
INTRODUCTION
Pseudogenes, a sub-class of long non-coding RNAs (lncRNAs) that developed from protein-coding genes (PCGs) but have lost the ability to produce proteins, have long been described as non-functional genomic relicts of evolution [1]. However, it is becoming clear that some of pseudogenes have important regulatory roles in cells. Far from being silent, pseudogenes participate in various biological activities, including being a part in the transcription process [2], or participating in the formation of small interfering RNA (siRNA) which regulated gene expression through RNA-interference pathway [3, 4]. Several studies also implicate dysregulation of pseudogenes as contributing factor in human cancer, with early example such as KRASIP [5].
Notably, an increasing number of studies describe pseudogenes that act as critical effectors in cancer progression [6]. For example, NANOG and OCT4 are essential transcription factors for the maintenance of pluripotency in embryonic stem cells [7, 8], while their pseudogenes, NANOGP1 and POU5F1P1, are aberrantly expressed in human cancers [9]. Poliseno et al. had shown that the pseudogene PTENP1 regulated the expression of tumor suppressor PTEN through binding microRNA and took part in tumor biological processes [10]. More recently, Florian et al. had provided an evidence that the BRAF pseudogene acted as a competitive endogenous RNA (ceRNA) and induced lymphoma in vivo [11]. These studies provide key insights into the potential role of pseudogenes in tumor biology. While intriguing, all of them are still limited in individual pseudogenes, and it is likely that more pseudogenes have roles in oncogenic programs. Therefore, it is essential to perform a systematic analysis across large patient sample cohorts to identify cancer-related pseudogenes. The idea was first explored in 13 cancer using RNA-Seq resource of 293 samples, revealing associations between pseudogene expression and cancer progression [12]. However, though pseudogenes has been reported to act as microRNA sponges that compete with mRNAs to attract microRNAs for interactions and influence the expression of mRNAs [13], the biological characteristic and clinical relevance of pseudogenes that function as ceRNAs remain unclear.
In order to systematically describe cancer-related pseudogenes that act as ceRNAs, here, compared with previous studies that identified ceRNA pairs [14–16], we developed a computational framework and gradually identified LUAD-related pseudogenes. We first obtained the RNA-seq transcript data of LUAD that made available from TCGA and selected positive pseudogene-mRNA interactions based on ceRNA hypothesis. These candidate pseudogenes were characterized in several ways, including transcript length, exon numbers, evolutionary conservation, neighboring gene analysis and co-methylation analysis. Then we inferred potential prognostic biomarker and small molecule drugs for LUAD treatment. Taken together, our study systematically characterized pseudogenes, provided a foundation for deeper understanding the role of LUAD-related pseudogenes and improved knowledge of tumor biology.
RESULTS
Identification of LUAD-related candidate pseudogenes that function as ceRNA
We constructed a framework to identify and analysis disease pseudogenes (Figure 1). First, we proposed a pipeline to gradually identify significant pseudogene-microRNA-mRNA triples. After processing the RNASeqV2 data of 576 LUAD samples, we obtained 729 pseudogenes and 16,610 mRNAs profiles respectively. Furthermore, based on the target information, we obtained 434,691 pseudogene-microRNA-mRNA triples. The mRNAs and microRNAs in those triples were LUAD-related which were selected from cancer databases. Recent studies had shown that the two microRNA sponges were more correlative with each other if they shared more microRNAs [14]. In order to identify candidate pseudogene-mRNA competing pairs, a hypergeometric test was used to compute the significance of shared microRNAs for each possible gene pair. All p-values were subject to FDR correction and 750 pseudogene-mRNA pairs with FDR < 0.05 were remained for further analysis. Moreover, in order to reduce the false positive rate of result, all the candidate pseudogene-mRNA pairs with Pearson Correlation Coefficient (PCC) ≥ 0.259 and p-adjusted < 0.05 were identified as ceRNA-ceRNA interactions. In total, 33 pseudogenes were identified as candidate LUAD-related genes (Supplementary Data 1). In addition, we found the mRNAs in candidate ceRNA pairs were enriched in several critical pathways, such as Jak-STAT signaling pathway, Adipocytokine signaling pathway, MicroRNA in cancer and One carbon pool by folate. These observations suggested that some of the candidate pseudogenes may be members of those signaling pathways and prompt cancer development.
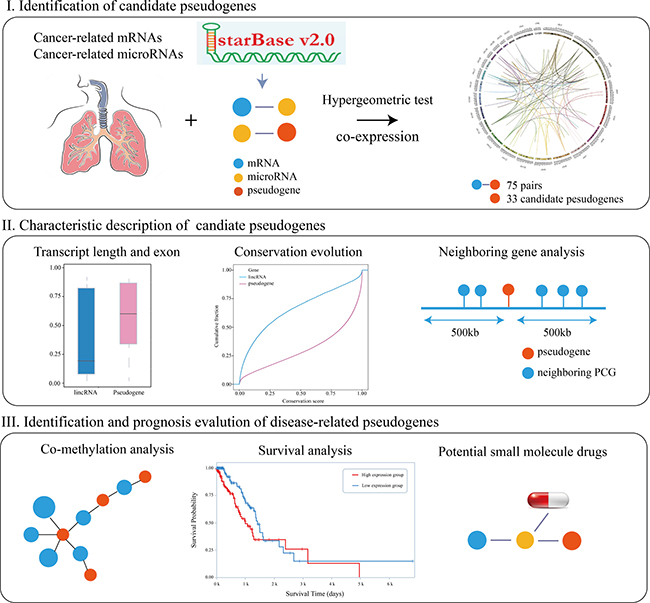
Figure 1: Work flow.
