
Introduction
Adjuvant RT is given to the breast cancer patients after conservation surgery and may be given to the chest wall after mastectomy to achieve better local tumor control thus improving survival of patients [1-3]. However, radioresistance impedes the anti-tumor effects of RT and could be attributed to BCSCs, since it has recently been shown that BCSCs are radioresistant[4, 5]; but radiation also can induce BCSCs, i.e., iBCSCs from nonstem breast cancer cells [6]. In order to significantly improve the efficacy and curability of RT for breast cancer, therefore, novel therapeutic approaches are urgently needed to not only eliminate radioresisitant pre-existing BCSCs, but also block the formation of radiation induced new BCSCs from nonstem BC cells. This study is a pre-clinical effort to develop such a new treatment, for rapid translation into clinical practice, that is scientific sound, simple, safe and economic.
Following the first discovery that mature adult cells can be reprogrammed to become pluripotent stem cells (iPSCs) by introduction of a set of four stemness factors (Oct4, SOX2, KLF4 and MYC) through retroviral transduction [7], it has been shown that cancer stem cells (CSCs) and relatively differentiated cancer cells are subject to bidirectional conversion [8]. Stemness gene-encoded transcription factors (TFs) play a central role in the determination of stem cell states [7], with specific transcriptional networks having essential roles in sustaining the growth and self-renewal of embryonic and neoplastic stem-like cells. The TFs Slug and SOX9 cooperatively determine mammary stem cell states in both normal and malignant cells [9]. Moreover, the overexpression in human cancers of reprogramming TFs correlates with tumor progression and poor prognosis [10]. IR has been shown to reprogram differentiated cancer cells into iBCSCs or liver CSCs through the re-expression or upexpression of the stemness genes Oct4/SOX2/Nanog/KLF4 and SOX2/OCT3/Oct4, respectively [6, 11].
There is compelling evidence showing that the cells expressing elevated levels of intracellular aldehyde dehydrogenase 1 (ALDH1) in human and mouse breast cancer are BCSCs [12, 13]. The ability of enhanced ALDH1 activity in these cells to eliminate genotoxic aldehydes contributes to their radio- and chemo-resistance. Research on the CSC area has been facilitated by the ability to routinely detect and isolate CSCs by flow cytometry analysis using the ALDEFLUOR reagent to measure their ALDH enzymatic activity [6, 14]. Consequently, ALDH activity is widely used as a CSC marker for many malignancies including leukemia, lung, liver, bone, colon, pancreatic, prostate, head and neck, bladder, thyroid, brain, and cervical cancer and melanoma [15]. Moreover, we demonstrated that ALDH1A1 is not only a marker but also a therapeutic target for CSCs [14, 16, 17].
DSF (Brand name: Antabuse) is an irreversible pan-ALDH inhibitor; it inhibits all the currently identified cytosolic and mitochondrial ALDH isoforms [18, 19], resulting in accumulation of, in particular, acetaldehyde, which causes unpleasant effects when alcohol is consumed. As a result, DSF has been an FDA approved drug for treatment of alcoholism since 1951 [20]. Studies have shown that in cells DSF converts to diethyldithiocarbamate (deDTC) and that two molecules of deDTC bind to one molecule of copper (Cu2+) to form the Cu[deDTC]2 complex (DSF/Cu) [21-23]. Cu2+ is an essential trace element for life [24] as it plays a crucial role in redox reactions and generation of reactive oxygen species (ROS) in human cells [25, 26]. It is known that DSF/Cu is an effective proteasome inhibitor resulting in inhibition of NF-κB [21, 27]. NF-κB is a key TF governing the activation of many genes involved in stress responses (e.g. IR), cell survival, apoptosis, inflammation, and radioresistance [28]. These NF-κB regulated stemness genes include ERBB2 [4], SOX9 [29], MYC [30] and WNT [31]. We have, therefore, investigated using human and mouse BC cell lines and a clinically relevant mouse model whether DSF/Cu can block in vitro and in vivo the IR-induced conversion of nonstem BC cells into iBCSCs via downregulation of the NF-κB-stemness gene pathway and enhance the efficacy of RT.
Results
DSF/Cu effectively depleted pre-existing BCSCs and radiation-induced BCSCs
Based on compelling evidence showing that elevated ALDH activity in human and mouse BC cells is a marker for BCSCs and iBCSCs [6, 12-14], in this study we have identified these cells by flow cytometry analysis of BC cells as ALDHbright cells, namely those ALDH+ cells with twice the mean fluorescence intensity (MFI) of the bulk ALDH+ cell population. We detected an increased percentage of BCSCs in vitro following fractionated irradiation (3.75 Gray (Gy) /day x 5 days) of BC cell lines MDA-MB-231 (2.4 fold), SUM149 (1.4 fold) and UACC-812 (4.6 fold) (Supplementary Fig. S1A). Within a range of doses of fractionated irradiation (1-5 Gy/day x 5 days), increased ALDHbright cells were detected in BC cell lines (Supplementary Fig. S1B). The increased percentage of BCSCs was caused by an increase in the absolute number of BCSCs accompanied by a 50.5% decrease in total cell number in irradiated cells vs. untreated cells, which indicates that IR induced the formation of new BCSCs or iBCSCs (Fig. 1A). The stem cell functional properties of these BCSCs and iBCSCs were further supported by in vitro formation of mammospheres (Fig. 1B) and increased tumorigenicity of the in vitro irradiated BC cells compared to untreated BC cells in mice (Fig. 1C). Treatment of cells with DSF/Cu effectively depleted pre-existing (before IR) BCSCs and iBCSCs (together referred to as BCSCs/iBCSCs) (Fig. 1A, B, C), including those induced by IR from nonstem ALDHneg cells, as evidenced using this cells isolated by fluorescence-activated cell sorting in vitro (Fig. 1D). In contrast, DSF/Cu or IR and DSF/Cu did not exhibit toxicity on normal human mammary epithelial cells as measured by cell growth and apoptosis assays (Supplementary Fig. S1C).
