

INTRODUCTION
Lung cancer is the leading cause of cancer-related mortality worldwide, and non-small-cell lung cancer (NSCLC) accounts for 80–85% of all lung cancer cases [1]. Epidermal growth factor receptor (EGFR) mutations (EGFRm+), such as exon 19 deletions and exon 21 L858R point mutations, occur in 10–35% of NSCLCs and confer sensitivity to EGFR tyrosine kinase inhibitors (TKIs) [2, 3]. EGFRm+ NSCLC patients benefit from treatment with first-generation EGFR TKIs (reversible, not mutant selective), such as erlotinib and gefitinib, which are the standard first-line therapy for these patients. However, most patients develop drug resistance within approximately one year of treatment due to various mechanisms [4, 5]. The secondary T790M point mutation in exon 20 contributes to approximately 60% of resistance cases. Additionally, about 30% of EGFRm+ patients show primary resistance to EGFR TKIs, and the factors involved in de novo resistance remain unidentified. Although the second-generation EGFR TKIs (irreversible, potent, not mutant selective), including afatinib, dacomitinib, and neratinib, exhibited promising preclinical activity against T790M mutants, dose-limiting toxicities in patients inhibited clinical success. Therefore, optimum treatments after disease progression resulting from acquired EGFR TKI resistance are not clearly defined. Switching EGFR TKI resistant patient treatment strategies to platinum-based chemotherapy is common practice. Fortunately, third-generation EGFR TKIs (mutant selective), such as osimertinib, rociletinib, and olmutinib, can overcome resistance resulting from T790M mutation [6–8]. For example, in 2015, osimertinib was approved for T790M-positive patients who had progressed on prior systemic therapy. Other treatment options aimed at EGFR TKI resistance include use of an EGFR TKI beyond progression, and chemotherapy plus an EGFR TKI. Since drug resistance to EGFR TKIs is a major clinical obstacle, and optimum treatment strategies are still elusive, this review focuses on anticancer agents and treatment options aimed at EGFR TKI resistant cases to better guide oncologists making clinical decisions.
MECHANISMS OF ACQUIRED RESISTANCE TO EGFR TKIS
Acquired resistance to EGFR TKIs in NSCLC patients is defined as disease progression after a period of clinical benefit, and is categorized into three clinical groups: central nervous system sanctuary progressive disease (PD); oligo-PD; and systemic PD [9, 10]. The mechanisms involved can include secondary mutations in EGFR, bypass or alternative pathway activation, and histological and phenotypic transformations (Table 1). Several acquired-resistance mechanisms and candidates could also act as both rational targets and biomarkers. The best studied of these is the T790M mutation in exon 20. Additionally, MET mutations and amplifications, and amplifications of the MET ligand are also potential predictive biomarkers for NSCLC patient treatment outcomes, and insulin-like growth factor-1 receptor (IGF-1R) is a biomarker for TKI resistance [11, 12].
Table 1: Main mechanisms involved in acquired resistance to EGFR TKIs
Mechanism |
Molecular alteration |
|---|---|
Secondary mutations in EGFR |
T790M |
A761T |
|
T854A |
|
L747S |
|
Bypass or alternative pathway activation |
MET amplification |
PIK3CA mutation |
|
HER2 amplification |
|
BRAF mutation |
|
AXL activation |
|
Histological and phenotypic transformation |
NSCLC to SCLC |
epithelial to mesenchymal |
Secondary mutations in EGFR
Secondary EGFR mutation is the most frequent mechanism of acquired resistance to EGFR TKIs. The secondary point mutation, T790M in exon 20, contributes to approximately 60% of drug resistance cases. [4, 5]. With the steric hindrance of the bulky methionine sidechain, T790M restores the kinase’s adenosine triphosphate (ATP) affinity back to wild type levels, eventually re-establishing ATP as the favored substrate rather than the EGFR TKI [13]. Other rare EGFR point mutations resulting in resistance include, A761T, T854A, and L747S [14–16].