Properties of candidate pseudogenes
We explored the transcript length and exon number of candidate pseudogenes, and compared these properties with all pseudogenes and lincRNAs. Transcripts for candidate pseudogenes were longer than those for all pseudogenes and lincRNAs were (Figure 2A, candidate pseudogenes versus all pseudogenes, p < 2.2e-16; candidate pseudogenes versus lincRNAs, p = 6.09e-06). Moreover, candidate pseudogenes had more exons per transcript than all pseudogenes and lincRNAs did (Figure 2B, candidate pseudogenes versus all pseudogenes, p < 2.2e-16; candidate pseudogenes versus lincRNAs, p = 4.505e-12). Previous study pointed out that the lncRNAs with long transcripts and a large number of exons may be involved in the function as microRNA sponges in biological processes [17, 18]. These results suggested that the candidate pseudogenes may also function as ceRNA and have key functions in cancer.
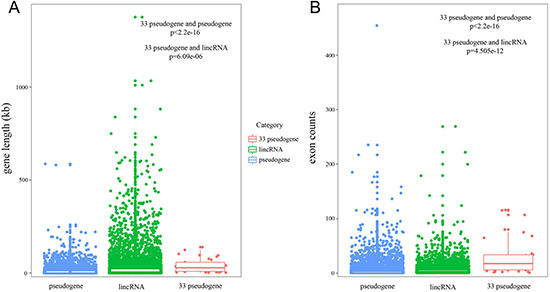
Figure 2: The properties of candidate pseudogenes. (A and B) The boxplot depicted the transcript length and exon number of genes respectively.
Candidate pseudogenes had high evolutionary conservation
Next, we evaluated the evolutionary conservation of candidate pseudogenes, all pseudogenes, lincRNAs, PCGs and neighboring PCGs respectively by using phastCons scores. The flanking PCGs within 500kb of pseudogenes were considered as neighboring PCGs. Our results showed that pseudogenes and lincRNAs exhibited poor evolutionary conservation relative to PCGs (Figure 3). We also found that all pseudogenes exhibited relatively higher evolutionary conservation than lincRNAs did (p < 2.2e-16). It can be explained by the fact that most intergenic transcripts show little or no evolutionary conservation [19]. Especially, candidate pseudogenes had a remarkably higher conservation than all pseudogenes did (p < 2.2e-16), perhaps because of its relatively high percentage of exons. The results indicated that the ceRNA pseudogenes may have important functions in biological process. Therefore, though losing the ability to produce proteins, it was still valuable to analyze the ceRNA pseudogenes.
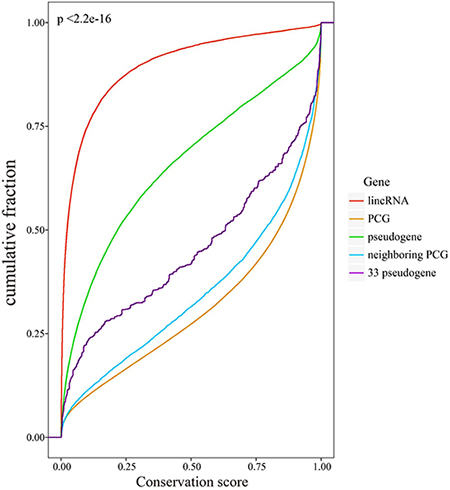
Figure 3: Evolutionary conservation analysis. Cumulative distribution of conservation scores of lincRNAs, pseudogenes, PCGs, candidate pseudogenes and neighboring PCGs.
Neighboring PCGs analysis
We next evaluated the potential roles of candidate pseudogenes in cancer. We calculated the PCC value between the candidate pseudogenes and their neighboring PCGs (Figure 4A and 4B). In network, the candidate pseudogenes were surrounded by disease-related genes. For example, MSTO2P was surrounded by 12 genes. LAMTOR2 complex regulated focal adhesion dynamics during cell migration [20] and CLK2 was proved to change in Alzheimer’s disease [21]. Furthermore, DAVID v6.8 was used to perform gene ontology (GO) terms and pathway enrichment for neighboring PCGs. The GO semantic annotation showed that neighboring PCGs were enriched in some functions that was related with development and progression of tumors such as autophagy and antigen processing (Figure 4C). The pathway enrichment of neighboring PCGs also revealed that they participated in cancer-related biology pathway such as viral carcinogenesis. The viral carcinogenesis pathway revealed the molecular mechanisms and the etiology of human disease [22], which may suggest that candidate pseudogenes were members of these pathway and affected the development and progression of cancer.
