
INTRODUCTION
microRNAs (miRNAs) are a class of conserved small non-coding RNAs that regulate gene expression post-transcriptionally; a list of abbreviations can be found in Supplementary Table 1 [1, 2]. miRNAs are involved in a host of biological processes, from growth and development to homeostasis and the immune response [3, 4]. The first miRNA, lin-4, was discovered in C. elegans by Victor Ambros’ and Gary Ruvkun’s laboratories in 1993 [2, 5]. It was not until 2000 when the second miRNA, let-7, was discovered [6]. This was important, as let-7 was the first miRNA to be conserved from model organisms to humans, greatly increasing the potential role of miRNAs in human biology [7]. Since 2000, the number of functional miRNAs in humans has changed, but recent literature suggests there are anywhere from 1,500–2,500 or more miRNAs in humans [3, 8–10]. The number and authenticity of candidate miRNAs are fiercely debated, as well as the mechanism of miRNA suppression of gene expression. miRNAs can suppress gene expression by promoting deadenylation and degradation of transcripts or by preventing efficient translation [11]. While the mechanism miRNAs utilize to repress gene expression is incompletely defined, miRNA biogenesis is more fully characterized.
Most miRNAs are transcribed by RNA polymerase II, contain a 5′ cap and 3′ poly A tail, and form a stem-loop structure known as a primary miRNA (pri-miRNA) [12, 13]. From here, the pri-miRNA is processed into a 60–70 nucleotide pre-miRNA by the nuclear enzyme Drosha [14]. Exportin 5 shuttles the pre-miRNA into the cytoplasm, where it is further processed into a mature miRNA [3]. Dicer is responsible for cleaving the stem-loop pre-miRNA into a miRNA duplex, containing the passenger and guide strands [15]. The guide (mature) strand is the strand that will ultimately incorporate into the RNA-induced silencing complex (RISC), while the passenger strand is rapidly degraded. This idea is evolving, as the passenger strand of some miRNAs have been shown to incorporate into the RISC and have a regulatory function, leading to the renaming of miRNAs as 5′ (5p) or 3′ (3p) [16]. Once incorporated into the RISC, the mature miRNA targets a transcript with incomplete complementarity mainly within the 3′ untranslated region (UTR), but can also target the 5′ UTR or exonic regions [11, 15]. miRNAs recognize their targets through their seed region, nucleotides 2–7, which bind to the target mRNA and suppress expression through translational stalling or transcript degradation [17, 18]. A single miRNA can have hundreds of targets, many of which generally have similar biological functions [19]. Depending on target mRNA expression patterns, miRNAs can have various effects in different cell types. Therefore, dysregulation of miRNA expression has been shown to have profound effects on disease initiation and progression.
One area of intense miRNA research is in cancer biology [20]. Depending on a miRNA’s expression, as well as its validated targets, it can be classified as an oncogenic miRNA or tumor suppressive miRNA. Oncogenic miRNAs are sometimes simply called oncomiRs, but the correct classification of an oncomiR is any miRNA dysregulated in cancer. Oncogenic miRNAs are generally overexpressed in cancer and target anti-proliferative, cell differentiation, and pro-apoptotic genes. Conversely, tumor suppressive miRNAs are generally expressed in lower levels in cancers compared to normal tissue and target pro-survival, cell cycle, and pro-proliferative genes [21]. To complicate matters, many miRNAs can be oncogenic in certain tumors and tumor suppressive in other cancers. Expression of a miRNA’s targetome fluctuates in different tumors; therefore the effect of the miRNA on cellular growth is dependent on expression of transcripts driving or suppressing tumor growth. A good example of this is miR-146a, which promotes tumor growth in breast cancer yet suppresses tumor growth in lung cancer [22–24]. This highlights the importance of understanding the function of each miRNA in different cancers, as expression and targets vary between and within tumor types.
One recently discovered miRNA identified as being misexpressed in multiple diseases is miR-708-5p. First classified as miR-708, miR-708 was more specifically identified as miR-708-5p, as the passenger strand (miR-708-3p) revealed potential biological function and incorporation into the RISC [25–30]. miR-708-5p has been implicated in many diseases, mainly cancer, but also neurodegeneration, cardiovascular disorders, and the immune response [31–35]. In this review, we will summarize the features and targets of miR-708-5p, its contribution to oncogenesis, and potential roles in regulating the tumor microenvironment (TME).
Discovery and regulation of expression
miR-708-5p was first identified in normal and cancerous cervical samples and has high sequence similarity to miR-28 [36, 37]. MIR708 is located on chromosome 11 (11q14.1) as a mirtron, a microRNA encoded within an intron of a protein-coding gene [38–40]. MIR708 is found within intron 1 of the ODZ4 gene, which encodes the transmembrane protein Teneurin Transmembrane Protein 4 (Tenm4) (Figure 1). Teneurins are a family of highly conserved proteins that function as type II transmembrane proteins and can be liberated from the plasma membrane to act as transcriptional regulators [41]. They are expressed mainly in the developing and adult central nervous system, where they are important for neuronal pathway connections and limb development [42]. During Tenm4 mRNA processing, pri-miR-708 is spliced out and further processed into pre-miR-708, which is then exported to the cytoplasm. The pre-miR-708 containing both -5p and -3p miRNAs is cleaved to create mature miR-708-5p, which can then be loaded into the RISC to suppress target genes [25–30]. miR-708-5p shows similar expression patterns as Tenm4 in certain tissues of mice, as both are highly expressed in the brain and eyes, with diminished expression in the heart, kidney, liver, lung, pancreas, and spleen [31]. miR-708-5p and Tenm4 expression patterns diverge in several mouse tissues, including the skeletal muscle and small intestine. In both instances, miR-708-5p is expressed at much higher levels than Tenm4 [31]. A similar pattern of miR-708-5p expression can be seen using the GTex portal (gtexportal.org), where miR-708-5p is not detected in immune cells, but is found in varying amounts in the reproductive tissues, secretory tissues, muscle, gastrointestinal, nervous, and respiratory systems. While dysregulated expression of miR-708-5p is found regularly in tumors, few researchers have studied Tenm4 in cancer. Although Tenm4 is frequently mutated in certain skin, uterus, colorectal, stomach and lymph node cancers (> 10%) and to a lesser extent in lung, esophagus, and cervix tumors (> 5%), the effects of these mutations have not been extensively studied (The Human Cancer Genome Atlas, https://cancergenome.nih.gov). One study found that while Tenm4 was expressed in ovarian cancer cell lines, tumors, and normal tissue, low Tenm4 expression was associated with decreased overall survival (OS) rates [43]. Decreased Tenm4 expression was not due to promoter methylation, and siRNAs targeting Tenm4 increased proliferation and resistance to cisplatin in ovarian cancer cells [43]. miR-708-5p is also expressed at lower amounts in ovarian cancers, hence Tenm4 and miR-708-5p may share tumor-suppressive functions in cancer [44]. Beyond this work, little is known of Tenm4′s contribution to tumorigenesis. Researchers have also not studied miR-708-5p and Tenm4 expressions together in the context of cancer. Therefore, while miR-708-5p’s expression varies significantly across tissue types, how it is transcriptionally regulated is poorly defined.
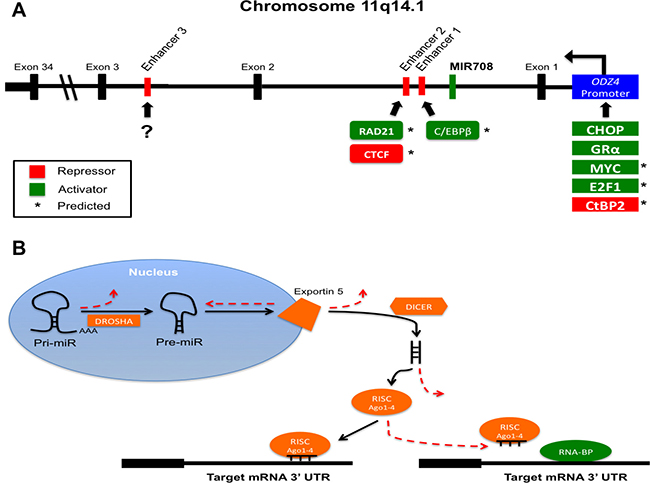
Figure 1: MiR-708-5p gene location and transcriptional and post-transcriptional regulation. (A) MiR708 (green) is located on chromosome 11 within intron 1 of ODZ4. Transcription runs right to left in the figure. Potential enhancers are found in red. Repressors (red) and activators (green) are designated by their predicted (*) or confirmed binding sites. (?) indicates unknown activator or repressor binding. (B) Illustration of various post-transcriptional mechanisms preventing miRNA maturation and function. Mutation or altered expression of DROSHA, Exportin 5, DICER, Ago1-4, or RNA-BPs can disrupt miRNA-mediated suppression of target genes. Solid black lines indicate normal, endogenous miRNA maturation and function. Dashed red lines represent steps where miRNA biogenesis and activity may be hindered in cancer cells.
To the best of our knowledge, there are only two confirmed transcriptional inducers of miR-708-5p, CCAAT enhancer-binding protein homologous protein (CHOP) and glucocorticoid receptor α (GRα) [31, 44]. CHOP is an important sensor in the unfolded protein response (UPR), which is initiated after an increase in endoplasmic reticulum (ER) stress due to accumulation or un- or mis-folded proteins. The UPR has a short-term cytoprotective role, while long-term ER stress induces CHOP expression, which acts as the switch from anti- to pro-apoptotic signaling [45, 46]. CHOP was shown to activate both Tenm4 and miR-708-5p in the brains and eyes of developing mice, where accumulation of rhodopsin in the ER activated the UPR and CHOP expression to stimulate miR-708-5p levels [31]. In turn, miR-708-5p targeted rhodopsin, suppressing its expression and resolving ER stress [31]. GRα is a cytoplasmic receptor that also acts as a transcription factor [47]. Upon glucocorticoid (GC) binding to the receptor, GRα undergoes a conformational change, causing dissociation of GRα from chaperone proteins and exposure of two nuclear localization signals [47]. Once in the nucleus, GRα binds to DNA glucocorticoid-response elements (GREs) to regulate gene transcription. In the case of miR-708-5p, GCs activate GRα, which in turn binds to the ODZ4 promoter, activating both Tenm4 and miR-708-5p transcriptional expression [44]. As GCs are prominent anti-inflammatory hormones, GC-mediated regulation of miR-708-5p would suggest a role in immune responses. Although these studies gave the first real insight into miR-708-5p transcriptional regulation, further research needs to be conducted to determine all regulators of miR-708-5p expression.
One possible glimpse into regulators of miR-708-5p expression can be obtained by examining transcriptional regulators of Tenm4. As Tenm4 and miR-708-5p share similar expression patterns across many tissues, revisiting transcription factor binding sites found within the ODZ4 promoter may identify miR-708-5p transcription factors as well. Two transcriptional activators, Myc and E2F1, have predicted binding sites within the ODZ4 promoter (Ensembl, ENSR00001770532, [48]). Myc and E2F transcription factor 1 (E2F1) have profound roles in cancer; both can promote tumor growth and also induce apoptosis, highlighting the complexity of transcription factor activities in cancer [49, 50]. Regardless, both transcription factors regulate genes that modulate cell growth, apoptosis, and metabolism, which could provide insight into potential miR-708-5p targets. C-terminal-binding protein 2 (CtBP2), a transcriptional co-repressor, also has a binding site within the ODZ4 promoter Ensembl, ENSR00001770532, [48]). Like Myc and E2F1, CtBP2 has both pro- and anti-tumorigenic roles depending on the specific tissue and target genes [51, 52]. H3K27me3 methylation of the ODZ4 promoter suppresses miR-708-5p expression in both chronic lymphoblastic leukemia (CLL) and breast cancer, highlighting multiple layers of regulation in suppressing miR-708-5p expression [53, 54]. Beyond the ODZ4 promoter, there is one confirmed and several predicted MIR708 enhancers. As seen in Figure 1A, enhancers 1-3 lie downstream of the MIR708 gene. Enhancer 1 is found closest to MIR708, while enhancer 3 is the farthest at +135.5 kb. Although ENCODE cannot predict potential transcription factor binding sites, this enhancer influences miR-708-5p expression (ENCODE, ENC11E079266.1, [33, 55]). While enhancer 3 was not sufficient to induce expression of a luciferase construct, when paired with the ODZ4 promoter, enhancer 3 intensified luciferase activity [33]. These findings were confirmed in CLL patients with low miR-708-5p expression, as H3K4me1 methylation of enhancer 3 contributed to decreased miR-708-5p levels beyond ODZ4 promoter methylation [33]. There are also two predicted enhancer sites downstream of MIR708, which we will refer to as enhancer 1 (+25.8 kb) and enhancer 2 (+30.7 kb) (ENCODE, ENC11E079375.1, [48, 55]). Enhancer 1 contains a binding site for CCAAT/enhancer-binding protein β (C/EBPβ), which is part of the same family of transcription factors as CHOP. Unlike CHOP, C/EBPβ has a pro-survival role in cells, and overexpression is associated with a subset of breast cancers [56]. In non-cancerous situations, C/EBPβ is important for neuronal production of acetylcholine, as well as macrophage production of cytokines such as interleukin (IL-4), IL-5, IL-6, and tumor necrosis factor-alpha (TNF-α) during inflammatory responses [57–62]. C/EBPβ also has a role in the UPR by decreasing TNF-α induced nuclear factor kappa-light-chain enhancer of activated B cells (NF-kB) expression [63]. Enhancer 2 contains binding sites for 11-zinc finger protein (CTCF) and RAD21 cohesion complex component (RAD21) (ENCODE, ENC11E079371.1, [55]). CTCF represses transcription by various mechanisms and also plays a role in alternative splicing [64]. RAD21 has been described to have a role in DNA repair and is part of the cohesin complex, an integral component of mitosis [65]. Whether C/EBPβ, CTCF, and RAD21 contribute to miR-708-5p expression, and if so, to what extent, remains a mystery. Regardless, these predictions help to provide insight into novel regulatory roles of miR-708-5p, possibly helping to define future targets and biological processes.
Beyond transcriptional regulation of MIR708, post-transcriptional mechanisms may be contributing to changes in mature miR-708-5p levels and activity in cancer cells. Researchers have found mutations and dysregulation of proteins essential for microRNA biogenesis and function [66]. As can be seen in Figure 1B, mutations and/or differential expression of DROSHA, Exportin 5, Dicer, and the core RISC proteins Argonaute 1–4 (Ago 1-4) can lead to aberrant miRNA processing in cancer cells [66]. Based on these data, it is important for researchers to measure pri-, pre-, and mature miRNA levels, as maturation can be influenced by changes in expression and activity of these proteins. Secondly, miRNAs frequently compete with RNA-binding proteins (RNA-BPs) for a target transcript, and RNA-BP expression and localization is altered in cancer (Figure 1B, [67, 68]). Therefore, a miRNA expressed equally in normal and cancerous cells may not be targeting transcripts in cancerous cell due to enhanced competition from RNA-BPs. Both of these post-transcriptional mechanisms further complicate an already intricate system, yet scientists should consider these aspects when performing miRNA-focused research in cancer.
Role in cancer
Hematological malignancies
Acute lymphoblastic leukemia
miR-708-5p has been most widely studied for its role as an oncogenic miRNA in acute lymphoblastic leukemia (ALL), which is divided into two major subtypes: B-cell and T-cell. B-cell ALLs, which make up about 70% of ALL cases, were found to have profoundly increased miR-708-5p expression in children compared to cluster of differentiation 34+ (CD34+) normal progenitors [69]. The same article discovered mixed-lineage leukemias (MLLs) had slightly increased miR-708-5p expression compared to normal progenitors, but the clinical significance of this finding was not established. The researchers did not see a difference between miR-708-5p expression and maturation status of the B-cell ALL patients, leading them to conclude it was a general feature of B-cell ALLs [69]. Two other groups confirmed that miR-708-5p was overexpressed in childhood B-cell ALL, but miR-708-5p overexpression was not associated with clinical prognostic markers [70, 71]. These findings were challenged by results indicating miR-708-5p expression was not associated with risk based on the Brazilian Childhood Leukemia Treatment Group Protocol [72]. miR-708-5p expression was increased in pre-B-cell ALL patients, unchanged in pro-B-cell ALL patients, and significantly lower in T-cell ALL patients compared to normal bone marrow [72]. These opposing results revealed a complex role of miR-708-5p in ALL, establishing the need to identify whether miR-708-5p is a marker for response, for relapse, or for survival in ALL.