Bypass or alternative pathway activation
MET signaling activates various pathways, such as PI3K/AKT, RAS-RAF-ERK1/2, and STAT3, promoting angiogenesis and tumor cell growth, survival, migration, invasion, and metastasis [11]. MET amplification, identified in 5–10% of resistant tumors, is the main mechanism by which EGFR inhibition is bypassed, allowing for drug resistance via ERBB3-mediated activation of PI3K/AKT downstream signaling [17]. Other bypass mechanisms include, PIK3CA mutation, HER2 amplification, BRAF (V600E, G469A) mutation, and AXL activation [18–20].
Histological and phenotypic transformation
After an initial response to EGFR TKIs, EGFRm+ NSCLC can transform to small cell lung cancer (SCLC) [18]. Epithelial to mesenchymal transition (EMT), which renders cells more migratory and invasive, is also observed in EGFR TKI-resistant tumor specimens [21]. EMT may be induced and controlled by multiple pathways, including Wnt, NFκB, and TGF-β signaling, as well as growth factors, such as FGF and EGF. Jakobsen, et al. found that erlotinib increased IGF1R activation, restoring PI3K/AKT and MAPK signaling, and triggering IGF1R-dependent EMT [22]. Alternatively, cancer cells treated with erlotinib may benefit from TGF-β signaling, which initiates EMT by inhibiting the miR-200 family, upregulating mesenchymal transcription factors and thus the EGFR repressor, MIG6, and restoring PI3K/AKT signaling. EMT is likely important in the development of NSCLC acquired EGFR TKI resistance, and influences TKI treatment response. EMT status may therefore be useful as a prognostic indicator and in treatment strategy decision-making in NSCLC.
CLINICAL STRATEGIES FOLLOWING EGFR TKI ACQUIRED RESISTANCE IN NSCLC
There are currently no standardized clinical strategies for NSCLC patients with EGFR TKI acquired resistance. Treatment options usually include chemotherapy, third generation EGFR TKIs, EGFR TKI treatment beyond progression, and chemotherapy plus an EGFR TKI (Figure 1).
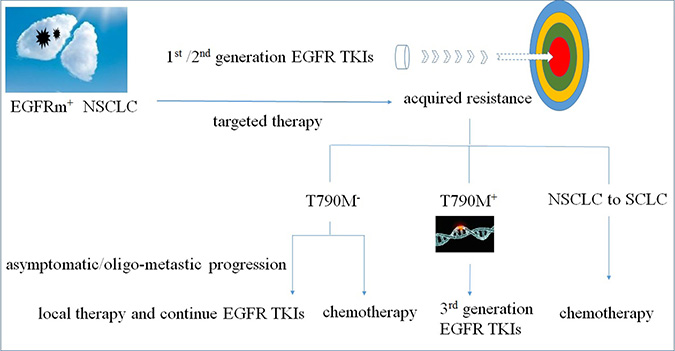
Figure 1: Treatment options for NSCLC patients with EGFR TKI acquired resistance.
Chemotherapy
When first-line EGFR TKI acquired resistance causes clinical progression, NSCLC patients are commonly switched onto platinum-based chemotherapeutics, which often provide palliative benefit. Second-line platinum-based combination therapies led to better overall survival (OS) than single agents or erlotinib, following the failure of first-line gefitinib treatment [23]. Second-line gemcitabine/platinum therapy combination treatment resulted in better OS in EGFRm+ patients (p = 0.035), but not in those with wild-type EGFR (p = 0.785) [23]. Additionally, chemotherapy can allow for simultaneous re-challenge with a TKI; previously arrested TKI-sensitive cells may repopulate more quickly than TKI-resistant cells, and may therefore be efficiently inhibited using TKIs [24].