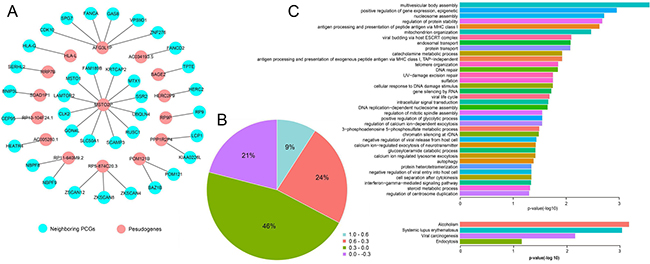
Figure 4: Analysis of neighboring PCGs. (A) Cis-acting network (PCC ≥ 0.6). (B) Pie chart. The proportion of PCC value between pseudogenes and their neighboring genes. (C) GO enrichment and KEGG pathways (p < 0.05, FDR < 0.05).
Construction of co-methylation network
Previous studies had demonstrated that DNA co-methylation suggested functional associations between gene pairs in cancers [23]. To further explore the relationship between candidate pseudogenes and LUAD-related mRNAs, we performed co-methylation analysis (Figure 5A). The co-methylated network contained 21 pseudogenes and 22 mRNAs (p < 0.05, PCC ≥ 0.2), and it may reveal the functional association between pseudogenes and their target mRNAs. For example, altered expression of EZH2 was proved to be associated with the risk of lung cancer [24]. In network, 4 pseudogenes (RRP7B, RPLP0P2, MSTO2P and AFG3L1P) co-methylated with EZH2, suggesting an involvement in the progression of lung cancer. Pak6 protein kinase is a novel effector of an atypical Rho family GTPase Chp/RhoV [25]and it co-methylated with pseudogene PLEKHM1P and RP11-480I12.5, indicating that the two pseudogenes may take part in cancer biological processes. In addition, 6 of the 21 pseudogenes were differentially-expressed (DE) genes (Supplementary Data 2). We further analyzed DNA methylation pattern of the 6 DE pseudogenes. The result showed that the methylation level of DE pseudogenes was lower than that of non-differentially expressed (NDE) pseudogenes (Figure 5B). Accumulating evidences indicated that hypomethylation was an important phenomenal characteristic. For example, a study proved that body-hypomethylated human genes were prone to cancer-associated dysregulation [26]. Therefore, it can be inferred that the DE pseudogenes played key roles in cancer processes and we selected them for further analysis. We also explored the methylation pattern of DE pseudogenes in tumor samples and normal samples, and the methylation level in normal samples was higher than that of tumor samples (Figure 5C).
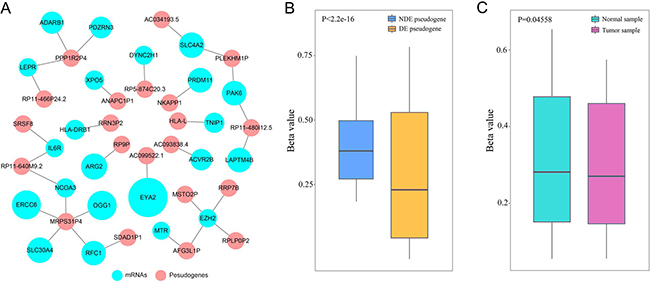
Figure 5: Co-methylation analysis. (A) Co-methylated network. (B) The boxplot depicted the methylation level between DE and NDE pseudogenes. (C) The boxplot depicted the methylation level between normal samples and tumor samples (7 differentially expressed pseudogenes).
Prognostic ceRNAs in LUAD
To explore the influence of the ceRNA triples on patient survival, we calculated the risk score for each pseudogene-microRNA-mRNA triple based on risk score of single factor regression analysis of each node, and divided patient samples into high-risk and low-risk groups by median, then drew Kaplan-Meier curves with R software. Cox p-value was used to evaluate the significance between expression of triples and overall survival. Log-Rank test p-value was used to test the significance between the two groups of patient samples. The result exhibited that the 3 triples (miR-21-5p-NKAPP1-PRDM11, miR-29c-3p-MSTO2P-EZH2 and miR-29c-3p-RPLP0P2-EZH2) were significantly associated with prognosis (p < 0.05, Figure 6). The high-risk group consisting of patients with high risk scores had lower survival time, which revealed that the high risk group was associated with the poor prognosis of LUAD. In addition, RPLP0P2 was proved to be associated with cell proliferation and adhesion in LUAD tumor cells [27]. It was suggested that the patient survival could be affected by the ceRNA pairs. These results indicated that the 3 ceRNA triples may serve as potential biomarkers of LUAD and contribute to the following treatment.
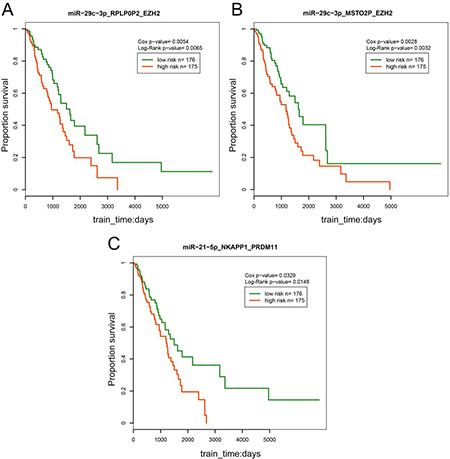
Figure 6: Survival analysis of the ceRNA triples.
