
INTRODUCTION
As a key structural component of the glomerular filtration barrier, glomerular podocytes play a critical role in maintaining the homeostasis of glomerular filtration rate and glomerular permselectivity [1-3]. Podocyte dysfunction or injury could be induced in a variety of acquired or congenital diseases by diverse immune or non-immune mediated mechanisms, such as podocytopathic effects of permeability factors generated following cellular or humoral immune reactions [4], direct podocyte toxicity caused by nephrotoxic substances [3], hemodynamics-elicited mechanical stress, oxidative damage, metabolic stress and others. Recently, a growing body of evidence suggests that podocytes, upon injury, may transdifferentiate and acquire de novo immune cell-like phenotypes [5-14]. By far, a number of immune molecules have been reported to be newly expressed in podocytes under diverse disease conditions, such as systemic autoimmune disease like lupus [9], primary glomerulopathies like membranous nephropathy or focal segmental glomerulosclerosis (FSGS) [15], or metabolic diseases like diabetes [16]. The de novo immune molecules consist of diverse proinflammatory cytokines, such as interleukin-1 a and β [5], and chemokines, like CC chemokine ligands (CCL) 2 and 5 [6, 7]. Besides, multiple immune cell surface marker proteins that are pivotal for mediating cell-mediated immune response, such as major histocompatibility complex (MHC) class II [8], dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin [9], and the costimulatory molecule CD80 [11], have also been shown in diseased podocytes. Moreover, lysosomal protease cathepsin L [12], which is primarily found in leukocytes and crucial for immune reactions [17], could be induced in stressed podocytes. The exact role of these newly acquired immune phenotypes in the pathogenesis of podocyte or glomerular injury remains unclear. But there is accumulating evidence suggesting that podocytes may behave as nonhematopoietic professional antigen-presenting cells that are able to trigger adaptive immune responses [10, 13-15]. On the other hand, many immune molecules, like the costimulatory molecule CD80 and lysosomal protease cathepsin L, function at the interface between innate/adaptive immunity and cytoskeletal remodeling [11, 18]. De novo expression of these immune molecules has been associated with impairment of cytoskeletal integrity in podocytes [11, 12, 18, 19], denoting that podocyte acquisition of these immune phenotypes may be pathogenic. Of note, nuclear factor (NF) kB signaling is commonly essential for expression of all immune molecules [20]. The NFkB-driven expression of numerous NFkB-dependent immune molecules is finely regulated by glycogen synthase kinase (GSK)3β [21, 22], which is a well-conserved, ubiquitous serine/threonine protein kinase originally characterized as one that regulates glucose metabolism [23] but lately implicated as an indispensable element for NFkB activation and proinflammatory responses [21, 24]. Thus, GSK3β may serve as a potential target for treating podocyte injury and glomerular disease.
GSK3β is a druggable target with several clinically available drugs known to possess GSK3β inhibitory activities [25]. Among these, valproate, a short branched-chain fatty acid and an FDA approved first line anticonvulsant and mood stabilizer, is a potent inhibitor of GSK3β that can effectively block the activity of GSK3β at concentrations similar to those attained clinically [26, 27]. It has been known for many years that valproate confers a renoprotective effect on diverse experimental kidney diseases, including Adriamycin nephropathy [28] and diabetic nephropathy [29, 30], resulting in prominent improvement in kidney function, proteinuria and renal histology, including diminished renal inflammation and podocyte and glomerular injury. However, the underlying mechanism is not clear. It remains uncertain whether valproate confers a direct podocyte protective effect and whether its inhibitory activity on GSK3β is implicated. This study employed an in vitro model of podocyte injury elicited by lipopolysaccharide (LPS), a prototypical pathogen-associated molecular pattern found on the outer membrane of Gram-negative bacteria [31], to determine the direct effect of valproate on podocyte injury and to decipher the role of GSK3β signaling. The in vitro findings were validated in a murine model of LPS-elicited albuminuria and podocyte injury.
RESULTS
Valproate mitigates podocyte acquisition of de novo immune phenotypes upon LPS injury
Conditionally immortalized murine podocytes were cultured and differentiated under nonpermissive condition [32]. LPS is a typical injurious stimulus that is able to elicit podocyte injury and dysfunction both in vivo [33] and in vitro [3]. Indeed, following exposure to LPS (100µg/ml) for 24 h, podocytes newly expressed evident immune phenotypes that were either absent or scant under basal conditions. These include MHC II, costimulatory molecule CD80, lysosomal protease cathepsin L and proinflammatory cytokines like CCL2 and 5, as shown by immunoblot analysis of cell lysates or conditioned media (Figure 1). This inducible effect of LPS on podocyte expression of all immune molecules was substantially mitigated by concomitant treatment with valproate in a dose-dependent fashion as evidenced by immunoblot analysis (Figure 1A) followed by densitometry (Figure 1B).
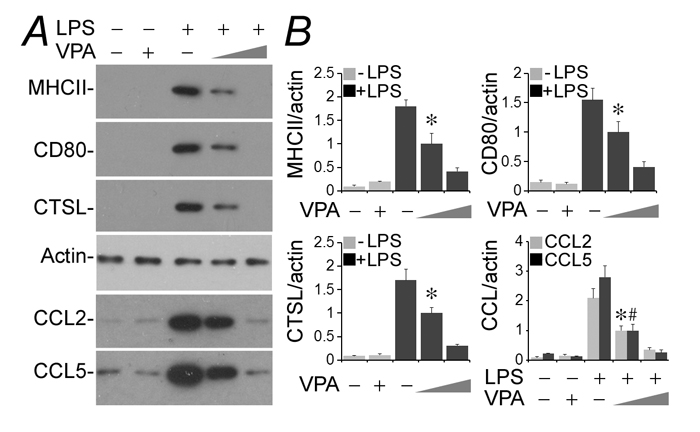
Figure 1: Valproate hinders podocyte acquisition of de novo immune phenotypes following LPS exposure. Immortalized murine podocytes were cultured and differentiated under nonpermissive conditions. Cells were injured with LPS (100µg/ml) or vehicle for 24 h in the presence or absence of sodium valproate (VPA; 0.5 or 1.5 mM). A. Cell lysates or conditioned media were collected and prepared for immunoblotting for indicated proteins. B. Immunoblots were subjected to densitometric analysis and arbitrary units were expressed respectively as immunoblot densitometric ratios of diverse molecules to actin as folds of a designated group. *P < 0.05 versus the same molecule in other groups; #P < 0.05 versus CCL5 levels in other groups (n = 3). CTSL, cathepsin L; LPS, lipopolysaccharide; VPA, sodium valproate.