Third-generation EGFR TKIs
EGFRm+ NSCLC patients can benefit from treatment with first-generation EGFR TKIs, including erlotinib and gefitinib [25, 26]. However, most patients develop drug resistance, driven in about 60% of cases by the T790M mutation [27, 28]. Second-generation EGFR TKIs cannot overcome T790M-mediated drug resistance due to nonselective inhibition of wild-type EGFR [6, 29, 30]. More recently, third-generation EGFR TKIs, including osimertinib, rociletinib, and olmutinib, have shown efficacy against the T790M mutation while sparing wild-type EGFR in vitro (Figure 2) [31, 32]. The third-generation EGFR TKIs represent a promising approach to overcoming T790M-mediated resistance to first- and second-generation EGFR TKIs in NCSLC patients.
Figure 2: Structures of third-generation EGFR TKIs.
Osimertinib
Osimertinib (AZD9291; AstraZeneca) exhibits 200-fold greater selectivity for the T790M/L858R EGFR than wild-type EGFR [33]. It has been hailed as a ‘‘breakthrough” compound with perfect objective response rate (ORR) in T790M-positive NSCLC patients who had progressed on first-generation EGFR TKIs [34]. Preclinical studies confirmed osimertinib antitumor activity and reduced activity against wild-type EGFR in cell lines and tumor xenograft and transgenic mouse models harboring EGFRm+ and EGFR T790M [35]. The osimertinib ORR in the phase I clinical trial was 51%. In patients with confirmed EGFR T790M (n = 127), osimertinib had an ORR of 61% and median progression-free survival (mPFS) of 9.6 months, while T790M-negative patients (n = 61) had an ORR of 21% and mPFS of 2.8 months. [36] A pooled analysis of two phase II studies found that osimertinib ORR was 66% in T790M patients. Median duration of remission (DOR) was 12.5 months, and mPFS was 11.0 months. At 12 months, 47.5% of patients were progression-free [37]. Osimertinib was approved in the US on November 13, 2015 for use in patients with metastatic EGFR T790M-positive NSCLC who have progressed with EGFR TKI therapy.
Rociletinib
Rociletinib (CO-1686) is another small-molecule, irreversibly binding, mutant selective TKI that targets commonly mutated forms of EGFR while sparing the wild-type protein [38]. In a phase I/II study of rociletinib in EGFRm+ NSCLC patients who had progressed during EGFR TKI therapy, ORR and mPFS were 59% and 13.1 months, respectively, in 46 T790M-positive patients, and 29% and 5.6 months, respectively, in 17 T790M-negative patients. Unlike for earlier generation EGFR TKIs, treatment side effects, including paronychia, rash, stomatitis, and diarrhea, were uncommon in rociletinib-treated patients. One patient experienced a grade 1 rash, and grade 1–2 diarrhea was observed in only 20% of patients [39].
Olmutinib
Like osimertinib and rociletinib, olmutinib (HM61713) is a third-generation EGFR TKI that selectively targets mutant EGFR. In a phase I/II study, olmutinib ORR was 58.8% and the disease control rate was 97.1% in 34 T790M-positive NSCLC patients [40].
Chemotherapy plus EGFR TKI
Patients with acquired resistance to EGFR TKIs may benefit from continued EGFR TKI therapy in combination with platinum-based chemotherapy. In a randomized, phase III, multicenter study, EGFRm+ patients with acquired resistance to gefitinib were randomly assigned (1:1) to either continued gefitinib treatment combined with pemetrexed/cisplatin chemotherapy or placebo plus pemetrexed/cisplatin chemotherapy. Ninety-eight (74%) patients progressed in the gefitinib plus chemotherapy group, and 107 (81%) progressed in the placebo plus chemotherapy group (hazard ratio = 0.86, 95% CI = 0.65–1.13; p = 0.27). mPFS was 5.4 months in both treatment groups (95% CI = 4.5–5.7 with gefitinib and 4.6–5.5 with placebo). The results demonstrated that continuing gefitinib with chemotherapy after disease progression with gefitinib did not prolong patient PFS [41]. Additionally, continued gefitinib treatment with chemotherapy may be detrimental to patient OS, although these findings require further study [41].