Potential small molecule drugs for LUAD treatment
In the pseudogene-microRNA-mRNA triples, the perturbation of microRNA expression can influence the expression level of pseudogenes and mRNAs. Moreover, several studies had verified that bioactive small molecules could regulated microRNA expression [28, 29]. Here, referring to a previous work [30], we combined the information that provided by SM2miR [31] with the module to infer potential small molecule drugs for LUAD treatment. In the triples, some of the potential drugs could up/down-regulate the microRNA expression and further down/up-regulate the expression of pseudogenes and contribute to the treatment of LUAD (The risk coefficient of miR-21was 0.382, so that we needed to down-regulate it; the risk coefficients of miR-29 were -0.303 and -0.381 respectively, thus we needed to up-regulate them). In miR-21-5p-NKAPP1-PRDM11, 5-aza-2’-deoxycytidine (5-Aza-CdR) could down-regulate the miR-21, and up-regulate the expression of corresponding pseudogene/mRNA. It was proved to be a potent inhibitor of DNA methylation for therapy of advanced non-small cell lung cancer [32]. Triptolide, a natural diterpenoid compound, was proved to be an inhibitor of lung inflammation. In addition, for miR-29c-3p-RPLP0P2-EZH2/miR-29c-3p-MSTO2P-EZH2 triples, Enoxacin can inhibit RNA helicase DHX9 in lung cancer and was an effective agent for lung cancer prevention and treatment [33]. Glucocorticoid could also up-regulate the expression of miR-29c and involve in important biological pathway [34] while it was not mentioned in LUAD. Therefore, we inferred that those small molecule drugs which regulated the expression of microRNAs in ceRNA triples, may serve as potential drugs for LUAD treatment (Figure 7 and Supplementary Data 3).
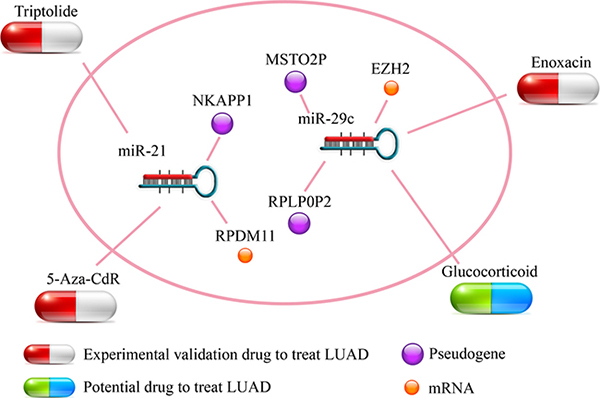
Figure 7: Potential small molecule drugs for LUAD treatment. The capsules represented experimental validation drugs and potential drug respectively. The nodes represented pseudogenes and LUAD-related mRNAs respectively.
DISCUSSION
Recently, pseudogenes have emerged as new players in tumor biology. However, the way of effectively identifying cancer-related pseudogenes that functions as ceRNA still remains unclear. Here, by establishing a novel computational framework we systematically explored the potential role of pseudogenes as microRNA sponges for LUAD. Notably, to ensure the accuracy of prediction power, positive pseudogene-mRNA interactions was identified by strict criteria: (i) both of the mRNA and microRNA in each triple must be verified by disease databases; (ii) each possible mRNA-pseudogene pair must significantly share common microRNAs which can interact with both of them (hypergeometric test, FDR < 0.05); (iii) only top correlated mRNA-pseudogene pairs, whose correlation coefficients were higher than the threshold of the 90th percentile of the corresponding overall correlation distribution (PCC ≥ 0.259), were regarded as candidate pseudogene-mRNA interactions. Moreover, DE analysis that has been used frequently in mRNA and microRNA research may not be suitable for identifying microRNA sponge interactions [35]. The reason is that the pseudogenes that are not DE may affect the amount of the microRNAs available for bindings [36]. Therefore, the DE analysis between normal samples and tumor samples of pseudogenes was performed after getting candidate pseudogenes. The candidate pseudogenes had relatively long transcripts and plenty of exons, suggesting they may have important functions. In addition, we observed that candidate pseudogenes had high evolutionary conservation, while previous studies had revealed that the evolutionary conservation of lncRNAs was poor [37]. It may suggest that ceRNA pseudogenes had highly evolutionary conservation and played important roles in biological progress. Although the existing classification of lncRNAs still faced with challenge [38], the above analysis suggested that pseudogenes may serve as an independent category because of the unique characteristic. Through neighboring PCGs analysis, we found candidate genes were surrounded by oncogenes, and they may be members of cancer pathways. Importantly, utilizing 450k DNA methylation data from TCGA, we found 21 pseudogenes co-methylated with mRNAs and 6 of them were differentially-expressed. We showed different methylation levels between DE and NDE pseudogenes in gene-body regions, which suggested methylation level may affect the expression of DE pseudogenes [39]. The 6 differentially-expressed pseudogenes co-expressed and co-methylated with experiment-validated LUAD-related mRNAs. Hence, we inferred they were potential LUAD-related pseudogenes. To infer clinical relevance of the LUAD-related pseudogenes, survival analysis was also taken into consideration. We found that the pseudogene RPLP0P2, one of the 6 potential LUAD-related genes, was reported to be associated with decreased cell proliferation and adhesion ability in LUAD [27, 40]. These analyses revealed that the 6 pseudogenes may be functional involved in tumorigenesis and implied a novel prognostic strategy for cancer treatment. At last, we provided a new perspective for the discovery of potential small molecule drugs and expected to find effective drugs for cancer treatment in the future.