Valproate attenuates podocyte shape changes and actin cytoskeleton disorganization associated with LPS-induced acquisition of de novo immune phenotypes
Evidence suggests that podocyte acquisition of de novo immune phenotypes is associated with cytoskeletal disarrangement and podocyte dysfunction [11, 12, 18]. As a matter of fact, a number of immune molecules, like MHC II [34], CD80 [10, 11] and cathepsin L [12, 18], function at the interface between innate/adaptive immunity and cytoskeletal remodeling and are known to cause podocyte cytoskeleton disorganization. Shown in Figure 2A, under basal conditions, podocytes exhibited as large, flat and arborized cells with abundant stress fibers in cytoplasm and well-developed processes, as demonstrated by phase contrast microscopy (Figure 2A). Labeling of filamentous actin (F-actin) with rhodamine-conjugated phalloidin revealed stretched cellular shape and intense phalloidin-labeled ventral stress fibers with long paralleled cortical stress fibers in normal podocytes (Figure 2A). The percentage of F-actin under fluorescent microscopy were analyzed (Figure 2B). Following LPS exposure, podocytes demonstrated prominent morphologic changes, marked by podocyte shrinkage and a spindle-like or asterlike cell shape, in parallel with drastic disruption of actin cytoskeleton that manifested as increased expression of cortical filaments, diminished ventral stress fibers, more transverse arcs, and sporadic short dorsal stress fibers (Figure 2A and 2B). This was concurrent with de novo expression of MHC II, CD80 and cathepsin L as described above. Valproate co-treatment strikingly prevented stress fiber disruption and aberrance in podocyte morphology (Figure 2A and 2B).
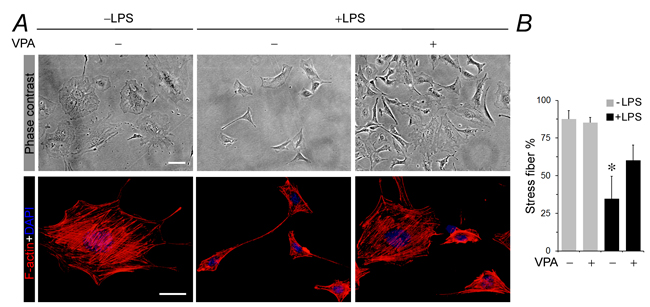
Figure 2: Valproate attenuates cellular shape changes, loss of stress fibers and disruption of actin cytoskeletal integrity in podocytes injured with LPS. Immortalized murine podocytes were cultured and differentiated under nonpermissive conditions. Cells were injured with LPS (100µg/ml) or vehicle for 24 h in the presence or absence of sodium valproate (VPA; 1.5 mM). A. Represetative phase contrast micrographs show podocyte shape changes. Podocytes were large, flat and arborized cells with evidently rich stress fibers in cytoplasm and well-developed processes under basal condition. Following LPS exposure, podocytes demonstrated prominent cytologic changes, marked by podocyte shrinkage and a spindle-like or asterlike cell shape. Valproate co-treatment strikingly prevented aberrance in podocyte morphology. Bar = 10µm. Alternatively, podocytes were processed for fluorescent labeling of cytoskeletal filamentous actin (F-actin) with rhodamine conjugated phalloidin and counterstained with 4’,6-diamidino-2-phenylindole (DAPI). Representative fluorescent microscopic images show changes in F-actin cytoskeleton. Bar = 10µm. B. The percentage of stress fibers under fluorescent microscopy were analyzed and estimated. *P < 0.05 versus other groups (n = 4). DAPI, 4’,6-diamidino-2-phenylindole; LPS, lipopolysaccharide; VPA, sodium valproate.
Valproate counteracts GSK3β overactivity and NFkB phosphorylation in LPS-injured podocytes
NFkB activation is a prerequisite for de novo expression of diverse immune molecules, whose transcription are all NFkB-dependent [20]. To discern if the valproate-abrogated podocyte de novo expression of immune phenotypes stems from a possible effect on NFkB activation, cell lysates were subjected to immunoblot analysis for phosphorylation of NFkB RelA/p65 at serine 467 residue, which plays a key role on specifying the expression of proinflammatory molecules involved in kidney injury and podocyte dysfunction [22, 35]. Shown in Figure 3A, LPS triggered considerable RelA/p65 phosphorylation and this effect was hindered by valproate in a dose dependent manner, accompanied with inhibition of GSK3β, a molecular target of valproate [26], as indicated by increased phosphorylation at the serine 9 inhibitory site. Previous data suggest that GSK3β is an indispensable element for NFkB activation [21]. In agreement, linear regression analysis illustrated an inverse correlation between p-GSK3β and p-RelA/p65 in podocytes (Figure 3B), namely a positive correlation between GSK3β overactivity and NFkB RelA/p65 activation.
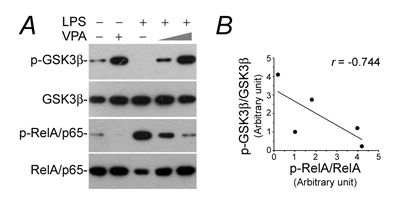
Figure 3: The LPS-elicited GSK3β overactivity and NFkB hyperactivation in podocytes is obliterated by valproate. Podocytes were treated as elaborated in Figure 1. A. Cell lysates were collected and prepared for immunoblotting for phosphorylated GSK3β at serine 9 residue, phosphorylated NFkB RelA/p65 at serine 467 residue and other indicated proteins. B. Immunoblots were subjected to densitometric analysis and arbitrary units of p-GSK3β and p-RelA were expressed respectively as immunoblot densitometric ratios of p-GSK3β/GSK3β and p-RelA/p65/RelA/p65 as folds of the control group. Linear regression analysis indicated a statistically significant (P < 0.05) inverse correlations between p-GSK3β and p-RelA/p65 in podocytes. LPS, lipopolysaccharide; VPA, sodium valproate.
