
INTRODUCTION
Lung cancer is the most common form of cancers with a high mortality [1]. Non-small-cell lung cancer (NSCLC) accounts for approximately 80% of lung cancers, in which adenocarcinoma accounts for about 38% [2]. Unfortunately, most NSCLC patients are diagnosed at advanced stage with a poor prognosis [3]. Epidermal growth factor receptor (EGFR) is overexpressed in most NSCLC and represents a major therapeutic target. Recently, the discovery of EGFR-tyrosine kinase inhibitors (TKIs) such as gefitinib and erlotinib led to great success in clinical treatment [4, 5]. EGFR tyrosine kinase inhibitor (TKI) treatment effectively improves survival of NSCLC patients with an EGFR mutation [6, 7].
Although EGFR-TKI treatment exhibited initial beneficial effects in most patients with NSCLC, these patients eventually developed resistance to EGFR-TKIs, with disease progression within approximately a year [8, 9]. Several mechanisms have been proposed for this resistance, including a secondary mutation in EGFR (such as T790M), amplification of the MET receptor tyrosine kinase, and other receptor tyrosine kinase overexpression.
PH domain leucine-rich-repeats protein phosphatase (PHLPP) belongs to a novel family of Ser/Thr protein phosphatases. There are two isoforms of PHLPP in its phosphatase family, PHLPP1 and PHLPP2, which share ~50% identity at the amino acid level. PHLPP negatively regulates PI3K/Akt and RAF/RAS/ERK signaling pathways [10, 11]. PI3K/Akt pathway is important in inhibiting apoptosis [12]. The RAF/RAS/ERK pathway contributes to proliferation differentiation and motility. EGFR activation triggers the Ras/Raf/Erk/MAPK, and stimulates the PI3K/Akt, pathways. Therefore, activation of proteins in PI3K/Akt pathway could also potentially lead to TKI resistance. Conversely, PHLPP as a negative regulator of PI3K/Akt and RAF/RAS/ERK pathways might slow down the process of resistance.
The aim of our study was to investigate the association between PHLPP expression and its effect on target-based therapies and acquired resistance to EGFR-TKI in lung adenocarcinoma.
RESULTS
Among the 75 patients with histologically proven lung adenocarcinoma, 33 (44%) were male and 42 (56%) were female. The median age was 60 years. All patients were treated with gefitinib or erlotinib and showed a complete response (CR), partial response (PR), or Stable disease (SD). The expression levels of PHLPP1 and PHLPP2 and p-Akt (S473) and p-Erk1/2 (Figure 1) in lung adenocarcinoma were assessed by immunohistochemistry. High (2–3 point) and low (0–1 point) expression levels of PHLPP1 were noted in 52 (69%) and 23 (30.67%) patients, respectively. Correlations between PHLPP1 abundance and clinicopathological characteristics were summarized in Table 1. High (2–3 point) and low (2–3 point) expression levels of PHLPP2 (2–3 point) were found in 46 (61.3%) and 29 (38.7%) patients, respectively. Correlations between PHLPP2 abundance and clinicopathological characteristics were summarized in Supplementary Table 1.
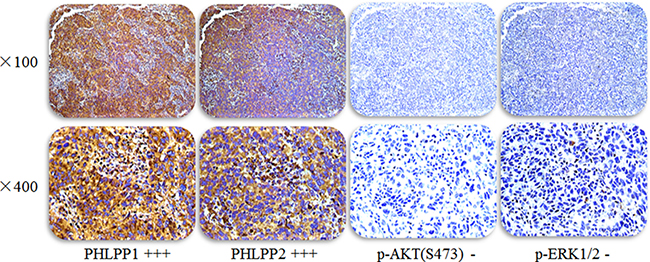
Figure 1: Representative images of PHLPP1 and PHLPP2 and p-AKT(S473) and p-ERK1/2 expression in the same lung adenocarcinoma patient with EGFR TKI treatment effectively. Staining in tissues was evaluated by three pathologists who were blinded to any clinical details related to the patients. The entire section was assessed at low (100×) (upper panels) and high (400×) power (below panels) magnification.