In recent years, a growing body of studies has focused on functional pseudogenes that played critical roles in human diseases. For example, Zheng et al. analyzed the pseudogene CYP4Z2P based on ceRNA hypothesis in breast cancer [41]. However, cancer-related pseudogenes may affect many genes in ceRNA network while they solely focus on single pseudogene or gene. Although the methods were all based on ceRNA hypothesis, our study had two advantages over them: (i) a strict computational pipeline was applied to identify candidate ceRNA pairs; (ii) the identified pseudogenes were differentially-expressed and co-expressed/methylated with experiment-verified LUAD-related gene. Certainly, as with any computational approach, our framework was limited by the quality and quantity of the input data. Further power is anticipated by including additional samples, complement of methylation probes, and better interaction networks. For example, since strict criteria were used in our study, few datasets was suitable for identifying cancer-related pseudogenes, which implies that our framework could be more accurate by the complement of data in the future.
In summary, we provided a framework to identify cancer-related pseudogenes and integrate them with genomic analysis. These candidate cancer-related pseudogenes could be further evaluated as potential therapeutic targets.
MATERIALS AND METHODS
Expression profiles of pseudogene and mRNA in LUAD
The RNA-seq V2 data of LUAD patient samples was obtained from TCGA project (http://cancergenome.nih.gov/) [42], including 517 tumor samples and 59 adjacent normal samples (Supplementary Table 1). GENCODE hg19 genome was used as a reference. The reads were mapped to the exons of mRNAs and pseudogenes. The pseudogenes/mRNAs that overlapped with mRNAs/pseudogenes were excluded. RPKM value was calculated to evaluate the expression levels of pseudogene and mRNA:
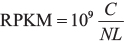
Where C is the number of mapped reads for pseudogene or mRNA, N is the number of total mapped reads, L is the length of the pseudogene or mRNA. To reduce false positive rate, pseudogenes or mRNAs with missing values in > 50% of the sample were removed [14]. Next, we added 0.00001 to the expression value of each gene and performed log2-transformed. In total, we obtained 729 pseudogenes and 16,610 mRNAs for further analysis.
Argonaute CLIP-supported microRNA-target interactions
Recently, several studies have reported that the use of cross-linking and Argonaute (Ago) immunoprecipitation coupled with high-throughput sequencing (CLIP-Seq) could identify endogenous genome-wide interaction maps for microRNAs [43, 44]. To investigate human microRNA-target regulatory relationships, the Human microRNA-mRNA interactions were collected from five prediction programs including TargetScan [45], PicTar [46], PITA [47], miRanda [48] and RNA22 [49] in starBase v2.0 [50]. By integrating the above databases, a total of 423,405 non-redundant microRNA-mRNA interactions were used in our study. The microRNA-pseudogene interactions were also collected from starBase v2.0, including 16,126 interactions pairs.
Collection of LUAD-associated mRNAs and microRNAs
Several database systems have proposed to provide a comprehensive resource of mRNAs and microRNAs dysregulation in various human diseases. LUAD-related mRNAs were collected from four databases, including COSMIC [51], OMIM [52], GAD [53] and Phenopedia-Genopedia database [54]. In addition, experimentally verified LUAD-related microRNAs were obtained from HMDD [55], miR2Diease [56], miREnvironment [57] and OncomiRDB [58].
Identification of potential LUAD-related pseudogenes
Having got the Ago CLIP-supported mRNA-microRNA and pseudogene-microRNA regulatory data, we performed a three-step pipeline to gradually identify LUAD-related pseudogenes that acted as microRNA sponges based on the ceRNA hypothesis. First, the pseudogene-microRNA-mRNA triples were obtained using predicted microRNA target information, in which all of the microRNAs and mRNAs were selected by disease database. Second, in order to identify competing pseudogene-mRNA interactions, a hypergeometric test was performed to evaluate the significance of shared microRNAs for each possible gene pair:
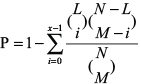
Where N is the total number of microRNAs which were associated with pseudogene or mRNA, M is the number of microRNAs interacting with this given pseudogene, L is the number of microRNAs interacting with this given mRNA, and x is the number of microRNAs that interact with both of them, respectively. The p-value and FDR correction less than 0.05 were used as the threshold [59]. Finally, in order to reduce the false positive rate of result, the pseudogene-mRNA pairs that P < 0.05, PCC ≥ 0.259 (75 pairs, 10% top correlated pseudogene-mRNA pairs, including 33 pseudogenes and 40 mRNAs) were considered to be potential pseudogene-mRNA interactions [16].
Evolutionary conservation analysis
We evaluate the evolutionary conservation of all pseudogenes, lincRNAs, PCGs, candidate pseudogenes and their neighboring PCGs. The evolutionary conservation was evaluated by 46-way phastCons vertebrate conserved elements from the UCSC Genome Browser website [60]. We considered a base as a unit and computed average phastCons scores for exons.
DNA methylation analysis
DNA methylation data of LUAD from Illumina Infinium Human Methylation 450 Beadchip (Infinium 450 k) arrays was obtained from TCGA, including 485,577 probes, 475 tumor samples and 32 normal samples (Supplementary Table 1). We then assigned the probes into the gene-body regions.