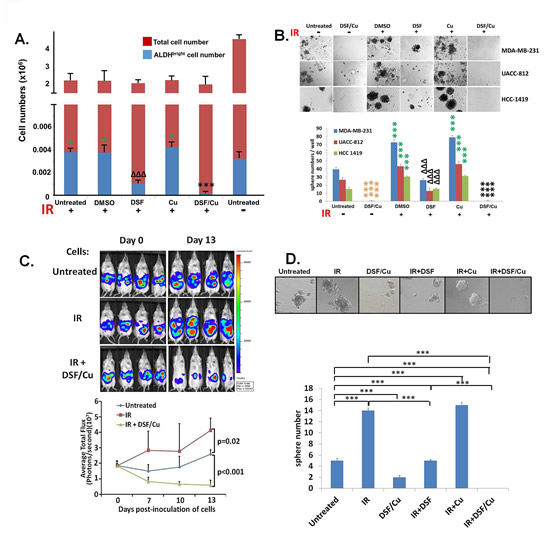
Figure 1: Depletion of BCSCs/iBCSCs by DSF/Cu as measured by decreased ALDHbright cells, mammosphere formation in vitro and tumorigenicity in vivo. Fraction-irradiated (3.75 Gy/day x 5 days) BC cells were cultured for 24 hours, followed by treatment with different agents for an additional 24 hours, as indicated. The total numbers of cell were counted using the Trypan blue exclusion method. The numbers of ALDHbright cells were calculated based on the total cell numbers and % of ALDHbright cells. * indicates P<0.05: IR or IR+DMSO or IR+Cu vs. untreated; *** indicates P<0.001: IR+DSF/Cu vs. any other treated or untreated; ΔΔΔ indicates P<0.001: IR+DSF vs. any other treated or untreated (A). Mammosphere formation assays were setup by seeding cells from fraction-irradiated human BC cell lines and culturing them for 14 days. Pictures were taken and mammospheres were quantified by counting mammosphere numbers per well on day 14. *** indicates P<0.001: IR+DMSO or IR+Cu vs. untreated; *** indicates P<0.001: IR+ DSF/Cu vs. any other treated or untreated except DSF/Cu; ΔΔΔ indicates P<0.001: IR+DSF vs. any other treated or untreated; *** indicates P<0.001: DSF/Cu vs. any other treated or untreated except IR+DSF/Cu (B). Three groups of 6 week old female NOD/SCID mice (n=4) were injected intraperitoneally (i.p.) with either untreated MDA-MB-231-luc cells (Untreated) or fraction-irradiated MDA-MB-231-luc cells and then cultured for 24 hours followed by an additional 24 hours of culture with (IR+DSF/Cu) or without (IR) treatment with DSF/Cu. Each group was administered 2x106 cells in 25μL serum- and drug-free RPMI 1640 + 25μL Matrigel/mouse. Tumor growth was monitored in each mouse every 3-4 days by whole animal bioluminescent imaging (BLI) and tumor burden, quantitated as photons/second. Increased tumorigenicity in IR treated and lack of tumorigenicity in IR+DSF/Cu treated, compared to untreated cells, were observed. P values for comparison between groups are shown (C). Using diethylaminobenzaldehyde (DEAB), an inhibitor of ALDH1/3 isoforms, to establish the baseline fluorescence as the gating reference standard of ALDHneg population, FACS sorted MDA-MB-231 ALDHneg cells were fraction-irradiated and then cultured for 24 hours, followed by treatment with different agents for an additional 24 hours, as indicated for mammosphere formation assays. *** indicates P<0.001(D). All experiments (presented in A, B, D) were performed in duplicate and repeated twice, and the means ± SD are shown.
The combination of in vitro treatment with IR and DSF/Cu induced more potent apoptosis of BC cells than either single treatment alone
We reasoned that the depletion of BCSCs/iBCSCs by DSF/Cu could be due to a combination of mechanisms: 1) induction of apoptosis and/or 2) obstruction of conversion of nonstem BC cells into iBCSCs. It is known that DSF/Cu is a potent inducer of apoptosis of BC cells through, at least partially, upregulation of the pro-apoptotic ROS- mitogen-activated protein kinases (MAPK) pathway [27]. We found evidence consistent with this, as p38 MAPK was upregulated and additionally, we found activation of the pro-survival AKT was inhibited in human BC UACC-812 cells treated with a combination of IR and DSF/Cu. These data strongly suggest increased apoptosis in BC cells exposed to this combinatorial treatment vs. DSF/Cu alone (Supplementary Fig. S2).
DSF/Cu blocked the IR-induced stemness via downregulation of the NF-κB-stemness gene pathway in vitro
As DSF/Cu has been shown to be an effective proteasome inhibitor, that can result in inhibition of NF-κB activity [21, 27]. We determined that DSF/Cu was able to markedly inhibit IR-induced upregulation of NF-κB (Supplementary Fig. S3). Studies have also shown that NF-κB plays a major role in regulating the expression of the stemness genes ERBB2 [4], SOX9 [29], MYC [30], and WNT [31]. In this regard, we found potential NF-κB binding sites in the promoter regions of WNT3 for human and mouse (Supplementary Table S1). However, the role of NF-κB in determining stem cell fates of irradiated BC cells has never been investigated. Using two human and one mouse BC cell lines, we detected significant increases in vitro of stemness gene expression of ERBB2, SOX9, and MYC at the mRNA and protein levels in irradiated cells (Fig. 2A, B and Supplementary Table S2). In vitro treatment of irradiated cells with DSF/Cu reduced the expression of these stemness genes at the mRNA as well as protein levels (Fig. 2A, B and Supplementary Table S2). Additionally, we showed that the inhibitory effect of DSF/Cu on stemness gene expression was NF-κB dependent, as treatment with either the NF-κB inhibitor(NF-κBi) IMD-0354 or siRNA knockdown of NF-κB (p65) expression displayed similar stemness inhibition as measured by gene expression and mammosphere formation (Fig. 2A-C). In contrast, treatment with the ROS inhibitor (ROSi) N-Acetyl-L-cysteine (NAC) did not. ROS did not seem to play a role in modulating the NF-κB- stemness gene pathway, since the ROS inhibitor did not impact the ability of either DSF/Cu, IMD-0354 or the NF-κB p65 siRNA to inhibit the expression of stemness genes (Fig. 2A, B).
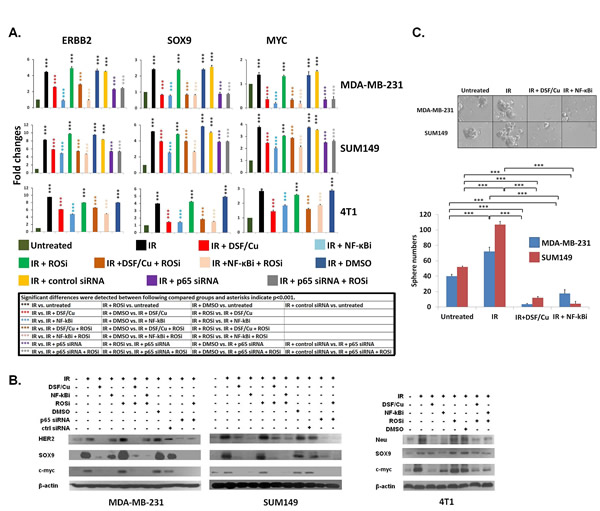
Figure 2: Inhibition by DSF/Cu of IR-induced stemness gene upregulation at the mRNA and protein levels of BC cells in vitro. Non-transfected BC, BC cells transfected with NF-κB p65 siRNA or control siRNA were fraction-irradiated (3.75 Gy/day x 5 days). Non-transfected, fraction-irradiated BC cells were cultured for 24 hours, followed by treatment with different agents as indicated for an additional 24 hours. siRNA transfected fraction-irradiated BC cells were cultured for 24 hours, followed by treatment with NAC for an additional 24 hours. The expression of ERBB2, SOX9 and MYC (genes named by HUGO gene database) was analyzed by real-time quantitative (q) RT-PCR. Gene expression values obtained from treated cells were compared with those obtained from untreated cells, which were standardized to a value of 1. The results of the comparison were expressed in terms of fold change ± SD and P values are shown (A). Treated cells were lysed and analyzed by Western blotting for expression of HER2/Neu, SOX9 and c-myc (proteins named by NCBI protein database) (B). Mammosphere formation assays were performed with cells treated as indicated. Pictures were taken on day 10 (SUM149) and on day 14 (MDA-MB-231). *** indicates P<0.001(C). The concentrations of NF-κBi and ROSi used were1 μM and 10 mM, respectively. All of the experiments were performed in duplicate and repeated twice, and the means ± SD are shown.