Ectopic expression of the constitutively active GSK3β blunts the podocyte protective activity of valproate
To substantiate if inhibitory phosphorylation of GSK3β at serine 9 plays a key role in transmitting the podoprotective activity of valproate, podocytes were subjected to transient transfection of plasmids encoding a hemagglutinin-conjugated wild-type GSK3β (WT) or mutant GSK3β (S9A), in which the regulatory serine 9 residue is replaced by alanine so that GSK3β becomes uninhabitable and constitutively active. Cells were then injured with LPS in the presence or absence of valproate. Shown by immunobot analysis, the LPS-induced NFkB activation (Figure 4A and 4B) and de novo expression of all immune phenotypes (Figure 4C and 4D) were apparently enhanced in podocytes expressing S9A as compared with those expressing WT, denoting that GSK3β overactivity amplifies the podocyte response to LPS. In accordance with the augmented NFkB activation and expression of all immune molecules in podocytes expressing S9A, the LPS-elicited actin cytoskeleton disorganization was sensitized in S9A-expressing cells as compared with that in WT-expressing cells (Figure 4E). This effect was quantified by analyses of the percentage of F-actin under fluorescent microscopy (Figure 4F). Valproate drastically hindered NFkB activation (Figure 4A and 4B) and podocyte de novo expression of immune molecules (Figure 4C and 4D) in WT-expressing following LPS exposure and mitigated the LPS impaired actin cytoskeleton integrity (Figure 4E and 4F). In contrast, the effect of valproate was largely blunted in S9A-expressing cells.
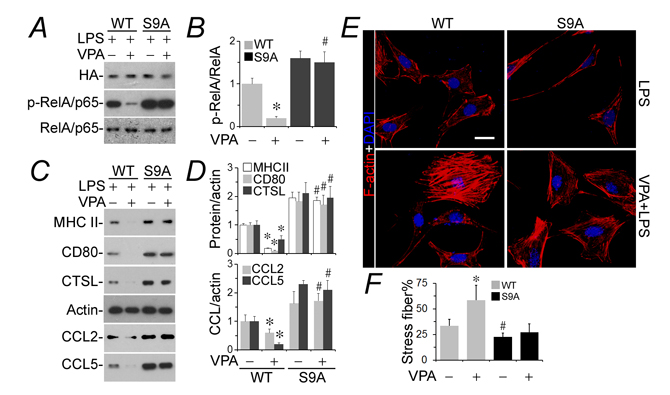
Figure 4: The podocyte protective activity of valproate is blunted in podocytes expressing a constitutively active mutant of GSK3β. Differentiated murine podocytes were subjected to liposome-mediated transient transfection with vectors encoding the HA-conjugated wild-type GSK3β (WT) or mutant GSK3β (S9A), in which the regulatory serine 9 residue is replaced by alanine so that GSK3β becomes uninhibitable. Cells were treated with LPS (100 µg/ml) for 24 h after transfection in the presence or absence of sodium valproate (VPA; 1.5 mM). A. Cell lysates were collected and prepared for immunoblotting for phosphorylated NFkB RelA/p65 at serine 467 residue or indicated molecules. B. Immunoblots were subjected to densitometric analysis and arbitrary units were expressed respectively as immunoblot densitometric ratios of p-NFkB RelA/p65 to NFkB RelA/p65 as folds of the control group. *P < 0.05 versus the level of the same molecule in the non-VPA treated WT group; #Not significant versus the level of the same molecule in the non-VPA treated S9A group (n = 3). C. Cell lysates or conditioned media were collected and prepared for immunoblotting for indicated immune molecules. D. Immunoblots were subjected to densitometric analysis and arbitrary units were expressed respectively as immunoblot densitometric ratios of diverse molecules to actin as folds of the control group. *P < 0.05 versus the level of the same molecule in the non-VPA treated WT group; #Not significant versus the level of the same molecule in the non-VPA-treated S9A group (n = 3). E. Cells were fixed and processed for fluorescent labeling of F-actin and counterstained with 4’,6-diamidino-2-phenylindole (DAPI). Represetative fluorescent microscopy shows changes in F-actin cytoskeleton. Bar = 10µm. F. The percentage of stress fibers under fluorescent microscopy were analyzed and estimated. *P < 0.05 versus non-VPA-treated WT cells; #P < 0.05 versus non-VPA-treated WT cells; #Not significant versus VPA-treated S9A cells (n = 4). CTSL, cathepsin L; DAPI, 4’,6-diamidino-2-phenylindole; LPS, lipopolysaccharide; HA, hemagglutinin; VPA, sodium valproate.
Valproate therapy mitigates podocyte acquisition of immune phenotypes and attenuates albuminuria in LPS-injured mice
To test if valproate is able to protect against podocyte injury in vivo, the murine model of LPS-elicited podocytopathy was employed. LPS has been reproducibly demonstrated by previous studies to cause acute podocyte injury and albuminuria in mice [11, 36, 37]. Indeed, 24 h after LPS injury, albuminuria was evident in our mice (Figure 5A), in parallel with signs of podocyte injury, marked by podocyte foot process effacement under transmission electron microscopy (Figure 5B and 5C). This was associated with GSK3β overactivity in glomeruli, reflected by reduced inhibitory phosphorylation of GSK3β as probed by immunoblot analysis of isolated glomeruli (Figure 6A and 6B). Consistent with in vitro findings, GSK3β overactivity in glomeruli resulted in NFkB RelA/p65 phosphorylation and activation (Figure 6A and 6B) as well as an induced glomerular expression of diverse podocytopathic immune molecules, including MHCII, CD80, cathepsin L and proinflammatory cytokines CCL2 and 5 (Figure 6C and 6D), as shown by immunoblot analysis of isolated glomeruli followed by densitometry. This was paralleled by prominent podocyte injury, marked by loss of podocyte marker proteins like synaptopodin (Figure 6C and 6D). Dual-color immunohistochemistry staining indicated that immune molecules, like CD80 and CCL2, were evidently detected in glomerular cells positive for podocyte-specific markers, such as synaptopodin and nephrin, thus indicative of a pattern of podocyte distribution (Figure 6E). Valproate treatment substantially attenuated albuminuria (Figure 5A) and prevented podocyte injury with significant improvement in podocyte injury and foot process effacement (Figure 5B and 5C). Mechanistically, valproate therapy diminished GSK3β overactivity in renal glomeruli in mice following LPS injury, mitigated NFkB activation (Figure 6A and 6B), and offset the LPS-elicited expression of diverse immune molecules in glomeruli and podocytes (Figure 6C~6E).