Recent research from multiple groups showed miR-708-5p has profound clinical relevance in ALL. First, miR-708-5p was overexpressed in ALL patients at diagnosis, decreased in those who achieved complete responses, but increased significantly in relapsed patients [73], suggesting an oncogenic and pro-relapse role of miR-708-5p in ALL. However, the same study found surprising data on relapse-free survival (RFS). Low miR-708-5p also increased the risk of relapse and high miR-708-5p expression correlated with increased RFS in patients at time of diagnosis [73]. To better understand the conflicting data above, researchers looked for other known markers of relapse and aggressiveness in ALL. It was found that patients who responded well to GC therapy had high miR-708-5p expression, while poor responders had low miR-708-5p expression [73]. There were also better response rates to chemotherapy in ALL patients with high miR-708-5p. Based on these data, patients were stratified into risk groups based on known risk factors such as age, response to therapy, cytogenetics, remission status, and survival rates. High-risk patients had lower miR-708-5p expression compared to low and intermediate-risk patients [73]. Further classification of ALL based on immunophenotype revealed increased miR-708-5p expression in common ALL and pre-B-cell ALL and decreased miR-708-5p in T-cell ALL [73]. T-cell ALL is classified as a more aggressive subtype, which could explain why miR-708-5p expression was low in the high-risk group. Finally, high miR-708-5p levels at time of diagnosis correlated with better OS rates in ALL patients [73]. In summary, miR-708-5p is overexpressed in common B-cell and pre-B-cell ALL, while T-cell ALL has decreased miR-708-5p levels. Whether miR-708-5p is a marker for high-risk patients remains controversial. Although there is agreement that miR-708-5p is overexpressed in some B-cell patients, its use as a marker for risk is dependent upon inclusion/exclusion of various risk factors. Lastly, although miR-708-5p is overexpressed in relapsed ALL patients, high miR-708-5p also correlated with increased OS, RFS, and response to therapy, potentially suggesting miR-708-5p as a tumor suppressor in ALL. These studies helped to identify the perplexing functions of miR-708-5p in ALL, but also highlight the need for better classification of ALL subtypes, as there are conflicting data between and within studies.
One way to better define the function of miR-708-5p in ALL is to validate its oncogenic or tumor suppressive targets. Previous work demonstrated that miR-708-5p targets forkhead box 3 (FOXO3), a transcription factor critical for hematopoietic stem cell self-renewal [73]. In ALL, FOXO3 has been shown to be a tumor suppressor and oncogene. FOXO3 is inactivated by oncogenic PI3K signaling, preventing it from activating pro-apoptotic genes such as fas ligand (FasL), Bcl-2-like protein 11 (BIM), and TNF-related apoptosis-inducing ligand (TRAIL), resulting in increased proliferation [74–76]. FOXO3 can also act as an oncogene by increasing PI3K activity to promote resistance and by maintaining leukemia-initiating cell (LIC) populations [77, 78]. miR-708-5p also targets CD38 in airway smooth muscle cells, resulting in decreased PI3K and MAPK signaling [35]. In ALL, CD38 is a poor prognosis marker, possibly helping to explain why high miR-708-5p expression increases OS rates [79]. miR-708-5p targeting of FOXO3 may also help to prevent resistance and relapse by stopping leukemia-initiating cell self-renewal. Conversely, miR-708-5p suppression of FOXO3 may prevent B-cell apoptosis, exacerbating B-cell proliferation. Whether miR-708-5p acts as an oncogenic miRNA or a tumor suppressor could be dependent on the pathways driving tumor growth in specific cancers. If CD38 is a major driver in a particular ALL patient, then it may be more likely miR-708-5p is a tumor suppressor in that cancer. Therefore, researchers must better understand the dichotomy of FOXO3 and identify novel miR-708-5p targets to more precisely define the function of miR-708-5p in ALL.
The opposing results found between and within studies is not an uncommon phenomenon in the field of miRNA biology. Inconsistencies may be due to a variety of mechanisms of action [80]. As is the case with FOXO3 in ALL, the expression of miR-708-5p and its targets may vary between LIC populations and the descendant cells that ultimately make up the vast majority of analyzed cancer cells. Examining the net effect of the miRNA has on various hallmarks of cancer by studying miRNA-induced changes in proliferation, invasion, survival, and apoptosis is critically important [80]. While these are part of tumor progression, there are also immune evasion, angiogenesis, and cross talk between cancer cells and the external environment. Lastly, expanding results to the clinic by looking at OS and RFS rate is also useful. Therefore, if a study focuses merely on miRNA-induced proliferation changes, the full effect of miR-708-5p in that cancer is not uncovered. There are also many subsets of ALL, many of which are not completely characterized. miR-708-5p may have differential effects in established subtypes and may also help scientists better delineate ALL subtypes in the future. Regardless, the diametric function of miR-708-5p in ALL remains a controversy and requires further investigation to fully resolve its oncogenic or tumor suppressive role.
Chronic lymphoblastic leukemia
Chronic lymphoblastic leukemia (CLL) is another major type of leukemia originating from lymphocytes. ALL and CLL are most easily distinguished by their growth characteristics and inflicted populations. CLL develops much more slowly compared to ALL, usually taking years before symptoms arise [81]. CLL cells generally are more mature than ALL cells, while ALL cells are more stem-like [81]. Unlike ALL, which is found mostly in children, CLL is diagnosed mainly in elderly patients, especially males [81]. miR-708-5p has not been as extensively studied in CLL as ALL, but current literature suggests it acts as a tumor suppressor in CLL. As in ALL, CD38 is a marker for aggressive, late stage, and overall poor prognosis in CLL patients [82]. Therefore, targeting of CD38by miR-708-5p would suggest a tumor suppressive role in CLL. miR-708-5p was shown to be expressed less in CLL patients compared to healthy volunteers [33]. Decreased miR-708-5p expression was attributed to a methylated downstream enhancer (enhancer 3, Figure 1A), which significantly suppressed miR-708-5p expression beyond promoter methylation alone in CLL patients. Methylation of enhancer 3 was also shown to be a marker for decreased OS rates in CLL patients [33, 54]. One pathway CLL cancers rely on is the NF-kB pathway [83]. Inhibitor of nuclear factor kappa-B kinase subunit beta (IKKβ), a NF-kB activator, is overexpressed in CLL patients, resulting in exacerbated NF-kB signaling [33]. miR-708-5p suppresses IKKβ expression in CLL by directly targeting the IKKβ 3′ UTR [33]. Pre-treatment of CLL cells with miR-708-5p prevented phorbol 12-myristate 13-acetate (PMA)/ionomycin induced NF-kB activity. The same study investigated other potential miR-708-5p targets in CLL and found NOTCH1 to be a putative target. NOTCH1, a proto-oncogene and prognostic marker, is commonly mutated or overexpressed in CLL patients [84]. Although results suggest a tumor suppressive function for miR-708-5p in CLL, studies must be performed in vitro and in vivo to determine the direct effect of miR-708-5p on CLL cell growth. As in ALL, further research is necessary before conclusions can be drawn regarding the function of miR-708-5p in CLL.
Other hematological malignancies
There is limited information on miR-708-5p in hematological cancers beyond leukemias. There are two other major hematological cancers: lymphomas and multiple myeloma. Mantle cell lymphoma (MCL), a B-cell lymphoma, is one subtype characterized as having high relapse rates, resulting in lower survival rates [81]. One study revealed MCL patients had increased miR-708-5p expression compared to normal B-cells [85]. Overexpression of miR-708-5p was not associated with any known prognostic markers, but 95% of MCL patients have a t(11;14)(q13;q32) translocation [86, 87]. This translocation occurs relatively close to the MIR708 gene, possibly contributing to the aberrant expression observed in these patients. This translocation also occurs in a subset of B-cell leukemias. It would be interesting to see if there is a correlation between miR-708-5p expression and t(11;14) in leukemias as observed for MCL. Outside of B-cell cancers, there is little known about miR-708-5p in myeloid cancers or multiple myeloma except for one biomarker study concluded that miR-708-5p expression was unchanged in multiple myeloma patients [88]. In conclusion, while many studies have investigated miR-708-5p in B-cell cancers, little research has been done on T-cell cancers, multiple myeloma, and myeloid cancers. miR-708-5p appears to have both tumor suppressing and tumor promoting functions in B-cell ALL, while evidence suggests miR-708-5p is a tumor suppressor miRNA in CLL. Further investigation of miR-708-5p and its targets will resolve the pro- or anti-cancerous behaviors of miR-708-5p in hematological malignancies.
Solid cancers
There is accumulating evidence that miRNAs can be both oncogenic and tumor suppressive. This contrast can exist in distinct cancer types, but can also occur within a tumor type. miR-708-5p is not immune to this phenomenon, as it suppresses or promotes tumor growth in a variety of cancers. In fact, miRNAs can target both oncogenes and tumor suppressors [89]. It is important to recognize that different tissues express different transcripts. Furthermore, the same protein can function differently in different tissues. Therefore, a miRNA must be classified as a tumor suppressor or oncogenic miRNA based on the transcripts it targets in a specific tissue or cell type. Researchers must look at the net effect a miRNA has by measuring the various phenotypic changes resulting from modulating expression of a miRNA. miR-708-5p has been shown to up- and down-regulate many genes in various cell types. Given the complexity of miRNA biology, it is hard to determine transcripts miR-708-5p directly regulates. To simplify the data presented in this review, direct miR-708-5p targets are transcripts containing a putative miR-708-5p binding site in their 3′ UTR that are suppressed at the RNA and protein level. More importantly, luciferase constructs containing the 3′ UTR of interest must be suppressed by miR-708-5p to be classified as direct. In contrast, miR-708-5p may increase or decrease mRNA and protein expression of indirect targets. Indirect targets are transcripts miR-708-5p regulates that may or may not contain a putative miR-708-5p binding site in their 3′ UTRs. Therefore, indirect targets may become direct targets after further experimentation, but direct targets cannot be reclassified as indirect targets. To help decipher miR-708-5p’s function in each cancer, Tables 1 and 2 show direct (1) and indirect (2) targets reported for miR-708-5p. These following sections will identify the pro- and anti-tumorigenic roles of miR-708-5p in solid cancers, highlighting the effect of miR-708-5p by tumor type. Please refer to Table 3 for a list of miR-708-5p’s function by tumor type.
Table 1: Direct miR-708-5p targets
Name of Target |
Cancer |
Function |
Role in Cancer |
Reference |
|---|---|---|---|---|
Rhodopsin |
N/A |
Light sensitivity |
N/A |
31 |
FOXO3 |
ALL |
Pro-apoptotic signaling, LIC maintainence |
Pro/anti-tumorigenic |
73 |
CD38 |
N/A |
immune checkpoint signaling |
Poor prognostic marker in leukemias |
35 |
IKKβ |
CLL |
NF-kB activator |
pro-tumorigenic |
33 |
Caspase-2 |
Bladder |
DNA repair & pro-apototic signaling |
anti-tumorigenic |
94 |
CDKN2B |
CRC |
Cell cyle inhibitor |
anti-tumorigenic |
98 |
NNAT |
Breast, Prostate |
Increased Ca2+ to promote ERK/FAK signaling |
pro-tumorigenic |
53, 164 |
CD276 |
Breast |
Inhibits T-cell activation |
pro-tumorigenic |
111 |
LSD1 |
Breast |
Demethylase |
pro-tumorigenic |
117 |
EYA3 |
Ewing's Sarcoma |
DNA repair |
pro-tumorigenic |
131 |
Rap1b |
Ovarian |
promotes integrin-mediated focal adhesion |
pro-tumorigenic |
44 |
CD44 |
Prostate |
various oncogenic pathways |
pro-tumorigenic |
160 |
AKT2 |
Prostate |
PI3K signaling |
pro-tumorigenic |
160 |
KPNA4 |
Prostate |
shuttles transcription factors into nucleus |
pro-tumorigenic |
165 |
BMI1 |
RCC |
DNA repair |
pro-tumorigenic |
175 |
ZEB2 |
RCC |
promotes EMT |
pro-tumorigenic |
175 |
Survivin |
RCC, Lung |
inhibitor of apoptosis |
pro-tumorigenic |
175, 193 |
cFLIPL |
RCC |
Suppresses apoptotic signaling |
pro-tumorigenic |
176 |
p21 |
Lung |
Suppresses apoptotic signaling |
pro-tumorigenic |
193 |
Table 2: Indirect miR-708-5p targets
Name of Target |
Cancer |
Regulates Expression |
Role in Cancer |
Reference |
|---|---|---|---|---|
TNF-α |
CLL |
down |
Inflammation |
33 |
CXCL1 |
CLL |
down |
Inflammation |
33 |
NFKBIA |
CLL |
down |
Inflammation |
33 |
AKT-1 |
GBM |
Down |
PI3K signaling |
137 |
Cyclin D1 |
GBM |
Down |
Cell cycle progression |
137 |
MMP-2 |
GBM, Lung |
Down |
promotes invasion |
137, 193 |
EZH2 |
GBM |
Down |
proliferation, DNA Repair |
137 |
BCL-2 |
GBM |
Down |
Anti-apoptotic |
137 |
PARP-1 |
GBM |
Down |
Proliferation, DNA Repair |
137 |
E-cadherin |
RCC |
Up |
inhibits invasion |
175 |
MCAM |
RCC |
Down |
EMT, invasion |
175 |
Fibronectin 1 |
RCC |
Down |
EMT, invasion |
175 |
TRAIL |
RCC |
Up |
Pro-apoptotic signaling |
175 |
TMEM88 |
Lung |
down |
Negative regulator of WNT signaling |
192 |
BCL2A1 |
Lung |
down |
Anti-apoptotic |
193 |
BCL2L2 |
Lung |
down |
Anti-apoptotic |
193 |
PIK3IP1 |
Lung |
up |
Suppresses PI3K signaling |
193 |
PHLDA3 |
Lung |
up |
Suppresses PI3K signaling |
193 |
INPPL1 |
Lung |
up |
Suppresses PI3K signaling |
193 |
MMP-9 |
Lung |
down |
promotes invasion |
193 |
VEGFC |
Lung |
down |
promotes invasion |
193 |
CADM1 |
Lung |
down |
promotes invasion |
193 |
CD117 |
Lung |
down |
cancer stem cell marker |
193 |
CD34 |
Lung |
down |
cancer stem cell marker |
193 |
OCT4 |
Lung |
down |
cancer stem cell marker |
193 |
ALDH1A2 |
Lung |
down |
cancer stem cell marker |
193 |
NANOG |
Lung |
down |
cancer stem cell marker |
193 |
Table 3: miR-708-5p function by cancer type
Cancer |
miR-708-5p Function |
|---|---|
Hematological |
|
ALL |
Both |
CLL |
Tumor Suppressor |
Multiple Myeloma |
Not Determined |
Solid |
|
Bladder |
Oncogenic |
Breast |
Tumor Suppressor |
Colorectal |
Oncogenic |
Ewing's Sarcoma |
Tumor Suppressor |
Glioblastoma Multiforme |
Tumor Suppressor |
Hepatocellular Carcinoma |
Tumor Suppressor |
Lung |
Both |
Ovarian |
Tumor Suppressor |
Prostate |
Tumor Suppressor |
Renal Cell Carcinoma |
Tumor Suppressor |
miR-708-5p as an oncogenic miRNA
Bladder cancer
Bladder cancer is comprised of a group of tumors originating from various cell types found within the bladder. Bladder cancer patients have high survival rates, with 5-year survival rates of 77%, although survival rates decrease dramatically if the tumor is diagnosed at an advanced stage [90, 91]. Therefore, it is crucial to identify biomarkers to better detect and treat bladder cancer. miR-708-5p is overexpressed in bladder urothelial carcinoma, the most common form of bladder cancer [92]. In fact, miR-708-5p is expressed at very low levels in normal bladder tissues, but bladder urothelial carcinoma samples have a 25-fold increase in miR-708-5p expression. This is in line with gtexportal.org’s RNA-seq data, which reports low miR-708-5p expression in normal bladder samples [93]. Researchers determined miR-708-5p’s contribution to bladder carcinoma progression is through targeting of caspase-2 [94]. Caspase-2 is the most highly conserved caspase and is important for DNA repair and apoptotic signaling, where it acts as an initiator and effector caspase [95, 96]. Consistent with its pro-apoptotic function, caspase-2 has been shown to promote cell death in a variety of tumors [97]. Given these data, researchers identified miR-708-5p as an anti-apoptotic miRNA in bladder carcinoma. A miR-708-5p inhibitor promoted apoptosis in vitro, and caspase-2 expression negatively correlated with miR-708-5p expression in bladder carcinoma patients [94]. Lastly, it was shown that a miR-708-5p inhibitor reduced tumor growth in a nude mouse model [94]. While this work helped to establish an oncogenic mechanism for miR-708-5p, miR-708-5p likely targets additional tumor suppressor genes in bladder cancer cells. Therefore, researchers should continue to discover novel miR-708-5p targets in bladder urothelial cancer and determine miR-708-5p’s contribution to survival, resistance, and aggressiveness in human tumors.