Table 1: PHLPP 1 and baseline characteristics of patients
No. of case |
PHLPP 1 expression |
P value |
||
|---|---|---|---|---|
High expression |
Low expression |
|||
Age (Median = 60 yeas) |
||||
≤ 60 years |
38 |
28 |
10 |
0.408 |
> 60 years |
37 |
24 |
13 |
|
Gender |
||||
Male |
33 |
20 |
13 |
0.146 |
Female |
42 |
32 |
10 |
|
Smoking Status |
||||
Ever-smoker |
19 |
10 |
9 |
0.068 |
Never-smoker |
56 |
42 |
14 |
|
EGFR-TKI |
||||
Erlotinib |
38 |
23 |
15 |
0.094 |
Gefitinib |
37 |
29 |
8 |
|
T |
||||
1 |
21 |
15 |
6 |
0.561 |
2 |
39 |
27 |
12 |
|
3 |
12 |
7 |
5 |
|
4 |
3 |
3 |
0 |
|
N |
||||
0 |
37 |
30 |
7 |
0.108 |
1 |
20 |
13 |
7 |
|
2 |
13 |
7 |
6 |
|
3 |
5 |
2 |
3 |
|
p-Akt (S473) |
||||
Positive |
14 |
4 |
10 |
0.001 |
Negative |
61 |
48 |
13 |
|
p-ERK 1/2 |
||||
Positive |
24 |
6 |
18 |
0.000 |
Negative |
51 |
46 |
5 |
|
EGFR |
||||
Mutation |
42 |
28 |
14 |
0.572 |
Unknown |
33 |
24 |
9 |
|
In Figure 2, Case 1, a 65-year-old smoker receiving gefitinib therapy was progressive free for 5 months. The patient did not join the medical donation project and died. Case 2 was a 67-year-old nonsmoking female with both gefitinib therapy who received free drugs from the medical donation project. She is still living and has been progression free for 28 months. Case 3 was a 57-year-old non-smoking female with Tarceva therapy who also received the drug from the medical donation project. She was progression free for 18 months, but died later. PHLPP protein expression was negatively correlated with p-Akt(S473) and p-ERK1/2.
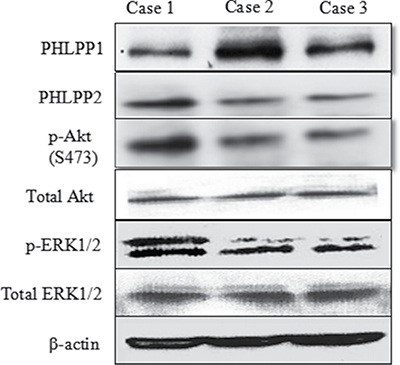
Figure 2: Representative images of Western blot of PHLPP and p-Akt and p-ERK in fresh tissue of three typical patients. Choose three typical cases include: case 1 without progress is less than half a year, case 2 with no progress is more than 2 years, and case 3 with no progress in 1 to 1.5 years. A negative correlation was noted between PHLPP protein expression with p-Akt and p-ERK in fresh tissue of patients.
The PFS and OS were independently correlated with PHLPP1 expression levels. The survival curves are shown in Figure 3. PFS and OS were significantly better in patients with high expression levels of PHLPP1 than those with low expression levels of PHLPP1 (p = 0.0050 and 0.0052). Patients with high expression levels of PHLPP1 showed significantly longer OS and higher PFS than those with low expression levels of PHLPP1 (36 months versus 19 months, 29 months versus 11 months respectively). In univariate and multivariate analyses, PHLPP1 was independently prognostic of PFS (Table 2 and Table 3) and OS (Supplementary Table 2 and Supplementary Table 3 ). PHLPP2 has no impact on either PFS or OS (Table 2 and Supplementary Table 2 and Supplementary Figure 1).
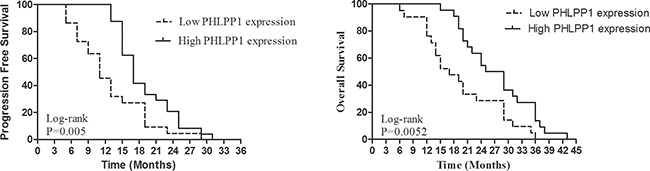
Figure 3: Kaplan-Meier PFS and OS curves of patients with high and low expression of PHLPP1. Patients with high expression of PHLPP1 showed significantly longer PFS and OS than those with low expression of PHLPP1 (14 months versus 7 months, 31 months versus 25 months, respectively).
