
INTRODUCTION
With the fast developments of computer-aided drug design (CADD) [1–5], currently a number of drug design approaches are developed, and several computer software packs [6, 7] are available that can speed up the discovery of new chemical and biological drugs in more efficient and economical procedure. However, so far we still have no perfect theories, ideal technologies, and faultless software tools that can guarantee complete success of the designed drugs due to the complicity of the interactions between medicinal drugs and their biological targets [8, 9]. The factors and parameters that may affect the bioactivities of drugs are not only from the structure of drug itself, but also from its biological target, coenzymes, and interaction environment [10, 11].
Principal component analysis (PCA) [12–14] is a useful tool that has been widely used in chemistry, biology, environment, and many fields of social science. The PCA approach has also been used in drug design for many years. Traditionally PCA is a single level and one direction prediction and analysis technique, described as the following equation
(1)
where are the physicochemical parameters of the i-th molecule, are the coefficients of molecular parameters, and wi is bioactivity of the i-th molecule [15, 16]. The bioactivity wi could be logarithm of IC50,i (pIC50,i=-logIC50,i), or binding free energy ΔG°i between drug and receptor.
After the coefficients of parameters are solved from the linear equations Eq.1 in a training set of drug candidates, the parameter coefficients can be used to predict the bioactivities of the designed or newly synthesized drug compounds,
(2)
where K is the total number of molecular parameters. Currently hundreds even thousands of molecular parameters are available for drug design [17, 18]. However for certain drug-receptor interaction system, these parameters are not equally important; actually too many parameters may cause the over correlation problem [19, 20]. In PCA technique only the principle components are selected to describe the bioactivities of drug molecules, and to predict the bioactivities of drug candidates.
In the present study, an improved principal component analysis method, the so-called two-level principal component analysis (2L-PCA), is proposed to deal with the extreme complexity and huge amount of parameters in drug design and discovery. In the 2L-PCA predictor, the 1st level is to deal with the physicochemical properties of drug molecules, and the 2nd level is to deal with the fragments of molecular structures. The proposed two-level model can not only significantly enhance the prediction power, but also yield more useful information for in-depth analysis.
According to Chou’s 5-step rule [21] that has been widely used by many investigators (see, e.g., [22–37]), to develop a really useful statistical predictor, one should consider the following five procedures: (1) benchmark dataset; (2) sample representation; (3) operation algorithm; (4) cross validation; (5) web-server. Below, let us describe how to deal with them one-by-one. However, to comply with the Journal’s rubric style, they are not exactly following the aforementioned order.
RESULTS AND DISCUSSION
As an example to show the advantage of 2L-PCA, we applied it for predicting the binding affinity of epitope-peptides with class I MHC molecules HLA-A*0201 [38, 39]. HLA-A*0201 is one of the most frequent class I alleles found in many different species and populations, which plays a critical role for antigen presentation in both viral antigens [40] and tumor antigens from a variety of cancers [41–44], and is expressed in approximately 50% of Caucasians population [45].
The epitope-peptides consist of nine amino acids [38, 39]. In the 2L-PCA study for the epitope-peptides, the nine side chains of the nine amino acids are the nine fragments. Eight physicochemical properties are used as the descriptors of the 20 natural amino acids. Four of them are the HMLP parameters [15, 16], describing the lipophilic character, hydrophilic character, surface area with lipophilic potential, and surface area with hydrophilic potential, respectively. The fifth property is the volume of amino acid side chains. The remaining three properties are the secondary structural potency indices of amino acids: the α-potency, β-potency, and coil-potency [46]. Listed in Table 1 are the eight physicochemical parameters of 20 amino acids used in this study.
Table 1: Eight physicochemical parametersa of 20 natural amino acid side chains
A.A. |
Lip |
Hyd |
SL (Å2) |
SH (Å2) |
Pα |
Pβ |
Pc |
V(Å3) |
|---|---|---|---|---|---|---|---|---|
Leu (L) |
1.2906 |
0.0000 |
84.5476 |
0.0000 |
1.21 |
1.30 |
0.68 |
166.7 |
Ile (I) |
1.1046 |
0.0000 |
88.6055 |
0.0000 |
1.08 |
1.60 |
0.66 |
166.7 |
Val (V) |
0.5324 |
0.0000 |
77.8108 |
0.0000 |
1.06 |
1.70 |
0.62 |
140.0 |
Phe (F) |
0.4412 |
-0.1195 |
105.7054 |
11.2472 |
1.13 |
1.38 |
0.71 |
189.9 |
Met (M) |
1.0768 |
-0.3068 |
70.3631 |
23.2299 |
1.45 |
1.05 |
0.58 |
162.9 |
Trp (W) |
0.8364 |
-0.4310 |
133.6980 |
14.8820 |
1.08 |
1.37 |
0.75 |
227.8 |
Ala (A) |
0.1744 |
0.0000 |
34.7760 |
0.0000 |
1.42 |
0.83 |
0.70 |
88.6 |
Cys (C) |
0.2479 |
-0.2402 |
23.5563 |
30.4540 |
0.70 |
1.19 |
1.18 |
108.5 |
Gly (G) |
0.0208 |
0.0000 |
3.7616 |
0.0000 |
0.57 |
0.75 |
1.50 |
60.1 |
Tyr (Y) |
0.4534 |
-0.5896 |
80.9646 |
42.7160 |
0.69 |
1.47 |
1.06 |
193.6 |
Thr (T) |
1.4265 |
-0.4369 |
46.7285 |
16.0490 |
0.83 |
1.19 |
1.07 |
116.1 |
Ser (S) |
0.2346 |
-0.6040 |
26.0681 |
15.9613 |
0.77 |
0.75 |
1.32 |
89.0 |
His (H) |
0.8124 |
-0.7766 |
82.1701 |
13.8631 |
1.00 |
0.87 |
1.06 |
153.2 |
Gln (Q) |
1.0036 |
-0.7211 |
70.0876 |
17.8662 |
1.11 |
1.10 |
0.86 |
143.9 |
Lys (K) |
1.4600 |
-0.6229 |
97.7144 |
8.0786 |
1.16 |
0.74 |
0.98 |
168.7 |
Asn (N) |
0.6396 |
-0.7211 |
50.5075 |
17.7804 |
0.67 |
0.89 |
1.35 |
117.7 |
Glu (E) |
1.0315 |
-0.9298 |
57.1582 |
25.5726 |
1.51 |
0.37 |
0.84 |
138.4 |
Asp (D) |
0.6058 |
-0.9298 |
37.4173 |
25.2736 |
1.01 |
0.54 |
1.20 |
111.1 |
Arg (R) |
1.2424 |
-1.4797 |
90.8008 |
35.3095 |
0.98 |
0.93 |
1.04 |
173.4 |
Pro (P) |
0.3226 |
0.0000 |
69.2297 |
0.0000 |
0.57 |
0.55 |
1.59 |
122.7 |
Lip: lipophilic index; Hyd: hydrophilic index; SL: lipophilic surface area; SH: hydrophilic surface area; Pα: potency of α-helix; Pβ: potency of β-band; Pc: potency of loop; V: volume of side chains.