Colorectal cancer
Colorectal cancer (CRC) is the fourth most common cancer in the United States, yet the second deadliest cancer [81]. Due to the frequency and risk of death of CRC patients, it is important to understand dysregulation of mechanisms that govern colorectal tumor growth. There are several published articles on miR-708-5p in CRC, which all conclude miR-708-5p is an oncogenic miRNA [98]. First, miR-708-5p is significantly overexpressed in colorectal tumors compared to adjacent normal tissue in 5 patients [98]. Although the sample size was small, a more comprehensive study with 19 patients reported similar results [99]. Consistent with an oncogenic function, anti-miR-708-5p suppressed proliferation and increased apoptosis when transfected in CRC cell lines [98]. Anti-miRs, also known as antagomirs, are miRNA inhibitors that bind to a specific miRNA and suppress its functions. A potential mechanism for the oncogenic function of miR-708-5p was shown through direct targeting of cyclin-dependent kinase inhibitor 2B (CDKN2B/p15INK4b). CDKN2B is a tumor suppressor that forms a complex with cyclin-dependent kinase4/6 (CDK4/6) to prevent cell cycle progression through cyclin D activation of CDK4/6 [100, 101]. CDKN2B is highly expressed in normal colon tissues but is suppressed in almost all colon tumors [102]. Although repression can be mediated through hypermethylation of the CDKN2B promoter, miR-708-5p suppressing CDKN2B provides an additional mechanism post-transcriptionally [98, 103]. To enhance the model that miR-708-5p is oncogenic miRNA in CRC, researchers found miR-708-5p displayed similar characteristics to miR-31, a well-defined oncogenic miRNA in CRC [104, 105]. These studies helped define the function of miR-708-5p in tumor progression through in vitro analysis, but in vivo studies still needed to be conducted.
miR-708-5p also functions in non-inflammatory and inflammatory induced colonic epithelial cell transformation [106]. This work employed two in vivo models to analyze colon tumorigenesis: APC(Min/+) or dextran sulfate induced colitis-associated tumors. The study revealed that miR-708-5p was overexpressed in tumors from both rodent models when compared to their non-cancerous controls [106]. Gene ontology (GO) analysis of predicted targets of miR-708-5p, miR-31, miR-315, and miR-135b (all of which were dysregulated in both rodent models) was performed to determine the pathways these miRNAs regulate. Putative targets of these miRNAs were enriched in TGF-β, MAPK, PI3K, and WNT signaling, all of which have established roles in colonic epithelial transformation [104, 105]. Although GO analysis was not performed on each miRNA individually, these results will help guide future research on the function of miR-708-5p in CRC transformation and progression. Researchers should consider testing the effect of anti-miR-708-5p on CRC initiation and progression in rodent models and expand research into the clinic.
miR-708-5p as a tumor suppressor
Breast cancer
Perhaps the strongest evidence for miR-708-5p as a tumor suppressor is found in breast cancer studies. While breast cancer patients have high survival rates due to early detection and improved therapies, aggressive subtypes and metastasis present challenges to effectively treating breast cancer. It was shown that miR-708-5p is highly expressed in normal breast cells, but is significantly lower in cancerous breast cells from primary tumors, and even more profoundly suppressed in metastatic breast tumor cells [53]. miR-708-5p expression was also lower in metastatic lesions compared to their paired primary tumors in human breast cancer patients, confirming in vitro data [53]. Decreased miR-708-5p expression was due to tri-methylation (H3K27me3) of chromatin in the miR-708-5p promoter by SUZ12 Polycomb Repressive Complex 2 Subunit (SUZ12), a key subunit of the Polycomb Repressive Complex 2 (PRC2). Restoration of miR-708-5p levels reduced the migratory potential of metastatic breast cancer cells, while primary tumor cells transduced with a miR-708-5p sponge showed increased migration [53]. The authors expanded these findings to mouse models where they found that miR-708-5p did not reduce primary tumor growth, but significantly inhibited metastasis, specifically to the lungs. It was determined that miR-708-5p suppresses migration by directly targeting neuronatin (NNAT), a modulator of intracellular Ca2+ [53, 107]. By suppressing expression of NNAT, intracellular Ca2+ levels were decreased, resulting in reduced activation of two pro-migratory signaling pathways, extracellular signal-regulated kinase (ERK) and focal adhesion kinase (FAK) [53, 108–110]. This work provided strong evidence for miR-708-5p as a tumor suppressor in breast cancer, particularly in preventing metastasis.
Building on these studies, it was shown that miR-708-5p directly targets the pro-metastatic gene CD276/B7-H3 [111]. CD276 is an immunoregulatory protein that inhibits T-cell activation, is overexpressed in breast cancer, and is associated with metastasis and advanced disease [112–115]. While CD276 is usually studied in cancer as an immune checkpoint molecule, it also been has a non-immunological role [116]. miR-708-5p suppressed CD276 expression in breast cancer cell lines [111], but this study did not examine phenotypic changes of miR-708-5p administration. Therefore, it is difficult to conclude what effect miR-708-5p suppression of CD276 has on breast cancer cells. Another study demonstrated that miR-708-5p reduced proliferation and invasion of breast cancer cells [117]. The researchers argued the anti-proliferative and anti-invasive effects of miR-708-5p could be attributed to its suppression of lysine-specific histone demethylase 1 (LSD1) [117]. LSD1 is a lysine demethylase that contributes to epigenetic regulation in normal and cancerous cells [118, 119]. LSD1 modulates expression of many genes; therefore there could be many mechanisms by which LSD1 can regulate proliferation and invasion in breast cancer cells. Regardless, direct suppression of LSD1 by miR-708-5p results in decreased proliferation and invasion of breast cancer cell lines [117]. These data, paired with miR-708-5p targeting of NNAT and CD276, provide strong evidence for miR-708-5p as a tumor suppressor in breast cancer through repression of invasion, proliferation, and potentially immune modulation.
Ewing’s sarcoma
Ewing’s sarcoma is a cancer of bone and soft tissue, usually manifesting in adolescents [120, 121]. It is believed to arise from mesenchymal cells with most patients having a 11;22 translocation resulting in fusion of the Ewing Sarcoma Breakpoint Region 1 (EWSR1) and Friend Leukemia Integration Transcription Factor 1 (FLI1) genes [122, 123]. The EWS/FLI1 fusion protein contains the transcriptional regulatory domain of EWSR1 paired with the FLI1 DNA binding domain to promote oncogenesis [124, 125]. While creating small molecule inhibitors to target EWS/FLI1 is an attractive therapeutic option, many obstacles have arisen, making normal drug development unachievable. Therefore, researchers are resorting to EWS/FLI1-regulated genes as therapeutic targets. Robin et al. discovered that EWS/FLI1 regulates expression of Eyes absent homolog 3 (EYA3), a transcription factor involved in development, DNA repair, and tumorigenesis [126–131]. Unexpectedly, EWS/FLI1 does not bind to the EYA3 promoter to induce EYA3 expression. Rather, EWS/FLI1 suppresses miR-708-5p expression, which results in increased EYA3 levels [131]. The authors also showed that miR-708-5p and EYA3 expressions were negatively correlated in Ewing’s sarcoma patients. Treating Ewing’s sarcoma cell lines with miR-708-5p mimic resulted in decreased DNA repair, increased apoptosis, and resensitization to chemotherapeutics [131]. Furthermore, Ewing’s sarcoma patients with low miR-708-5p paired with high EYA3 expression had worse RFS rates [131]. Given these data, the authors concluded that EWS/FLI1 mediated-suppression of miR-708-5p led to increased EYA3 expression in Ewing’s sarcoma, allowing the cancer cells to more efficiently repair chemotherapeutic-induced DNA damage. This ultimately leads to enhanced risk of relapse or poor response to therapy. While the authors did not provide a mechanism for how EWS/FLI1 suppresses miR-708-5p expression, there may be clues in the literature to help explain this phenomenon.
First, miR-708-5p is predicted to target the FLI1 3′ UTR (microrna.org). Given FLI1 is on the 3′ end of the fusion protein, the transcript should contain the FLI1 3′ UTR. It would be interesting to test if miR-708-5p suppresses EWS/FLI1 expression, as this could provide a therapeutic approach to overcome the issues with small molecule inhibitors. Second, CHOP, a transcriptional activator of miR-708-5p, is also commonly mutated or translocated in Ewing’s sarcoma [132, 133]. Translocation frequently creates a fusion protein, which may contribute to miR-708-5p dysregulation. This would make sense given CHOP’s annotated role as a DNA-damage response protein [134]. While chemotherapeutic-induced DNA damage may stimulate CHOP in normal cells, mutated or fused CHOP proteins may not activate miR-708-5p expression, resulting in increased EYA3 expression, efficient DNA repair, and cancer cell survival. It would be worthwhile to investigate whether CHOP’s mutational status correlates with miR-708-5p expression in Ewing’s sarcoma. Although these statements are speculative and must be experimentally tested, they fit well into the established tumor suppressive function of miR-708-5p in Ewing’s sarcoma.
Glioblastoma multiforme
Glioblastoma Multiforme (GBM) is the most common type of brain tumor, originating from glial cells [81]. Glial cells are non-neuronal cells in the brain that help maintain neuronal homeostasis and function. The cancerous glial cells in GBM arise from astrocytes or oligodendrocytes. Astrocytes are responsible for supporting endothelial cell homeostasis, neuronal function, and response to injury, while oligodendrocytes insulate axons through creation and maintenance of the myelin sheath [135, 136]. There is a single report that miR-708-5p is a tumor suppressive miRNA in GBM, as miR-708-5p was expressed less in GBM cell lines compared to normal glial cells [137]. The same study showed that forced overexpression of miR-708-5p in two GBM cell lines caused decreased survival, invasion, and proliferation [137]. The authors investigated possible mechanisms underlying these phenotypic changes and found miR-708-5p decreased expression of RAC-alpha serine/threonine-protein kinase 1 (AKT1), cyclin D1, matrix metalloproteinase-2 (MMP-2), enhancer of zeste homolog 2 (EZH2), B-cell lymphoma 2 (BCL-2), and poly[ADP-ribose] polymerase 1 (PARP-1) [137]. All of these proteins have profound pro-oncogenic roles in survival (AKT1, BCL-2, PARP-1) [138, 139], proliferation (cyclin D1, AKT1, EZH2) [140], DNA repair (EZH2) [141], and invasion (MMP-2) [142]. Cyclin D1, MMP-2, and EZH2 are predicted miR-708-5p targets, but AKT1, BCL-2, and PARP-1 are not. miR-708-5p may have multiple points of direct and indirect suppression in GBM oncogenic signaling, as cyclin D1 expression is induced through Ras signaling, which also activates PI3K/AKT signaling [143, 144]. EZH2 is part of the pro-oncogenic PRC2, of which SUZ12 is also a member [145]. As stated earlier, SUZ12 suppresses miR-708-5p expression in breast cancer [53]. In GBM, overexpression of EZH2 may lead to suppression of miR-708-5p through increased PRC2 activity. miR-708-5p suppression may liberate cyclin D1, MMP-2, and EZH2 from miR-708-5p-mediated regulation, propagating a pro-tumorigenic phenotype by directly increasing AKT1, BCL-2, and PARP-1 expression. As miR-708-5p is predicted to target EZH2, it would be intriguing to see whether EZH2 overexpression results in decreased expression of miR-708-5p, or if another mechanism is first suppressing miR-708-5p. Although current data suggest miR-708-5p acts as a tumor suppressor in GBM, these data highlight the need for more research to be conducted on miR-708-5p to define direct targets in GBM.
Hepatocellular carcinoma
Hepatocellular carcinoma (HCC), more commonly referred to as liver cancer, is the sixth most prevalent cancer in the world [146]. While it is a fairly rare cancer, HCC prevalence has increased 3-fold over the last 20 years [146]. Increased incidence paired with a less than 20% 5-year survival rate highlights the need to better understand and treat HCC [81]. The function of miR-708-5p in HCC is continuing to be defined, as it was first found to have decreased expression in HCC tumors compared to paired non-HCC tissue in humans [147]. Low miR-708-5p expression was a prognostic marker for late-stage HCC as determined by Edmondson-Steiner grading and Tumor Node Metastasis (TNM) staging [147]. These findings were expanded to see what phenotypic effects miR-708-5p overexpression would have in two HCC cell lines (HepG2 and SMMC-7221). They determined miR-708-5p overexpression decreased migration and invasion of the HCC cell lines compared to a negative control miRNA [147]. This research was supported by work showing that miR-708-5p expression was lower in hepatitis B-associated HCC tumors [148]. This is especially important, given increased chronic hepatitis B/C infection has been associated with the increase in HCC prevalence over the last two decades [146].
Contrary to previous studies, there is evidence for miR-708-5p to function as an oncogenic miRNA in HCC. Researchers showed that miR-708-5p was overexpressed in persistent preneoplastic liver lesions (pPNLs) compared to surrounding normal liver tissue in rats [149]. Treatment with β-Ionone, a suppressor of pPNL growth, decreased miR-708-5p levels [149]. miR-708-5p expression also negatively correlated with two tumor suppressors, metalloproteinase inhibitor 3 (Timp3) and metastasis suppressor protein 1 (Mtss1) levels, leading the authors to conclude miR-708-5p acts as a transformative promoter in liver cells [149]. This conclusion is oversimplified, as both Timp3 and Mtss1 do not always act as tumor suppressors in HCC. Timp3 inhibits MMPs important for cell migration and has tumor suppressive roles in HCC, yet Timp3-/- mice have decreased incidences of HCC compared to wild-type mice after exposure to carcinogens [150–152]. Mtss1 overexpression is also associated with metastasis and poor prognosis of hepatitis B-related HCCs [153]. Therefore, it may not suitable to use Timp3 and Mtss1 as markers for transformation in HCC given the pro- and anti-tumorigenic functions of both proteins. The question remains, why is miR-708-5p overexpressed in pPNLs? Does miR-708-5p expression change during pPNL transformation into HCC? Is miR-708-5p differentially expressed between preneoplastic lesions? Does differential miR-708-5p expression contribute or suppress transformation? Regardless, it would be insightful to know how miR-708-5p expression differs from preoneoplastic lesions to late stage HCC samples.