Combined EGFR signaling pathway blockade
Targeting several levels of EGFR signaling is another strategy employed against acquired resistance to EGFR TKIs. Afatinib combined with the monoclonal anti-EGFR antibody, cetuximabhas, demonstrated robust clinical activity and a reasonable safety profile in patients with acquired gefitinib or erlotinib resistance, both with and without T790M mutations [42]. As previously discussed, bypass or alternative pathway activation can contribute to acquired resistance to EGFR TKIs. Thus, treatment strategies that both maintain inhibition of EGFR signaling and inhibit bypass signaling may be efficacious against resistance. EGFR TKIs combined with the aPI3K inhibitor, buparlisib (BKM120), or MET inhibitors, such as cabozantinib, tivantinib, or INC280, are presently under investigation for use in resistant NSCLC cases [42, 43].
TKIs beyond progression
Discontinuing TKI treatment may lead to a disease flare [45]. In cases of isolated progression, such as in the central nervous system (CNS), local therapy plus targeted therapy may be efficacious in some patients [46, 47]. In patients with CNS metastasis, controlling brain lesions with radiotherapy is important for maintaining or improving quality of life and for prolonging survival [48]. The phase II, single-arm, open-label ASPIRATION (Asian Pacific trial of Tarceva as first-line in EGFR mutation) study investigated post-progression continued erlotinib therapy in NSCLC patients with activating EGFR mutations [49]. In this study, patients received erlotinib until progression, after which erlotinib was continued if deemed appropriate. Patients who received post-progression erlotinib therapy had longer PFS, longer time from best overall response to progression, fewer new lung lesions, and better performance status at progression. The ASPIRATION trial showed that in clinically asymptomatic patients, maintaining EGFR TKI therapy as long as possible is a viable treatment option.
To effectively provide the best treatment options for each patient, the mechanisms underlying resistance onset must be better understood. Analyses of tumor samples from NSCLC patients with acquired EGFR TKI resistance could reveal molecular mechanisms of resistance. Thus, performing a biopsy at the time of acquired resistance may inform treatment decisions [50, 51]. Because procedures used to obtain tumor tissues are often invasive, analysis of circulating plasma cell-free DNA (cfDNA), which is released from tumors or circulating tumor cells and can be collected noninvasively, could provide tumor cell genetic information in many cases [52, 53].
Immunotherapy
Interest in immunotherapies for NSCLC treatment continues to grow. Preclinical evidence suggests that programmed death-ligand 1 (PD-L1) expression in EGFRm+ NSCLC may be constitutively driven by EGFR signaling. Preclinical EGFRm+ lung cancer models treated with PD-L1 antibodies exhibited reduced tumor growth and increased survival [54]. However, clinical data suggest that anti-PD1 immunotherapy may not be effective in EGFRm+ NSCLC.
CONCLUSIONS
Optimum treatment strategies for NSCLC patients with acquired resistance to EGFR TKIs are currently unclear. This review summarized anticancer agents and clinical strategies employed following EGFR TKI acquired resistance onset in NSCLC patients, emphasizing therapeutic advances with improved outcomes. Current common practice is to switch to platinum-based chemotherapy following resistance onset. However, third-generation EGFR TKIs may overcome T790M-mediated resistance to first- and second-generation EGFR TKIs in NCSLC patients. In asymptomatic/oligo-metastic progression cases, localized therapies followed by continuation of EGFR TKI treatment may be a viable strategy.
ACKNOWLEDGMENTS AND FUNDING
This work was supported by the National Natural Science Foundation of China (81371678).
CONFLICTS OF INTEREST
The author declares no conflicts of interest.
REFERENCES
1. Zhang H. Apatinib for molecular targeted therapy in tumor. Drug Des Devel Ther. 2015; 9:6075–6081.
2. Russo A, Franchina T, Ricciardi GR, Picone A, Ferraro G, Zanghì M, Toscano G, Giordano A, Adamo V. A decade of EGFR inhibition in EGFR-mutated non small cell lung cancer (NSCLC): Old successes and future perspectives. Oncotarget. 2015; 6:26814–26825. https://doi.org/10.18632/oncotarget.4254.