In this study the HMLP parameters were used to describe the lipophilicity and hydrophilicity of molecular fragments. In peptides the HMLP parameters of the 20 natural amino acid side chains are available from literatures. However, the HMLP parameters of common chemical molecular fragments have to be derived using complicated calculations. In such cases other hydrophobic parameters can be used, e.g., the atom-based hydrophobic parameters in [47].
To reduce computational time, the cross validation in this study was performed via the independent dataset test [48], as described as follows. The sequences and experimental binding affinities of the 90 peptides were used as the training dataset to train the model, while those of the 40 peptides taken from [49] as the independent dataset to test the model. Actually, such 40 peptides had also been compiled in a series of publications [41, 42, 50–55]. The logarithms (pIC50) of IC50 were used as the bioactivity, because they are related to the changes in the free binding energy [55, 56]. Listed in Table 2 are the sequences and the experimental pIC50 of the peptides used in the training set. The binding strength of the 90 training peptides and 40 testing peptides covers the low, intermediate, and high affinity. The following two criteria were applied in the choice of the testing peptides: (1) the range of binding affinities in the testing dataset should not exceed the range of affinities in the training set; (2) the amino acid at each position in the testing dataset should also be present at that position in the training set of peptides. These two conditions make the 130 peptides to be the ideal benchmark dataset for 2L-PCA method.
Table 2: Amino acid sequences and experimental and predicted bioactivities of 90 MHC-I peptides in the training set
No. |
Peptide |
Expt |
Pred |
pIC50 |
No. |
Peptide |
Expt |
Pred |
pIC50 |
|---|---|---|---|---|---|---|---|---|---|
1 |
VALVGLFVL |
5.148 |
5.7543 |
-0.6063 |
46 |
VVMGTLVAL |
7.174 |
7.3163 |
-0.1423 |
2 |
GTLVALVGL |
5.342 |
5.9368 |
-0.5948 |
47 |
YLEPGPVTI |
7.187 |
7.1654 |
0.0216 |
3 |
LQTTIHDII |
5.501 |
5.8143 |
-0.3133 |
48 |
GLSRYVARL |
7.248 |
7.4620 |
-0.2131 |
4 |
SLHVGTQCA |
5.842 |
6.1580 |
-0.3160 |
49 |
LLAQFTSAI |
7.301 |
7.4302 |
-0.1292 |
5 |
ALPYWNFAT |
5.869 |
6.6416 |
-0.7726 |
50 |
VLLDYQGML |
7.328 |
7.5911 |
-0.2631 |
6 |
SLNFMGYVI |
5.881 |
5.9560 |
-0.0750 |
51 |
YLEPGPVTV |
7.342 |
7.4078 |
-0.0658 |
7 |
NLQSLTNLL |
6.000 |
6.6992 |
-0.6992 |
52 |
ILSPFMPLL |
7.3470 |
7.1400 |
0.2070 |
8 |
FVTWHRYHL |
6.025 |
5.7230 |
0.3020 |
53 |
YLSPGPVTA |
7.383 |
7.5610 |
-0.1780 |
9 |
DPKVKQWPL |
6.176 |
5.7407 |
0.4354 |
54 |
IIDQVPFSV |
7.398 |
7.6528 |
-0.2548 |
10 |
ITSQVPFSV |
6.196 |
6.5888 |
-0.3928 |
55 |
SVYDFFVWL |
7.444 |
7.3654 |
0.0786 |
11 |
ALAKAAAAI |
6.211 |
6.2433 |
-0.0323 |
56 |
ITWQVPFSV |
7.463 |
7.4417 |
0.0213 |
12 |
GLGQVPLIV |
6.301 |
6.5651 |
-0.2641 |
57 |
ITYQVPFSV |
7.480 |
7.6613 |
-0.1813 |
13 |
MLDLQPETT |
6.335 |
6.8570 |
-0.5220 |
58 |
GLYSSTVPV |
7.481 |
7.6303 |
-0.1493 |
14 |
LLSSNLSWL |
6.342 |
6.3502 |
-0.0082 |
59 |
VMGTLVALV |
7.553 |
7.2369 |
0.3161 |
15 |
GLACHQLCA |
6.380 |
6.0594 |
0.3206 |
60 |
LLLCLIFLL |
7.585 |
7.1406 |
0.4444 |
16 |
LIGNESFAL |
6.415 |
7.0559 |
-0.6409 |
61 |
SLDDYNHLV |
7.585 |
7.1764 |
0.4086 |
17 |
ALAKAAAAV |
6.419 |
6.4857 |
-0.0667 |
62 |
VLIQRNPQL |
7.644 |
6.9473 |
0.6967 |
18 |
LLAVGATKV |
6.477 |
6.5115 |
-0.0344 |
63 |
SLYADSPSV |
7.658 |
7.7106 |
-0.0526 |
19 |
ALAKAAAAL |
6.511 |
6.2262 |
0.2848 |
64 |
ILSQVPFSV |
7.699 |
7.6472 |
0.0518 |
20 |
WILRGTSFV |
6.556 |
6.9084 |
-0.3524 |
65 |
IMDQVPFSV |
7.719 |
8.0305 |
-0.3115 |
21 |
IISCTCPTV |
6.580 |
6.6649 |
-0.0849 |
66 |
QLFEDNYAL |
7.764 |
7.4713 |
0.2927 |
22 |
FLGGTPVCL |
6.623 |
6.8756 |
-0.2526 |
67 |
ALMDKSLHV |
7.770 |
7.5250 |
0.2450 |
23 |
ALIHHNTHL |
6.623 |
6.7908 |
-0.1677 |
68 |
YAIDLPVSV |
7.796 |
7.6075 |
0.1885 |
24 |
NLSWLSLDV |
6.639 |
6.0466 |
0.5924 |
69 |