To estimate the methylation level of a given probe, we used the beta value: the ratio of intensities between methylated and unmethylated alleles. The beta value was obtained from the level 3 Infinium 450k data in TCGA; the corresponding p-value of each probe was obtained from level 2 Infinium 450k data. We only used the beta values with significant detection p-values (p < 0.05) in calculations to avoid using the missing data [61]. The average value of probes within a gene was regarded as methylated value.
Survival analysis
The clinical information of LUAD patient samples was obtained from TCGA. Cox regression analysis was used to evaluate the correlation between survival time and pseudogene expression. The risk ratio was used to calculate the risk score for each sample. Then these sample were divided into high-risk and low-risk group based on the mid-value of risk score [62]. The Kaplan-Meier survival method was used to evaluate the influence of the pseudogene for patient prognosis. The Log-Rank test p-value was used to test the significance of correlation between two groups of patients (p-value < 0.05). The Cox p-value was used to evaluate the significant correlation between the overall survival and genes (p-value < 0.05).
Statistical analyses
The p-value and FDR correction less than 0.05 were used as the threshold in the hypergeometric test. The pseudogenes that fold change > 1.5 and FDR < 0.01 were considered as DE pseudogenes. Functional enrichments of mRNAs were consisted on the Fisher’s exact test (two-tailed) implemented by DAVID v6.8 (https://david-d.ncifcrf.gov/) [63]. Wilcox rank sum test was used to test the significance between two groups of data (p < 0.05).
Abbreviations
LUAD: lung adenocarcinoma; PCGs: protein-coding genes; ceRNA: competitive endogenous RNA; siRNA: small interfering RNA; DE: differentially expressed; NDE: non-differentially expressed; PCC: pearson correlation coefficient
Author contributions
YX designed the study, YW performed the research and wrote the manuscript. YW, ZC, CW, and YZ analyzed the data. KL revised the manuscript. All authors reviewed the manuscript.
CONFLICTS OF INTEREST
The authors declared that they have no conflicts of interest.
FUNDING
This work was supported by the National Natural Science Foundation of China (grant numbers 81372492 and 81673036).
REFERENCES
1. Harrison PM, Hegyi H, Balasubramanian S, Luscombe NM, Bertone P, Echols N, Johnson T, Gerstein M. Molecular fossils in the human genome: identification and analysis of the pseudogenes in chromosomes 21 and 22. Genome Res. 2002; 12:272–280.
2. Hawkins PG, Morris KV. Transcriptional regulation of Oct4 by a long non-coding RNA antisense to Oct4-pseudogene 5. Transcription. 2010; 1:165–175.
3. Tam OH, Aravin AA, Stein P, Girard A, Murchison EP, Cheloufi S, Hodges E, Anger M, Sachidanandam R, Schultz RM, Hannon GJ. Pseudogene-derived small interfering RNAs regulate gene expression in mouse oocytes. Nature. 2008; 453:534–538.
4. Watanabe T, Totoki Y, Toyoda A, Kaneda M, Kuramochi-Miyagawa S, Obata Y, Chiba H, Kohara Y, Kono T, Nakano T, Surani MA, Sakaki Y, Sasaki H. Endogenous siRNAs from naturally formed dsRNAs regulate transcripts in mouse oocytes. Nature. 2008; 453:539–543.
5. Poliseno L. Pseudogenes: newly discovered players in human cancer. Sci Signal. 2012; 5:re5.
6. Pink RC, Wicks K, Caley DP, Punch EK, Jacobs L, Carter DR. Pseudogenes: pseudo-functional or key regulators in health and disease? RNA. 2011; 17:792–798.
7. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006; 126:663–676.
8. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007; 131:861–872.
9. Cantz T, Key G, Bleidissel M, Gentile L, Han DW, Brenne A, Scholer HR. Absence of OCT4 expression in somatic tumor cell lines. Stem Cells. 2008; 26:692–697.
10. Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010; 465:1033–1038.
11. Karreth FA, Reschke M, Ruocco A, Ng C, Chapuy B, Leopold V, Sjoberg M, Keane TM, Verma A, Ala U, Tay Y, Wu D, Seitzer N, et al. The BRAF pseudogene functions as a competitive endogenous RNA and induces lymphoma in vivo. Cell. 2015; 161:319–332.
12. Kalyana-Sundaram S, Kumar-Sinha C, Shankar S, Robinson DR, Wu YM, Cao X, Asangani IA, Kothari V, Prensner JR, Lonigro RJ, Iyer MK, Barrette T, Shanmugam A, et al. Expressed pseudogenes in the transcriptional landscape of human cancers. Cell. 2012; 149:1622–1634.
13. Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014; 505:344–352.
14. Xu J, Li Y, Lu J, Pan T, Ding N, Wang Z, Shao T, Zhang J, Wang L, Li X. The mRNA related ceRNA-ceRNA landscape and significance across 20 major cancer types. Nucleic Acids Res. 2015; 43:8169–8182.
15. Chen X, Yan CC, Luo C, Ji W, Zhang Y, Dai Q. Constructing lncRNA functional similarity network based on lncRNA-disease associations and disease semantic similarity. Sci Rep. 2015; 5:11338.
16. Zhou M, Wang X, Shi H, Cheng L, Wang Z, Zhao H, Yang L, Sun J. Characterization of long non-coding RNA-associated ceRNA network to reveal potential prognostic lncRNA biomarkers in human ovarian cancer. Oncotarget. 2016; 7:12598–12611. https://doi.org/10.18632/oncotarget.7181.