3. Zhang Q, Wang Z, Guo J, Liu L, Han X, Li M, Fang S, Bi X, Tang N, Liu Y. Comparison of single-agent chemotherapy and targeted therapy to first-line treatment in patients aged 80 years and older with advanced non-small-cell lung cancer. Onco Targets Ther. 2015; 8:893–898.
4. Ercan D, Choi HG, Yun CH, Capelletti M, Xie T, Eck MJ, Gray NS, Jänne PA. EGFR Mutations and Resistance to Irreversible Pyrimidine-Based EGFR Inhibitors. Clin Cancer Res. 2015; 21:3913–3923.
5. Villadolid J, Ersek JL, Fong MK, Sirianno L, Story ES. Management of hyperglycemia from epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) targeting T790M-mediated resistance. Transl Lung Cancer Res. 2015; 4:576–583.
6. Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, Orme JP, Finlay MR, Ward RA, Mellor MJ, Hughes G, Rahi A, Jacobs VN, et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014; 4:1046–1061.
7. Jänne PA, Yang JC, Kim DW, Planchard D, Ohe Y, Ramalingam SS, Ahn MJ, Kim SW, Su WC, Horn L, Haggstrom D, Felip E, Kim JH, et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med. 2015; 372:1689–1699.
8. Sequist LV, Soria JC, Goldman JW, Wakelee HA, Gadgeel SM, Varga A, Papadimitrakopoulou V, Solomon BJ, Oxnard GR, Dziadziuszko R, Aisner DL, Doebele RC, Galasso C, et al. Rociletinib in EGFR-mutated non-small-cell lung cancer. N Engl J Med. 2015; 372:1700–1709.
9. Jackman D, Pao W, Riely GJ, Engelman JA, Kris MG, Jänne PA, Lynch T, Johnson BE, Miller VA. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J Clin Oncol. 2010; 28:357–360.
10. Gandara DR, Li T, Lara PN, Kelly K, Riess JW, Redman MW, Mack PC. Acquired resistance to targeted therapies against oncogene-driven non-small-cell lung cancer: approach to subtyping progressive disease and clinical implications. Clin Lung Cancer. 2014; 15:1–6.
11. Pérez-Ramírez C, Cañadas-Garre M, Jiménez-Varo E, Faus-Dáder MJ, Calleja-Hernández MÁ. MET: a new promising biomarker in non-small-cell lung carcinoma. Pharmacogenomics. 2015; 16:631–647.
12. Peled N, Wynes MW, Ikeda N, Ohira T, Yoshida K, Qian J, Ilouze M, Brenner R, Kato Y, Mascaux C, Hirsch FR. Insulin-like growth factor-1 receptor (IGF-1R) as a biomarker for resistance to the tyrosine kinase inhibitor gefitinib in non-small cell lung cancer. Cell Oncol (Dordr). 2013; 36:277–288.
13. Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK, Meyerson M, Eck MJ. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci USA. 2008; 105:2070–2075.
14. Balak MN, Gong Y, Riely GJ, Somwar R, Li AR, Zakowski MF, Chiang A, Yang G, Ouerfelli O, Kris MG, Ladanyi M, Miller VA, Pao W. Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin Cancer Res. 2006; 12:6494–6501.
15. Bean J, Riely GJ, Balak M, Marks JL, Ladanyi M, Miller VA, Pao W. Acquired resistance to epidermal growth factor receptor kinase inhibitors associated with a novel T854A mutation in a patient with EGFR-mutant lung adenocarcinoma. Clin Cancer Res. 2008; 14:7519–7525.
16. Costa DB, Halmos B, Kumar A, Schumer ST, Huberman MS, Boggon TJ, Tenen DG, Kobayashi S. BIM mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung cancers with oncogenic EGFR mutations. PLoS Med. 2007; 4:1669–1680.
17. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007; 316:1039–1043.