Ovarian cancer
Ovarian cancer is a common cancer that afflicts women and is usually diagnosed at late stage, resulting in high rates of metastasis [154, 155]. Mature miR-708-5p levels are lower in ovarian cancers compared to normal tissue [44]. miR-708-5p expression also correlated with stage, as patients in late stage disease (grade III-IV) had lower miR-708-5p expression compared to early stage (I-II) [44]. This result was replicated using the human ovarian primary tumor cell line (SKOV-3) and its daughter lung metastatic cell line (SKOV-I6iv). Unsurprisingly, ovarian cancer patients with high miR-708-5p expression had longer OS and RFS [44]. Restoration of miR-708-5p expression in ovarian cancer cell lines suppressed migration and invasion, while miR-708-5p overexpression dramatically reduced metastasis to the lungs of mice engrafted with ovarian cancer cells in vivo [44]. The authors then proceeded to test the effects of GCs on miR-708-5p expression, as ALL patients with high miR-708-5p expression responded well to GC therapy [73]. GCs are commonly used in conjunction with chemotherapies to alleviate side effects but also have anti-tumorigenic functions themselves [156, 157]. Two GCs, dexamethasone and prednisolone, increased miR-708-5p expression through GRα in ovarian cancer cells [44]. GC-induced miR-708-5p expression resulted in decreased Ras-related protein Rap-1b (Rap1b) mRNA and protein expression. It was demonstrated that miR-708-5p-mediated reduction in migration and invasion was through directly targeting Rap1b in ovarian cancer cells [44]. Rap1b promotes integrin-mediated focal adhesion through phosphorylation of FAK and Paxillin (Pax) [158, 159]. GC induction of miR-708-5p decreased Rap1b expression, causing a reduction in pFAK and pPAX, leading to loss of integrin-mediated focal adhesion and invasion. While there is only a single report examining miR-708-5p in ovarian cancer, the authors provided strong evidence for miR-708-5p as a tumor suppressor in ovarian cancer.
Prostate cancer
Prostate cancer is the second most common cancer in men but has a high survival rate due to early detection and efficacious therapeutics [81]. Prostate cancer treatments still pose risks and have lasting side effects, both of which can significantly impair a survivor’s quality of life. Late-stage prostate tumors dramatically decrease survival rates due to metastasis, highlighting the need to discover novel therapeutic options to treat late-stage and metastatic prostate cancer. Work on prostate cancer revealed miR-708-5p might modulate relapse and metastasis [160]. First, researchers discovered miR-708-5p was expressed less in prostate cancers, and low miR-708-5p expression was associated with poor-survival, tumor progression, and reoccurrence [160]. miR-708-5p overexpression in prostate cancer cells decreased cell viability, migration, and invasion while simultaneously inducing apoptosis in vitro. It was also shown that miR-708-5p administration suppressed prostate tumor growth in vivo [160]. In particular, miR-708-5p expression was suppressed in CD44+ prostate cancer cell populations [160]. CD44 is a cell surface glycoprotein important for intercellular communication and cell movement [161]. CD44+ cancer cells display an aggressive phenotype, and CD44 is a cancer stem cell marker in prostate cancer [162, 163]. Interestingly, miR-708-5p directly targets CD44 [160]. As CD44 undergoes alternative splicing and post-translational modifications, designing small molecule inhibitors or antibodies as therapies have been difficult. miR-708-5p avoids these obstacles, as it suppresses CD44 expression by targeting the CD44 3′ UTR, thus targeting all alternative splice variants and suppressing expression prior to post-translational modification. Therefore, using miR-708-5p as a therapeutic in prostate cancer may be more efficacious than other CD44 targeting agents.
The anti-tumorigenic role of miR-708-5p in prostate cancer was not solely due to CD44, as miR-708-5p also targets RAC-beta serine/threonine-protein kinase (AKT2), NNAT, and karyopherin importin subunit alpha-4 (KPNA4) in prostate cancer [160, 164, 165] (Figure 2A, 2C). KPNA4 is an importin that shuttles transcription factors into the nucleus. Specifically, it has been shown to translocate two oncogenic transcription factors, NF-kB and the intracellular domain of NOTCH [166–168]. miR-708-5p-mediated reduction in migration was determined to be through suppression of KPNA4 [165]. Although the authors did not further test miR-708-5p-induced phenotypic changes, they did show that KPNA4 knockdown led to decreased bone metastases in vivo [165]. Mechanistically, suppression of KPNA4 inhibited TNF-α pro-metastatic signaling by preventing NF-kB translocation into the nucleus [165]. Beyond the direct effect of KPNA4 suppression on prostate cancer cell growth, KPNA4 knockdown reduced M2 tumor-associated macrophage (TAM) infiltration into tumors in vivo by reducing chemokine (C-C motif) ligand 2 (CCL2) and chemokine (C-C motif) ligand 8 (CCL8) expression in prostate tumor cells [165]. M2 TAMs are pro-tumorigenic as they release pro-angiogenic, immunosuppressive, and pro-growth signals into the TME [169]. This profound role of KPNA4 in prostate cancer makes the use of miR-708-5p as a therapeutic enticing. It would be worthwhile to investigate whether miR-708-5p treatments would replicate the KPNA4 knockdown results, as it would solidify a novel class of therapeutics in prostate cancer.
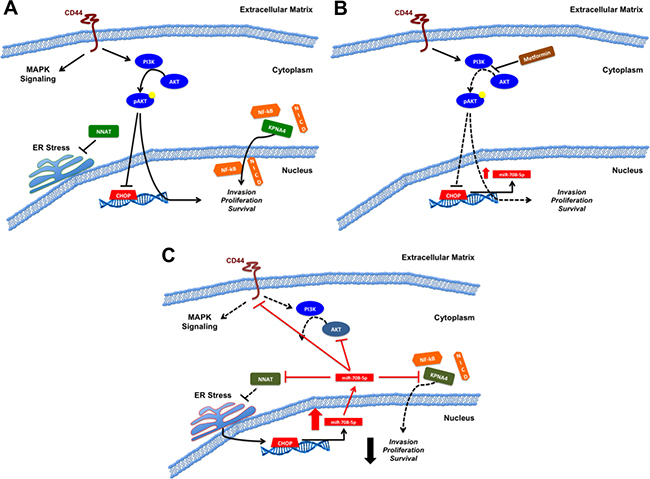
Figure 2: Model of miR-708-5p in prostate cancer. (A) Pro-oncogenic signaling of CD44, NNAT, the PI3K pathway, and KPNA4. CD44 activates MAPK and PI3K signaling pathways, potentially suppressing CHOP expression. NNAT mitigates ER stress while KPNA4 shuttles pro-oncogenic transcription factors (NF-kB, NICD) into the nucleus. Collectively, CD44, NNAT, the PI3K pathway, and KPNA4 promote invasion, proliferation, and survival in prostate cancer. (B) Proposed metformin anti-tumorigenic activities. Metformin increases miR-708-5p expression, possibly by inhibiting PI3K signaling, which in turn derepresses CHOP. Increased CHOP expression potentially promotes miR-708-5p expression in prostate cancer cells. (C) Restoration of miR-708-5p (exogenous source/long term metformin treatment) suppresses various pro-oncogenic signaling pathways. miR-708-5p directly targets KPNA4 to suppress transcription factor shuttling. miR-708-5p suppression of NNAT results in exacerbated ER stress. ER stress may further propagate CHOP and miR-708-5p expression through the UPR. PI3K signaling is mitigated through miR-708-5p targeting of CD44 and AKT2. miR-708-5p activities culminate with decreased invasion, proliferation, and survival in prostate cancer cells. Black solid lines indicate activation (arrows) or suppression (blocks) while red solid lines indicate miR-708-5p targeting. Dotted lines represent loss of signaling within a pathway.
While miR-708-5p was previously shown to target NNAT in breast cancer [53], the miR-708-5p/NNAT axis was expanded to prostate cancer [164]. The authors discovered that metformin, an anti-diabetes drug, induced apoptosis in prostate cancer by increasing miR-708-5p expression [164]. Metformin, which enters the mitochondria and blocks oxidative phosphorylation, has been shown to reduce prostate cancer risk, is anti-tumorigenic, and can be used as to treat metastatic prostate cancer [170–172]. Through an unexplained mechanism, metformin promoted miR-708-5p expression, which in turn suppressed NNAT levels, resulting in ER stress-induced apoptosis [164]. Metformin is proposed to exert its anti-tumorigenic effects by suppressing PI3K pro-survival signaling, which ultimately results in increased apoptosis of prostate cancer cells [173] (Figure 2B). This model may need to be expanded to include miR-708-5p. While the proposed mechanism does not explain an initial increase (~2-3 fold) in miR-708-5p levels 24 hours after metformin treatment, early activation of miR-708-5p expression may be from inhibition of PI3K signaling, as PI3K/AKT signaling suppresses CHOP expression [174]. Metformin derepression of CHOP may result in a small increase in miR-708-5p expression, which then suppresses NNAT expression, amplifying ER stress to further promote CHOP expression. CHOP in turn further propagates miR-708-5p expression to promote pro-apoptotic signaling. Beyond this positive feedback loop, miR-708-5p suppression of AKT2further impedes PI3K pathway signaling. Therefore, miR-708-5p may induce apoptosis through at least two mechanisms by amplifying ER stress through NNAT suppression and by decreasing pro-survival PI3K signaling through AKT2 repression. A proposed schematic describing the function of miR-708-5p in prostate cancer is shown in Figure 2. These results highlight miR-708-5p as a profound tumor suppressive in prostate cancer and lay the foundation for further analysis of miR-708-5p as a novel therapeutic to treat prostate cancer.
Renal cell carcinoma
In renal cell carcinoma (RCC), miR-708-5p suppresses tumorigenicity by targeting multiple pro-survival genes [175, 176]. It was revealed that miR-708-5p has reduced expression in RCC tumor samples, and low expression is associated with advanced tumors [175]. miR-708-5p restoration resulted in decreased cell viability, migration, invasion, and proliferation while also inducing apoptosis in RCC cell lines [175, 176]. miR-708-5p treatment in xenograft rodent models reduced tumor burden and synergistically suppressed tumor growth when used in combination with the chemotherapeutic doxorubicin [175, 176].
Mechanistically, researchers identified several miR-708-5p targets in RCC. It was shown miR-708-5p directly targets the 3′ UTR of Zinc finger E-box-binding homeobox 2 (ZEB2) and Polycomb complex protein BMI-1 (BMI1) [175]. ZEB2 is a transcription factor that promotes epithelial-mesenchymal transition (EMT) by suppressing cellular adhesion proteins such as E-cadherin [177]. EMT is an important mechanism cancer cells seize to invade and metastasize. Transfection of miR-708-5p in RCC cells suppressed ZEB2 levels, resulting in decreased expression of the EMT markers melanoma cell adhesion molecule (MCAM) and fibronectin 1, while also increasing E-cadherin expression [175]. The second miR-708-5p target, BMI1, is an oncogene that promotes cell cycle progression and DNA double-strand break repair [178]. This partially defines how miR-708-5p acts synergistically with doxorubicin, as doxorubicin causes DNA double-strand breaks, which in turn cannot be repaired efficiently due to miR-708-5p suppression of BMI1.
Although BMI1 suppression helps explain the anti-proliferative properties of miR-708-5p, the ability of miR-708-5p to induce apoptosis independent of doxorubicin suggests alternative pro-survival targets. In fact, miR-708-5p targets two anti-apoptotic genes, baculoviral inhibitor of apoptosis repeat-containing 5 (survivin), and cellular FLICE-like inhibitory protein (cFLIP) [175, 176]. Survivin is a member of the inhibitor of apoptosis (IAP) family of proteins that impede caspase-mediated cell death [179]. cFLIP is alternatively spliced into long (cFLIPL) or short (cFLIPS) forms and acts as a pro-survival gene by preventing death-inducing signaling complex (DISC) formation [180]. Beyond targeting apoptosis suppressors, miR-708-5p activates the pro-apoptotic cytokine TNF-related apoptosis-inducing ligand (TRAIL) [175]. cFLIP protects cells against TRAIL-mediated cell death, therefore miR-708-5p promotes apoptosis by inducing TRAIL expression while also suppressing cFLIP, a negative regulator of TRAIL. Surprisingly, miR-708-5p had no affect on expression of cFLIPS in RCC cells [176]. The authors concluded that this was due to additional cytosine base insertions in the miR-708-5p binding site within the cFLIPS 3′ UTR, eliminating miR-708-5’s ability to target the 3′ UTR [176]. cFLIPS promotes necroptosis, a programmed form of necrosis, which elicits a strong pro-inflammatory response [181, 182]. This is in contrast to apoptosis, which suppresses immune responses. Therefore, it is plausible that miR-708-5p indirectly induces an inflammatory response in RCC tumors, which would be beneficial for tumor clearance beyond a direct tumor killing function for miR-708-5p. It would be interesting to test miR-708-5p’s effects in an immunocompetent rodent model to determine its effect on necroptosis-induced inflammation. Taken together, these findings help define miR-708-5p as a tumor suppressor in RCC, yet more work needs to be completed before identifying its use as a potential therapy in RCC.
Controversy
Lung cancer
Lung cancer is the second most common cancer in Americans, but more importantly, the deadliest [183, 184]. Each year, $13.4 billion is spent on lung cancer care in the United States, yet late detection and ineffective therapies result in low survival rates [185, 186]. Lung cancer is a broad term encompassing many different diseases, the most common of which is non-small cell lung cancer (NSCLC), making up 80–85% of all lung cancers [187]. Within NSCLC, there are two major sub-groups, adenocarcinomas and squamous cell carcinomas (SCCs). Adenocarcinomas, which arise from mucus secreting cells in the lung, comprise 40% of all lung cancers, and are associated with smoking [81]. Squamous cell lung cancers emerge from squamous cells that line the lung’s airways, are highly associated with smoking, and comprise 30% of all lung cancers [81]. Although treated as a single disease in the clinic, the morphology, location, and mutations found in adenocarcinomas and SCCs are different [188]. Therefore, it is crucial to better identify differences between these subtypes to improve therapeutic options and define novel targets.
Studies on the function of miR-708-5p in lung cancer have drawn various, sometimes conflicting conclusions. The first study on miR-708-5p in lung cancer investigated differential miRNA expression profiles in NSCLC Stage I patients whose tumor did or did not reoccur. The authors discovered that miR-708-5p was expressed in both groups, but miR-708-5p was more highly expressed in recurrent patients [189]. These NSCLC patients were not divided by subtype (adenocarcinoma, SCC, bronchioloalveolar), so it is impossible to determine whether this was a broad feature of NSCLC patients or subtype-specific. Further research revealed that miR-708-5p overexpression in NSCLC might be subtype-specific by demonstrating that miR-708-5p was overexpressed in SCC patients [190]. The authors also found that miR-708-5p was overexpressed in the sputum of Stage I SCC patients [190]. Sputum is the phlegm coughed up by SCC patients, which researchers are studying as a less invasive biomarker source in lung cancer. Clinically, these results suggest miR-708-5p could be used as a potential biomarker to detect SCC. This concept was strengthened as it was confirmed miR-708-5p is overexpressed in SCC tumors compared to adenocarcinomas [191]. These authors did not compare either SCC or adenocarcinoma tumors to healthy lung tissue, so it is impossible to resolve if miR-708-5p expression is lower in adenocarcinomas compared to healthy tissue or the degree of overexpression in SCC. Regardless, miR-708-5p appears to be a plausible biomarker in differentiating SCC from adenocarcinomas.