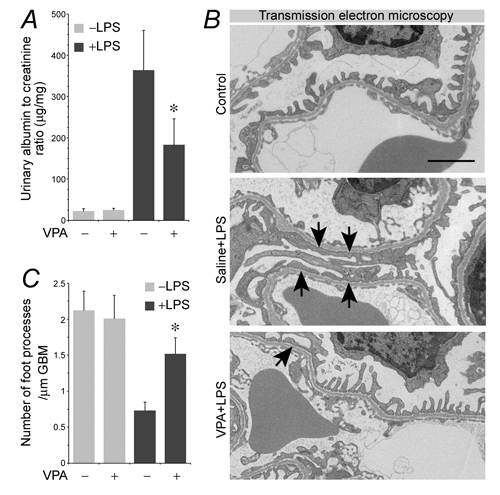
Figure 5: Valproate protects against podocyte injury and albuminuria in LPS-injured mice. Mice were treated with sodium valproate (200mg/kg) or saline (100µl) followed by LPS (200µg) injury or vehicle treatment. Mice were followed for 24 h. A. Urine was collected and processed for urine albumin ELISA assay followed by adjustment for urine creatinine concentrations. *P < 0.05 versus non-VPA-treated and LPS-injured mice (n = 6). B. Transmission electron microscopy of kidney glomeruli demonstrated marked foot process effacement (black arrows) in LPS-injured mice. This effect was prevented by valproate treatment. C. Morphometric analysis of the number of foot processes per µm glomerular basement membrane (GBM) revealed by electron microscopy (right panel). Bar = 2 µm.*P < 0.05 versus other groups (n = 6). LPS, lipopolysaccharide; VPA, sodium valproate.
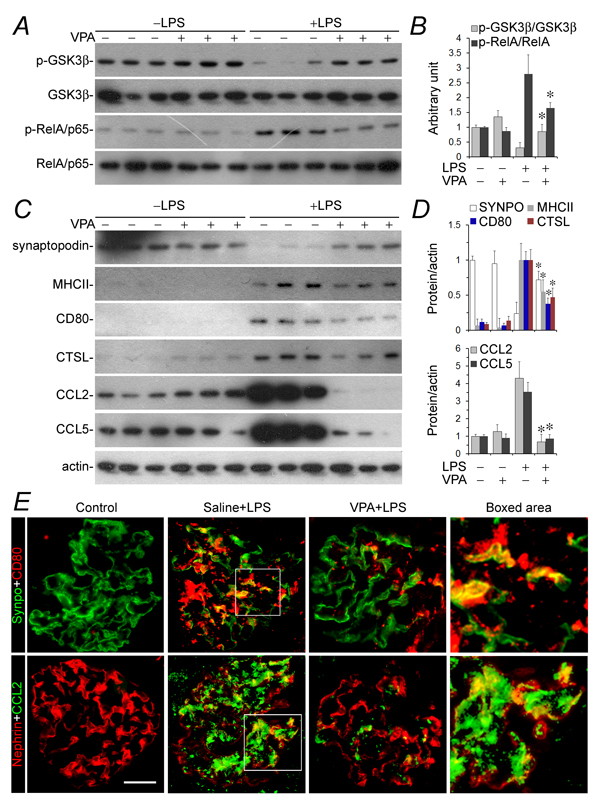
Figure 6: GSK3β overactivity and NFkB activation in glomeruli in LPS-injured mice were mitigated after valproate therapy, resulting in a blunted podocyte acquisition of immune phenotypes and podocytopathy. Mice were treated as stated in Figure 5 and mouse kidneys were procured for further examination. A. and C. Isolated glomeruli were processed for immunoblot analysis for diverse molecules. B. and D. Immunoblots were subjected to densitometric analysis and arbitrary units were expressed respectively as immunoblot densitometric ratios of diverse molecules to actin as folds of the control group. *P < 0.05 versus the level of the same molecule in LPS-injured mice without VPA treatment (n = 6). E. Frozen sections of mouse kidneys were processed for dual color fluorescent immunohistochemistry staining for indicated immune molecules and podocyte marker proteins. Representative microscopic images were shown. Bar = 50 µm. CTSL, cathepsin L; LPS, lipopolysaccharide; SYNPO, synaptopodin; VPA, sodium valproate.
DISCUSSION
Valproate, an FDA approved drug, has been used for almost 50 years as the first line anti-convulsant for the treatment of epilepsy and also as a mood-stabilizer to control bipolar affective disorder [38], [39]. Exposure to valproate can trigger a multitude of cellular responses that may be responsible for its clinical efficacy and multiple actions in diverse organ systems [38]. Burgeoning evidence recently supports a protective activity of valproate in diverse somatic diseases, including kidney disease [28-30]. Although a number of cell signaling transducers, including inositol, histone deacetylase (HDAC), GSK3β and others, have been implicated as molecular targets of valproate [38], the underlying mechanisms responsible for the beneficial effect of valproate in diverse experimental diseases are largely unknown. The present study for the first time provided evidence in support of a direct podocyte protective effect of valproate that is likely mediated via intercepting the GSK3β facilitated NFkB activation and the ensuing NFkB-dependent de novo acquisition of immune phenotypes.
Our finding that valproate possesses a podocyte protective activity both in vitro in cultured podocytes and in vivo in murine models of LPS podocytopathy is congruent to several recent pre-clinical observations suggesting that valproate exerts a glomerular protective and proteinuria reducing effect [28-30]. Indeed, in rats with streptozotocin-elicited diabetes [29], oral treatment with low or high dose of valproic acid attenuated physiologic signs of diabetic nephropathy, including kidney dysfunction and albuminuria, and improved histologic evidence of glomerular injury, such as glomerular matrix accumulation and enlargement, glomerular cell apoptosis, and ultrastructural lesions in podocytes, marked by podocyte foot process effacement. In agreement, in another study on murine models of Adriamycin nephropathy [28], both preventive and rescue treatment with valproic acid were able to drastically attenuate urinary excretion of albumin and ameliorate renal histologic injuries, including glomerulosclerosis, renal inflammation, fibrosis and podocyte detachment and foot process effacement. Although both studies demonstrated an appealing renoprotective property of valproate, the underlying mechanism is unknown. Of note, podocyte injury and proteinuria could be a bystander phenomenon caused by systemic factors ensuing metabolic, hematopoietic or immune dysregulations [40]. Correction of diabetes or systemic hyperglycemia per se is known to improve and even reverse glomerular damage and podocyte injury [41]. Likewise, adoptive transfer of myeloid-derived suppressor cells has been shown to be sufficient to attenuate podocyte injury, glomerulopathy and proteinuria in experimental glomerulopathies [42]. At least, there is evidence suggesting an anti-diabetic effect of valproate that is able to correct hyperglycemia in diabetic models [43]. Moreover, valproate is known to have a potent immunoregulatory activity that can lead to profound alternations in immune response in both animal models and in humans [44]. Given these confounding effects of valproate on immune system or on diabetes, it remains to be clarified if the anti-protenuric and glomerular protective activity of valproate is attributable to an effect on podocyte autonomous injury. Our present study took advantage of the in vitro and in vivo models of the LPS-inflicted podocytopathy in the absence of any systemic cues, like hyperglycemia or immune factors, and unequivocally demonstrated a protective effect of valproate on podocyte autonomous injury.