DSF blocked IR-induced stemness in vivo resulting in inhibition of both primary tumor growth and spontaneous lung metastasis
To determine the impact of IR on stemness in primary breast tumors, and whether DSF/Cu can counteract IR-induced stemness in vivo, we used the 4T1 cell line-derived syngeneic BALB/c mouse tumor model, which closely resembles human breast cancer and forms spontaneous lung metastases [32]. Consistent with our in vitro findings (Fig. 2A, B), IR increased stemness genes ERBB2, SOX9, and MYC and WNT3 mRNA (1.21-4.52 fold) and protein expression (Fig. 3B-D, Supplementary Table S3). Additionally, the number of mammospheres formed from tumors obtained from IR-treated mice was higher than the number obtained from tumors of mice treated with vehicle, as measured using tumors obtained from these groups of mice one day after the treatment was completed (day 15) and at the end of the experiment (day 29). The increase at day 15 was 61.0%, and at day 29, it was 84.2% (Fig. 3E, F). Although IR yielded a modest reduction in primary tumor volume compared to the vehicle control (20.1%) (Fig. 3A), strikingly, the high level of IR-induced stemness correlated with a 51.7% greater nodular area of spontaneous lung metastases compared to the vehicle control (Fig. 4A). However, we found that the combination of IR and DSF was significantly more effective at suppressing primary tumor growth compared to IR alone (74.2%), DSF alone (71.1%) or vehicle (79.4% ) treated mice on day 29 (Fig. 3A). It should be noted that in contrast to in vitro experiments, that because a high level of endogenous Cu2+ exists in tumor environments, exogenous Cu2+ was not needed in these in vivo-based experiments for DSF efficacy [21, 33]. The higher potency anti-tumor effect of the combinatorial therapy of the mice was associated with a marked reduction of stemness gene expression and mammosphere formation in primary tumors compared to that of mice treated with either IR or DSF alone or vehicle (Fig. 3B-F). On day 15, the combination of IR and DSF already resulted in a reduction of stemness mRNA expression (4.13-6.25 fold) and mammospheres (87.2%) compared to tumors treated with IR alone (Fig. 3B, D, E, Supplementary Table S3). This effect was sustained until day 29, when the tumor diameter of control mice reached the upper allowable limit of 1.5 cm (Fig. 3C, D, F, Supplementary Table S3). The decreased expression of stemness genes (1.92-5.25 fold) was directly related to the functional properties of BCSCs, as noted by the markedly reduced ability of the treated tumor cells to form mammospheres (87.2%) (Fig. 3C, D, F, Supplementary Table S3). Relative to mice treated with vehicle, DSF-treated non-irradiated mice also exhibited a reduction of stemness gene expression (1.88-6.24 fold on day 15; 0-2.73 fold on day 29) and decreased mammosphere formation (60.2% on day 15; 64.8% on day 29) (Fig. 3B-F and Supplementary Table S3). Clearly, the combination of IR and DSF eliminated the majority of BCSCs/iBCSCs in vivo. Consequently, the size of spontaneous lung metastatic nodules was reduced 89.6% in DSF-treated irradiated mice and 20.4% in DSF treated non-irradiated mice, compared to mice treated with vehicle (Fig. 4A). In sharp contrast, as previously noted, there was a 51.7% increase in the size of spontaneous lung metastatic nodules in irradiated mice compared to mice treated with vehicle (Fig. 4A). Toxicity was not observed in the mice enrolled in this study using body weight as an indicator of drug-induced toxicity (Fig. 4B) [34].
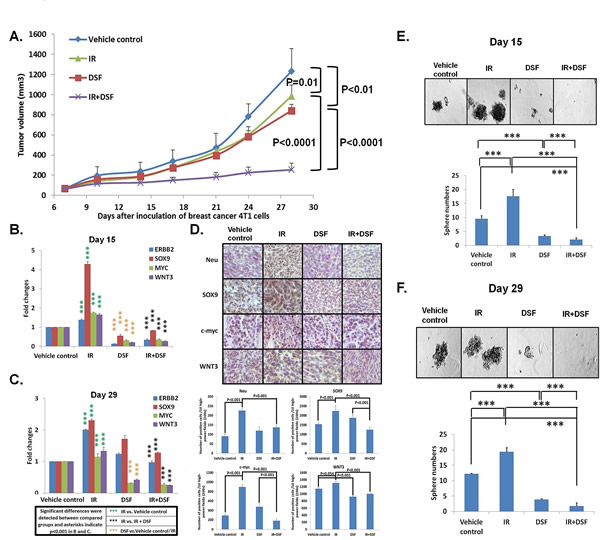
Figure 3: IR + DSF is more potent than either DSF or IR alone in targeting 4T1 tumors in mice. Mouse 4T1 cells were injected subcutaneously (s.c.) into the right hind leg of 28 mice. On day 7, when the tumors reached 5 mm in diameter, mice were assigned in a stratified randomized manner to 4 groups (n=7 per group) and the treatments were initiated. Group 1 (Vehicle control) was treated orally with the vehicle control olive oil. Group 2 (IR) was administered a single dose of IR (20Gy) on day 10. Group 3 (DSF) was treated orally with DSF once daily for 8 days. Group 4 (IR+DSF) was treated orally with DSF, once daily for 8 days and mice were administered a single dose of IR on day 10. Tumor sizes were monitored twice per week. Mean tumor volumes of each group ± SD and P values for comparison between groups are shown (A). On day 15, two mice from each group were sacrificed and their tumors (Day 15 tumors) were collected for analysis of intratumoral levels of stemness gene expression. Total RNA was extracted from fresh 4T1 tumors and analyzed for mRNA of stemness genes by quantitative real-time qRT-PCR. Gene expression values obtained from treated tumors were compared with those obtained from untreated tumors, which were standardized to a value of 1. The results of the comparison were expressed in terms of fold change. The experiments were performed in duplicate and the means of fold change ± SD and P values are shown (B). The remaining 5 mice from each group were sacrificed on day 29 and tumors (Day 29 tumors) were collected for analysis of stemness genes using the same method as described above. The experiments were performed in duplicate and the means of fold change ± SD and P values are shown (C). Tumors from all mice collected on both days 15 and 29 were immunohistochemically (IHC) stained for protein products of stemness genes. Staining results of Neu, SOX9, c-myc and WNT3 were evaluated and expressed as positive cells in 10 high-power (200x) fields.P values for comparison between groups are shown (D). Day 15 tumors (n=2 per group) were digested to test intratumoral BCSCs/iBCSCs using mammosphere formation assays. The experiments were performed in duplicate and the means ± SD are shown. *** indicates P<0.001 (E). Day 29 tumors (n=5 per group) were digested for analysis of intratumoral BCSCs/iBCSCs using the same method as described above. The experiments were performed in duplicate and the means ± SD are shown. *** indicates P<0.001 (F).