FVWLHYYSV |
7.824 |
8.1149 |
-0.2909 |
25 |
YMIMVKCWM |
6.663 |
6.6427 |
0.02035 |
70 |
MLGTHTMEV |
7.845 |
7.3180 |
0.5270 |
26 |
VLQAGFFLL |
6.682 |
7.0412 |
-0.3592 |
71 |
LLFGYPVYV |
7.886 |
8.0253 |
-0.1393 |
27 |
GTLGIVCPI |
6.714 |
6.5233 |
0.1907 |
72 |
ILKEPVHGV |
7.921 |
7.5915 |
0.3295 |
28 |
VILGVLLLI |
6.785 |
7.4728 |
-0.6878 |
73 |
YLMPGPVTV |
7.932 |
7.9139 |
0.0181 |
29 |
VTWHRYHLL |
6.793 |
6.5597 |
0.2333 |
74 |
WLDQVPFSV |
7.939 |
7.9514 |
-0.0124 |
30 |
PLLPIFFCL |
6.796 |
7.5217 |
-0.7257 |
75 |
KTWGQYWQV |
7.955 |
7.6934 |
0.2616 |
31 |
TLGIVCPIC |
6.815 |
5.9499 |
0.8651 |
76 |
ALMPLYACI |
8.000 |
7.4383 |
0.5617 |
32 |
CLTSTVQLV |
6.832 |
7.1061 |
-0.2741 |
77 |
YLAPGPVTA |
8.032 |
7.6408 |
0.3912 |
33 |
ILLLCLIFL |
6.845 |
6.7815 |
0.0635 |
78 |
YLYPGPVTV |
8.051 |
8.3112 |
-0.2602 |
34 |
FAFRDLCIV |
6.886 |
6.6689 |
0.2171 |
79 |
LLMGTLGIV |
8.097 |
7.6769 |
0.4201 |
35 |
FLEPGPVTA |
6.898 |
7.4940 |
-0.5960 |
80 |
YLWPGPVTV |
8.125 |
8.0916 |
0.0334 |
36 |
ALAKAAAAA |
6.947 |
6.8081 |
0.1389 |
81 |
FLLTRILTI |
8.149 |
7.8796 |
0.2694 |
37 |
LMAVVLASL |
6.954 |
7.4908 |
-0.5368 |
82 |
GLLGWSPQA |
8.237 |
8.2184 |
0.0185 |
38 |
YVITTQHWL |
6.983 |
6.3410 |
0.6420 |
83 |
ILYQVPFSV |
8.310 |
8.7197 |
-0.4097 |
39 |
LLCLIFLLV |
6.996 |
7.5015 |
-0.5055 |
84 |
GILTVILGV |
8.347 |
7.8414 |
0.5056 |
40 |
ITAQVPFSV |
7.020 |
6.6685 |
0.3515 |
85 |
NMVPFFPPV |
8.398 |
8.0854 |
0.3126 |
41 |
YLEPGPVTL |
7.058 |
7.1483 |
-0.0903 |
86 |
ILDQVPFSV |
8.481 |
7.6904 |
0.7906 |
42 |
YTDQVPFSV |
7.066 |
7.0742 |
-0.0082 |
87 |
YLFPGPVTA |
8.495 |
8.3473 |
0.1477 |
43 |
NLYVSLLLL |
7.114 |
6.9769 |
0.1371 |
88 |
YLDQVPFSV |
8.638 |
8.1326 |
0.5054 |
44 |
ILHNGAYSL |
7.127 |
7.3493 |
-0.2223 |
89 |
ILFQVPFSV |
8.699 |
8.4335 |
0.2655 |
45 |
SIISAVVGI |
7.159 |
7.3048 |
-0.1458 |
90 |
ILWQVPFSV |
8.770 |
8.5002 |
0.2698 |
Statistical indices:
R=0.887132 R2= 0.787003 RES=0.366873 SEE=0.038672.
The iterative 2L-PCA technique described in Method section is used for the binding affinity study of peptides based on the sequences and experimental data listed in Table 2. The initial coefficient values of fragment parameters were assigned to 1, implying that all fragment parameters are equally important. Shown in Figure 1 are the curves of correlation coefficients R vs iterations, where the curve Ra is for the iteration of coefficients , and the curve Rb is for the iterations of coefficients . The average fitting error Q between the calculated bioactivities and the experimental bioactivities of peptides are shown in Figure 2, where Qa is for iteration and Qb for iteration. It has been observed that, after 10 to 12 iterations, the iterative result converged smoothly. The converged prediction coefficient sets and are given in Table 3. In the iterative solution precedure the correlation coefficien increases from the first value RA(1)=0.4167 to the converged value RA(98)=0.8871, and the prediction residue decreases from the first value QA (1)=0.7223 to the converged value QA(98)=0.0387.
Table 3: Prediction coefficients of eight physicochemical properties and nine residue positions obtained from the training set of MHC-I peptides
No. |
Property |
Coefficient |
No. |
Position |
Coefficient |
|---|---|---|---|---|---|
(Residue) |
|||||
1 |
Lip |
-0.02445 |
1 |
R1 |
2.53268 |
2 |
Hyd |
0.19258 |
2 |
R2 |
8.36712 |
3 |
SL |
-0.00212 |
3 |
R3 |
3.06856 |
4 |
SH |
0.00348 |
4 |
R4 |
-4.89559 |
5 |
Pα |
0.15367 |
5 |
R5 |
3.12686 |
6 |
Pβ |
0.07823 |
6 |
R6 |
2.45367 |
7 |
Pc |
0.19764 |
7 |
R7 |
1.24669 |
8 |
Vol |
0.00366 |
8 |
R8 |
-3.50416 |
-- |
-- |
-- |
9 |
R9 |
-3.79249 |
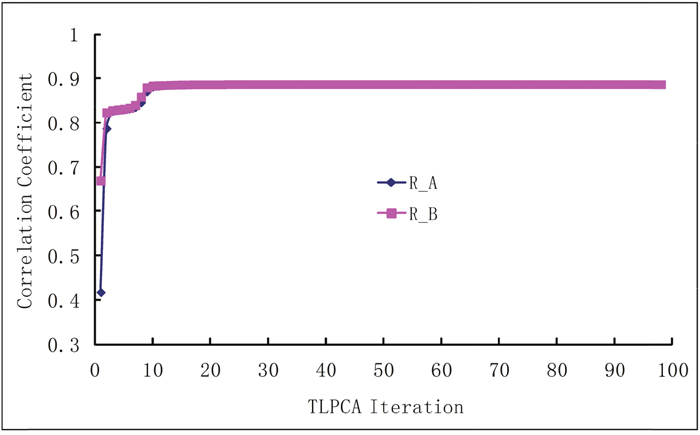
Figure 1: The correlation coefficients between experimental and predicted bioactivities increase with the iterations. Ra is the correlation coefficient in the iterative procedure for of the physicochemical properties, and Rb is the correlation coefficient in the iterative procedure for of the molecular fragments.