17. Wang P, Ning S, Zhang Y, Li R, Ye J, Zhao Z, Zhi H, Wang T, Guo Z, Li X. Identification of lncRNA-associated competing triplets reveals global patterns and prognostic markers for cancer. Nucleic Acids Res. 2015; 43:3478–3489.
18. Liu D, Yu X, Wang S, Dai E, Jiang L, Wang J, Yang Q, Yang F, Zhou S, Jiang W. The gain and loss of long noncoding RNA associated-competing endogenous RNAs in prostate cancer. Oncotarget. 2016; 7:57228–57238. https://doi.org/10.18632/oncotarget.11128.
19. Ponjavic J, Ponting CP, Lunter G. Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs. Genome Res. 2007; 17:556–565.
20. Schiefermeier N, Scheffler JM, de Araujo ME, Stasyk T, Yordanov T, Ebner HL, Offterdinger M, Munck S, Hess MW, Wickstrom SA, Lange A, Wunderlich W, Fassler R, et al. The late endosomal p14-MP1 (LAMTOR2/3) complex regulates focal adhesion dynamics during cell migration. J Cell Biol. 2014; 205:525–540.
21. Glatz DC, Rujescu D, Tang Y, Berendt FJ, Hartmann AM, Faltraco F, Rosenberg C, Hulette C, Jellinger K, Hampel H, Riederer P, Moller HJ, Andreadis A, et al. The alternative splicing of tau exon 10 and its regulatory proteins CLK2 and TRA2-BETA1 changes in sporadic Alzheimer's disease. J Neurochem. 2006; 96:635–644.
22. Butel JS. Viral carcinogenesis: revelation of molecular mechanisms and etiology of human disease. Carcinogenesis. 2000; 21:405–426.
23. Akulenko R, Helms V. DNA co-methylation analysis suggests novel functional associations between gene pairs in breast cancer samples. Hum Mol Genet. 2013; 22:3016–3022.
24. Yoon KA, Gil HJ, Han J, Park J, Lee JS. Genetic polymorphisms in the polycomb group gene EZH2 and the risk of lung cancer. J Thorac Oncol. 2010; 5:10–16.
25. Shepelev MV, Korobko IV. Pak6 protein kinase is a novel effector of an atypical Rho family GTPase Chp/RhoV. Biochemistry (Mosc). 2012; 77:26–32.
26. Mendizabal I, Zeng J, Keller TE, Yi SV. Body-hypomethylated human genes harbor extensive intragenic transcriptional activity and are prone to cancer-associated dysregulation. Nucleic Acids Res. 2017; 45:4390–4400.
27. Chen J, Hu L, Chen J, Wu F, Hu D, Xu G, Zhu P, Wang Y. Low expression lncRNA RPLP0P2 is associated with poor prognosis and decreased cell proliferation and adhesion ability in lung adenocarcinoma. Oncol Rep. 2016; 36:1665–1671.
28. Meng F, Wang J, Dai E, Yang F, Chen X, Wang S, Yu X, Liu D, Jiang W. Psmir: a database of potential associations between small molecules and miRNAs. Sci Rep. 2016; 6:19264.
29. Wang J, Meng F, Dai E, Yang F, Wang S, Chen X, Yang L, Wang Y, Jiang W. Identification of associations between small molecule drugs and miRNAs based on functional similarity. Oncotarget. 2016; 7:38658–38669.
30. Liu D, Yu X, Wang S, Dai E, Jiang L, Wang J, Yang Q, Yang F, Zhou S, Jiang W. The gain and loss of long noncoding RNA associated-competing endogenous RNAs in prostate cancer. Oncotarget. 2016; 7:57228–57238. https://doi.org/10.18632/oncotarget.11128.
31. Liu X, Wang S, Meng F, Wang J, Zhang Y, Dai E, Yu X, Li X, Jiang W. SM2miR: a database of the experimentally validated small molecules' effects on microRNA expression. Bioinformatics. 2013; 29:409–411.
32. Momparler RL, Ayoub J. Potential of 5-aza-2'-deoxycytidine (Decitabine) a potent inhibitor of DNA methylation for therapy of advanced non-small cell lung cancer. Lung Cancer. 2001; 34:S111–5.
33. Cao S, Sun R, Wang W, Meng X, Zhang Y, Zhang N, Yang S. RNA helicase DHX9 may be a therapeutic target in lung cancer and inhibited by enoxacin. Am J Transl Res. 2017; 9:674–682.
34. Lu KD, Radom-Aizik S, Haddad F, Zaldivar F, Kraft M, Cooper DM. Glucocorticoid receptor expression on circulating leukocytes differs between healthy male and female adults. J Clin Transl Sci. 2017; 1:108–114.
35. Jiang W, Jia P, Hutchinson KE, Johnson DB, Sosman JA, Zhao Z. Clinically relevant genes and regulatory pathways associated with NRASQ61 mutations in melanoma through an integrative genomics approach. Oncotarget. 2015; 6:2496–2508. https://doi.org/10.18532/oncotarget.2954.