Table 2: Univariate analysis of PFS in patients with lung adenocarcinoma treated with EGFR-TKI
Variables |
HR |
95% CI |
p value |
|
|---|---|---|---|---|
Age |
≤ 60 years vs. > 60years (Ref.) |
0.858 |
0.479–1.534 |
0.605 |
Gender |
Female vs. Male (Ref) |
0.617 |
0.346–1.101 |
0.103 |
Smoking Status |
Never-smoker vs. Ever-smoker (Ref) |
0.448 |
0.244–0.824 |
0.010 |
EGFR-TKI |
Erlotinib vs. Gefitinib (Ref) |
0.763 |
0.422–1.381 |
0.372 |
T |
1 and 2 vs. 3 and 4 (Ref) |
1.508 |
0.764–2.975 |
0.236 |
N |
0 and 1 vs. 2 and 3 (Ref) |
2.421 |
1.380–4.248 |
0.002 |
PHLPP1 expression |
High expression vs. Low expression (Ref) |
0.210 |
0.116–1.381 |
0.000 |
PHLPP2 expression |
High expression vs. Low expression (Ref) |
1.032 |
0.566–1.880 |
0.919 |
pAKT (S473) expression |
High expression vs. Low expression (Ref) |
2.542 |
1.333–0.380 |
0.005 |
pERK 1/2 expression |
High expression vs. Low expression (Ref) |
2.444 |
1.359–4.393 |
0.003 |
Table 3: Multivariate analysis of PFS in patients with lung adenocarcinoma treated with EGFR-TKI
Variables |
HR |
95% CI |
p value |
|
|---|---|---|---|---|
Smoking Status |
Never-smoker vs. Ever-smoker (Ref) |
0.488 |
0.236–1.007 |
0.052 |
N |
0 and 1 vs. 2 and 3 (Ref) |
1.488 |
0.825–2.683 |
0.187 |
PHLPP1 expression |
High expression vs. Low expression (Ref) |
0.232 |
0.123–0.438 |
0.000 |
pAKT (S473) expression |
High expression vs. Low expression (Ref) |
1.439 |
0.634–3.266 |
0.384 |
pERK 1/2 expression |
High expression vs. Low expression (Ref) |
0.666 |
0.339–1.310 |
0.239 |
DISCUSSION
Specific EGFR-TKIs, such as gefitinib and erlotinib, are the first line medication for advanced NSCLC. However, published studies have revealed that the clinical response to EGFR tyrosine kinase inhibitors varies in patient cohorts. Treatment with gefitinib was associated with significantly greater survival for patients with refractory advanced NSCLC in non-smokers or Asians [14]. Additionally, gefitinib provided survival benefit in a subgroup of NSCLC patients with adenocarcinoma histology and any degree of skin rash following therapy [15]. Probably, EGFR somatic mutation is the most effective molecular predictor for EGFR-TKIs’ responsiveness and efficacy. The EGFR mutations are present in four exons (18–21 exons) of the tyrosine kinase domain of EGFR. EGFR mutation frequency vary in patients’ ethics, smoking status and histology [16–17]. It has been shown that EGFR-TKI therapeutic efficacy in advanced NSCLC with EGFR mutations can reach 60–80% and PFS can be longer than one year in many large clinical studies [9, 18–20]. However, acquired EGFR-TKI resistance is still inevitable although the precise underlying mechanisms remain unclear [21].
To find effective biological factors associated with EGFR-TKIs responsiveness and to provide the most direct and valuable guidance for clinicians to make decisions on EGFR-TKIs therapy has been a big challenge. It has been reported that 50–60% of cases with the resistance to EGFR-TKIs were due to a second T790M mutation [22] and that 5–22% resulted from gene amplification of mesenchymal-epithelial transition (MET) [23]. Other factors such as small-cell lung cancer transformation, hepatocyte growth factor (HGF) overexpression, Gas6-Axl activation, Loss of phosphatase and pensin homolog (PTEN), epithelial–mesenchymal transition (EMT) and activating mutations in phosphatidylinositol-4,5-bisphosphate 3- kinase (PI3K) have also been reported to associate with acquired EGFR-TKI resistance [24–29].