How valproate protects against podocyte injury has been barely examined before. Valproate is a well-established quintessential inhibitor of GSK3β [26, 27], a kinase that has been implicated in podocyte autonomous injury via integrating multiple podocytopathic cellular pathways [45], including proinflammatory NFkB activation and the ensuing de novo expression of NFkB-dependent immune molecules in podocytes [22, 45]. The mode of action by which valproate blocks GSK3β activity is still a matter of debate, but predominant evidence points to an induced inhibitory phosphorylation of GSK3β at the serine 9 residue [27], a mechanism that is also adopted by lithium, another well characterized GSK3β inhibitor. In support of this, in valproic acid-associated fetal cardiac teratogenicity, the severity of the defect is correlated to the phosphorylation state of GSK3β serine 9 [46]. Moreover, in neuroblastoma cells, valproate treatment activated Akt and subsequently augmented inhibitory phosphorylation of GSK3β [27]. In line with these reports, our present study found that the beneficial activity of valproate was offset in podocytes ectopically expressing a mutant GSK3β, in which the regulatory serine 9 residue is changed to alanine so that GSK3β became uninhibitable and constitutively active, entailing that inhibitory phosphorylation is likely essential for the protective effect of valproate. In vivo, in LPS-injured mice, valproate treatment prevented albuminuria and this effect was associated with GSK3β inhibition. LPS is also able to induce acute kidney injury (AKI). Inhibition of GSK3β has been shown to confer a renoprotective action in various models of AKI. Provided the inhibitory effect of valproate on GSK3β, it is highly possible that valproate treatment may also be able to improve kidney dysfunction and AKI in the LPS-injured mice. This merits further studies. In addition to GSK3β, valproate may also target other cell signaling molecules in podocytes. Admittedly, accumulating data pointed to valproate as an inhibitor of HDACs [47]. In consistency, the aforementioned two studies on the effect of valproic acid in experimental diabetic [29] or Adriamycin [28] nephropathy also demonstrated association between kidney protection and increased histone acetylation in kidney cells and thus assumed that inhibition of HDAC might mediate glomerular protection by valproic acid. Such conclusions are, nevertheless, arbitrary and inconclusive because no causal relationship was established in both studies [28, 29]. Latest research work did uncover that a couple of members of HDAC family may play a direct pathogenic role in podocyte injury [48, 49]. These include HDAC4 [48] and HDAC9 [49], which belong to the class II HDACs family. However, valproate, at clinically achievable concentrations, is a typical inhibitor of the class I HDACs [50, 51]. In order to suppress the activity of class II HDACs, valproate requires more than 20 times the clinical concentrations [51]. Thus, it remains doubtful if HDAC inhibition mediates the direct podocyte protective effect of valproate. Our study was not designed and thus unable to determine whether inhibition of HDACs also contributes, at least in part to, the podoprotective effect of valproate. But in S9A-expressing podocytes, the protective efficacy of valproate was abolished. This finding apparently implies that GSK3β is likely the key, if not the sole, target of valproate that is principally responsible for valproate’s podocytoprotective activity. Even if HDAC inhibition is implicated in the podocyte protective effect of valproate, it might be distal or downstream to GSK3β inhibition, as there is evidence suggesting that multiple HDACs [52-54], including HDAC4 [53] are direct targets of GSK3β and their activities and expression levels are under the control of GSK3β.
In summary, our study for the first time indicates that valproate directly protects against podocyte injury and hampers podocyte acquisition of de novo immune phenotypes via intercepting GSK3β-facilitated NFkB activation. In murine models of LPS podocytopathy, valproate therapy ameliorates podocyte injury and albuminuria. Our study may pave the way to develop a novel, feasible and affordable therapeutic modality based on valproate for the treatment of proteinuric glomerulopathies.
Materials and Methods
Cell culture
Conditionally immortalized mouse podocytes in culture (courtesy of Dr. Stuart Shankland, University of Washington, Seattle, WA) [55] between passages 21 and 25 were used. Podocytes were cultured in RPMI 1640 medium (Invitrogen) supplemented with 10% FBS in a humidified incubator with 5% CO2. The cells were cultured at 33 °C with 50 units/ml recombinant mouse interferon-g (Millipore,Billerica, MA) on collagen-coated plastic Petri dishes and were transferred to a 37 °C incubator without interferon-g to induce differentiation for 14 days. Podocytes were pretreated with valproic acid (sodium salt, Sigma, St. Louis, USA; 0.5 or 1.5 mM) or vehicle for 30 min and then stimulated with 100 µg/ml LPS (serotype: E. coli 0111: B4, Sigma) or saline for 24 h.
Transient transfection
The eukaryotic expression vectors encoding the hemagglutinin (HA) conjugated constitutively active (S9A) GSK3β mutant (S9A-GSK3β-HA/pcDNA3) or wild-type (WT) GSK3β (WT-GSK3β-HA/pcDNA3) were provided by Dr. Johnson (Birmingham, AL) [55]. Transient transfection was carried out by using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) as described previously [55]. In brief, podocytes were cultured under permissive conditions at 50~70% confluence in the absence of antibiotics. The plasmid-Lipofectamine 2000 complexes were prepared and applied to proliferating podocytes for transfection. The ratio of Lipofectamine 2000 to vectors was optimized by a series of pilot experiments for each study until the best transfection efficiency or the best gene-silencing efficiency was achieved. After transfection, the cells were cultured under nonpermissive conditions in normal growth medium for 48 h before transfection efficiency was assessed by immunocytochemistry staining or immunoblot analysis for target molecules. Cells were then subjected to LPS, vehicle or other treatments.