To complicate matters further, increased miR-708-5p expression is observed in adenocarcinomas from never-smokers [192]. Furthermore, the authors concluded that high miR-708-5p expressing tumors were associated with lower survival in the same group of patients [192]. These results were replicated in SCC tumors, which had similar overexpression, although miR-708-5p was not associated with changes in survival rates in SCC [192]. In contrast to these findings, Wu et al. concluded that high miR-708-5p in NSCLC (SCC and adenocarcinoma) tumors improved survival rates [193]. They also found that highly invasive NSCLC tumors had lower miR-708-5p expression [193]. Based on these studies, there is significant disagreement on the pro-oncogenic or tumor suppressive activities of miR-708-5p in lung cancer. Therefore, it is crucial to better identify subtypes that differentially express miR-708-5p to more fully elucidate the exact function of miR-708-5p in lung cancer. A schematic detailing the various functions of miR-708-5p reported in lung cancer subtypes can be found in Figure 3. While these studies highlighted miR-708-5p’s clinical relevance, the effect of miR-708-5p modulation in lung cancer cells remained unanswered.
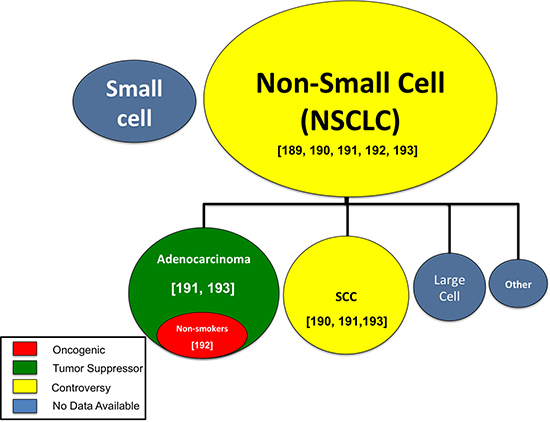
Figure 3: MiR-708-5p research by lung cancer subtype. The oncogenic (red), tumor suppressive (green), controversial (yellow), or unknown (blue) function of miR-708-5p in various lung cancer subtypes. Circle size represents relative abundance of each subtype. [Numbers] identify citations relevant to each lung cancer subtype.
As with the studies in tumor tissues, analysis of miR-708-5p has yielded contrasting results in lung cancer cell lines. It was first observed that increased miR-708-5p expression in H1299 lung cancer cells compared to normal BEAS2B cells [192]. Transient transfection or stable induction of miR-708-5p increased normal (BEAS2B) and NSCLC cell line (H1299 & A549) proliferation, while a miR-708-5p antagomir decreased H1299 proliferation [192]. Increasing miR-708-5p levels in both A549 and H1299 resulted in increased invasion [192]. Mechanistic studies revealed miR-708-5p was suppressing target transmembrane protein 88 (TMEM88), a negative regulator of WNT signaling [192, 194] (Figure 4). While the authors did not determine if TMEM88 expression was negatively correlated with miR-708-5p expression, they did show that high TMEM88 increased survival rates in adenocarcinomas of never smokers. They also failed to demonstrate whether miR-708-5p directly targets the TMEM88 3′ UTR, as they did not repeat luciferase experiments with a mutated miR-708-5p predicted binding site. This may be due to the fact the TMEM88 3′ UTR does not contain a predicted miR-708-5p binding site (microrna.org, targetscan, miRDB). This does not mean that miR-708-5p does not regulate TMEM88 expression, but this regulation may not be direct. The authors also did not test the effect of miR-708-5p on TMEM88 protein expression; rather they only measured differences in mRNA levels. Lastly, TMEM88 localization is crucial for determining its function in NSCLC. While membrane-bound TMEM88 is tumor suppressor by repressing WNT signaling, cytoplasmic localization is oncogenic and correlates with lower survival, metastasis, and late stage disease [195]. A549 and H1299 lung cancer cells have high levels of cytoplasmic TMEM88, which did not contribute to proliferation, but did promote metastasis in vivo [195]. If miR-708-5p directly targets TMEM88, is it suppressing both membrane-bound and cytoplasmic TMEM88 equally? One protein may have a longer half-life, altering the effect of miR-708-5p on TMEM88’s pro- or anti-tumorigenic functions. Also, if miR-708-5p is repressing TMEM88 proteins equally, does loss of membrane-bound TMEM88 expression have a more/less profound effect on tumor cell phenotype? Based on these data, it is impossible to conclude whether miR-708-5p pro-oncogenic effects on lung cancer cells are through TMEM88.
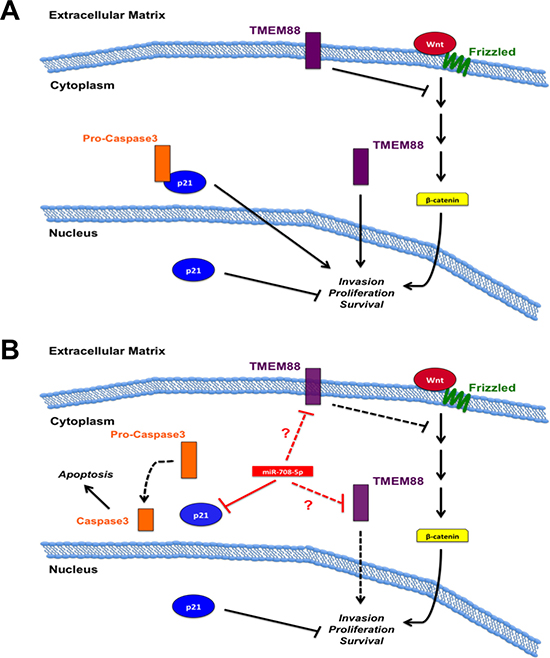
Figure 4: miR-708-5p model in lung cancer. (A) TMEM88 and p21 oncogenic and tumor suppressive mechanisms in lung cancer. Membrane bound TMEM88 suppresses pro-oncogenic WNT signaling, while cytoplasmic TMEM88 promotes tumorigenesis. Cytoplasmic p21 sequesters pro-caspase 3 to hinder pro-apoptotic signaling while nuclear p21 acts as a tumor suppressort. (B) Function of miR-708-5p on TMEM88 and p21 signaling. miR-708-5p decreases cytoplasmic p21 levels while not altering nuclear p21 levels. Deregulation of pro-caspase 3 allows for apoptotic signaling to proceed, while nuclear p21’s tumor suppressive functions remain intact. miR-708-5p may directly or indirectly suppress TMEM88. If miR-708-5p represses membrane-bound TMEM88, regulation of WNT signaling is lost, resulting in increased pro-oncogenic signaling. miR-708-5p repression of pro-tumorigenic cytoplasmic TMEM88 would result in decreased invasion, proliferation, and survival. Black solid lines indicate activation (arrows) or suppression (blocks) while red lines indicate miR-708-5p direct (solid) or unknown (dotted, ?) targeting. Dotted black lines represent signaling inhibition.
In contrast to previous studies, there is evidence that suggests miR-708-5p is a tumor suppressive miRNA in NSCLC [193]. These authors found human adenocarcinoma and SCC tumors with a highly invasive phenotype expressed significantly less miR-708-5p than non-invasive tumors [193]. These data were supplemented by dividing multiple lung cancer lines into low or high metastatic groups based on their migratory potential using a wound-healing assay. The authors found the high metastatic group (A549, H1299, PG, H226, H1703) expressed significantly less miR-708-5p than the low metastatic group (QG56, H520, H2170, 95C) [193]. In contrast to previous findings [192], this study reported decreased migration and invasion in A549 and H1299 cells after transient transfection of miR-708-5p [193]. miR-708-5p also induced apoptosis in A549 and H1299 cells, while an antagomir increased invasion in lung cancer cell lines (QG56, 95C) with low metastatic potential [193]. There was a significant increase in metastases in mice injected with QG56 cells containing a miR-708-5p sponge compared to a control sponge [193]. The miRNA sponge works by sequestering a specific endogenous miRNA, thereby preventing the miRNA from suppressing its targets. miR-708-5p also showed it could be used as a novel therapeutic agent in lung cancer as it decreased tumor growth in nude mice with A549 injected tumors [193]. It was determined that miR-708-5p suppressed cyclin-dependent kinase inhibitor 1 (p21) expression and cytoplasmic localization [193] (Figure 4). By being localized to the nucleus, p21 loses its pro-tumorigenic functions while simultaneously increasing its tumor suppressive activities. The ability of miR-708-5p to suppress cytoplasmic p21 localization was shown to be through decreased pAKT levels [193]. pAKT phosphorylates p21 to promote shuttling to the cytoplasm [196]. miR-708-5p reduced AKT phosphorylation by decreasing PI3K pathway targets while simultaneously increasing expression of PI3K pathway suppressors [193]. Therefore, loss of pAKT prevented p21 from being phosphorylated, resulting in nuclear localization. miR-708-5p treatment in H1299, A549, and PG cells decreased levels of PI3K-related transcripts (survivin, Bcl-2-related protein A1 [BCL2A1], and Bcl-like protein 2 [BCL2L2]), while also increasing mRNA expression of PI3K signal suppressors (Phosphoinositide-3-kinase-interacting protein 1 [PIK3IP1], Pleckstrin Homology Like Domain Family A Member 3 [PHLDA3], and SH2-domain containing Phosphatidylinositol-3,4,5-trisphosphate 5-phosphatase 2 [INPPL1]) [193]. miR-708-5p treatment of the same cells also increased E-cadherin mRNA expression while simultaneously decreasing the expression of pro-metastatic genes (MMP-2, MMP-9, Vascular endothelial growth factor C [VEGFC], and Cell adhesion molecule 1 [CADM1]) [193]. Lastly, miR-708-5p suppressed expression of various cancer stem cell markers (CD117, CD34, CD44, octamer-binding transcription factor 4 [OCT4], Aldehyde Dehydrogenase 1 Family Member A2 [ALDH1A2], and NANOG) in A549 and PG cells [193]. It has yet to be determined if miR-708-5p-mediated regulation of these transcripts is direct or indirect. Regardless, these results suggest miR-708-5p suppresses multiple hallmarks of cancer, which is crucial for improving treatment efficacy. Together, this provides a strong case for miR-708-5p as a tumor suppressive miRNA in NSCLC cancer.
Many questions remain: Does miR-708-5p expression alter with lung cancer stage? Are there other biomarkers miR-708-5p can be paired with to improve identification of lung cancer subtypes? Does miR-708-5p expression correlate with response to various chemotherapies? Why is miR-708-5p increased in some lung tumors while decreased in others? Are these tumors being mischaracterized as adenocarcinomas or SCCs? Lastly, how can miR-708-5p have opposing phenotypic effects in the same cell lines in two different studies? As these questions are answered, researchers will gain a better understanding of the precise function of this dynamic miRNA in lung cancer.
miR-708-5p in inflammation
There is limited research on the function of miR-708-5p beyond it effects on cancer cells, but some recent research may provide insights into novel functions of miR-708-5p in cancer beyond the cancer cell itself. Many cell types comprise the tumor stroma including immune cells, fibroblasts, and endothelial cells, all of which have diverse roles in supporting tumor growth. Immune surveillance actively targets cancer cells to prevent tumor establishment, and immunocompromised people are more susceptible to cancer, highlighting the importance of a functional immune system in fighting tumor growth [197, 198]. Cancer cells also manipulate immune cells to support tumor progression and suppress an immune response. Recent work by multiple groups has uncovered miR-708-5p may regulate immune cells. Chronically inflamed colonic cells from mice had increased miR-708-5p expression, and HEK293T cells pre-treated with miR-708-5p followed by TNF-α stimulation resulted in mitigation of NF-kB-dependent genes expressions such as TNF-α, chemokine (C-X-C motif) ligand 1 (CXCL1), and NF-kappa-B inhibitor alpha (NFKBIA) [33, 106]. Given that miR-708-5p directly targets the NF-kB activator IKKβ, miR-708-5p may act as a negative regulator of TNF-α-induced NF-kB signaling. The mechanism for miR-708-5p induction was not explained but these observations provide the foundation for further research. In breast cancer cells, it was shown that miR-708-5p not only targeted CD276, an immune checkpoint molecule, but also decreased phosphorylated signal transducer and activator of transcription 3 (pSTAT3) levels [111]. STAT3 is a potent activator of immunosuppression through induction of anti-inflammatory cytokine production and immunosuppressive cell recruitment to inhibit Th1 responses [199]. Whether miR-708-5p regulation of STAT3 phosphorylation is through CD276 or another pathway remains to be determined. As miR-708-5p shows both pro- and anti-inflammatory qualities, it would be interesting to further test exactly the function of this miRNA in immune cells. In addition, several miRNAs have been shown to modulate TME cell composition and cytokine profiles [200]. Therefore, exploring miR-708-5p beyond its direct effect on cancer cell physiology has significant merit.
Researchers investigated differential miRNA expression in peripheral blood mononuclear cells (PBMCs) in normal women who later developed breast cancer. They found that miR-708-5p expression was diminished in women who developed breast cancer compared to women who did not [201]. The authors did not study the subset of PBMCs that showed reduced miR-708-5p levels. The most abundant PBMC population is generally T-cells followed by an equal abundance of B-cells, natural killer cells, and monocytes. Defining which cell type, or combination of cell types, have lower miR-708-5p expression would have value. Importantly, does decreased miR-708-5p expression in PBMCs affect immune surveillance capabilities? Building on this work, it was shown that miR-708-5p expression was decreased in alveolar macrophages of smokers compared to healthy volunteers [32]. Interestingly, these same macrophages displayed an “anti-M1” phenotype, meaning they had decreased expression of pro-inflammatory markers such as CXCL9, CXCL10, and CXCL11 [32]. The authors determined that miR-708-5p was not transcriptionally downregulated; rather sumoylation of DICER resulted in decreased pre-miR-708 processing, resulting in lower mature miR-708-5p levels [202]. Although not studied, DICER defects most likely resulted in the global miRNA profile of alveolar macrophages. The importance of miR-708-5p loss in these macrophages has not been established; therefore it is impossible to conclude the contribution of miR-708-5p to an anti-M1 phenotype. Whether this phenotype contributes to lung tumorigenesis is unknown, but given the importance of M1 macrophages as tumor suppressing cells in lung cancer, a loss of functional M1 macrophages may allow unchecked tumor progression [203] Experiments testing the response of the “anti-M1” macrophages to pro- and anti-inflammatory cytokines would be revealing. Finally, restoring mature miR-708-5p expression in these macrophages and measuring phenotypic changes may reveal intriguing results. Although studies on miR-708-5p in inflammation are not abundant, the current literature suggests miR-708-5p may have prominent functions in immune cells.
CONCLUSIONS
Current studies on miR-708-5p highlight its emerging activities as a miRNA involved in multiple aspects of oncogenesis. miR-708-5p is oncogenic or tumor suppressive in various tumor types, both solid and hematological. Translational work shows that miR-708-5p has potential uses as a biomarker, therapeutic target, or therapeutic in the clinic. Beyond the cancer cell, studies are revealing a novel function for miR-708-5p in immune responses. These investigations could uncover a function for miR-708-5p in regulating the composition and activities of fibroblasts, immune, endothelial, and other cells within the TME. Although not presented in this review, miR-708-5p is being studied in many other diseases, including neurological and cardiovascular illnesses [35, 204–212]. As researchers continue to discover novel miR-708-5p targets in normal and disease states, the comprehensive function of miR-708-5p in cell biology will be characterized. This line of investigation may one day lead to improved diagnosis, treatment, and outcome for patients afflicted with cancer or other debilitating diseases.
ACKNOWLEDGMENTS
We would like to thank past and present members of the Lutz lab for their insight and feedback. We would also like to thank friends and family of the Lutz lab for their unwavering support. We apologize to the researchers whose work we did not cite due to the narrowed focus of miR-708-5p in cancer.
CONFLICTS OF INTEREST
None.
FUNDING
We would like to thank our funding sources, the American Heart Association (Grant-In-Aid 15GRNT23240019) and NIH R01 (1R01AR069044-01A1) for funding our work.