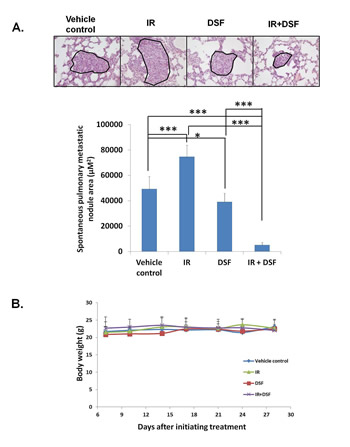
Figure 4: IR+DSF shows potent inhibition of spontaneous lung metastasis counteracting the incremental effect of IR. As described in Fig. 3, at the time of sacrifice (on day 29), lungs were harvested from the mice (n=5 per group) and formalin-fixed, paraffin-embedded lung tissues were H&E stained. Representative images are shown at a 200x magnification. The areas of spontaneous pulmonary metastatic nodules in each section were quantified from 10 randomly selected high-power 200x fields. Means ± SD are shown. *** indicates P<0.001 and *indicates P<0.05(A). Whole body weight of each mouse, a marker for toxicity, was measured twice per week. Means ± SD are shown (B).
Discussion
RT uses high-energy radiation to kill cancer cells and shrink tumors and is one of the major treatments for breast cancer and other types of cancer with curative or palliative intent [35]. However, radioresistant pre-existing BCSCs, and as our paper describes, the presence of radiation-induced BCSCs are challenging for a beneficial outcome of RT as BCSCs are the root of cancer initiation, progression, and metastasis [36, 37]. In an effort to significantly improve the efficacy of RT for breast cancer, we investigated and identified a way to target radioresistant pre-existing BCSCs and block the formation of radiation induced BCSCs with DSF/Cu. Our study shown here not only confirmed independently the finding of radiation induces BCSCs, first reported by F Pajonk’s group [6], but also identified the role of the NF-κB-mediated stemness gene pathway in the generation of IR-induced stem cells from nonstem BC cells. In addition, we found that IR had only a modest effect on controlling primary tumor growth at the same time promoting spontaneous lung metastasis in a syngeneic mouse model system. This surprising and provocative finding was confirmed repeatedly in in vivo experiments using the same mouse model (Fig. 4A, B and data not shown). The significant increase in lung metastasis in IR treated mice compared to vehicle treated mice is likely attributed to IR-induced stemness (Fig. 3B-F). Nevertheless, it is noteworthy that the inhibition of primary tumor growth may also be a contributing factor to significant decrease in lung metastasis in IR and DSF treated mice compared to those treated with either IR or DSF alone (Fig. 3A, Fig. 4A). When comparing IR and DSF with DSF alone, it was shown that the products of the four stemness genes at the mRNA level in day 15 tumors were comparable in both groups but markedly reduced compared to those detected in tumors of mice treated with IR. However, in day 29 tumors, mRNA expression of c-myc and WNT3 were reduced in both groups of mice relative to the IR group, but ERBB2 and SOX9 mRNA levels were significantly reduced only in the tumors of mice treated with IR and DSF (Fig.3B, C). Of the four stemness gene products analyzed in day 15 and 29 tumors at the protein level, the expression of SOX-9 and c-myc, but not ERBB2 and WNT3, was significantly lower in the mice treated with the IR and DSF than DSF alone. Furthermore, the number of mammospheres of tumors treated with IR and DSF, however, was significantly lower than that of tumors treated with DSF alone (Fig. 3E, F). Therefore, it seems that SOX9 and c-myc might play more dominant roles than ERBB2 and WNT3 in determining tumor stemness in this tumor model.
Through investigation of the mechanism underling the inhibition of stemness gene expression, we found that DSF/Cu and NF-κB inhibitors, which both result in a blockade of NF-κB signaling, prevented the formation of iBCSCs. The downregulation of NF-κB signaling by DSF/Cu or the NF-κB inhibitor or NF-κB p65 siRNA knockdown significantly blocked IR-induced expression of stemness genes ERBB2, SOX9, MYC and WNT3 (Fig. 2A, B). As a result of these events, markedly decreased or depleted BCSCs/iBCSCs were detected using in vitro and in vivo-based assays, namely ALDH activity as a marker (Fig. 1A), mammosphere formation (Fig. 1B, D, Fig. 2C ), tumorigenicity in vivo (Fig. 1C) , mammosphere formation of digested primary tumor cells (Fig. 3E, F) and spontaneous lung metastasis (Fig. 4A). The information from our study contributes to our understanding of the bidirectional conversion between iBCSCs and nonstem BC cells and introduces the novel concept of blocking the formation of iBCSCs as a result of IR. Although we focused only on the NF-κB- stemness gene pathway involving stemness genes ERBB2, SOX9, and MYC and WNT3 in our present study, we suspect other stemness genes or pathways may be involved as well in the process of IR-induced stemness [6, 11]. It will be well worth pursuing studies on this aspect of the BCSCs/iBCSCs challenge to RT.
RT plays a significant role in the management of early stage, locoregionally advanced, and metastatic breast cancer [38]. The combinatorial approach of RT with DSF/Cu, in which Cu2+ is endogenously present in tumors, we have described here provides a means to not only induce apoptosis of radioresistant pre-existing BCSCs, but also prevent the conversion of nonstem BC cells into radiation-induced BCSCs. It is worth noting that although addition of Cu2+ to DSF is definitely needed for in vitro depletion of BCSCs/iBCSCs (Fig. 1A, B, D) [27], exogenous copper should not be administered in conjunction with clinical use of DSF given the fact that many tumors including breast cancer tissues contain high levels of copper compared to normal tissues [39-44] and the beneficial effect of the copper depleting agent tetrathiomolybdate (TM) on reducing relapse in breast cancer patients[45]. Indeed, our in vivo data confirmed that exogenous copper gluconate administered orally (1mg/kg /day) with DSF(50 mg/kg/day) and IR (20Gy, once) was significantly less effective than DSF and IR without copper gluconate in inhibiting primary tumor growth and lung metastasis in the 4T1 mouse model system (data not shown). Therefore, one should be cautious on designing a clinical trial for cancer treatment using DSF and copper together.