Animal studies
Animal studies were approved by the institutional Animal Care and Use Committees, and they conform to the US Department of Agriculture regulations and the NIH’s Guide for human care and use of Laboratory Animals. Male FVB mice aged 8 weeks were randomized to each of the following treatments. Sodium valproate (200mg/kg) or saline (100µl) was given via intraperitoneal injection based on previous reports [28] 1 h before intraperitoneal injection of saline (100µl) or LPS (200µg; serotype: E. coli 0111:B4, Sigma) [36]. Mice were followed for 24 h before they were killed and the kidneys resected for further investigation. Urine was collected 24 h after LPS injection. Six mice were randomly assigned to each group.
Urine analyses
Urine albumin levels were measured using mouse albumin ELISA quantitation kit (Bethyl Laboratories, Montgomery, TX, USA). Urine creatinine was determined by a creatinine assay kit (BioAssay Systems, Hayward, CA, USA). To rule out the influence of glomerular filtration rate on urinary excretion of albumin, urine albumin levels were adjusted by urine creatinine levels and presented as urine albumin-to-creatinine ratios.
Transmission electron microscopy
For electronmicroscopy, kidney cortical tissues were cut into small pieces (1 mm3), fixed with 2.5% glutaraldehyde, and embedded in Epon 812 (Polysciences Inc.,Warrington, PA). Transmission electron micrographs were obtained using an EM-10 microscope (Zeiss) operated at 60 kV. The number of foot processes per 10 μm of glomerular basement membrane (GBM) in different electron microscopic fields was measured. The result was divided by the total length of the GBM and expressed as the number per micrometer of GBM length as previously described [55].
Immunofluorescent staining
Cultured cells or frozen kidney cryostat sections were fixed and processed for fluorescent staining. For dual color fluorescent immunohistochemistry staining, samples were stained with primary antibodies and followed by applying the Alexa Fluor-conjugated secondary antibodies (Invitrogen). As a negative control, the primary antibodies were replaced by preimmune IgG from the same species; no staining occurred. Finally, samples were counterstained with 4’,6-diamidino-2-phenylindole (DAPI) and mounted with Vectashield mounting medium (Vector Laboratories, Burlingame, CA, USA). For fluorescence microscopy, all sections were analyzed at the same time to exclude artifacts due to variable decay of the fluorochrome. Sections were examined using an Olympus fluorescence microscope equipped with a Spot II digital camera. For dual-color staining, images were acquired sequentially to avoid dye interference. ImageJ software was used for post processing of the images, e.g. scaling, merging, and colocalization analysis.
Fluorescent labeling of cytoskeletal filamentous actin in podocytes
Following various treatments, podocytes in culture were fixed in 4% paraformaldehyde in PBS and permeabilized filamentous actin (F-actin) was stained by incubation at 4°C with rhodamine conjugated phalloidin (Cytoskeleton Inc., Denver, CO). Then, cells were counterstained with 4’,6-diamidino-2-phenylindole (DAPI) and mounted with Vectashield mounting medium. Images were documented using an Olympus fluorescence microscope equipped with a Spot II digital camera. The percentage of F-actin or stress fibers under fluorescent microscopy were analyzed and quantitated as described elsewhere [56].
Glomerular isolation
Glomerular isolation was conducted as reported elsewhere [22]. In brief, mice were anesthetized and kidney perfused with 5ml of phosphate-buffered saline containing 8×107 Dynabeads M-450 (Dynal Biotech ASA, Oslo, Norway). After perfusion, the kidneys were resected and kidney cortices minced into 1-mm3 pieces and digested in collagenase A (1 mg/ml, Sigma) at 37oC for 30 min with gentle shaking. The tissue was pressed gently through a 100 μm cell strainer (BD Falcon, Bedford, MA, USA) and glomeruli containing Dynabeads were then gathered using a magnetic particle concentrator. An aliquot (1:1500) of the glomerular isolate was visualized under a microscope to ensure that the sample contained fewer than five tubular fragments per x200 field. Most isolated glomeruli (80%) were decapsulated, which was similar to what had been reported previously [55].
Western immunoblot
Cultured cells or kidney tissues were lysed in Radioimmunoprecipitation assay (RIPA) buffer supplemented with protease inhibitors. Cell lysates and conditioned media were subjected to immunoblot analysis as previously described [35]. The antibodies against MHC II, cathepasin L, and actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA), those against CD80, CCL2 and CCL5 from R&D Systems (Minneapolis, MN, USA), and that against p-GSK3β, GSK3, p-RelA/p65 and RelA/p65 from Cell Signaling Technology (Danvers, MA, USA).
Statistical analyses
For immunoblot analysis, bands were scanned and the integrated pixel density was determined using a densitometer and the ImageJ analysis program. All data are expressed as mean ± SD. Unless otherwise indicated, all experimental observations were repeated three times. Statistical analysis of the data from multiple groups was performed by repeated measures ANOVA followed by Fisher’s Least Significant Difference (LSD) tests. Data from two groups were compared by Student’s t-test. Linear regression analysis was applied to examine possible relationships between two parameters. P < 0.05 was considered significant.
Authors’ contributions
Z.L. and R.G. conceived the study, and developed the study design and protocol with contributions from all authors. P.W., S.Z., M.L. and Y.G. performed the experiments. P.W., S.Z., M.L. and Y.G. extracted the data and undertook the analysis, supervised by Z.L. and R.G., and overseen by all authors. P.W., S.Z. and Y.G. prepared the figures. All authors contributed to the data interpretation. R.G. drafted the paper and all authors contributed to revisions and approved the final version.
Acknowledgments
The research work of the authors was supported in part by the key program of Joint Funds of the National Natural Science Foundation of China U1604284, the National key R&D program of China (2016YFC1305404), the Natural Science Foundation of China 81670663, the Medical Science and Technology Foundation of Henan Province (grant 201601002), the Foundation for Health, and the U.S. National Institutes of Health grant DK092485. The funders had no role in the design and conduct of this study, collection and interpretation of the data, or preparation and approval of the manuscript.
Conflicts of interest
The authors declare no conflicts of interest.