18. Ohashi K, Sequist LV, Arcila ME, Moran T, Chmielecki J, Lin YL, Pan Y, Wang L, de Stanchina E, Shien K, Aoe K, Toyooka S, Kiura K, et al. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc Natl Acad Sci USA. 2012; 109:E2127–2133.
19. Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, Abdel-Rahman M, Wang X, Levine AD, Rho JK, Choi YJ, Choi CM, Kim SW, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet. 2012; 44:852–860.
20. Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger S, Cosper AK, Akhavanfard S, Heist RS, Temel J, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011; 3:75ra26.
21. Suda K, Tomizawa K, Fujii M, Murakami H, Osada H, Maehara Y, Yatabe Y, Sekido Y, Mitsudomi T. Epithelial to mesenchymal transition in an epidermal growth factor receptor-mutant lung cancer cell line with acquired resistance to erlotinib. J Thorac Oncol. 2011; 6:1152–1161.
22. Jakobsen KR, Demuth C, Sorensen BS, Nielsen AL. The role of epithelial to mesenchymal transition in resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Transl Lung Cancer Res. 2016; 5:172–182.
23. Wu JY, Shih JY, Yang CH, Chen KY, Ho CC, Yu CJ, Yang PC. Second-line treatments after first-line gefitinib therapy in advanced nonsmall cell lung cancer. Int J Cancer. 2010; 126:247–255.
24. Hata A, Katakami N, Yoshioka H, Takeshita J, Tanaka K, Nanjo S, Fujita S, Kaji R, Imai Y, Monden K, Matsumoto T, Nagata K, Otsuka K, et al. Rebiopsy of non-small cell lung cancer patients with acquired resistance to epidermal growth factor receptor-tyrosine kinase inhibitor: Comparison between T790M mutation-positive and mutation-negative populations. Cancer. 2013; 119:4325–4332.
25. Tiseo M, Bartolotti M, Gelsomino F, Bordi P. Emerging role of gefitinib in the treatment of non-small-cell lung cancer (NSCLC). Drug Des Devel Ther. 2010; 4:81–98.
26. Zhang Q, Wang Z, Guo J, Liu L, Han X, Li M, Fang S, Bi X, Tang N, Liu Y. Comparison of single-agent chemotherapy and targeted therapy to first-line treatment in patients aged 80 years and older with advanced non-small-cell lung cancer. Onco Targets Ther. 2015; 8:893–898.
27. Villadolid J, Ersek JL, Fong MK, Sirianno L, Story ES. Management of hyperglycemia from epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) targeting T790M-mediated resistance. Transl Lung Cancer Res. 2015; 4:576–583.
28. Ercan D, Choi HG, Yun CH, Capelletti M, Xie T, Eck MJ, Gray NS, Jänne PA. EGFR Mutations and Resistance to Irreversible Pyrimidine-Based EGFR Inhibitors. Clin Cancer Res. 2015; 21:3913–3923.
29. Sequist LV, Yang JC, Yamamoto N, O’Byrne K, Hirsh V, Mok T, Geater SL, Orlov S, Tsai CM, Boyer M, Su WC, Bennouna J, Kato T, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013; 31:3327–3334.
30. Ramalingam SS, Blackhall F, Krzakowski M, Barrios CH, Park K, Bover I, SeogHeo D, Rosell R, Talbot DC, Frank R, Letrent SP, Ruiz-Garcia A, Taylor I, et al. Randomized phase II study of dacomitinib (PF-00299804), an irreversible pan-human epidermal growth factor receptor inhibitor, versus erlotinib in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2012; 30:3337–3344.
31. Pirker R. Third-generation epidermal growth factor receptor tyrosine kinase inhibitors in advanced nonsmall cell lung cancer. Curr Opin Oncol. 2016; 28:115–121.
32. Greig SL. Osimertinib: First Global Approval. Drugs. 2016; 76:263–273.
33. Jiang T, Zhou C. Clinical activity of the mutant-selective EGFR inhibitor AZD9291 in patients with EGFR inhibitor-resistant non-small cell lung cancer. Transl Lung Cancer Res. 2014; 3:370–372.