In summary, our study offers a solid foundation for an immediate and practicable strategy of increasing the efficacy and curability of radiation therapy for breast cancer patients using a long established, safe and low cost drug DSF. Moreover, our study i) introduces the novel concept of blocking iBCSCs through targeting the NF-κB-stemness gene pathway, ii) reports the finding that IR, delivered locally to the primary mammary tumors, promotes distant lung metastasis in mice, and iii) contributes to a paradigm shift in understanding of the importance of targeting CSCs and blocking the formation of radiation induced CSCs in RT of cancers, and therefore leading to the next generation of radiotherapy.
Materials and Methods
Cell culture
The human BC cell lines MDA-MB-231, MDA-MB-231-luc-D3H1, SUM149, UACC-812 and mouse BC cell line 4T1 were cultured in RPMI 1640 medium (Mediatech) supplemented with 2 mmol/L L-glutamine (Mediatech) and 10% fetal calf serum (FCS; Atlanta Biologicals). This cell culture medium is referred to as complete medium. Normal human mammary epithelial cells were purchased from Lifeline Cell Technology and cultured using MammaryLife Medium Complete Kit (Lifeline Cell Technology). All cells were cultured at 37°C in a 5% CO2 atmosphere. MDA-MB-231 and SUM149 cell lines were acquired from the Duke Comprehensive Cancer Center Cell Culture Facility. UACC-812 and 4T1 cell lines were purchased from the American Type Culture Collection (ATCC). The MDA-MB-231-luc-D3H1 cell line was obtained from Xenogen Corporation.
Chemical reagents and antibodies
Tetraethylthiuram disulfide (disulfiram, DSF), Copper Chloride or Copper(II) D-gluconate (Cu), NF-κB inhibitor IMD-0354, and ROS inhibitor NAC were purchased from Sigma-Aldrich. DSF and IMD-0354 were reconstituted in DMSO for all in vitro experiments. DSF was reconstituted in olive oil for in vivo experiments. Cu was reconstituted in distilled water and NAC was reconstituted in RPMI 1640 medium for all experiments.
Antibodies for Western blotting: human and mouse HER2/ERBB2 (#2242) (1:1000 dilution)-, human and mouse c-myc (#9402) (1:1000 dilution)-, human cleaved PARP (#9541) (1:1000 dilution), human phosphorylated (p)-p38 MAPK (#9211) (1:1000 dilution)-, human p38 MAPK (#9212) (1:1000 dilution)-, human p-AKT (#9271) (1:1000 dilution), human AKT (#4685) (1:1000 dilution), and human and mouse β-actin (#4970) (1:2000 dilution)-specific rabbit monoclonal antibodies (mAbs) and goat anti-rabbit IgG, HRP-linked antibody (#7074) (1:2000) were purchased from Cell Signaling Technology. Human and mouse SOX9-specific rabbit antibody (Ab) (ab26414) (1μg/mL) was purchased from Abcam. All antibodies were diluted in Tris Buffered Saline with 0.1% Tween® 20 (TBST) containing 5% nonfat dry milk plus 2% bovine serum albumin (BSA). All dilutions were prepared immediately before use.
Antibodies for IHC staining: mouse Neu-specific rabbit Ab (#sc-284) (1:250 dilution) and mouse SOX9-specific rabbit Ab (#sc-20095) (1:200 dilution) were purchased from Santa Cruz Biotechnology. Mouse c-myc-specific rabbit mAb (#9402) (1:500 dilution) was purchased from Cell Signaling Technology. Mouse WNT3-specific rabbit Ab (#ab32249) (1μg/mL) was purchased from Abcam. All antibodies were diluted in TBST containing 5% normal horse serum and 1% BSA. All dilutions were prepared immediately before use. D-Luciferin Firefly, potassium salt was purchased from Xenogen. The Dual-Glo™ Luciferase Assay System, pGL4.32[luc2P/NF-κB-RE/Hygro]vector, and pRL-SV40 vector were purchased from Promega.
Animals
Six week old NOD/SCID female mice were purchased from Taconic and 6 week old BALB/c female mice were purchased from the Massachusetts General Hospital COX7 animal facility. All the animal studies have been approved by the Institutional Animal Care and Use Committee.
Flow cytometry and cell sorting
Cells were collected and were then incubated with ALDEFLUOR® reagent (Stem Cell Technologies), with or without the ALDH inhibitor DEAB according to the manufacturer’s instructions. ALDHneg cells were sorted using a BD Biosciences FACSAria II cell sorter.
Irradiation (IR)
In vitro fractionated IR was performed on cells plated in 6-well plates at a density of 2x105 cells/ well in 2 mL of complete medium at 3.75Gy daily for 5 days unless otherwise specified. In vitro a single dose of IR was performed on cells plated in 6-well plates at a density of 2x105 cells/ well in 2 mL of complete medium at 10Gy. In vivo, a single dose of 20Gy was delivered locally to each mouse tumor. The Mark I Model 30 Cesium Irradiator (JL Shepherd and Associates) was used for all IR experiments in this study.
Mammosphere formation
Cells were plated in 6-well plates at a density of 2x105 cells/ well in 2 mL of complete medium and received fractionated irradiation. Twenty-four hours after irradiation, cells were treated with DSF/Cu (2.5 µM/1 µM) or 1 µM NF-κB inhibitor IMD0354 for an additional 24 hours. Mammosphere formation was performed by seeding the cells (1000 cells/ well) in a 24-well ultra-low adherent plate (Corning Incorporated) in 500 μL of mixed medium containing 32% MethoCult medium, 20% MammoCult basal human medium with a final concentration of 2% MammoCult proliferation supplements (STEMCELL Technologies), and 48% DMEM supplemented with final concentrations of 100 pg/mL EGF, 50 ng/mL bFGF, 5 ng/mL stem cell factor, 1x10-6 M hydrocortisone, and 5 μg/mL insulin. The cells were cultured at 37°C in a 5% O2 and 5% CO2 humidified atmosphere for 14 days. Mammosphere pictures were taken using a Zeiss Inverted Fluorescence Microscope on day 10 or 14. Numbers of mammosphere were count on day 14. Mammosphere formation was also performed by seeding (1000 cells/ well) in a 24-well ultra-low adherent plate of the Fluorescence Activated Cell Sorting (FACS) sorted ALDHneg cells or cells from a single cell suspension collected from a disaggregated tumor.
Transfection with siRNA
MDA-MB-231 and SUM149 cells were plated in 12-well plates at a density of 4x104 cells/ well and 3.2x104 cells/ well in 0.5mL of complete medium, respectively. Cells were transfected with 100 nmol Signalsilence NF-κB p65 siRNA (#6261; Cell signaling Technology) using the X-tremeGENE siRNA Transfection Reagent (Roche) following the manufacturer’s instructions. SignalSilence Control siRNA (#6201; Cell Signaling technology) was used as a control. Cells were then incubated for 48 hours before initiating specified treatments.