REFERENCES
1. Pavenstadt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev. 2003; 83: 253-307.
2. Mundel P, Shankland SJ. Podocyte biology and response to injury. J Am Soc Nephrol. 2002; 13: 3005-15.
3. Shankland SJ. The podocyte’s response to injury: role in proteinuria and glomerulosclerosis. Kidney Int. 2006; 69: 2131-47.
4. Shankland SJ, Pollak MR. A suPAR circulating factor causes kidney disease. Nat Med. 2011; 17: 926-7.
5. Niemir ZI, Stein H, Dworacki G, Mundel P, Koehl N, Koch B, Autschbach F, Andrassy K, Ritz E, Waldherr R, Otto HF. Podocytes are the major source of IL-1 alpha and IL-1 beta in human glomerulonephritides. Kidney Int. 1997; 52: 393-403.
6. Lee EY, Chung CH, Khoury CC, Yeo TK, Pyagay PE, Wang A, Chen S. The monocyte chemoattractant protein-1/CCR2 loop, inducible by TGF-beta, increases podocyte motility and albumin permeability. Am J Physiol Renal Physiol. 2009; 297: F85-94.
7. Valino-Rivas L, Gonzalez-Lafuente L, Sanz AB, Ruiz-Ortega M, Ortiz A, Sanchez-Nino MD. Non-canonical NFkappaB activation promotes chemokine expression in podocytes. Sci Rep. 2016; 6: 28857.
8. Coers W, Brouwer E, Vos JT, Chand A, Huitema S, Heeringa P, Kallenberg CG, Weening JJ. Podocyte expression of MHC class I and II and intercellular adhesion molecule-1 (ICAM-1) in experimental pauci-immune crescentic glomerulonephritis. Clin Exp Immunol. 1994; 98: 279-86.
9. Cai M, Zhou T, Wang X, Shang M, Zhang Y, Luo M, Xu C, Yuan W. DC-SIGN expression on podocytes and its role in inflammatory immune response of lupus nephritis. Clin Exp Immunol. 2016; 183: 317-25.
10. Reiser J, Mundel P. Danger signaling by glomerular podocytes defines a novel function of inducible B7-1 in the pathogenesis of nephrotic syndrome. J Am Soc Nephrol. 2004; 15: 2246-8.
11. Reiser J, von Gersdorff G, Loos M, Oh J, Asanuma K, Giardino L, Rastaldi MP, Calvaresi N, Watanabe H, Schwarz K, Faul C, Kretzler M, Davidson A, et al. Induction of B7-1 in podocytes is associated with nephrotic syndrome. J Clin Invest. 2004; 113: 1390-7.
12. Sever S, Altintas MM, Nankoe SR, Moller CC, Ko D, Wei C, Henderson J, del Re EC, Hsing L, Erickson A, Cohen CD, Kretzler M, Kerjaschki D, et al. Proteolytic processing of dynamin by cytoplasmic cathepsin L is a mechanism for proteinuric kidney disease. J Clin Invest. 2007; 117: 2095-104.
13. Banas MC, Banas B, Hudkins KL, Wietecha TA, Iyoda M, Bock E, Hauser P, Pippin JW, Shankland SJ, Smith KD, Stoelcker B, Liu G, Grone HJ, et al. TLR4 links podocytes with the innate immune system to mediate glomerular injury. J Am Soc Nephrol. 2008; 19: 704-13.
14. Goldwich A, Burkard M, Olke M, Daniel C, Amann K, Hugo C, Kurts C, Steinkasserer A, Gessner A. Podocytes are nonhematopoietic professional antigen-presenting cells. J Am Soc Nephrol. 2013; 24: 906-16.
15. Tipping PG. Are podocytes passive or provocative in proteinuric glomerular pathology? J Am Soc Nephrol. 2008; 19: 651-3.
16. Fiorina P, Vergani A, Bassi R, Niewczas MA, Altintas MM, Pezzolesi MG, D’Addio F, Chin M, Tezza S, Ben Nasr M, Mattinzoli D, Ikehata M, Corradi D, et al. Role of podocyte B7-1 in diabetic nephropathy. J Am Soc Nephrol. 2014; 25: 1415-29.
17. Conus S, Simon HU. Cathepsins and their involvement in immune responses. Swiss Med Wkly. 2010; 140: w13042.
18. Mundel P, Reiser J. Proteinuria: an enzymatic disease of the podocyte? Kidney Int. 2010; 77: 571-80.
19. Welsh GI, Saleem MA. The podocyte cytoskeleton—key to a functioning glomerulus in health and disease. Nat Rev Nephrol. 2012; 8: 14-21.
20. Hayden MS, West AP, Ghosh S. NF-kappaB and the immune response. Oncogene. 2006; 25: 6758-80.
21. Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature. 2000; 406: 86-90.
22. Bao H, Ge Y, Peng A, Gong R. Fine-tuning of NFkappaB by glycogen synthase kinase 3beta directs the fate of glomerular podocytes upon injury. Kidney Int. 2015; 87: 1176-90.
23. Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003; 116: 1175-86.
24. Radinger M, Kuehn HS, Kim MS, Metcalfe DD, Gilfillan AM. Glycogen synthase kinase 3beta activation is a prerequisite signal for cytokine production and chemotaxis in human mast cells. J Immunol. 2010; 184: 564-72.
25. Cohen P, Goedert M. GSK3 inhibitors: development and therapeutic potential. Nat Rev Drug Discov. 2004; 3: 479-87.
26. Chen G, Huang LD, Jiang YM, Manji HK. The mood-stabilizing agent valproate inhibits the activity of glycogen synthase kinase-3. J Neurochem. 1999; 72: 1327-30.
27. De Sarno P, Li X, Jope RS. Regulation of Akt and glycogen synthase kinase-3 beta phosphorylation by sodium valproate and lithium. Neuropharmacology. 2002; 43: 1158-64.
28. Van Beneden K, Geers C, Pauwels M, Mannaerts I, Verbeelen D, van Grunsven LA, Van den Branden C. Valproic acid attenuates proteinuria and kidney injury. J Am Soc Nephrol. 2011; 22: 1863-75.
29. Khan S, Jena G, Tikoo K, Kumar V. Valproate attenuates the proteinuria, podocyte and renal injury by facilitating autophagy and inactivation of NF-kappaB/iNOS signaling in diabetic rat. Biochimie. 2015; 110: 1-16.