PHLPP negatively regulates PI3K/Akt [10] and RAF/RAS/ERK in cancer cells [11]. As demonstrated previously, both Akt and ERK contribute to carcinogenesis, chemoresistance [30, 31] and the tolerance of chemoradiotherapy in lung cancer [32]. Several recent studies have provided strong evidence that PHLPP serves as an important tumor suppressor in cancers such as colon cancer, glioblastoma and prostate cancer [33–35]. We previously reported the high expression rate (23.4%) of PHLPP in lung adenocarcinoma. Additionally, PHLPP expression is significantly correlated with differentiation stage and local tumor T-stage in lung adenocarcinoma, whereas low PHLPP expression is associated with poor prognosis in patients with resected lung adenocarcinoma [13]. In the present study, however, the high expression rate of PHLPP1 (2–3 point) reached 69.3%. One possibility is that two studies used different PHLPP antibodies. One was mainly to detect its membrane expression (Abcam), whereas another was to detect its cytoplasm expression (ProteinTech). Another possibility is that all patients in the present study were all EGFR TKI treating sensitive, with a better prognosis than patients in our previous study.
We previously found that PHLPP expression significantly correlated with survival time in lung adenocarcinoma patients [13]. The current study directly examined the association between PHLPP1 expression and the efficacy of target therapy in human lung adenocarcinoma. We further demonstrated that the efficacy of target therapy was better in patients with high expression of PHLPP1. Additionally, the time of acquired resistance to EGFR-TKI in NSCLC was much longer in patients with high expression of PHLPP1. Of note, PHLPP1 still inversely correlated with the expression of p-Akt and/or p-ERK in human lung adenocarcinoma tissues.
High expression of PHLPP1 in lung adenocarcinoma highly correlated with longer survival. The molecular mechanism responsible for the correlation between PHLPP1 expression with the outcome of NSCLC patients receiving EGFR-TKIs is unclear. One possible explanation is that both PI3K/Akt and RAF/RAS/ERK pathways are the downstream signaling molecules of EGFR signaling, which is associated with the occurrence and development of lung cancer. Also, it has been reported that PHLPP isozymes set the amplitude of receptor tyrosine kinase (RTK) signaling by serving as regulators of RTK transcription [36]. Therefore, PHLPP as a tumor suppressor shows an even broader role. It may be a potential prognostic marker for screening patients for target therapy.
In conclusion, our study suggests that high levels of PHLPP1 might predict a better survival of target therapy and a longer time before acquired resistance to EGFR-TKI in lung adenocarcinoma patients. PHLPP1 might be a potential prognostic marker to screen patients for target therapy. To the best of our knowledge, this study for the first time demonstrates the relationship between efficacy of EGFR-TKI target therapy and PHLPP1 expression. However, it is a single center study and the number of subjects studied is limited. Therefore, future larger multi-center trials with larger sample sizes may be necessary to generalize the results.
MATERIALS AND METHODS
Patients
We recruited 75 patients diagnosed with advanced lung adenocarcinoma and having received treatment with a single agent EGFR-TKI and exhibiting objective therapeutic benefit from 2008 to 2012 at Taizhou Hospital of Zhejiang Province in China. The Eastern Cooperative Oncology Group (ECOG) performance status scores of the recruited patients were from 0 to 2. All patients had adequate organ function as defined as a white blood cell (WBC) counts > 4 × 109/dl (absolute granulocyte > 2 × 109/dl), platelet > 100 × 109/dl, normal liver function tests and serum creatinine level < 1.4 mg/dl, and no other severe comorbid conditions. Patient stage was redetermined according to the TNM Staging System of AJCC (7th version, 2009). All patients had distant metastases (M1). All patients gave informed consent. This study was granted by the Ethics Committee of Taizhou Hospital and tissue specimen acquisition was performed in agreement with institutional guidelines.