REFERENCES
1. Ambros V. The functions of animal microRNAs. Nature. 2004; 431:350–5.
2. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004; 116:281–97.
3. Ameres SL, Zamore PD. Diversifying microRNA sequence and function. Nat Rev Mol Cell Biol. 2013; 14:475–88.
4. Lindsay MA. microRNAs and the immune response. Trends Immunol. 2008; 29:343–51.
5. Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993; 75:843–54.
6. Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, Horvitz HR, Ruvkun G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000; 403:901–6.
7. Almeida MI, Reis RM, Calin GA. MicroRNA history: discovery, recent applications, and next frontiers. Mutat Res. 2011; 717:1–8.
8. Chou CH, Chang NW, Shrestha S, Hsu SD, Lin YL, Lee WH, Yang CD, Hong HC, Wei TY, Tu SJ, Tsai TR, Ho SY, Jian TY, et al. miRTarBase 2016: updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res. 2016; 44:D239–47.
9. Friedlander MR, Lizano E, Houben AJ, Bezdan D, Banez-Coronel M, Kudla G, Mateu-Huertas E, Kagerbauer B, Gonzalez J, Chen KC, LeProust EM, Marti E, Estivill X. Evidence for the biogenesis of more than 1,000 novel human microRNAs. Genome Biol. 2014; 15:R57.
10. Chiang HR, Schoenfeld LW, Ruby JG, Auyeung VC, Spies N, Baek D, Johnston WK, Russ C, Luo S, Babiarz JE, Blelloch R, Schroth GP, Nusbaum C, et al. Mammalian microRNAs: experimental evaluation of novel and previously annotated genes. Genes Dev. 2010; 24:992–1009.
11. Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet. 2015; 16:421–33.
12. Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, Kim VN. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004; 23:4051–60.
13. Cai X, Hagedorn CH, Cullen BR. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA. 2004; 10:1957–66.
14. Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Radmark O, Kim S, Kim VN. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003; 425:415–9.
15. Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. 2014; 15:509–24.
16. Mah SM, Buske C, Humphries RK, Kuchenbauer F. miRNA*: a passenger stranded in RNA-induced silencing complex? Crit Rev Eukaryot Gene Expr. 2010; 20:141–8.
17. Williams AE. Functional aspects of animal microRNAs. Cell Mol Life Sci. 2008; 65:545–62.
18. Eulalio A, Huntzinger E, Nishihara T, Rehwinkel J, Fauser M, Izaurralde E. Deadenylation is a widespread effect of miRNA regulation. RNA. 2009; 15:21–32.
19. Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003; 115:787–98.
20. Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer. 2006; 6:259–69.
21. Zhang B, Pan X, Cobb GP, Anderson TA. microRNAs as oncogenes and tumor suppressors. Dev Biol. 2007; 302:1–12.
22. Cornett AL, Lutz CS. Regulation of COX-2 expression by miR-146a in lung cancer cells. RNA. 2014; 20:1419–30.
23. Chen G, Umelo IA, Lv S, Teugels E, Fostier K, Kronenberger P, Dewaele A, Sadones J, Geers C, De Greve J. miR-146a inhibits cell growth, cell migration and induces apoptosis in non-small cell lung cancer cells. PLoS One. 2013; 8:e60317.
24. Sandhu R, Rein J, D’Arcy M, Herschkowitz JI, Hoadley KA, Troester MA. Overexpression of miR-146a in basal-like breast cancer cells confers enhanced tumorigenic potential in association with altered p53 status. Carcinogenesis. 2014; 35:2567–75.
25. Bar M, Wyman SK, Fritz BR, Qi J, Garg KS, Parkin RK, Kroh EM, Bendoraite A, Mitchell PS, Nelson AM, Ruzzo WL, Ware C, Radich JP, et al. MicroRNA discovery and profiling in human embryonic stem cells by deep sequencing of small RNA libraries. Stem Cells. 2008; 26:2496–505.
26. Zhu JY, Pfuhl T, Motsch N, Barth S, Nicholls J, Grasser F, Meister G. Identification of novel Epstein-Barr virus microRNA genes from nasopharyngeal carcinomas. J Virol. 2009; 83:3333–41.
27. Stark MS, Tyagi S, Nancarrow DJ, Boyle GM, Cook AL, Whiteman DC, Parsons PG, Schmidt C, Sturm RA, Hayward NK. Characterization of the Melanoma miRNAome by Deep Sequencing. PLoS One. 2010; 5:e9685.
28. Witten D, Tibshirani R, Gu SG, Fire A, Lui WO. Ultra-high throughput sequencing-based small RNA discovery and discrete statistical biomarker analysis in a collection of cervical tumours and matched controls. BMC Biol. 2010; 8:58.
29. Kuchen S, Resch W, Yamane A, Kuo N, Li Z, Chakraborty T, Wei L, Laurence A, Yasuda T, Peng S, Hu-Li J, Lu K, Dubois W, et al. Regulation of microRNA expression and abundance during lymphopoiesis. Immunity. 2010; 32:828–39.
30. Meunier J, Lemoine F, Soumillon M, Liechti A, Weier M, Guschanski K, Hu H, Khaitovich P, Kaessmann H. Birth and expression evolution of mammalian microRNA genes. Genome Res. 2013; 23:34–45.
31. Behrman S, Acosta-Alvear D, Walter P. A CHOP-regulated microRNA controls rhodopsin expression. J Cell Biol. 2011; 192:919–27.
32. Graff JW, Powers LS, Dickson AM, Kim J, Reisetter AC, Hassan IH, Kremens K, Gross TJ, Wilson ME, Monick MM. Cigarette smoking decreases global microRNA expression in human alveolar macrophages. PLoS One. 2012; 7:e44066.
33. Baer C, Oakes CC, Ruppert AS, Claus R, Kim-Wanner SZ, Mertens D, Zenz T, Stilgenbauer S, Byrd JC, Plass C. Epigenetic silencing of miR-708 enhances NF-kappaB signaling in chronic lymphocytic leukemia. Int J Cancer. 2015; 137:1352–61.
34. Vela EM, Kasoji MD, Wendling MQ, Price JA, Knostman KA, Bresler HS, Long JP. MicroRNA expression in mice infected with seasonal H1N1, swine H1N1 or highly pathogenic H5N1. J Med Microbiol. 2014; 63:1131–42.
35. Dileepan M, Jude JA, Rao SP, Walseth TF, Panettieri RA, Subramanian S, Kannan MS. MicroRNA-708 regulates CD38 expression through signaling pathways JNK MAP kinase and PTEN/AKT in human airway smooth muscle cells. Respir Res. 2014; 15:107.
36. Lui WO, Pourmand N, Patterson BK, Fire A. Patterns of known and novel small RNAs in human cervical cancer. Cancer Res. 2007; 67:6031–43.
37. Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M, Lin C, Socci ND, Hermida L, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007; 129:1401–14.
38. Berezikov E, Chung WJ, Willis J, Cuppen E, Lai EC. Mammalian mirtron genes. Mol Cell. 2007; 28:328–36.
39. Okamura K, Hagen JW, Duan H, Tyler DM, Lai EC. The mirtron pathway generates microRNA-class regulatory RNAs in Drosophila. Cell. 2007; 130:89–100.
40. Ruby JG, Jan CH, Bartel DP. Intronic microRNA precursors that bypass Drosha processing. Nature. 2007; 448:83–6.
41. Tucker RP, Chiquet-Ehrismann R. Teneurins: a conserved family of transmembrane proteins involved in intercellular signaling during development. Dev Biol. 2006; 290:237–45.
42. Leamey CA, Sawatari A. The teneurins: new players in the generation of visual topography. Semin Cell Dev Biol. 2014; 35:173–9.
43. Graumann R, Di Capua GA, Oyarzun JE, Vasquez MA, Liao C, Branes JA, Roa I, Casanello P, Corvalan AH, Owen GI, Delgado I, Zangemeister-Wittke U, Ziegler A. Expression of teneurins is associated with tumor differentiation and patient survival in ovarian cancer. PLoS One. 2017; 12:e0177244.
44. Lin KT, Yeh YM, Chuang CM, Yang SY, Chang JW, Sun SP, Wang YS, Chao KC, Wang LH. Glucocorticoids mediate induction of microRNA-708 to suppress ovarian cancer metastasis through targeting Rap1B. Nat Commun. 2015; 6:5917.
45. Chakrabarti A, Chen AW, Varner JD. A review of the mammalian unfolded protein response. Biotechnol Bioeng. 2011; 108:2777–93.
46. Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006; 7:880–5.
47. Oakley RH, Cidlowski JA. The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. J Allergy Clin Immunol. 2013; 132:1033–44.
48. Aken BL, Ayling S, Barrell D, Clarke L, Curwen V, Fairley S, Fernandez Banet J, Billis K, Garcia Giron C, Hourlier T, Howe K, Kahari A, Kokocinski F, et al. The Ensembl gene annotation system. Database (Oxford). 2016; 2016.
49. Engelmann D, Putzer BM. The dark side of E2F1: in transit beyond apoptosis. Cancer Res. 2012; 72:571–5.
50. Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol. 1999; 19:1–11.
51. Winklmeier A, Poser I, Hoek KS, Bosserhoff AK. Loss of full length CtBP1 expression enhances the invasive potential of human melanoma. BMC Cancer. 2009; 9:52.
52. Paliwal S, Ho N, Parker D, Grossman SR. CtBP2 Promotes Human Cancer Cell Migration by Transcriptional Activation of Tiam1. Genes Cancer. 2012; 3:481–90.
53. Ryu S, McDonnell K, Choi H, Gao D, Hahn M, Joshi N, Park SM, Catena R, Do Y, Brazin J, Vahdat LT, Silver RB, Mittal V. Suppression of miRNA-708 by polycomb group promotes metastases by calcium-induced cell migration. Cancer Cell. 2013; 23:63–76.
54. Baer C, Claus R, Frenzel LP, Zucknick M, Park YJ, Gu L, Weichenhan D, Fischer M, Pallasch CP, Herpel E, Rehli M, Byrd JC, Wendtner CM, et al. Extensive promoter DNA hypermethylation and hypomethylation is associated with aberrant microRNA expression in chronic lymphocytic leukemia. Cancer Res. 2012; 72:3775–85.
55. Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74.
56. Zahnow CA. CCAAT/enhancer-binding protein beta: its role in breast cancer and associations with receptor tyrosine kinases. Expert Rev Mol Med. 2009; 11:e12.
57. Kovacs KA, Steinmann M, Magistretti PJ, Halfon O, Cardinaux JR. C/EBPbeta couples dopamine signalling to substance P precursor gene expression in striatal neurones. J Neurochem. 2006; 98:1390–9.
58. Robert I, Sutter A, Quirin-Stricker C. Synergistic activation of the human choline acetyltransferase gene by c-Myb and C/EBPbeta. Brain Res Mol Brain Res. 2002; 106:124–35.
59. Natsuka S, Akira S, Nishio Y, Hashimoto S, Sugita T, Isshiki H, Kishimoto T. Macrophage differentiation-specific expression of NF-IL6, a transcription factor for interleukin-6. Blood. 1992; 79:460–6.
60. Davydov IV, Krammer PH, Li-Weber M. Nuclear factor-IL6 activates the human IL-4 promoter in T cells. J Immunol. 1995; 155:5273–9.
61. van Dijk TB, Baltus B, Raaijmakers JA, Lammers JW, Koenderman L, de Groot RP. A composite C/EBP binding site is essential for the activity of the promoter of the IL-3/IL-5/granulocyte-macrophage colony-stimulating factor receptor beta c gene. J Immunol. 1999; 163:2674–80.
62. Greenwel P, Tanaka S, Penkov D, Zhang W, Olive M, Moll J, Vinson C, Di Liberto M, Ramirez F. Tumor necrosis factor alpha inhibits type I collagen synthesis through repressive CCAAT/enhancer-binding proteins. Mol Cell Biol. 2000; 20:912–8.
63. Nakajima S, Hiramatsu N, Hayakawa K, Saito Y, Kato H, Huang T, Yao J, Paton AW, Paton JC, Kitamura M. Selective abrogation of BiP/GRP78 blunts activation of NF-kappaB through the ATF6 branch of the UPR: involvement of C/EBPbeta and mTOR-dependent dephosphorylation of Akt. Mol Cell Biol. 2011; 31:1710–8.
64. Ong CT, Corces VG. CTCF: an architectural protein bridging genome topology and function. Nat Rev Genet. 2014; 15:234–46.
65. Losada A. Cohesin in cancer: chromosome segregation and beyond. Nat Rev Cancer. 2014; 14:389–93.
66. Lin S, Gregory RI. MicroRNA biogenesis pathways in cancer. Nat Rev Cancer. 2015; 15:321–33.
67. Jens, M, Rajewsky N. Competition between target sites of regulators shapes post-transcriptional gene regulation. Nat Rev Genet. 2015; 16:113–26.
68. Audic Y, Hartley RS. Post-transcriptional regulation in cancer. Biol Cell. 2004; 96:479–98.
69. Schotte D, Chau JC, Sylvester G, Liu G, Chen C, van der Velden VH, Broekhuis MJ, Peters TC, Pieters R, den Boer ML. Identification of new microRNA genes and aberrant microRNA profiles in childhood acute lymphoblastic leukemia. Leukemia. 2009; 23:313–22.
70. Candia J, Cherukuri S, Guo Y, Doshi KA, Banavar JR, Civin CI, Losert W. Uncovering low-dimensional, miR-based signatures of acute myeloid and lymphoblastic leukemias with a machine-learning-driven network approach. Converg Sci Phys Oncol. 2015; 1. [Epub ahead of print] http://doi.org/10.1088/2057-1739/1/2/025002.
71. Duyu M, Durmaz B, Gunduz C, Vergin C, Yilmaz Karapinar D, Aksoylar S, Kavakli K, Cetingul N, Irken G, Yaman Y, Ozkinay F, Cogulu O. Prospective evaluation of whole genome microRNA expression profiling in childhood acute lymphoblastic leukemia. Biomed Res Int. 2014; 2014:967585.
72. de Oliveira JC, Scrideli CA, Brassesco MS, Yunes JA, Brandalise SR, Tone LG. MiR-708-5p is differentially expressed in childhood acute lymphoblastic leukemia but not strongly associated to clinical features. Pediatr Blood Cancer. 2015; 62:177–8.
73. Han BW, Feng DD, Li ZG, Luo XQ, Zhang H, Li XJ, Zhang XJ, Zheng LL, Zeng CW, Lin KY, Zhang P, Xu L, Chen YQ. A set of miRNAs that involve in the pathways of drug resistance and leukemic stem-cell differentiation is associated with the risk of relapse and glucocorticoid response in childhood ALL. Hum Mol Genet. 2011; 20:4903–15.
74. Miyamoto K, Araki KY, Naka K, Arai F, Takubo K, Yamazaki S, Matsuoka S, Miyamoto T, Ito K, Ohmura M, Chen C, Hosokawa K, Nakauchi H, et al. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell. 2007; 1:101–12.
75. Ausserlechner MJ, Salvador C, Deutschmann A, Bodner M, Viola G, Bortolozzi R, Basso G, Hagenbuchner J, Obexer P. Therapy-resistant acute lymphoblastic leukemia (ALL) cells inactivate FOXO3 to escape apoptosis induction by TRAIL and Noxa. Oncotarget. 2013; 4:995–1007. http://doi.org/10.18632/oncotarget.953.
76. Tothova Z, Kollipara R, Huntly BJ, Lee BH, Castrillon DH, Cullen DE, McDowell EP, Lazo-Kallanian S, Williams IR, Sears C, Armstrong SA, Passegue E, DePinho RA, et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007; 128:325–39.
77. Hui RC, Gomes AR, Constantinidou D, Costa JR, Karadedou CT, Fernandez de Mattos S, Wymann MP, Brosens JJ, Schulze A, Lam EW. The forkhead transcription factor FOXO3a increases phosphoinositide-3 kinase/Akt activity in drug-resistant leukemic cells through induction of PIK3CA expression. Mol Cell Biol. 2008; 28:5886–98.