Western blotting analysis
Cells were plated in 6-well plates at a density of 2x105 cells/ well in 2 mL of complete medium and were treated as indicated. Cells were collected and lysed in lysis buffer (10 mM Tris–HCl (pH 8.2), 1% NP40, 1 mM EDTA, 0.1% BSA, 150 mM NaCl) containing 1/50 (vol/vol) of a protease inhibitor cocktail (Calbiochem), or proteins were extracted from frozen mouse tumors by homogenization in the presence of 1 mL of lysis buffer. Western blotting for signaling-related proteins and proteins of stemness genes was carried out as described [46]. The investigators who analyzed the samples from the tumors were blinded to the type of treatment received by the mice.
Cell proliferation and MTT assay
Cells were plated in triplicate in 96-well plates at a density of 2.5x103 cells/ well in 100 uL of complete medium and were treated as indicated. Cell proliferation was evaluated by an MTT assay at the indicated time point. MTT assays were carried out as described [46].
Real-Time qRT-PCR
Cells were plated in 6-well plates at a density of 2x105 cells/ well in 2 mL of complete medium and were treated as indicated. Total RNA from all in vitro experimental samples or frozen mouse tumors was isolated using TRIzol (Life Technologies), and was reverse transcribed using M-MLV reverse transcriptase (Life Technologies) according to the manufacturer’s instructions. All samples within each experiment were reverse transcribed at the same time. The resulting cDNA was diluted to a concentration of 100 ng/mL in nuclease-free water and was stored in aliquots at –20°C until use. Real-time qPCR using cDNA as a template with SYBR green detection was performed using a LightCycler 96 system (Roche) with FastStart SYBR Green Master Mix (Roche) and primers for each gene as shown in Supplementary Table S4. Appropriate no-RT and non-template controls were included in each 96-well PCR reaction, and dissociation analysis was performed at the end of each run to confirm the specificity of the reaction. Each gene expression level was normalized to mRNA levels of the housekeeping gene r18S. Fold changes of each gene’s expression was compared to that of the untreated sample using the 2(-Delta Delta C(T)) method [47].
Dual-Glo luciferase assay
MDA-MB-231 cells were plated in 12-well plates at a density of 4x104 cells/ well in 0.5 mL of complete medium and cultured for 24 hours, the pGL4.32[luc2P/NF-3ba;B-RE/Hygro] and pRL-SV40 vectors were co-transfected into adherent cells. After 24 hours, the transfected cells were irradiated with a single dose of 10 Gy and were then treated with DSF/Cu (2.5 µM/1 µM) or 1 µM IMD-0354 for 24 hours. A dual luciferase assay was then performed with these cells using the Dual-Glo luciferase assay kit according to the manufacturer’s instructions. NF-κB luciferase activity, normalized to Renilla luciferase activity, was expressed relative to that of the untreated cells, which was set at 1.0.
In vivo tumorigenicity assay
In vitro, MDA-MB-231-luc-D3H1 cells were fraction-irradiated at 3.75 Gy/day for 5 days and were cultured for 24 hours followed by an additional 24 hours with or without the treatment of DSF/Cu (2.5 µM/1 µM). Three groups of NOD/SCID mice (n=4) were injected i.p. with either untreated MDA-MB-231-luc-D3H1 cells, irradiated MDA-MB-231-luc-D3H1 cells treated with DSF/Cu, or only irradiated MDA-MB-231-luc-D3H1 cells. The same number of cells was used for each group, with 2x106 cells in 25 μL RPMI 1640 and 25 μL Matrigel injected i.p. in each mouse. Tumorigenicity measured as tumor growth was monitored in each mouse every 3-4 days by bioluminescence image (BLI) and tumor burden quantitated as photons/second.
In vivo syngeneic tumor model
Mouse BC 4T1 cells (5x105 cells/mouse) were implanted subcutaneously in the thigh of right hind leg of BLAB/c mice. Body weight and tumor volume were measured twice per week. Tumor volume was measured by digital calipers. Treatments were initiated on day 7 when the tumor developed and had an approximate diameter of 5 mm. Mice were divided into 4 groups (n=7) using a stratified randomization strategy, such that the difference of mean tumor volumes was not statistically significant between each group (P>0.5). Mice in group 1 were treated with olive oil (vehicle control, 200 µL) orally from day 7 to day 14. Mice in group 2 were treated with a 20Gy irradiation on day 10. Mice in group 3 were treated with oral administration of DSF (50 mg/kg/day) from day 7 to day 14. Mice in group 4 were treated with a 20Gy irradiation on day 10 and oral administration of DSF (50 mg/kg/day) from day 7 to day 14. All oral administration was given by an oral gavage using an 18 gauge plastic feeding tube (Solomon Scientific). When tumor diameters from untreated mice reached 1.5 cm, all mice were sacrificed. Primary tumors and lungs were collected for further analysis.
Samples preparations from primary mouse tumors
For mammosphere formation assay: a non-necrotic primary tumor tissue specimen from each mouse was collected at the time of sacrifice, minced into 3x3 mm pieces and digested with Collagenase IV (1mg/mL PBS)(Worthington Biochemical Corp.) at 37°C for 1 hour. A single cell suspension was obtained by filtering the digested tumor through a 40μM cell strainer.
For total RNA and protein extractions: a non-necrotic primary tumor tissue specimen from each mouse was collected at the time of sacrifice, minced (3x3 mm) and stored at -80°C for total RNA or protein extraction.
For IHC staining: the remaining non-necrotic tumor tissue specimen from each mouse was collected at the time of sacrifice and formalin-fixed and paraffin-embedded (FFPE). FFPE blocks were sectioned using MICROM HM325 Rotary Microtome (Thermo Fisher Scientific Inc).
Analysis of areas of lung metastasis
At the time of sacrifice, mouse lungs were collected as described above for histological analysis of lung metastasis. Four micron-thick formalin-fixed paraffin-embedded (FFPE) sections of mouse lung tissue from treated BALB/c mice bearing 4T1 cell tumors were stained with Mayer’s Hematoxylin (Sigma-Aldrich) and Eosin Y solution (Sigma-Aldrich) according to the manufacturer’s instructions. The areas of the metastatic nodules in 10 randomly selected high-power fields (200x magnification) per section were measured and calculated by the SPOT Advanced Imaging software (Diagnostic Instruments). The research fellow who analyzed the lungs was blinded to the type of treatment received by the mice.
IHC staining
Four micron-thick tissue sections were deparaffinized and subjected to antigen retrieval. Following incubation with blocking buffer, slides were incubated with anti-Neu, -SOX9, -c-myc, and -WNT3 Abs overnight. Sections were then rinsed with TBST, followed by 45-minute incubation with the EnVision™+ Dual Link System-HRP goat anti-rabbit Ig (Dako). The detection of staining signals was performed with the DAB Peroxidase Substrate Kit from Dako, and the sections were counterstained with hematoxylin. Staining results of stemness gene protein were analyzed by two investigators who were blinded to the type of treatment received by the mice, by counting the positive cells in 10 randomly selected high-power (200x) fields. Complete membrane staining was considered as positive for Neu expression; nuclear staining was considered as positive for SOX9 and c-myc expression, and cell membrane and cytoplasm staining were considered as positive for WNT3 expression.