36. Le TD, Zhang J, Liu L, Li J. Computational methods for identifying miRNA sponge interactions. Brief Bioinform. 2016.
37. Xu J, Bai J, Zhang X, Lv Y, Gong Y, Liu L, Zhao H, Yu F, Ping Y, Zhang G, Lan Y, Xiao Y, Li X. A comprehensive overview of lncRNA annotation resources. Brief Bioinform. 2016.
38. St Laurent G, Wahlestedt C, Kapranov P. The Landscape of long noncoding RNA classification. Trends Genet. 2015; 31:239–251.
39. Szafranski P, Dharmadhikari AV, Brosens E, Gurha P, Kolodziejska KE, Zhishuo O, Dittwald P, Majewski T, Mohan KN, Chen B, Person RE, Tibboel D, de Klein A, et al. Small noncoding differentially methylated copy-number variants, including lncRNA genes, cause a lethal lung developmental disorder. Genome Res. 2013; 23:23–33.
40. Xu G, Chen J, Pan Q, Huang K, Pan J, Zhang W, Chen J, Yu F, Zhou T, Wang Y. Long noncoding RNA expression profiles of lung adenocarcinoma ascertained by microarray analysis. PLoS One. 2014; 9:e104044.
41. Zheng L, Li X, Meng X, Chou J, Hu J, Zhang F, Zhang Z, Xing Y, Liu Y, Xi T. Competing endogenous RNA networks of CYP4Z1 and pseudogene CYP4Z2P confer tamoxifen resistance in breast cancer. Mol Cell Endocrinol. 2016; 427:133–142.
42. Cancer Genome Atlas Research Network, Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013; 45:1113–1120.
43. Helwak A, Kudla G, Dudnakova T, Tollervey D. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell. 2013; 153:654–665.
44. Zhang C, Darnell RB. Mapping in vivo protein-RNA interactions at single-nucleotide resolution from HITS-CLIP data. Nat Biotechnol. 2011; 29:607–614.
45. Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005; 120:15–20.
46. Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, Rajewsky N. Combinatorial microRNA target predictions. Nat Genet. 2005; 37:495–500.
47. Kertesz M, Iovino N, Unnerstall U, Gaul U, Segal E. The role of site accessibility in microRNA target recognition. Nat Genet. 2007; 39:1278–1284.
48. Betel D, Koppal A, Agius P, Sander C, Leslie C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 2010; 11:R90.
49. Miranda KC, Huynh T, Tay Y, Ang YS, Tam WL, Thomson AM, Lim B, Rigoutsos I. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell. 2006; 126:1203–1217.
50. Li JH, Liu S, Zhou H, Qu LH, Yang JH. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014; 42:D92–D97.
51. Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, Kok CY, Jia M, De T, et al. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015; 43:D805–D811.
52. Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Hamosh A. OMIM.org: Online Mendelian Inheritance in Man (OMIM(R)), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015; 43:D789–D798.
53. Becker KG, Barnes KC, Bright TJ, Wang SA. The genetic association database. Nat Genet. 2004; 36:431–432.
54. Yu W, Clyne M, Khoury MJ, Gwinn M. Phenopedia and Genopedia: disease-centered and gene-centered views of the evolving knowledge of human genetic associations. Bioinformatics. 2010; 26:145–146.
55. Li Y, Qiu C, Tu J, Geng B, Yang J, Jiang T, Cui Q. HMDD v2.0: a database for experimentally supported human microRNA and disease associations. Nucleic Acids Res. 2014; 42:D1070–D1074.
56. Jiang Q, Wang Y, Hao Y, Juan L, Teng M, Zhang X, Li M, Wang G, Liu Y. miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009; 37:D98–D104.
57. Yang Q, Qiu C, Yang J, Wu Q, Cui Q. miREnvironment database: providing a bridge for microRNAs, environmental factors and phenotypes. Bioinformatics. 2011; 27:3329–3330.
58. Wang D, Gu J, Wang T, Ding Z. OncomiRDB: a database for the experimentally verified oncogenic and tumor-suppressive microRNAs. Bioinformatics. 2014; 30:2237–2238.
59. Chen X. Predicting lncRNA-disease associations and constructing lncRNA functional similarity network based on the information of miRNA. Sci Rep. 2015; 5:13186.
60. Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, Weinstock GM, Wilson RK, Gibbs RA, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005; 15:1034–1050.
61. Kamoun A, Idbaih A, Dehais C, Elarouci N, Carpentier C, Letouze E, Colin C, Mokhtari K, Jouvet A, Uro-Coste E, Martin-Duverneuil N, Sanson M, Delattre JY, et al. Integrated multi-omics analysis of oligodendroglial tumours identifies three subgroups of 1p/19q co-deleted gliomas. Nat Commun. 2016; 7:11263.
62. Li Y, Xu J, Chen H, Bai J, Li S, Zhao Z, Shao T, Jiang T, Ren H, Kang C, Li X. Comprehensive analysis of the functional microRNA-mRNA regulatory network identifies miRNA signatures associated with glioma malignant progression. Nucleic Acids Res. 2013; 41:e203.
63. Sherman BT, Huang da W, Tan Q, Guo Y, Bour S, Liu D, Stephens R, Baseler MW, Lane HC, Lempicki RA. DAVID Knowledgebase: a gene-centered database integrating heterogeneous gene annotation resources to facilitate high-throughput gene functional analysis. BMC Bioinformatics. 2007; 8:426.