78. Naka K, Hoshii T, Muraguchi T, Tadokoro Y, Ooshio T, Kondo Y, Nakao S, Motoyama N, Hirao A. TGF-beta-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature. 2010; 463:676–80.
79. Chiaretti S, Zini G, Bassan R. Diagnosis and subclassification of acute lymphoblastic leukemia. Mediterr J Hematol Infect Dis. 2014; 6:e2014073.
80. Svoronos AA, Engelman DM, Slack FJ. OncomiR or Tumor Suppressor? The Duplicity of MicroRNAs in Cancer. Cancer Res. 2016; 76:3666–70.
81. National Institutes of Health US. N.C.I. SEER Cancer Statistics Review, 1975-2011. 2011. https://seer.cancer.gov/archive/csr/1975_2011/.
82. Durig J, Naschar M, Schmucker U, Renzing-Kohler K, Holter T, Huttmann A, Duhrsen U. CD38 expression is an important prognostic marker in chronic lymphocytic leukaemia. Leukemia. 2002; 16:30–5.
83. Furman RR, Asgary Z, Mascarenhas JO, Liou HC, Schattner EJ. Modulation of NF-kappa B activity and apoptosis in chronic lymphocytic leukemia B cells. J Immunol. 2000; 164:2200–6.
84. Rossi D, Rasi S, Fabbri G, Spina V, Fangazio M, Forconi F, Marasca R, Laurenti L, Bruscaggin A, Cerri M, Monti S, Cresta S, Fama R, et al. Mutations of NOTCH1 are an independent predictor of survival in chronic lymphocytic leukemia. Blood. 2012; 119:521–9.
85. Iqbal J, Shen Y, Liu Y, Fu K, Jaffe ES, Liu C, Liu Z, Lachel CM, Deffenbacher K, Greiner TC, Vose JM, Bhagavathi S, Staudt LM, et al. Genome-wide miRNA profiling of mantle cell lymphoma reveals a distinct subgroup with poor prognosis. Blood. 2012; 119:4939–48.
86. Barouk-Simonet E, Andrieux J, Copin MC, Grardel-Duflos N, Huyghe P, Patte JH, Preudhomme C, Quesnel B, Lai JL. TPA stimulation culture for improved detection of t(11;14)(q13;q32) in mantle cell lymphoma. Ann Genet. 2002; 45:165–8.
87. Li JY, Gaillard F, Moreau A, Harousseau JL, Laboisse C, Milpied N, Bataille R, Avet-Loiseau H. Detection of translocation t(11;14)(q13;q32) in mantle cell lymphoma by fluorescence in situ hybridization. Am J Pathol. 1999; 154:1449–52.
88. Moussay E, Wang K, Cho JH, van Moer K, Pierson S, Paggetti J, Nazarov PV, Palissot V, Hood LE, Berchem G, Galas DJ. MicroRNA as biomarkers and regulators in B-cell chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2011; 108:6573–8.
89. Shenouda SK, Alahari SK. MicroRNA function in cancer: oncogene or a tumor suppressor? Cancer Metastasis Rev. 2009; 28:369–78.
90. Advanced Bladder Cancer (ABC) Meta-analysis Collaboration. Adjuvant chemotherapy in invasive bladder cancer: a systematic review and meta-analysis of individual patient data Advanced Bladder Cancer (ABC) Meta-analysis Collaboration. Eur Urol. 2005; 48:189–199. discussion 199–201.
91. Advanced Bladder Cancer (ABC) Meta-analysis Collaboration. Neoadjuvant chemotherapy in invasive bladder cancer: update of a systematic review and meta-analysis of individual patient data advanced bladder cancer (ABC) meta-analysis collaboration. Eur Urol. 2005; 48:202–5. discussion 205–6.
92. Song T, Xia W, Shao N, Zhang X, Wang C, Wu Y, Dong J, Cai W, Li H. Differential miRNA expression profiles in bladder urothelial carcinomas. Asian Pac J Cancer Prev. 2010; 11:905–11.
93. Consortium GT. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013; 45:580–5.
94. Song T, Zhang X, Zhang L, Dong J, Cai W, Gao J, Hong B. miR-708 promotes the development of bladder carcinoma via direct repression of Caspase-2. J Cancer Res Clin Oncol. 2013; 139:1189–98.
95. Krumschnabel G, Manzl C, Villunger A. Caspase-2: killer, savior and safeguard--emerging versatile roles for an ill-defined caspase. Oncogene. 2009; 28:3093–6.
96. Krumschnabel G, Sohm B, Bock F, Manzl C, Villunger A. The enigma of caspase-2: the laymen’s view. Cell Death Differ. 2009; 16:195–207.
97. Jelinek M, Balusikova K, Kopperova D, Nemcova-Furstova V, Sramek J, Fidlerova J, Zanardi I, Ojima I, Kovar J. Caspase-2 is involved in cell death induction by taxanes in breast cancer cells. Cancer Cell Int. 2013; 13:42.
98. Lei SL, Zhao H, Yao HL, Chen Y, Lei ZD, Liu KJ, Yang Q. Regulatory roles of microRNA-708 and microRNA-31 in proliferation, apoptosis and invasion of colorectal cancer cells. Oncol Lett. 2014; 8:1768–1774.
99. Piepoli A, Tavano F, Copetti M, Mazza T, Palumbo O, Panza A, di Mola FF, Pazienza V, Mazzoccoli G, Biscaglia G, Gentile A, Mastrodonato N, Carella M, et al. Mirna expression profiles identify drivers in colorectal and pancreatic cancers. PLoS One. 2012; 7:e33663.
100. Malumbres M, Ortega S, Barbacid M. Genetic analysis of mammalian cyclin-dependent kinases and their inhibitors. Biol Chem. 2000; 381:827–38.
101. Jeffrey PD, Tong L, Pavletich NP. Structural basis of inhibition of CDK-cyclin complexes by INK4 inhibitors. Genes Dev. 2000; 14:3115–25.
102. Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, Olsson I, Edlund K, Lundberg E, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015; 347:1260419.
103. Nieminen TT, Shoman S, Eissa S, Peltomaki P, Abdel-Rahman WM. Distinct genetic and epigenetic signatures of colorectal cancers according to ethnic origin. Cancer Epidemiol Biomarkers Prev. 2012; 21:202–11.
104. Olaru AV, Selaru FM, Mori Y, Vazquez C, David S, Paun B, Cheng Y, Jin Z, Yang J, Agarwal R, Abraham JM, Dassopoulos T, Harris M, et al. Dynamic changes in the expression of MicroRNA-31 during inflammatory bowel disease-associated neoplastic transformation. Inflamm Bowel Dis. 2011; 17:221–31.
105. Slaby O, Svoboda M, Fabian P, Smerdova T, Knoflickova D, Bednarikova M, Nenutil R, Vyzula R. Altered expression of miR-21, miR-31, miR-143 and miR-145 is related to clinicopathologic features of colorectal cancer. Oncology. 2007; 72:397–402.
106. Necela BM, Carr JM, Asmann YW, Thompson EA. Differential expression of microRNAs in tumors from chronically inflamed or genetic (APC(Min/+)) models of colon cancer. PLoS One. 2011; 6:e18501.
107. Lin HH, Bell E, Uwanogho D, Perfect LW, Noristani H, Bates TJ, Snetkov V, Price J, Sun YM. Neuronatin promotes neural lineage in ESCs via Ca(2+) signaling. Stem Cells. 2010; 28:1950–60.
108. Huang C, Jacobson K, Schaller MD. MAP kinases and cell migration. J Cell Sci. 2004; 117:4619–28.
109. Yang S, Zhang JJ, Huang XY. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell. 2009; 15:124–34.
110. Swanson KD, Reigh C, Landreth GE. ATP-stimulated activation of the mitogen-activated protein kinases through ionotrophic P2X2 purinoreceptors in PC12 cells. Difference in purinoreceptor sensitivity in two PC12 cell lines. J Biol Chem. 1998; 273:19965–71.
111. Nygren MK, Tekle C, Ingebrigtsen VA, Makela R, Krohn M, Aure MR, Nunes-Xavier CE, Perala M, Tramm T, Alsner J, Overgaard J, Nesland JM, Borgen E, et al. Identifying microRNAs regulating B7-H3 in breast cancer: the clinical impact of microRNA-29c. Br J Cancer. 2014; 110:2072–80.
112. Collins M, Ling V, Carreno BM. The B7 family of immune-regulatory ligands. Genome Biol. 2005; 6:223.
113. Leitner J, Klauser C, Pickl WF, Stockl J, Majdic O, Bardet AF, Kreil DP, Dong C, Yamazaki T, Zlabinger G, Pfistershammer K, Steinberger P. B7-H3 is a potent inhibitor of human T-cell activation: No evidence for B7-H3 and TREML2 interaction. Eur J Immunol. 2009; 39:1754–64.
114. Liu C, Liu J, Wang J, Liu Y, Zhang F, Lin W, Gao A, Sun M, Wang Y, Sun Y. B7-H3 expression in ductal and lobular breast cancer and its association with IL-10. Mol Med Rep. 2013; 7:134–8.
115. Arigami T, Narita N, Mizuno R, Nguyen L, Ye X, Chung A, Giuliano AE, Hoon DS. B7-h3 ligand expression by primary breast cancer and associated with regional nodal metastasis. Ann Surg. 2010; 252:1044–51.
116. Yuan H, Wei X, Zhang G, Li C, Zhang X, Hou J. B7-H3 over expression in prostate cancer promotes tumor cell progression. J Urol. 2011; 186:1093–9.
117. Ma L, Ma S, Zhao G, Yang L, Zhang P, Yi Q, Cheng S. miR-708/LSD1 axis regulates the proliferation and invasion of breast cancer cells. Cancer Med. 2016; 5:684–92.
118. Amente S, Lania L, Majello B. The histone LSD1 demethylase in stemness and cancer transcription programs. Biochim Biophys Acta. 2013; 1829:981–6.
119. Ancelin K, Syx L, Borensztein M, Ranisavljevic N, Vassilev I, Briseno-Roa L, Liu T, Metzger E, Servant N, Barillot E, Chen CJ, Schule R, Heard E. Maternal LSD1/KDM1A is an essential regulator of chromatin and transcription landscapes during zygotic genome activation. Elife. 2016; 5.
120. Maheshwari AV, Cheng EY. Ewing sarcoma family of tumors. J Am Acad Orthop Surg. 2010; 18:94–107.
121. Burchill SA. Ewing’s sarcoma: diagnostic, prognostic, and therapeutic implications of molecular abnormalities. J Clin Pathol. 2003; 56:96–102.
122. May WA, Lessnick SL, Braun BS, Klemsz M, Lewis BC, Lunsford LB, Hromas R, Denny CT. The Ewing’s sarcoma EWS/FLI-1 fusion gene encodes a more potent transcriptional activator and is a more powerful transforming gene than FLI-1. Mol Cell Biol. 1993; 13:7393–8.
123. May WA, Gishizky ML, Lessnick SL, Lunsford LB, Lewis BC, Delattre O, Zucman J, Thomas G, Denny CT. Ewing sarcoma 11;22 translocation produces a chimeric transcription factor that requires the DNA-binding domain encoded by FLI1 for transformation. Proc Natl Acad Sci U S A. 1993; 90:5752–6.
124. Tanaka K, Iwakuma T, Harimaya K, Sato H, Iwamoto Y. EWS-Fli1 antisense oligodeoxynucleotide inhibits proliferation of human Ewing’s sarcoma and primitive neuroectodermal tumor cells. J Clin Invest. 1997; 99:239–47.
125. Mateo-Lozano S, Gokhale PC, Soldatenkov VA, Dritschilo A, Tirado OM, Notario V. Combined transcriptional and translational targeting of EWS/FLI-1 in Ewing’s sarcoma. Clin Cancer Res. 2006; 12:6781–90.
126. Cook PJ, Ju BG, Telese F, Wang X, Glass CK, Rosenfeld MG. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature. 2009; 458:591–6.
127. Li CM, Guo M, Borczuk A, Powell CA, Wei M, Thaker HM, Friedman R, Klein U, Tycko B. Gene expression in Wilms’ tumor mimics the earliest committed stage in the metanephric mesenchymal-epithelial transition. Am J Pathol. 2002; 160:2181–90.
128. Behbakht K, Qamar L, Aldridge CS, Coletta RD, Davidson SA, Thorburn A, Ford HL. Six1 overexpression in ovarian carcinoma causes resistance to TRAIL-mediated apoptosis and is associated with poor survival. Cancer Res. 2007; 67:3036–42.
129. Grifone R, Demignon J, Houbron C, Souil E, Niro C, Seller MJ, Hamard G, Maire P. Six1 and Six4 homeoproteins are required for Pax3 and Mrf expression during myogenesis in the mouse embryo. Development. 2005; 132:2235–49.
130. Xu PX, Zheng W, Huang L, Maire P, Laclef C, Silvius D. Six1 is required for the early organogenesis of mammalian kidney. Development. 2003; 130:3085–94.
131. Robin TP, Smith A, McKinsey E, Reaves L, Jedlicka P, Ford HL. EWS/FLI1 regulates EYA3 in Ewing sarcoma via modulation of miRNA-708, resulting in increased cell survival and chemoresistance. Mol Cancer Res. 2012; 10:1098–108.
132. Cleary ML. Oncogenic conversion of transcription factors by chromosomal translocations. Cell. 1991; 66:619–22.
133. Trancau IO. Chromosomal translocations highlighted in Primitive Neuroectodermal Tumors (PNET) and Ewing sarcoma. J Med Life. 2014; 7 Spec No. 3:44–50.
134. Friedman AD. GADD153/CHOP, a DNA damage-inducible protein, reduced CAAT/enhancer binding protein activities and increased apoptosis in 32D c13 myeloid cells. Cancer Res. 1996; 56:3250–6.
135. Kimelberg HK, Nedergaard M. Functions of astrocytes and their potential as therapeutic targets. Neurotherapeutics. 2010; 7:338–53.
136. Bradl M, Lassmann H. Oligodendrocytes: biology and pathology. Acta Neuropathol. 2010; 119:37–53.
137. Guo P, Lan J, Ge J, Nie Q, Mao Q, Qiu Y. miR-708 acts as a tumor suppressor in human glioblastoma cells. Oncol Rep. 2013; 30:870–6.
138. Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat Rev Cancer. 2010; 10:293–301.
139. Sun M, Wang G, Paciga JE, Feldman RI, Yuan ZQ, Ma XL, Shelley SA, Jove R, Tsichlis PN, Nicosia SV, Cheng JQ. AKT1/PKBalpha kinase is frequently elevated in human cancers and its constitutive activation is required for oncogenic transformation in NIH3T3 cells. Am J Pathol. 2001; 159:431–7.
140. Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer. 2011; 11:558–72.
141. Kim KH, Roberts CW. Targeting EZH2 in cancer. Nat Med. 2016; 22:128–34.
142. Roomi MW, Monterrey JC, Kalinovsky T, Rath M, Niedzwiecki A. Patterns of MMP-2 and MMP-9 expression in human cancer cell lines. Oncol Rep. 2009; 21:1323–33.
143. Mittnacht S, Paterson H, Olson MF, Marshall CJ. Ras signalling is required for inactivation of the tumour suppressor pRb cell-cycle control protein. Curr Biol. 1997; 7:219–21.
144. Kennedy AL, Morton JP, Manoharan I, Nelson DM, Jamieson NB, Pawlikowski JS, McBryan T, Doyle B, McKay C, Oien KA, Enders GH, Zhang R, Sansom OJ. et al. Activation of the PIK3CA/AKT pathway suppresses senescence induced by an activated RAS oncogene to promote tumorigenesis. Mol Cell. 2011; 42:36–49.