Statistical analysis
For in vivo tumor volumes, estimation of the treatment group specific mean responses over time and their comparisons were performed by using longitudinal general linear mixed effects model with compound symmetry error covariance structure. The subject level intercepts were random, and both time effect and time x treatments interactions were considered fixed effects. The longitudinal treatment effects were compared by comparing the treatment specific slopes by linear contrast. For all other data analysis, averages, standard deviations, and unpaired t-tests were calculated using Microsoft Excel. In vitro data shown represent the mean ± SD of the results obtained in at least three independent experiments. In vivo data shown represent the mean ± SD of the results obtained in each group. Differences between groups were considered significant when the P value was < 0.05.
Conflict of Interest Statement
The authors declare that they have no competing financial interests.
Author Contributions
Conception and design: X. Wang and Y. Wang.
Development of methodology: Y. Wang and X. Wang.
Acquisition of data: Y. Wang, W. Li, J. Cong, N. Zhang, S. S. Patel, F. Sabbatino, X. Liu and Y. Qi.
Analysis and interpretation of data: X. Wang, Y. Wang and H. Lee.
Writing, review and/or revision of the manuscript: X. Wang, Y. Wang , S. S. Patel. and A.B. DeLeo.
Study supervision: X. Wang.
Technical advice: P. Huang, M. W. Epperly, A. Ly, E. F. Brachtel.
Conceptual advice: A. Taghian, J. Li, A. B. DeLeo, S. Ferrone and C. R. Ferrone.
Grant Support
This study was supported by NIH grant RO3 CA141086 (X.W.), Susan G. Komen Postdoctoral Fellowship KG111486 (Y.W.) and NIH grants R01-1CA138188 (S. F.), 2P50CA121973-06 (S. F.) and 7R01CA110249-06 (S. F.), a Fondazione Umberto Veronesi Post Doctoral Fellowship awarded by the Fondazione Umberto Veronesi (F. S.).
References
1. American Cancer Society. (2013). Breast cancer radiation therapy. http://www.cancer.org/cancer/breastcancer/detailedguide/breast-cancer-treating-radiation
2. National Collaborating Centre for Cancer (UK). Early and Locally Advanced Breast Cancer: Diagnosis and Treatment [Internet]. Cardiff (UK): National Collaborating Centre for Cancer (UK); 2009 Feb. (NICE Clinical Guidelines, No. 80.) 6, Adjuvant radiotherapy. Available from: http://www.ncbi.nlm.nih.gov/books/NBK11634/.
3. Clarke M, Collins R, Darby S, Davies C, Elphinstone P, Evans E, Godwin J, Gray R, Hicks C, James S, MacKinnon E, McGale P, McHugh T, Peto R, Taylor C, Wang Y, et al. Effects of radiotherapy and of differences in the extent of surgery for early breast cancer on local recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005; 366(9503):2087-2106.
4. Duru N, Fan M, Candas D, Menaa C, Liu HC, Nantajit D, Wen Y, Xiao K, Eldridge A, Chromy BA, Li S, Spitz DR, Lam KS, Wicha MS and Li JJ. HER2-associated radioresistance of breast cancer stem cells isolated from HER2-negative breast cancer cells. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012; 18(24):6634-6647.
5. Phillips TM, McBride WH and Pajonk F. The response of CD24(-/low)/CD44+ breast cancer-initiating cells to radiation. Journal of the National Cancer Institute. 2006; 98(24):1777-1785.
6. Lagadec C, Vlashi E, Della Donna L, Dekmezian C and Pajonk F. Radiation-induced reprogramming of breast cancer cells. Stem Cells. 2012; 30(5):833-844.
7. Takahashi K and Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006; 126(4):663-676.
8. Li Y and Laterra J. Cancer stem cells: distinct entities or dynamically regulated phenotypes? Cancer Research. 2012; 72(3):576-580.
9. Guo W, Keckesova Z, Donaher JL, Shibue T, Tischler V, Reinhardt F, Itzkovitz S, Noske A, Zurrer-Hardi U, Bell G, Tam WL, Mani SA, van Oudenaarden A and Weinberg RA. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell. 2012; 148(5):1015-1028.
10. Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A and Weinberg RA. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nature Genetics. 2008; 40(5):499-507.
11. Ghisolfi L, Keates AC, Hu X, Lee DK and Li CJ. Ionizing radiation induces stemness in cancer cells. PloS One. 2012; 7(8):e43628.
12. Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, Schott A, Hayes D, Birnbaum D, Wicha MS and Dontu G. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem cell. 2007; 1(5):555-567.
13. Luo M, Fan H, Nagy T, Wei H, Wang C, Liu S, Wicha MS and Guan JL. Mammary epithelial-specific ablation of the focal adhesion kinase suppresses mammary tumorigenesis by affecting mammary cancer stem/progenitor cells. Cancer Research. 2009; 69(2):466-474.
14. Visus C, Wang Y, Lozano-Leon A, Ferris RL, Silver S, Szczepanski MJ, Brand RE, Ferrone CR, Whiteside TL, Ferrone S, DeLeo AB and Wang X. Targeting ALDH(bright) human carcinoma-initiating cells with ALDH1A1-specific CD8(+) T cells. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011; 17(19):6174-6184.
15. Marcato P, Dean CA, Giacomantonio CA and Lee PW. Aldehyde dehydrogenase: its role as a cancer stem cell marker comes down to the specific isoform. Cell Cycle. 2011; 10(9):1378-1384.
16. Visus C, Ito D, Amoscato A, Maciejewska-Franczak M, Abdelsalem A, Dhir R, Shin DM, Donnenberg VS, Whiteside TL and DeLeo AB. Identification of human aldehyde dehydrogenase 1 family member A1 as a novel CD8+ T-cell-defined tumor antigen in squamous cell carcinoma of the head and neck. Cancer Research. 2007; 67(21):10538-10545.
17. Khoury T, Ademuyiwa FO, Chandrasekhar R, Jabbour M, Deleo A, Ferrone S, Wang Y and Wang X. Aldehyde dehydrogenase 1A1 expression in breast cancer is associated with stage, triple negativity, and outcome to neoadjuvant chemotherapy. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2012; 25(3):388-397.
18. Eneanya DI, Bianchine JR, Duran DO and Andresen BD. The actions of metabolic fate of disulfiram. Annual Review of Pharmacology Toxicology. 1981; 21:575-596.
19. Lam JP, Mays DC and Lipsky JJ. Inhibition of recombinant human mitochondrial and cytosolic aldehyde dehydrogenases by two candidates for the active metabolites of disulfiram. Biochemistry. 1997; 36(44):13748-13754.