Immunohistochemistry
The presence of PHLPP and p-AKT and p-ERK in lung tissues was assessed by immunohistochemistry (IHC) as we previously reported [13]. Antibody information is: PHLPP1 (1:200, 22789-1-AP, ProteinTech Group, Chicago, USA), PHLPP2 (1:200, 25244-1-AP, ProteinTech Group, Chicago, USA), p-AKT(S473) (1:200, BS4007, Bioworld Technology, MN, USA) and p-ERK 1/2(BS5016, 1:200, Bioworld Technology, MN, USA). Three pathologists, who were blind to all clinical and biological data, performed and analyzed IHC. A four tier system (level 0–3: negative = 0, weakest = 1, moderate = 2, strong = 3) was used to grade the intensity of staining [14]. The group with low PHLPP low expression had 0–1 point, and the PHLPP high expression group was scored as 2–3 point. To confirm the specificity of IHC, two types of negative controls were used, which included substituting rabbit non-immune IgG for primary antibodies, and omitting primary antibodies to rule out potential non specificity of secondary antibodies. Positive staining was confirmed in normal colonic mucosa slide which was used as a positive control.
Western blotting
Fresh tissue was collected prior to targeted therapy and was snap-frozen in liquid nitrogen and stored at – 80ºC. Three typical cases were chosen: case 1 without progress in less than half a year, case 2 with no progress in more than 2 years, and case 3 with no progress in 1 to 1.5 years. Tissues were extracted in a lysis buffer (pH7.4, 50 mM Tris-base, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100). Proteins extracted from the tissues were loaded onto 10% SDSPAGE and electrophoretically transferred to PVDF membranes (Millipore, USA). After blocking with 5% non-fat dry milk for 1 hour, membranes were washed in PBS containing 0.2% Tween-20 and then incubated in the corresponding primary antibodies overnight at 4ºC. The membranes were subsequently incubated with Peroxidase-conjugated Affinipure Goat Anti-Rabbit IgG Fc fragment (Dako, USA) for 1 hour at room temperature. Immunoreactivity was visualized using Biomax film (Kodak, USA) for 1–5 min in the dark room after incubation of membranes in an enhanced chemiluminescence reagent (ECL Plus Western Blotting Detection Systems, Amersham Biosciences, USA).
Patient follow-up
All patients received initial treatment with a single administration of EGFR-TKI and exhibited objective clinical benefit from the treatment. Acquired resistance was defined if patients experienced disease progression during continuous EGFR-TKI treatment. Each patients’ record included age, sex, smoking status, the medicine type (gefitinib or erlotinib), date of diagnosis, date of acquired resistance, and time of follow-up which was every 3 months.
Statistics
Statistical analyses were performed using SPSS for Windows (version 21.0). Chi-squared test was used for count data. Progression-free survival (PFS) and overall survival (OS) were estimated using the Kaplan-Meier method. PFS was defined from the date of gefitinib or erlotinib initiation until the date of first documented progression. OS was defined from the date of gefitinib or erlotinib initiation until the date of death. P-value < 0.05 represents statistical significance.
Abbreviations
Non-small-cell lung cancer (NSCLC); epidermal growth factor receptor (EGFR); tyrosine kinase inhibitors (TKIs); PH domain leucine-rich-repeats protein phosphatase (PHLPP); mesenchymal-epithelial transition (MET); Phosphatase and Tensin Homolog (PTEN); epithelial–mesenchymal transition (EMT); phosphatidylinositol-4,5-bisphosphate 3- kinase (PI3K); receptor tyrosine kinase (RTK).
Author contributions
YY Xie, Dongqing Lv, MH Ye, HH Yang participated in the design and case selection of this study. YY Xie, W Wang, HH Yang performed the experiments. HH Yang and W Wang performed the data analysis. HH Yang and XF Chen edited the manuscript while YY Xie, XF Chen and HH Yang drafted the manuscript. All authors read and approved the final manuscript.
ACKNOWLEDGMENTS
None.
CONFLICTS OF INTEREST
None.
FUNDING
The work was supported by Zhejiang Provincial Medicine and Health Foundation (2013KYA225 and 2015KYA240) and Science and Technology Agency of Taizhou City (121KY08), China.
