujo M, Lietzau M, Knaack H, Wrenzycki C, Hoedemaker M, Piechotta M. The somatotropic axis during the physiological estrus cycle in dairy heifers--Effect on hepatic expression of GHR and SOCS2. J Dairy Sci. 2015; 98:2409–2418.
112. Basrai HS, Christie KJ, Turbic A, Bye N, Turnley AM. Suppressor of Cytokine Signaling-2 (SOCS2) Regulates the Microglial Response and Improves Functional Outcome after Traumatic Brain Injury in Mice. PloS One. 2016; 11:e0153418.
113. Bisgaard LS, Mogensen CK, Rosendahl A, Cucak H, Nielsen LB, Rasmussen SE, Pedersen TX. Bone marrow-derived and peritoneal macrophages have different inflammatory response to oxLDL and M1/M2 marker expression-implications for atherosclerosis research. Sci Rep. 2016; 6:35234.
114. Essandoh K, Li Y, Huo J, Fan GC. MiRNA-Mediated Macrophage Polarization and its Potential Role in the Regulation of Inflammatory Response. Shock. 2016; 46:122–131.
115. Wang Z, Xu L, Hu Y, Huang Y, Zhang Y, Zheng X, Wang S, Wang Y, Yu Y, Zhang M, Yuan K, Min W. miRNA let-7b modulates macrophage polarization and enhances tumor-associated macrophages to promote angiogenesis and mobility in prostate cancer. Sci Rep. 2016; 6:25602.
116. Wang S, Zhang C, Li J, Niyazi S, Zheng L, Xu M, Rong R, Yang C, Zhu T. Erythropoietin protects against rhabdomyolysis-induced acute kidney injury by modulating macrophage polarization. Cell Death Dis. 2017; 8:e2725.
117. Sang Y, Brichalli W, Rowland RR, Blecha F. Genome-wide analysis of antiviral signature genes in porcine macrophages at different activation statuses. PloS One. 2014; 9:e87613.
118. Hallowell RW, Collins SL, Craig JM, Zhang Y, Oh M, Illei PB, Chan-Li Y, Vigeland CL, Mitzner W, Scott AL, Powell JD, Horton MR. mTORC2 signalling regulates M2 macrophage differentiation in response to helminth infection and adaptive thermogenesis. Nat Commun. 2017; 8:14208.
119. McCormick SM, Heller NM. Regulation of Macrophage, Dendritic Cell, and Microglial Phenotype and Function by the SOCS Proteins. Front Immunol. 2015; 6:549.
120. Yang R, Xu X, Li H, Chen J, Xiang X, Dong Z, Zhang D. p53 induces miR199a-3p to suppress SOCS7 for STAT3 activation and renal fibrosis in UUO. Sci Rep. 2017; 7:43409.
121. Chikuma S, Kanamori M, Mise-Omata S, Yoshimura A. Suppressors of cytokine signaling: Potential immune checkpoint molecules for cancer immunotherapy. Cancer Sci. 2017; 108:574–580.