34. Yosaatmadja Y, Silva S, Dickson JM, Patterson AV, Smaill JB, Flanagan JU, McKeage MJ, Squire CJ. Binding mode of the breakthrough inhibitor AZD9291 to epidermal growth factor receptor revealed. J Struct Biol. 2015; 192:539–544.
35. Zhang H. Osimertinib making a breakthrough in lung cancer targeted therapy. Onco Targets Ther. 2016; 9:5489–5493.
36. Jänne PA, Yang JC, Kim DW, Planchard D, Ohe Y, Ramalingam SS, Ahn MJ, Kim SW, Su WC, Horn L, Haggstrom D, Felip E, Kim JH, et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med. 2015; 372:1689–1699.
37. Yang J, Ramalingam SS, Jänne PA, Cantarini M, Mitsudomi T. LBA2_PR: Osimertinib (AZD9291) in pre-treated pts with T790M-positive advanced NSCLC: updated Phase 1 (P1) and pooled Phase 2 (P2) results. J Thorac Oncol. 2016; 11:S152–153.
38. Walter AO, Sjin RT, Haringsma HJ, Ohashi K, Sun J, Lee K, Dubrovskiy A, Labenski M, Zhu Z, Wang Z, Sheets M, St Martin T, Karp R, et al. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov. 2013; 3:1404–1415.
39. Sequist LV, Soria JC, Goldman JW, Wakelee HA, Gadgeel SM, Varga A, Papadimitrakopoulou V, Solomon BJ, Oxnard GR, Dziadziuszko R, Aisner DL, Doebele RC, Galasso C, et al. Rociletinib in EGFR-mutated non-small-cell lung cancer. N Engl J Med. 2015; 372:1700–1709.
40. Park K, Lee JS, Lee KH, Kim JH, Min YJ, Cho JY, Han JY, Kim BS, Kim JS, Lee DH, Kang JH, Cho EK, Jang IJ, et al. Updated safety and efficacy results from phase I/II study of HM61713 in patients (pts) with EGFR mutation positive non-small cell lung cancer (NSCLC) who failed previous EGFR-tyrosine kinase inhibitor (TKI). J Clin Oncol (Meeting Abstracts). 2015; 33:8084.
41. Soria JC, Wu YL, Nakagawa K, Kim SW, Yang JJ, Ahn MJ, Wang J, Yang JC, Lu Y, Atagi S, Ponce S, Lee DH, Liu Y, et al. Gefitinib plus chemotherapy versus placebo plus chemotherapy in EGFR-mutation-positive non-small-cell lung cancer after progression on first-line gefitinib (IMPRESS): a phase 3 randomisedtrial. Lancet Oncol. 2015; 16:990–998.
42. Noto A, De Vitis C, Roscilli G, Fattore L, Malpicci D, Marra E, Luberto L, D’Andrilli A, Coluccia P, Giovagnoli MR, Normanno N, Ruco L, Aurisicchio L, et al. Combination therapy with anti-ErbB3 monoclonal antibodies and EGFR TKIs potently inhibits non-small cell lung cancer. Oncotarget. 2013; 4:1253–1265. https://doi.org/10.18632/oncotarget.1141.
43. Lara MS, Holland WS, Chinn D, Burich RA, Lara PN Jr, Gandara DR, Kelly K, Mack PC. Preclinical Evaluation of MET Inhibitor INC-280 With or Without the Epidermal Growth Factor Receptor Inhibitor Erlotinib in Non-Small-Cell Lung Cancer. Clin Lung Cancer. 2017; 18:281–285.
44. Janjigian YY, Smit EF, Groen HJ, Horn L, Gettinger S, Camidge DR, Riely GJ, Wang B, Fu Y, Chand VK, Miller VA, Pao W. Dual inhibition of EGFR with afatinib and cetuximab in kinase inhibitor-resistant EGFR-mutant lung cancer with and without T790M mutations. Cancer Discov. 2014; 4:1036–1045.