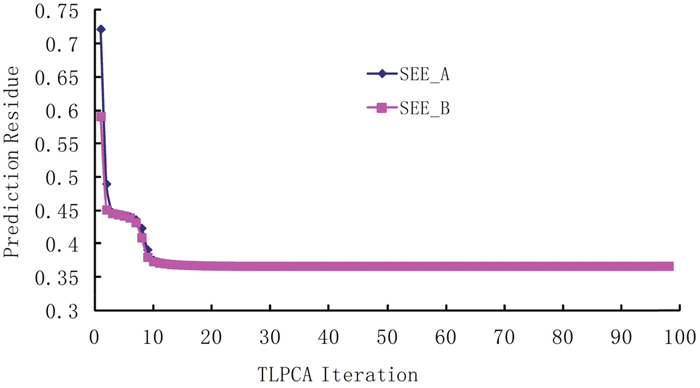
Figure 2: The residue between predicted bioactivities and experimental bioactivities in the iterative procedure. The Q is the average square root of the summation of squared differences between predicted bioactivities and experimental bioactivities. Qa is for iteration and Qb is for iteration.
The predicted pIC50 of the 40 queried peptides in the testing set are given in Table 4, which were predicted using the coefficients of properties and of fragments based on the eight physicochemical parameters and the nine fragments (amino acid side chains). The diversity of the peptides in the training set is very important for the prediction power of TLPC, especially for the residue positions at which we want to make prediction. It is expected that, with more experimental data available, the predictive power of 2L-PCA will be further improved. Actually, 30 prediction servers for human MHC-I peptide molecules were evaluated in a review article [57]. Among the 30 existing servers, 16 were ranked as the first class that provided the most accurate prediction results for MHC-I peptide molecules with the correlation coefficients ranging from r = 0.55 to r = 0.87. It has been shown in this study that the prediction correlation coefficient yielded by our 2L-PCA method is r = 0.868, being ranked around the very top of the first class.
Table 4: Amino acid sequences and experimental and predicted bioactivities of 40 MHC-I peptides in the testing set
No. |
Sequence |
Expt |
Pred |
pIC50 |
No. |
Sequence |
Expt |
Pred |
pIC50 |
|---|---|---|---|---|---|---|---|---|---|
1 |
LLGCAANWI |
5.301 |
5.1708 |
0.1302 |
21 |
ITFQVPFSV |
7.179 |
7.3750 |
-0.1960 |
2 |
SAANDPIFV |
5.342 |
4.8592 |
0.4828 |
22 |
FTDQVPFSV |
7.212 |
6.8379 |
0.3741 |
3 |
TTAEEAAGI |
5.380 |
5.4678 |
-0.0878 |
23 |
RLMKQDFSV |
7.342 |
7.5681 |
-0.2261 |
4 |
LTVILGVLL |
5.580 |
5.3216 |
0.2584 |
24 |
KLHLYSHPI |
7.352 |
6.6450 |
0.7070 |
5 |
HLLVGSSGL |
5.792 |
6.4811 |
-0.6891 |
25 |
ITMQVPFSV |
7.398 |
7.2641 |
0.1340 |
6 |
GIGILTVIL |
6.000 |
5.7321 |
0.2679 |
26 |
KIFGSLAFL |
7.478 |
6.7818 |
0.6962 |
7 |
TVILGVLLL |
6.072 |
5.4662 |
0.6058 |
27 |
ALVGLFVLL |
7.585 |
7.3852 |
0.1998 |
8 |
WTDQVPFSV |
6.145 |
6.8930 |
-0.7480 |
28 |
YLSPGPVTV |
7.642 |
7.2387 |
0.4033 |
9 |
AIAKAAAAV |
6.176 |
6.4480 |
-0.2720 |
29 |
GLYSSTVPV |
7.699 |
7.6303 |
0.0687 |
10 |
ILTVILGVL |
6.419 |
7.0160 |
-0.5970 |
30 |
YLYPGPVTA |
7.772 |
8.6335 |
-0.8615 |
11 |
AVAKAAAAV |
6.495 |
5.9131 |
0.5819 |
31 |
YLAPGPVTV |
7.818 |
7.3184 |
0.4996 |
12 |
ILDEAYVMA |
6.623 |
7.4445 |
-0.8215 |
32 |
VVLGVVFGI |
7.845 |
7.4509 |
0.3941 |
13 |
LLWFHISCL |
6.682 |
6.3594 |
0.3226 |
33 |
MMWYWGPSL |
7.921 |
7.4007 |
0.5203 |
14 |
TLDSQVMSL |
6.793 |
7.2566 |
-0.4636 |
34 |
ILAQVPFSV |
7.939 |
7.7270 |
0.2120 |
15 |
HLYQGCQVV |
6.832 |
7.6799 |
-0.8479 |
35 |
FLLSLGIHL |
8.053 |
8.1578 |
-0.1048 |
16 |
QLFHLCLII |
6.886 |
7.6475 |
-0.7615 |
36 |
ILMQVPFSV |
8.125 |
8.3225 |
-0.1975 |
17 |
ITDQVPFSV |
6.947 |
6.6320 |
0.3150 |
37 |
YLFPGPVTV |
8.237 |
8.0249 |
0.2121 |
18 |
ALCRWGLLL |
7.000 |
7.2766 |
-0.2766 |
38 |
YLMPGPVTA |
8.367 |
8.2363 |
0.1307 |
19 |
NLGNLNVSI |
7.119 |
7.0974 |
0.02160 |
39 |
YLWPGPVTA |
8.495 |
8.4140 |
0.0810 |
20 |
HLYSHPIIL |
7.131 |
7.5663 |
-0.4353 |
40 |
FLDQVPFSV |
8.658 |
7.8964 |
0.7616 |
Statistical indices:
R=0.867872 R2= 0.753202 RES=0.469728 SEE=0.074271.