REFERENCES
1. Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010; 60:277–300.
2. Travis WD, Brambilla E, Noguchi M, Nicholson AG, Geisinger KR, Yatabe Y, Beer DG, Powell CA, Riely GJ, Van Schil PE, Garg K, Austin JH, Asamura H, et al. International association for the study of lung cancer/american thoracic society/european respiratory society international multidisciplinary classification of lung adenocarcinoma. J Thorac Oncol. 2011; 6:244–85.
3. Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008; 359:1367–80.
4. Pao W, Kris MG, Iafrate AJ, Ladanyi M, Janne PA, Wistuba, II, Miake-Lye R, Herbst RS, Carbone DP, Johnson BE, Lynch TJ. Integration of molecular profiling into the lung cancer clinic. Clin Cancer Res. 2009; 15:5317–22.
5. Sequist LV, Bell DW, Lynch TJ, Haber DA. Molecular predictors of response to epidermal growth factor receptor antagonists in non-small-cell lung cancer. J Clin Oncol. 2007; 25:587–95.
6. Yang CH, Yu CJ, Shih JY, Chang YC, Hu FC, Tsai MC, Chen KY, Lin ZZ, Huang CJ, Shun CT, Huang CL, Bean J, Cheng AL, et al. Specific EGFR mutations predict treatment outcome of stage IIIB/IV patients with chemotherapy-naive non-small-cell lung cancer receiving first-line gefitinib monotherapy. J Clin Oncol. 2008; 26:2745–53.
7. Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I, Fujita Y, Okinaga S, Hirano H, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010; 362:2380–8.
8. Rosell R, Moran T, Queralt C, Porta R, Cardenal F, Camps C, Majem M, Lopez-Vivanco G, Isla D, Provencio M, Insa A, Massuti B, Gonzalez-Larriba JL, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med. 2009; 361:958–67.
9. Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, Nishiwaki Y, Ohe Y, Yang JJ, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009; 361:947–57.
10. Gao T, Furnari F, Newton AC. PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell. 2005; 18:13–24.
11. Li X, Stevens PD, Liu J, Yang H, Wang W, Wang C, Zeng Z, Schmidt MD, Yang M, Lee EY, Gao T. PHLPP is a negative regulator of RAF1, which reduces colorectal cancer cell motility and prevents tumor progression in mice. Gastroenterology. 2014; 146:1301–12 e1–10.
12. Brognard J, Clark AS, Ni Y, Dennis PA. Akt/protein kinase B is constitutively active in non-small cell lung cancer cells and promotes cellular survival and resistance to chemotherapy and radiation. Cancer Res. 2001; 61:3986–97.
13. Lv D, Yang H, Wang W, Xie Y, Hu W, Ye M, Chen X. High PHLPP expression is associated with better prognosis in patients with resected lung adenocarcinoma. BMC Cancer. 2015; 15:687.
14. Thatcher N, Chang A, Parikh P, Rodrigues Pereira J, Ciuleanu T, von Pawel J, Thongprasert S, Tan EH, Pemberton K, Archer V, Carroll K. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer). Lancet 2005; 366:1527–37.
15. Dudek AZ, Kmak KL, Koopmeiners J, Keshtgarpour M. Skin rash and bronchoalveolar histology correlates with clinical benefit in patients treated with gefitinib as a therapy for previously treated advanced or metastatic non-small cell lung cancer. Lung Cancer. 2006; 51:89–96.
16. Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004; 304:1497–1500.
17. Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004; 350:2129–39.
18. Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Tsurutani J, Seto T, Satouchi M, Tada H, Hirashima T, Asami K, Katakami N, Takada M, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010; 11:121–8.
19. Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, Zhang S, Wang J, Zhou S, Ren S, Lu S, Zhang L, Hu C, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011; 12:735–42.
20. Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, Palmero R, Garcia-Gomez R, Pallares C, Sanchez JM, Porta R, Cobo M, Garrido P, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012; 13:239–46.
21. Qiao X, Zhang Y, Wang J, Nong J, Li X, Yang X, Lv J, Zhang H, Qin N, Zhang Q, Yue W, Zhang S. Subsequent treatment of epidermal growth factor receptor-tyrosine kinase inhibitor failure in patients with advanced lung adenocarcinoma. Thorac Cancer. 2015; 6:678–86.
22. Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005; 352:786–92.
23. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007; 316:1039–43.
24. Ludovini V, Bianconi F, Pistola L, Chiari R, Minotti V, Colella R, Giuffrida D, Tofanetti FR, Siggillino A, Flacco A, Baldelli E, Iacono D, Mameli MG, et al. Phosphoinositide-3-kinase catalytic alpha and KRAS mutations are important predictors of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in patients with advanced non-small cell lung cancer. J Thorac Oncol. 2011; 6:707–15.
25. Yano S, Wang W, Li Q, Matsumoto K, Sakurama H, Nakamura T, Ogino H, Kakiuchi S, Hanibuchi M, Nishioka Y, Uehara H, Mitsudomi T, Yatabe Y, et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res. 2008; 68:9479–87.
26. Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, Abdel-Rahman M, Wang X, Levine AD, Rho JK, Choi YJ, Choi CM, Kim SW, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet. 2012; 44:852–60. https://doi.org/10.1038/ng.2330.
27. Yamamoto C, Basaki Y, Kawahara A, Nakashima K, Kage M, Izumi H, Kohno K, Uramoto H, Yasumoto K, Kuwano M, Ono M. Loss of PTEN expression by blocking nuclear translocation of EGR1 in gefitinib-resistant lung cancer cells harboring epidermal growth factor receptor-activating mutations. Cancer Res. 2010; 70:8715–25.
28. Suda K, Tomizawa K, Fujii M, Murakami H, Osada H, Maehara Y, Yatabe Y, Sekido Y, Mitsudomi T. Epithelial to mesenchymal transition in an epidermal growth factor receptor-mutant lung cancer cell line with acquired resistance to erlotinib. J Thorac Oncol. 2011; 6:1152–61.
29. Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger S, Cosper AK, Akhavanfard S, Heist RS, Temel J, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011; 3:75ra26.
30. McCubrey JA, Steelman LS, Abrams SL, Bertrand FE, Ludwig DE, Basecke J, Libra M, Stivala F, Milella M, Tafuri A, Lunghi P, Bonati A, Martelli AM. Targeting survival cascades induced by activation of Ras/Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways for effective leukemia therapy. Leukemia. 2008; 22:708–22.
31. Katso R, Okkenhaug K, Ahmadi K, White S, Timms J, Waterfield MD. Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis, and cancer. Annu Rev Cell Dev Biol. 2001; 17:615–75.
32. David O, Jett J, LeBeau H, Dy G, Hughes J, Friedman M, Brody AR. Phospho-Akt overexpression in non-small cell lung cancer confers significant stage-independent survival disadvantage. Clin Cancer Res. 2004; 10:6865–71.
33. Liu J, Weiss HL, Rychahou P, Jackson LN, Evers M, Gao T. Loss of PHLPP expression in colon cancer: Role in proliferation and tumorigenesis. Oncogene. 2009; 28:994–1004.
34. Chen M, Pratt CP, Zeeman ME, Schultz N, Taylor BS, O’Neill A, Castillo-Martin M, Nowak DG, Naguib A, Grace DM, Murn J, Navin N, Atwal GS, et al. Identification of PHLPP1 as a tumor suppressor reveals the role of feedback activation in PTEN-mutant prostate cancer progression. Cancer Cell. 2011; 20:173–186.
35. Molina JR, Agarwal NK, Morales FC, Hayashi Y, Aldape KD, Cote G, Georgescu MM. PTEN, NHERF1 and PHLPP form a tumor suppressor network that is disabled in glioblastoma. Oncogene. 2012; 31:1264–1274.
36. Reyes G, Niederst M, Cohen-Katnelson K, Stender JD, Kunkel MT, Chen M, Brognard J, Sierecki E, Gao T, Nowak DG, Trotman LC, Glass CK, Newton AC. Pleckstrin homology domain leucine-rich repeat protein phosphatases set the amplitude of receptor tyrosine kinase output. Proc Natl Acad Sci U S A. 2014; 111:E3957–65.