30. Sun XY, Qin HJ, Zhang Z, Xu Y, Yang XC, Zhao DM, Li XN, Sun LK. Valproate attenuates diabetic nephropathy through inhibition of endoplasmic reticulum stressinduced apoptosis. Mol Med Rep. 2016; 13: 661-8.
31. Ranf S. Immune Sensing of Lipopolysaccharide in Plants and Animals: Same but Different. PLoS Pathog. 2016; 12: e1005596.
32. Shankland SJ, Pippin JW, Reiser J, Mundel P. Podocytes in culture: past, present, and future. Kidney Int. 2007; 72: 26-36.
33. Pippin JW, Brinkkoetter PT, Cormack-Aboud FC, Durvasula RV, Hauser PV, Kowalewska J, Krofft RD, Logar CM, Marshall CB, Ohse T, Shankland SJ. Inducible rodent models of acquired podocyte diseases. Am J Physiol Renal Physiol. 2009; 296: F213-29.
34. Delaguillaumie A, Marin-Esteban V, Setterblad N, Leh LJ, Assier E, Gelin C, Charron D, Galy A, Mooney N. Contrasting cytoskeletal regulation of MHC class II peptide presentation by human B cells or dendritic cells. Eur J Immunol. 2008; 38: 1096-105.
35. Gong R, Rifai A, Ge Y, Chen S, Dworkin LD. Hepatocyte growth factor suppresses proinflammatory NFkappaB activation through GSK3beta inactivation in renal tubular epithelial cells. J Biol Chem. 2008; 283: 7401-10.
36. Faul C, Donnelly M, Merscher-Gomez S, Chang YH, Franz S, Delfgaauw J, Chang JM, Choi HY, Campbell KN, Kim K, Reiser J, Mundel P. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med. 2008; 14: 931-8.
37. Yu H, Kistler A, Faridi MH, Meyer JO, Tryniszewska B, Mehta D, Yue L, Dryer S, Reiser J. Synaptopodin Limits TRPC6 Podocyte Surface Expression and Attenuates Proteinuria. J Am Soc Nephrol. 2016; 27: 3308-19.
38. Monti B, Polazzi E, Contestabile A. Biochemical, molecular and epigenetic mechanisms of valproic acid neuroprotection. Curr Mol Pharmacol. 2009; 2: 95-109.
39. Perucca E. Pharmacological and therapeutic properties of valproate: a summary after 35 years of clinical experience. CNS Drugs. 2002; 16: 695-714.
40. Patrakka J, Tryggvason K. New insights into the role of podocytes in proteinuria. Nat Rev Nephrol. 2009; 5: 463-8.
41. Fioretto P, Mauer M. Reversal of diabetic nephropathy: lessons from pancreas transplantation. J Nephrol. 2012; 25: 13-8.
42. Li L, Zhang T, Diao W, Jin F, Shi L, Meng J, Liu H, Zhang J, Zeng CH, Zhang MC, Liang S, Liu Y, Zhang CY, et al. Role of Myeloid-Derived Suppressor Cells in Glucocorticoid-Mediated Amelioration of FSGS. J Am Soc Nephrol. 2015; 26: 2183-97.
43. Khan S, Kumar S, Jena G. Valproic acid reduces insulin-resistance, fat deposition and FOXO1-mediated gluconeogenesis in type-2 diabetic rat. Biochimie. 2016; 125: 42-52.
44. Beghi E, Shorvon S. Antiepileptic drugs and the immune system. Epilepsia. 2011 (Suppl 3); 52: 40-4.
45. Li C, Ge Y, Dworkin L, Peng A, Gong R. The beta isoform of GSK3 mediates podocyte autonomous injury in proteinuric glomerulopathy. J Pathol. 2016; 239: 23-35.
46. Yu WH, Ho YL, Huang PT, Chu SL, Tsai HJ, Liou HH. The Phosphorylation State of GSK3beta Serine 9 Correlated to the Development of Valproic Acid-Associated Fetal Cardiac Teratogenicity, Fetal VPA Syndrome, Rescued by Folic Acid Administration. Cardiovasc Toxicol. 2016; 16: 34-45.
47. Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem. 2001; 276: 36734-41.
48. Wang X, Liu J, Zhen J, Zhang C, Wan Q, Liu G, Wei X, Zhang Y, Wang Z, Han H, Xu H, Bao C, Song Z, et al. Histone deacetylase 4 selectively contributes to podocyte injury in diabetic nephropathy. Kidney Int. 2014; 86: 712-25.
49. Liu F, Zong M, Wen X, Li X, Wang J, Wang Y, Jiang W, Guo Z, Qi H. Silencing of Histone Deacetylase 9 Expression in Podocytes Attenuates Kidney Injury in Diabetic Nephropathy. Sci Rep. 2016; 6: 33676.
50. Van Beneden K, Mannaerts I, Pauwels M, Van den Branden C, van Grunsven LA. HDAC inhibitors in experimental liver and kidney fibrosis. Fibrogenesis Tissue Repair. 2013; 6: 1.
51. Gurvich N, Tsygankova OM, Meinkoth JL, Klein PS. Histone deacetylase is a target of valproic acid-mediated cellular differentiation. Cancer Res. 2004; 64: 1079-86.
52. Bardai FH, D’Mello SR. Selective toxicity by HDAC3 in neurons: regulation by Akt and GSK3beta. J Neurosci. 2011; 31: 1746-51.
53. Cernotta N, Clocchiatti A, Florean C, Brancolini C. Ubiquitin-dependent degradation of HDAC4, a new regulator of random cell motility. Mol Biol Cell. 2011; 22: 278-89.
54. Chen S, Owens GC, Makarenkova H, Edelman DB. HDAC6 regulates mitochondrial transport in hippocampal neurons. PLoS One. 2010; 5: e10848.
55. Xu W, Ge Y, Liu Z, Gong R. Glycogen synthase kinase 3beta dictates podocyte motility and focal adhesion turnover by modulating paxillin activity: implications for the protective effect of low-dose lithium in podocytopathy. Am J Pathol. 2014; 184: 2742-56.
56. Dai R, Lin Y, Liu H, Rao J, Zhai Y, Zha X, Fang X, Xu H. A vital role for Angptl3 in the PAN-induced podocyte loss by affecting detachment and apoptosis in vitro. BMC Nephrol. 2015; 16: 38.