2L-PCA neither needs knowing the exact comformations of the peptides nor needs aligning the peptides according to a template. The two steps are necessary but quite difficult for CoMFA [58, 59] and CoMSIA [60, 61] owing to that there are numerous possible conformations for peptides and that the experimental crystal structure for serving as a template is often not available. 2L-PCA method provides an alternate way for design of the chemical drugs and peptide drugs.
The eigenvalues and contributions of physicochemical properties and amino acid positions in peptides are summarized in Table 5 and shown in Figure 3. In Table 5 the eigenvalues are normalized. The eigenvalue portion of the first three property eigenvectors is almost 100%, and the eigenvalue portion of the first eigenvector alone is larger than 99%. Most contributions are made by the three properties: side chain volume (Vol), lipophilic surface area (SL), and hydrophilic surface area (SH), as shown in Figure 3b and Table 5. The contributions of other 5 properties seem very small. The eigenvalue of the first peptide position eigenvector is larger than 98%. In Table 5 the contributions of the nine amino acid positions are different. However the differences are not big, implying that all positions are almost equally important. The detailed computation results are given in Supplementary Information 1.
Table 5: Eigenvalues and contributions of physicochemical properties and amino acid positions in training set of peptides
Physicochemical properties |
Positions (fragments) |
||||||
|---|---|---|---|---|---|---|---|
No. |
Eigenvaluea |
Property |
Contribution |
No. |
Eigenvaluea |
Property |
Contribution |
1 |
0.99118 |
Lip |
0.00004 |
1 |
0.98873 |
Residue-1 |
0.12126 |
2 |
0.00641 |
Hyd |
0.00000 |
2 |
0.00300 |
Residue-2 |
0.12043 |
3 |
0.00240 |
SL |
0.20595 |
3 |
0.00249 |
Residue-3 |
0.1130 |
4 |
0.00005 |
SH |
0.00534 |
4 |
0.00213 |
Residue-4 |
0.09871 |
5 |
0.00004 |
Pα |
0.00004 |
5 |
0.00106 |
Residue-5 |
0.1046 |
6 |
0.00003 |
Pβ |
0.00005 |
6 |
0.00094 |
Residue-6 |
0.10804 |
7 |
0.00002 |
Pc |
0.00002 |
7 |
0.0007 |
Residue-7 |
0.11931 |
8 |
0.00001 |
Vol |
0.78857 |
8 |
0.00058 |
Residue-8 |
0.10186 |
-- |
-- |
-- |
-- |
9 |
0.00037 |
Residue-9 |
0.11278 |
a Eigenvalues are normalized.
b The positions of peptides are equal to the fragments of molecules.
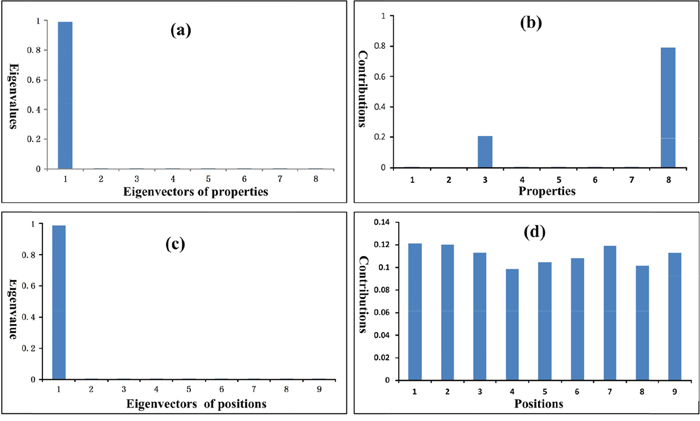
Figure 3: Eigenvalues and contributions of properties and peptide positions. (a) The eigenvalues of property eigenvectors. (b) The contributions of properties to the eigenvalues. The volumes (Vol) and hydrophobic surface areas (SL) of amino acid side chains make the largest contributions. (c) The eigenvalues of peptide position eigenvectors. (d) The contributions of peptide positions to the eigenvalues. The contributions of all nine amino acid positions are almost equally important.
We are often facing two kinds of challenges in theoretical prediction for drug design: one is over-correlation problem, and the other is lack of information and explanation for the predicted results. The over-correlation problem is caused by large amount of parameters used in the prediction model, which may yield quite good correlation results in self-consistency test [62, 63], but very poor predicted results in independent dataset test owing to the high dimensional disaster [19] or “curse of dimensionality” problem. To solve this problem, the pseudo amino acid composition (PseAAC) was introduced [64]. Ever since then, the concept of PseAAC or the general PseAAC [21] has been widely used in drug development and biomedicine [65, 66] and nearly all the areas of computational proteomics (see, e.g., [67] as well as a long list of references cited in [68, 69]). Actually, the physicochemical properties used here can be regarded as some optimal pseudo components [70]. It is through such a PseAAC approach to remove the trivial parameters (or reduce the feature vector’s dimension) and grasp the key ones. Besides, the traditional prediction methods fail to provide a good explanation for the predicted results; i.e., how do the physicochemical properties and the structural changes affect the bioactivities? In contrast to that, the proposed “2L-PCA” method can provide more information about the impact of the physicochemical properties and molecular fragments to the bioactivities of drug candidates.
MATERIALS AND METHODS
In practical drug design and development, usually the basic structure of drug candidates keep constant, only small modifications are made on several fragments. The structure parameters of the entire molecules cannot clearly describe the detailed characters of the small changes at individual fragments or substitutes. In the 2L-PCA model the molecular structures are separated into several fragments, and are described by a set of fragment parameters. An example of molecular structure and its fragments is shown in Figure 4A. The idea of molecular fragments also can be applied to the peptide drugs, in which each side chain of an amino acid is a fragment, as shown in Figure 4B.