20. Johansson B. A review of the pharmacokinetics and pharmacodynamics of disulfiram and its metabolites. Acta psychiatrica Scandinavica Supplementum. 1992; 369:15-26.
21. Chen D, Cui QC, Yang H and Dou QP. Disulfiram, a clinically used anti-alcoholism drug and copper-binding agent, induces apoptotic cell death in breast cancer cultures and xenografts via inhibition of the proteasome activity. Cancer Research. 2006; 66(21):10425-10433.
22. Doytcheva MA and Jeliazkova BG. Structure of copper(II) dithiocarbamate mixed-ligand complexes and their photoreactivities in alcohols. Spectrochimica acta Part A, Molecular and biomolecular spectroscopy. 2004; 60(6):1299-1305.
23. Cen D, Brayton D, Shahandeh B, Meyskens FL, Jr. and Farmer PJ. Disulfiram facilitates intracellular Cu uptake and induces apoptosis in human melanoma cells. Journal of Medicinal Chemistry. 2004; 47(27):6914-6920.
24. Watts DL. The Nutritional Relationships of Copper. Journal of Orthomolecular Medicine. 1989; 4(2):10.
25. Nishikawa T, Lee IS, Shiraishi N, Ishikawa T, Ohta Y and Nishikimi M. Identification of S100b protein as copper-binding protein and its suppression of copper-induced cell damage. The Journal of Biological Chemistry. 1997; 272(37):23037-23041.
26. Tisato F, Marzano C, Porchia M, Pellei M and Santini C. Copper in diseases and treatments, and copper-based anticancer strategies. Medicinal Research Reviews. 2010; 30(4):708-749.
27. Yip NC, Fombon IS, Liu P, Brown S, Kannappan V, Armesilla AL, Xu B, Cassidy J, Darling JL and Wang W. Disulfiram modulated ROS-MAPK and NFkappaB pathways and targeted breast cancer cells with cancer stem cell-like properties. British Journal of Cancer. 2011; 104(10):1564-1574.
28. Ahmed KM and Li JJ. NF-kappa B-mediated adaptive resistance to ionizing radiation. Free Radical Biology & Medicine. 2008; 44(1):1-13.
29. Sun L, Mathews LA, Cabarcas SM, Zhang X, Yang A, Zhang Y, Young MR, Klarmann KD, Keller JR and Farrar WL. Epigenetic Regulation of SOX9 by the NF-kappaB Signaling Pathway in Pancreatic Cancer Stem Cells. Stem Cells. 2013; 31(8):1454-1466.
30. La Rosa FA, Pierce JW and Sonenshein GE. Differential regulation of the c-myc oncogene promoter by the NF-kappa B rel family of transcription factors. Molecular and Cellular Biology. 1994; 14(2):1039-1044.
31. Schwitalla S, Fingerle AA, Cammareri P, Nebelsiek T, Goktuna SI, Ziegler PK, Canli O, Heijmans J, Huels DJ, Moreaux G, Rupec RA, Gerhard M, Schmid R, Barker N, Clevers H, Lang R, et al. Intestinal Tumorigenesis Initiated by Dedifferentiation and Acquisition of Stem-Cell-like Properties. Cell. 2013; 152(1-2):25-38.
32. Pulaski B and Ostrand-Rosenberg S. (2000). Mouse 4T1 breast tumor model. In: Coligan J, Bierer B, Margulies D, Shevach E, Strober W and Coico R, eds. Current Protocols in Immunology. (New York: John Wiley), pp. 20.22.21–20.22.16.
33. Rae C, Tesson M, Babich JW, Boyd M, Sorensen A and Mairs RJ. The role of copper in disulfiram-induced toxicity and radiosensitization of cancer cells. Journal of nuclear medicine : official publication, Society of Nuclear Medicine. 2013; 54(6):953-960.
34. Cao S, Durrani FA, Toth K and Rustum YM. Se-methylselenocysteine offers selective protection against toxicity and potentiates the antitumour activity of anticancer drugs in preclinical animal models. British Journal of Cancer. 2014; 110(7):1733-1743.
35. Lawrence TS, Ten Haken RK and Giaccia A. (2008). Principles of Radiation Oncology. In: DeVita VTJ, Lawrence TS and Rosenberg SA, eds. Cancer: Principles and Practice of Oncology. (Philadelphia: Lippincott Williams and Wilkins).
36. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ and Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proceedings of the national academy of sciences of the United States of America. 2003; 100(7):3983-3988.
37. Owens TW and Naylor MJ. Breast cancer stem cells. Frontiers in Physiology. 2013; 4:225.
38. McCloskey SA, Lee SP and Steinberg ML. Roles and types of radiation in breast cancer treatment: early breast cancer, locoregionally advanced, and metastatic disease. Current Opinion in Obstetrics & Gynecology. 2011; 23(1):51-57.
39. Kuo HW, Chen SF, Wu CC, Chen DR and Lee JH. Serum and tissue trace elements in patients with breast cancer in Taiwan. Biological Rrace Element Research. 2002; 89(1):1-11.
40. Rizk SL and Sky-Peck HH. Comparison between concentrations of trace elements in normal and neoplastic human breast tissue. Cancer Research. 1984; 44(11):5390-5394.
41. Huang YL, Sheu JY and Lin TH. Association between oxidative stress and changes of trace elements in patients with breast cancer. Clinical Biochemistry. 1999; 32(2):131-136.
42. Turecky L, Kalina P, Uhlikova E, Namerova S and Krizko J. Serum ceruloplasmin and copper levels in patients with primary brain tumors. Klinische Wochenschrift. 1984; 62(4):187-189.
43. Diez M, Arroyo M, Cerdan FJ, Munoz M, Martin MA and Balibrea JL. Serum and tissue trace metal levels in lung cancer. Oncology. 1989; 46(4):230-234.
44. Nayak SB, Bhat VR, Upadhyay D and Udupa SL. Copper and ceruloplasmin status in serum of prostate and colon cancer patients. Indian Journal of Physiology and Pharmacology. 2003; 47(1):108-110.
45. Jain S, Cohen J, Ward MM, Kornhauser N, Chuang E, Cigler T, Moore A, Donovan D, Lam C, Cobham MV, Schneider S, Hurtado Rua SM, Benkert S, Mathijsen Greenwood C, Zelkowitz R, Warren JD, et al. Tetrathiomolybdate-associated copper depletion decreases circulating endothelial progenitor cells in women with breast cancer at high risk of relapse. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2013; 24(6):1491-1498.
46. Yu L, Favoino E, Wang Y, Ma Y, Deng X and Wang X. The CSPG4-specific monoclonal antibody enhances and prolongs the effects of the BRAF inhibitor in melanoma cells. Immunologic Research. 2011; 50(2-3):294-302.
47. Livak KJ and Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001; 25(4):402-408.