145. Simon JA, Lange CA. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat Res. 2008; 647:21–9.
146. Waghray A, Murali AR, Menon KN. Hepatocellular carcinoma: From diagnosis to treatment. World J Hepatol. 2015; 7:1020–9.
147. Li G, Yang F, Xu H, Yue Z, Fang X, Liu J. MicroRNA-708 is downregulated in hepatocellular carcinoma and suppresses tumor invasion and migration. Biomed Pharmacother. 2015; 73:154–9.
148. Wang W, Zhao LJ, Tan YX, Ren H, Qi ZT. Identification of deregulated miRNAs and their targets in hepatitis B virus-associated hepatocellular carcinoma. World J Gastroenterol. 2012; 18:5442–53.
149. Furtado KS, de Oliveira Andrade F, Campos A, Rosim MP, Vargas-Mendez E, Henriques A, De Conti A, Scolastici C, Barbisan LF, Carvalho RF, Moreno FS. beta-ionone modulates the expression of miRNAs and genes involved in the metastatic phenotype of microdissected persistent preneoplastic lesions in rats submitted to hepatocarcinogenesis. Mol Carcinog. 2017; 56:184–196.
150. Brew K, Dinakarpandian D, Nagase H. Tissue inhibitors of metalloproteinases: evolution, structure and function. Biochim Biophys Acta. 2000; 1477:267–83.
151. Fan H, Chen L, Zhang F, Quan Y, Su X, Qiu X, Zhao Z, Kong KL, Dong S, Song Y, Chan TH, Guan XY. MTSS, a novel target of DNA methyltransferase 3B, functions as a tumor suppressor in hepatocellular carcinoma. Oncogene. 2012; 31:2298–308.
152. Defamie V, Sanchez O, Murthy A, Khokha R. TIMP3 controls cell fate to confer hepatocellular carcinoma resistance. Oncogene. 2015; 34:4098–108.
153. Huang XY, Huang ZL, Xu B, Chen Z, Re TJ, Zheng Q, Tang ZY, Huang XY. Elevated MTSS1 expression associated with metastasis and poor prognosis of residual hepatitis B-related hepatocellular carcinoma. J Exp Clin Cancer Res. 2016; 35:85.
154. Hennessy BT, Coleman RL, Markman M. Ovarian cancer. Lancet. 2009; 374:1371–82.
155. Cannistra SA. Cancer of the ovary. N Engl J Med. 2004; 351:2519–29.
156. Mattern J, Buchler MW, Herr I. Cell cycle arrest by glucocorticoids may protect normal tissue and solid tumors from cancer therapy. Cancer Biol Ther. 2007; 6:1345–54.
157. Coleman RE. Glucocorticoids in cancer therapy. Biotherapy. 1992; 4:37–44.
158. Boettner, B, Van Aelst L. Control of cell adhesion dynamics by Rap1 signaling. Curr Opin Cell Biol. 2009; 21:684–93.
159. Kinbara K, Goldfinger LE, Hansen M, Chou FL, Ginsberg MH. Ras GTPases: integrins’ friends or foes? Nat Rev Mol Cell Biol. 2003; 4:767–76.
160. Saini S, Majid S, Shahryari V, Arora S, Yamamura S, Chang I, Zaman MS, Deng G, Tanaka Y, Dahiya R. miRNA-708 control of CD44(+) prostate cancer-initiating cells. Cancer Res. 2012; 72:3618–30.
161. Spring FA, Dalchau R, Daniels GL, Mallinson G, Judson PA, Parsons SF, Fabre JW, Anstee DJ. The Ina and Inb blood group antigens are located on a glycoprotein of 80,000 MW (the CDw44 glycoprotein) whose expression is influenced by the In(Lu) gene. Immunology. 1988; 64:37–43.
162. Patrawala L, Calhoun T, Schneider-Broussard R, Li H, Bhatia B, Tang S, Reilly JG, Chandra D, Zhou J, Claypool K, Coghlan L, Tang DG. Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells. Oncogene. 2006; 25:1696–708.
163. Yan Y, Zuo X, Wei D. Concise Review: Emerging Role of CD44 in Cancer Stem Cells: A Promising Biomarker and Therapeutic Target. Stem Cells Transl Med. 2015; 4:1033–43.
164. Yang J, Wei J, Wu Y, Wang Z, Guo Y, Lee P, Li X. Metformin induces ER stress-dependent apoptosis through miR-708-5p/NNAT pathway in prostate cancer. Oncogenesis. 2015; 4:e158.
165. Yang J, Lu C, Wei J, Guo Y, Liu W, Luo L, Fisch G, Li X. Inhibition of KPNA4 attenuates prostate cancer metastasis. Oncogene, 2017; 36:2868–78.
166. Sachan N, Mishra AK, Mutsuddi M, Mukherjee A. The Drosophila importin-alpha3 is required for nuclear import of notch in vivo and it displays synergistic effects with notch receptor on cell proliferation. PLoS One. 2013; 8:e68247.
167. Mak P, Li J, Samanta S, Mercurio AM. ERbeta regulation of NF-kB activation in prostate cancer is mediated by HIF-1. Oncotarget. 2015; 6:40247–54. http://doi.org/10.18632/oncotarget.5377.
168. Agrawal T, Gupta GK, Agrawal DK. Calcitriol decreases expression of importin alpha3 and attenuates RelA translocation in human bronchial smooth muscle cells. J Clin Immunol. 2012; 32:1093–103.
169. Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014; 41:49–61.
170. Babcook MA, Shukla S, Fu P, Vazquez EJ, Puchowicz MA, Molter JP, Oak CZ, MacLennan GT, Flask CA, Lindner DJ, Parker Y, Daneshgari F, Gupta S. Synergistic simvastatin and metformin combination chemotherapy for osseous metastatic castration-resistant prostate cancer. Mol Cancer Ther. 2014; 13:2288–302.
171. Preston MA, Riis AH, Ehrenstein V, Breau RH, Batista JL, Olumi AF, Mucci LA, Adami HO, Sorensen HT. Metformin use and prostate cancer risk. Eur Urol. 2014; 66:1012–20.
172. Clyne M. Prostate cancer: metformin--the new wonder drug? Nat Rev Urol. 2014; 11:366.
173. Kasznicki J, Sliwinska A, Drzewoski J. Metformin in cancer prevention and therapy. Ann Transl Med. 2014; 2:57.
174. Hyoda K, Hosoi T, Horie N, Okuma Y, Ozawa K, Nomura Y. PI3K-Akt inactivation induced CHOP expression in endoplasmic reticulum-stressed cells. Biochem Biophys Res Commun. 2006; 340:286–90.
175. Saini S, Yamamura S, Majid S, Shahryari V, Hirata H, Tanaka Y, Dahiya R. MicroRNA-708 induces apoptosis and suppresses tumorigenicity in renal cancer cells. Cancer Res. 2011; 71:6208–19.
176. Kim EA, Kim SW, Nam J, Sung EG, Song IH, Kim JY, Kwon TK, Lee TJ. Inhibition of c-FLIPL expression by miRNA-708 increases the sensitivity of renal cancer cells to anti-cancer drugs. Oncotarget. 2016; 7:31832–46. http://doi.org/10.18632/oncotarget.7149.
177. Vandewalle C, Comijn J, De Craene B, Vermassen P, Bruyneel E, Andersen H, Tulchinsky E, Van Roy F, Berx G. SIP1/ZEB2 induces EMT by repressing genes of different epithelial cell-cell junctions. Nucleic Acids Res. 2005; 33:6566–78.
178. Siddique HR, Saleem M. Role of BMI1, a stem cell factor, in cancer recurrence and chemoresistance: preclinical and clinical evidences. Stem Cells. 2012; 30:372–8.
179. Fukuda S, Pelus LM. Survivin, a cancer target with an emerging role in normal adult tissues. Mol Cancer Ther. 2006; 5:1087–98.
180. Safa AR, Pollok KE. Targeting the Anti-Apoptotic Protein c-FLIP for Cancer Therapy. Cancers (Basel). 2011; 3:1639–71.
181. Safa AR. Roles of c-FLIP in Apoptosis, Necroptosis, and Autophagy. J Carcinog Mutagen. 2013; suppl 6.
182. Pasparakis, M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015; 517:311–20.
183. American Cancer Society. Cancer facts & figures. Atlanta, GA: American Cancer Society. https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2016.html.
184. World Health Organization. The top 10 Causes of Death. 2014. http://www.who.int/mediacentre/factsheets/fs310/en/index4.html.
185. Mariotto AB, Yabroff KR, Shao Y, Feuer EJ, Brown ML. Projections of the cost of cancer care in the United States: 2010-2020. J Natl Cancer Inst. 2011; 103:117–28.
186. Howlader N, Noone AM, Krapcho M, Neyman N, Aminou R, Waldron W, Altekruse SF, Kosary CL, Ruhl J, Tatalovich Z, Cho H, Mariotto A, Eisner MP, et al. 2012 Lung Cancer Facts. Lung Cancer Alliance, 2012. http://www.lungcanceralliance.org/assets/docs/media/LCA%20Funding%20Fact%20Sheet%202012.pdf.
187. Kenfield SA, Wei EK, Stampfer MJ, Rosner BA, Colditz GA. Comparison of aspects of smoking among the four histological types of lung cancer. Tob Control. 2008; 17:198–204.
188. Rekhtman N, Ang DC, Sima CS, Travis WD, Moreira AL. Immunohistochemical algorithm for differentiation of lung adenocarcinoma and squamous cell carcinoma based on large series of whole-tissue sections with validation in small specimens. Mod Pathol. 2011; 24:1348–59.
189. Patnaik SK, Kannisto E, Knudsen S, Yendamuri S. Evaluation of microRNA expression profiles that may predict recurrence of localized stage I non-small cell lung cancer after surgical resection. Cancer Res. 2010; 70:36–45.
190. Xing L, Todd NW, Yu L, Fang H, Jiang F. Early detection of squamous cell lung cancer in sputum by a panel of microRNA markers. Mod Pathol. 2010; 23:1157–64.
191. Molina-Pinelo S, Gutierrez G, Pastor MD, Hergueta M, Moreno-Bueno G, Garcia-Carbonero R, Nogal A, Suarez R, Salinas A, Pozo-Rodriguez F, Lopez-Rios F, Agullo-Ortuno MT, Ferrer I, et al. MicroRNA-dependent regulation of transcription in non-small cell lung cancer. PLoS One. 2014; 9:e90524.
192. Jang JS, Jeon HS, Sun Z, Aubry MC, Tang H, Park CH, Rakhshan F, Schultz DA, Kolbert CP, Lupu R, Park JY, Harris CC, Yang P, et al. Increased miR-708 expression in NSCLC and its association with poor survival in lung adenocarcinoma from never smokers. Clin Cancer Res. 2012; 18:3658–67.
193. Wu X, Liu T, Fang O, Dong W, Zhang F, Leach L, Hu X, Luo Z. MicroRNA-708-5p acts as a therapeutic agent against metastatic lung cancer. Oncotarget. 2016; 7:2417–32. http://doi.org/10.18632/oncotarget.6594.
194. Palpant NJ, Pabon L, Rabinowitz JS, Hadland BK, Stoick-Cooper CL, Paige SL, Bernstein ID, Moon RT, Murry CE. Transmembrane protein 88: a Wnt regulatory protein that specifies cardiomyocyte development. Development. 2013; 140:3799–808.
195. Zhang X, Yu X, Jiang G, Miao Y, Wang L, Zhang Y, Liu Y, Fan C, Lin X, Dong Q, Han Q, Zhao H, Han Y, et al. Cytosolic TMEM88 promotes invasion and metastasis in lung cancer cells by binding DVLS. Cancer Res. 2015; 75:4527–37.
196. Zhou BP, Liao Y, Xia W, Spohn B, Lee MH, Hung MC. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat Cell Biol. 2001; 3:245–52.
197. Swann JB, Smyth MJ. Immune surveillance of tumors. J Clin Invest. 2007; 117:1137–46.
198. Engels EA, Biggar RJ, Hall HI, Cross H, Crutchfield A, Finch JL, Grigg R, Hylton T, Pawlish KS, McNeel TS, Goedert JJ. Cancer risk in people infected with human immunodeficiency virus in the United States. Int J Cancer. 2008; 123:187–94.
199. Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007; 7:41–51.
200. Kuninty PR, Schnittert J, Storm G, Prakash J. MicroRNA Targeting to Modulate Tumor Microenvironment. Front Oncol. 2016; 6:3.
201. Chang CW, Wu HC, Terry MB, Santella RM. microRNA Expression in Prospectively Collected Blood as a Potential Biomarker of Breast Cancer Risk in the BCFR. Anticancer Res. 2015; 35:3969–77.
202. Gross TJ, Powers LS, Boudreau RL, Brink B, Reisetter A, Goel K, Gerke AK, Hassan IH, Monick MM. A microRNA processing defect in smokers’ macrophages is linked to SUMOylation of the endonuclease DICER. J Biol Chem. 2014; 289:12823–34.
203. Yuan A, Hsiao YJ, Chen HY, Chen HW, Ho CC, Chen YY, Liu YC, Hong TH, Yu SL, Chen JJ, Yang PC. Opposite Effects of M1 and M2 Macrophage Subtypes on Lung Cancer Progression. Sci Rep. 2015; 5:14273.
204. Xu S, Zhang R, Niu J, Cui D, Xie B, Zhang B, Lu K, Yu W, Wang X, Zhang Q. Oxidative stress mediated-alterations of the microRNA expression profile in mouse hippocampal neurons. Int J Mol Sci. 2012; 13:16945–60.
205. Deng S, Zhao Q, Zhou X, Zhang L, Bao L, Zhen L, Zhang Y, Fan H, Liu Z, Yu Z. Neonatal Heart-Enriched miR-708 Promotes Differentiation of Cardiac Progenitor Cells in Rats. Int J Mol Sci. 2016; 17.
206. Girardot M, Pecquet C, Boukour S, Knoops L, Ferrant A, Vainchenker W, Giraudier S, Constantinescu SN. miR-28 is a thrombopoietin receptor targeting microRNA detected in a fraction of myeloproliferative neoplasm patient platelets. Blood. 2010; 116:437–45.
207. Rothman AM, Arnold ND, Pickworth JA, Iremonger J, Ciuclan L, Allen RM, Guth-Gundel S, Southwood M, Morrell NW, Thomas M, Francis SE, Rowlands DJ, Lawrie A. MicroRNA-140-5p and SMURF1 regulate pulmonary arterial hypertension. J Clin Invest. 2016; 126:2495–508.
208. Dileepan M, Sarver AE, Rao SP, Panettieri RA Jr, Subramanian S, Kannan MS. MicroRNA Mediated Chemokine Responses in Human Airway Smooth Muscle Cells. PLoS One. 2016; 11:e0150842.
209. Forstner AJ, Hofmann A, Maaser A, Sumer S, Khudayberdiev S, Muhleisen TW, Leber M, Schulze TG, Strohmaier J, Degenhardt F, Treutlein J, Mattheisen M, Schumacher J, et al. Genome-wide analysis implicates microRNAs and their target genes in the development of bipolar disorder. Transl Psychiatry. 2015; 5:e678.
210. Fiorentino A, O’Brien NL, Sharp SI, Curtis D, Bass NJ, McQuillin A. Genetic variation in the miR-708 gene and its binding targets in bipolar disorder. Bipolar Disord. 2016; 18:650–656.
211. Bot AM, Debski KJ, Lukasiuk K. Alterations in miRNA levels in the dentate gyrus in epileptic rats. PLoS One. 2013; 8:e76051.
212. Lau P, Bossers K, Janky R, Salta E, Frigerio CS, Barbash S, Rothman R, Sierksma AS, Thathiah A, Greenberg D, Papadopoulou AS, Achsel T, Ayoubi T, et al. Alteration of the microRNA network during the progression of Alzheimer’s disease. EMBO Mol Med. 2013; 5:1613–34.