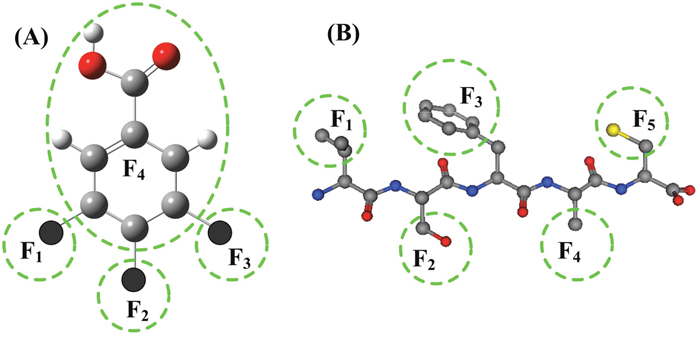
Figure 4: Illustration of molecular fragments. (A) The structural fragments in neuraminidase (NA) of influenza virus A inhibitors. The molecular structure is divided into 4 fragments according to the substitutes being investigated. The fragments F1, F2 and F3 are three substituent groups, and the fragment F4 is the remaining part of the molecular parent. (B) In short peptides each side chain of amino acid residue is a fragment.
General 3D equation of 2L-PCA
In the 2L-PCA prediction model the bioactivity wi of molecule i is the summation of contributions Δgi,l from all molecular fragments; i.e.,
(3)
where Δgi,l is the contribution of fragment l to the bioactivity wi of molecule i, bl is the prediction coefficient of fragment l, and L is the total number of molecular fragments. The contribution Δgi,l of fragment l is the summation of the contributions from all physicochemical properties of fragment l, namely
(4)
where xi,l,k is the physicochemical property k of fragment l in molecule i, ak is the prediction coefficient of physicochemical property k, and K is the total number of physicochemical properties.
Inserting the Eq.4 into Eq.3 we get the general equation of 2L-PCA prediction model as given by
(5)
where N is the total number of molecular samples. Eq.5 can be expressed in vector and matrix form as given below
(6)
where XN,L,K is the three dimensional (3D) data matrix of molecular parameters, WN is the bioactivity column vector of molecular samples, BL is the coefficient vector of fragments, and AK is the coefficient vector of physicochemical properties.
2D equations of properties and fragments
The general three-dimensional 2L-PCA equation of Eq.6 can be reduced to two 2D equations with the following algebra operations,
(7)
where HN,L is the 2D data matrix of molecular fragments. Substituting HN,L into Eq.6, we obtain the following fragment 2D equation
(8)
Likewise, the property 2D equation can also be expressed as
(9)
and
(10)
where FN,K is the 2D data matrix of physicochemical properties.
Algebra solutions of property and fragment 2D equations
The fragment 2D equation Eq.8 and the property 2D equation Eq.10 can be solved using the standard algebra method. Both sides of the fragment 2D equation of Eq.8 are multiplied with the transposed matrix HtN,L from left, it follows that
(11)
and
(12)
Thus, we get the following symmetrically square matrix equation of fragments
(13)
Since the fragment square matrix equation of Eq.13 is multiplied by its inverse matrix U-1L,L, the prediction coefficients BL for the fragments are obtained, as given below
(14)
where the inverse matrix U-1L,L can be obtained by solving the eigen equation [71] [48] of UL,L, namely the equation
(15)
meaning
(16)
where ΨL,L is the eigenvectors and αL is the eigenvalues of fragment square matrix UL,L[72, 73].
Similarly, left-multiplying both sides of property 2D equation of Eq.10 with FtN,K, we have
(17)
and
(18)
From Eqs.17-18, we get the following square matrix equation of properties
(19)
Multiplying Equation Eq.19 with the inverse matrix V-1K,K, will give the solution of property prediction coefficients AK; i.e.
(20)
Thus, the inverse matrix V-1K,K is obtained by solving the eigen equation of property square matrix VK,K:
(21)
and
(22)
where ΦK,K is the eigen-vectors and βk is the eigen-values of the property square matrix VK,K.
Iterative solution of 2L-PCA equations
In the training dataset for drug candidates the two prediction coefficients set AK and BL in the 2L-PCA general equation Eq.6 are solved in an iterative procedure [74, 75]. Firstly the initial fragment coefficients B(0)L are assigned to 1 {bi=1, i=1,2…,L}, implying all fragments are equally important. The initial B(0)L are used in the property 2D equations Eq (9) and (10), thus the first solution of property coefficients A(1)K is obtained by solving the eigen-equations Eq.17-20. Then the property coefficients A(1)K are used in the fragment equations Eqs.7-8, and the first solution of fragment coefficients B(1)L are obtained by solving eigen-equations Eq.11-14. In the next iterative cycle the B(1)L is used to find the A(2)K. Above iterative procedure is repeated for n times, until to reaching a threshold value ε; i.e.,
(23)
The bioactivities of designed drugs and newly synthesized drug candidates are predicted using the converged coefficients and as given below
(24)
Illustrated in Figure 5 is the iterative solution procedure for the 2L-PCA predictor.
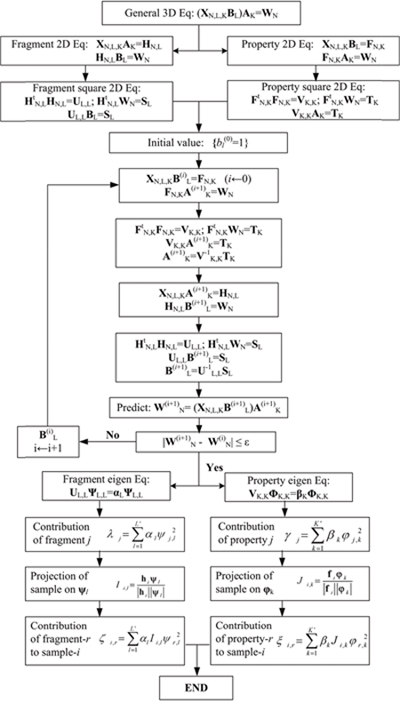
Figure 5: The iterative algebra solution procedure of the solution of 2L-PCA prediction model for the two sets of coefficients and , where N is the number of molecular samples, L is the number of fragments in molecules, L′ is the principal number of fragments, K is the number of physicochemical properties, and K′ is the principal number of properties.