45. Chaft JE, Oxnard GR, Sima CS, Kris MG, Miller VA, Riely GJ. Disease flare after tyrosine kinase inhibitor discontinuation in patients with EGFR-mutant lung cancer and acquired resistance to erlotinib or gefitinib: implications for clinical trial design. Clin Cancer Res. 2011; 17:6298–6303.
46. Weickhardt AJ, Scheier B, Burke JM, Gan G, Lu X, Bunn PA Jr, Aisner DL, Gaspar LE, Kavanagh BD, Doebele RC, Camidge DR. Local ablative therapy of oligoprogressive disease prolongs disease control by tyrosine kinase inhibitors in oncogene-addicted non-small-cell lung cancer. J Thorac Oncol. 2012; 7:1807–1814.
47. Gan GN, Weickhardt AJ, Scheier B, Doebele RC, Gaspar LE, Kavanagh BD, Camidge DR. Stereotactic radiation therapy can safely and durably control sites of extra-central nervous system oligoprogressive disease in anaplastic lymphoma kinase-positive lung cancer patients receiving crizotinib. Int J Radiat Oncol Biol Phys. 2014; 88:892–898.
48. Shukuya T, Takahashi T, Naito T, Kaira R, Ono A, Nakamura Y, Tsuya A, Kenmotsu H, Murakami H, Harada H, Mitsuya K, Endo M, Nakasu Y, et al. Continuous EGFR-TKI administration following radiotherapy for non-small cell lung cancer patients with isolated CNS failure. Lung Cancer. 2011; 74:457–461.
49. Park K, Yu CJ, Kim SW, Lin MC, Sriuranpong V, Tsai CM, Lee JS, Kang JH, Chan KC, Perez-Moreno P, Button P, Ahn MJ, Mok T. First-Line Erlotinib Therapy Until and Beyond Response Evaluation Criteria in Solid Tumors Progression in Asian Patients With Epidermal Growth Factor Receptor Mutation-Positive Non-Small-Cell Lung Cancer: The ASPIRATION Study. JAMA Oncol. 2016; 2:305–312.
50. Kawamura T, Kenmotsu H, Taira T, Omori S, Nakashima K, Wakuda K, Ono A, Naito T, Murakami H, Mori K, Nakajima T, Ohde Y, Endo M, et al. Rebiopsy for patients with non-small-cell lung cancer after epidermal growth factor receptor-tyrosine kinase inhibitor failure. Cancer Sci. 2016; 107:1001–1005.
51. Hasegawa T, Sawa T, Futamura Y, Horiba A, Ishiguro T, Marui T, Yoshida T. Feasibility of Rebiopsy in Non-Small Cell Lung Cancer Treated with Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors. Intern Med. 2015; 54:1977–1980.
52. Zhou Q, Yang JJ, Chen ZH, Zhang XC, Yan HH, Xu CR, Su J, Chen HJ, Tu HY, Zhong WZ, Yang XN, Wu YL. Serial cfDNA assessment of response and resistance to EGFR-TKI for patients with EGFR-L858R mutant lung cancer from a prospective clinical trial. J Hematol Oncol. 2016; 9:86.
53. Seki Y, Fujiwara Y, Kohno T, Takai E, Sunami K, Goto Y, Horinouchi H, Kanda S, Nokihara H, Watanabe S, Ichikawa H, Yamamoto N, Kuwano K, et al. Picoliter-Droplet Digital Polymerase Chain Reaction-Based Analysis of Cell-Free Plasma DNA to Assess EGFR Mutations in Lung Adenocarcinoma That Confer Resistance to Tyrosine-Kinase Inhibitors. Oncologist. 2016; 21:156–164.
54. Santabarbara G, Maione P, Rossi A, Palazzolo G, Gridelli C. Novel immunotherapy in the treatment of advanced non-small cell lung cancer. Expert Rev Clin Pharmacol. 2016; 9:1571–1581.