Principal component analysis of properties and fragments
The property eigenvectors are orthogonal and normalized; i.e.,
(25)
and
(26)
where the term φ2j,k is the component of the j-th property in the k-th eigen-vector φk. The first K′ property eigen-vectors are the principal components whose eigen-values are larger than a threshold (e.g., ε=90% or 95%); i.e.,
(27)
The total contribution γj of the j-th property to the bioactivity of molecular samples in training set is defined as the following summation,
(28)
The property eigen-vectors span an orthogonal multiple space, in which a drug molecule Pi is a vector, and its projection Ji,k on the k-th property-eigenvector φk is calculated by
(29)
where fi is the i-th row vector of the property matrix FNK of Eq.9. In the projection Ji,k of molecular sample Pi on the k-th property-eigenvector jk the component of the r-th property is αkφ2r,k, therefore the total contribution of r-th property to the sample Pi is the summation of components from all principal property eigenvectors, namely
(30)
Similarly, the fragment eigenvectors ψl span an L-dimensional orthogonal space. The first L′ fragment eigenvectors are the principal components. The total contribution factor λj of the j-th fragment to the bioactivity of peptide set is given by
(31)
In the same way the projection Ii,l of sample Pi on the l-th fragment-eigenvector ψl can be calculated by
(32)
where hi is the i-th row vector of the fragment matrix HNL of Eq.7. In the projection Ii,l of molecule Pi on the l-th fragment-eigenvector φl the component of the r-th fragment is αlψ2r,l, therefore the total contribution of r-th fragment to the sample Pi is the summation of components from all principal fragment eigenvectors; i.e.,
(33)
Web-server
As pointed out in [76], user-friendly and publicly accessible web-servers represent the future direction for developing practically more useful predictors or any computational tools. Actually, user-friendly web-servers as given in a series of recent publications [23–25, 30, 32, 34–36, 69, 70, 77–91] will significantly enhance the impacts by attracting the broad experimental scientists [66, 92]. We will do our best to establish a web-server for 2L-PCA as soon as possible. Once it has been done, an announcement will be made thorough a publication or our webpage.
CONCLUSION
The 2L-PCA predictor proposed in this paper is a very useful tool for drug design. Its advantages can be summarized as follows. (1) With 2L-PCA, the molecular structures of drug candidates can be separated into several fragments described by physicochemical parameters of the molecular fragments, thus the small modifications on individual fragments can be clearly shown. (2) Its two prediction coefficient sets of properties and of fragments can be solved in an iterative procedure, which possesses self-learning ability and information feed-back function in certain degree, and hence greatly promoting the prediction power of 2L-PCA. (3) It possesses the information from both of the structures of molecular fragments and the physicochemical properties, able to significantly improve the drug candidates in both the structure and property. (4) Its elegant algebra solution procedure will be very useful for further enhancing the ability of principal component analysis (PCA).
ACKNOWLEDGMENTS
The authors are very much indebted to the eight anonymous reviewers, whose constructive comments are very helpful for strengthening the presentation of this paper. This work was supported by grants from the National Science Foundation of China (NSFC http://www.nsfc.gov.cn/) under the contract numbers 31360207, 31370716, and 31400079, Guangxi Science and Technology Development Projects (no. 14123001-19, 1425008-2-22, and 15104001-1), Guangxi President Academic Foundation under number 16449-02, and Science foundation Project of Guangxi Academy of Sciences (No.15YJ22SW01). We thank the National Supper Computing Center (NSCC www.nscc-tj.gov.cn) in Tianjin for the valuable help in the modeling calculations using TH-1A super computer.
CONFLICTS OF INTEREST
The authors declare no conflicting interest.
REFERENCES
1. Chou KC, Wei DQ, Zhong WZ. Binding mechanism of coronavirus main proteinase with ligands and its implication to drug design against SARS. (Erratum: ibid., 2003; 310: 675). Biochem Biophys Res Commun. 2003; 308:148-151.
2. Jorgensen WL. The many roles of computation in drug discovery. Science. 2004; 303:1813-1818.
3. Schneider G, Fechner U. Computer-based de novo design of drug-like molecules. Nat Rev Drug Discov. 2005; 4:649-663.
4. Tollenaere JP. The role of structure-based ligand design and molecular modelling in drug discovery. Pharm World Sci. 1996; 18:56-62.
5. Chou KC. Structural bioinformatics and its impact to biomedical science. Curr Med Chem. 2004; 11:2105-2134.
6. Ferrara P, Gohlke H, Price DJ, Klebe G, Brooks CL 3rd. Assessing scoring functions for protein-ligand interactions. J Med Chem. 2004; 47:3032-3047.
7. Yu S, Gao S, Gan Y, Zhang Y, Ruan X, Wang Y, Yang L, Shi J. QSAR models for predicting octanol/water and organic carbon/water partition coefficients of polychlorinated biphenyls. SAR QSAR Environ Res. 2016; 27:249-263.
8. Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov. 2004; 3:935-949.
9. Greer J, Erickson JW, Baldwin JJ, Varney MD. Application of the three-dimensional structures of protein target molecules in structure-based drug design. J Med Chem. 1994; 37:1035-1054.
10. Lengauer T, Rarey M. Computational methods for biomolecular docking. Curr Opin Struct Biol. 1996; 6:402-406.
11. Meng EC, Shoichet BK, Kuntz ID. Automated docking with grid-based energy evaluation. JComput Chem. 1992; 13:505-524.
12. Du QS, Jiang ZQ, He WZ, Li DP. Amino acid principal component analysis (AAPCA) and its applications in protein structural class prediction. J Biomol Struct Dyn. 2006; 23:635-640.
13. Andrecut M. Parallel GPU implementation of iterative PCA algorithms. J Comput Biol. 2009; 16:1593-1599.
14. Warmuth MK, Kuzmin D. Randomized online PCA algorithms with regret bounds that are logarithmic in the dimension. J Mach Learn Res. 2008; 9:2287-2320.
15. Du Q, Mezey PG. Heuristic molecular lipophilicity potential (HMLP): a 2D-QSAR study to LADH of molecular family pyrazole and derivatives. J Comput Chem. 2005; 26:461-470.
16. Du QS, Li DP, He WZ. Heuristic molecular lipophilicity potential (HMLP): lipophilicity and hydrophilicity of amino acid side chains. J Comput Chem. 2006; 27:685-692.
17. Kawashima S, Pokarowski P, Pokarowska M, Kolinski A, Katayama T, Kanehisa M. AAindex: amino acid index database, progress report 2008. Nucleic Acids Res. 2008; 36:D202-D205.
18. Kawashima S, Ogata H, Kanehisa M. AAindex: amino acid index database. Nucleic Acids Res. 1999; 27:368-369.
19. Wang T, Yang J, Shen HB. Predicting membrane protein types by the LLDA algorithm. Protein Pept Lett. 2008; 15:915-921.
20. Ding H, Deng EZ, Yuan LF, Liu L, Lin H. iCTX-type: a sequence-based predictor for identifying the types of conotoxins in targeting ion channels. Biomed Res Int. 2014; 2014:286419.
21. Chou KC. Some remarks on protein attribute prediction and pseudo amino acid composition. J Theor Biol. 2011; 273:236-247.
22. Xu Y, Shao XJ, Wu LY, Deng NY. iSNO-AAPair: incorporating amino acid pairwise coupling into PseAAC for predicting cysteine S-nitrosylation sites in proteins. PeerJ. 2013; 1:e171.
23. Chen W, Feng PM, Lin H. iRSpot-PseDNC: identify recombination spots with pseudo dinucleotide composition. Nucleic Acids Res. 2013; 41:e68.
24. Lin H, Deng EZ, Ding H. iPro54-PseKNC: a sequence-based predictor for identifying sigma-54 promoters in prokaryote with pseudo k-tuple nucleotide composition. Nucleic Acids Res. 2014; 42:12961-12972.
25. Liu B, Fang L, Long R, Lan X. iEnhancer-2L: a two-layer predictor for identifying enhancers and their strength by pseudo k-tuple nucleotide composition. Bioinformatics. 2016; 32:362-369.
26. Jia J, Liu Z, Xiao X, Liu B. iSuc-PseOpt: identifying lysine succinylation sites in proteins by incorporating sequence-coupling effects into pseudo components and optimizing imbalanced training dataset. Anal Biochem. 2016; 497:48-56.
27. Liu Z, Xiao X, Yu DJ, Jia J, Qiu WR. pRNAm-PC: predicting N-methyladenosine sites in RNA sequences via physical-chemical properties. Anal Biochem. 2016; 497:60-67.
28. Jia J, Liu Z, Xiao X. pSuc-Lys: predict lysine succinylation sites in proteins with PseAAC and ensemble random forest approach. J Theor Biol. 2016; 394:223-230.
29. Meher PK, Sahu TK, Saini V, Rao AR. Predicting antimicrobial peptides with improved accuracy by incorporating the compositional, physico-chemical and structural features into Chou's general PseAAC. Sci Rep. 2017; 7:42362.
30. Cheng X, Zhao SG, Xiao X. iATC-mISF: a multi-label classifier for predicting the classes of anatomical therapeutic chemicals. Bioinformatics. 2017; 33:341-346.
31. Khan M, Hayat M, Khan SA, Iqbal N. Unb-DPC: identify mycobacterial membrane protein types by incorporating un-biased dipeptide composition into Chou's general PseAAC. J Theor Biol. 2017; 415:13-19.
32. Liu B, Wang S, Long R. iRSpot-EL: identify recombination spots with an ensemble learning approach. Bioinformatics. 2017; 33:35-41.
33. Rahimi M, Bakhtiarizadeh MR, Mohammadi-Sangcheshmeh A. OOgenesis_Pred: a sequence-based method for predicting oogenesis proteins by six different modes of Chou's pseudo amino acid composition. J Theor Biol. 2017; 414:128-136.
34. Cheng X, Zhao SG, Xiao X. iATC-mHyb: a hybrid multi-label classifier for predicting the classification of anatomical therapeutic chemicals. Oncotarget. 2017; 8:58494-58503. https://doi.org/10.18632/oncotarget.17028.
35. Qiu WR, Jiang SY, Xu ZC. iRNAm5C-PseDNC: identifying RNA 5-methylcytosine sites by incorporating physical-chemical properties into pseudo dinucleotide composition. Oncotarget. 2017; 8:41178-41188. https://doi.org/10.18632/oncotarget.17104.
36. Su Q, Lu W, Du D, Chen F, Niu B. Prediction of the aquatic toxicity of aromatic compounds to tetrahymena pyriformis through support vector regression. Oncotarget. 2017; 8:49359-49369. https://doi.org/10.18632/oncotarget.17210.
37. Liu B, Yang F. 2L-piRNA: a two-layer ensemble classifier for identifying piwi-interacting RNAs and their function. Mol Ther Nucleic Acids. 2017; 7:267-277.
38. Middleton D, Williams F, Hamill MA, Meenagh A. Frequency of HLA-B alleles in a Caucasoid population determined by a two-stage PCR-SSOP typing strategy. Hum Immunol. 2000; 61:1285-1297.
39. Grundschober C, Sanchez-Mazas A, Excoffier L, Langaney A, Jeannet M, Tiercy JM. HLA-DPB1 DNA polymorphism in the Swiss population: linkage disequilibrium with other HLA loci and population genetic affinities. Eur J Immunogenet. 1994; 21:143-157.
40. McMichael AJ, Parham P, Brodsky FM, Pilch JR. Influenza virus-specific cytotoxic T lymphocytes recognize HLA-molecules. Blocking by monoclonal anti-HLA antibodies. J Exp Med. 1980; 152:195s-203s.
41. Schendel DJ, Gansbacher B, Oberneder R, Kriegmair M, Hofstetter A, Riethmuller G, Segurado OG. Tumor-specific lysis of human renal cell carcinomas by tumor-infiltrating lymphocytes. I. HLA-A2-restricted recognition of autologous and allogeneic tumor lines. J Immunol. 1993; 151:4209-4220.
42. Rivoltini L, Kawakami Y, Sakaguchi K, Southwood S, Sette A, Robbins PF, Marincola FM, Salgaller ML, Yannelli JR, Appella E. Induction of tumor-reactive CTL from peripheral blood and tumor-infiltrating lymphocytes of melanoma patients by in vitro stimulation with an immunodominant peptide of the human melanoma antigen MART-1. J Immunol. 1995; 154:2257-2265.
43. Rongcun Y, Salazar-Onfray F, Charo J, Malmberg KJ, Evrin K, Maes H, Kono K, Hising C, Petersson M, Larsson O, Lan L, Appella E, Sette A, et al. Identification of new HER2/neu-derived peptide epitopes that can elicit specific CTL against autologous and allogeneic carcinomas and melanomas. J Immunol. 1999; 163:1037-1044.
44. Parkhurst MR, Fitzgerald EB, Southwood S, Sette A, Rosenberg SA, Kawakami Y. Identification of a shared HLA-A*0201-restricted T-cell epitope from the melanoma antigen tyrosinase-related protein 2 (TRP2). Cancer Res. 1998; 58:4895-4901.
45. Peoples GE, Goedegebuure PS, Smith R, Linehan DC, Yoshino I, Eberlein TJ. Breast and ovarian cancer-specific cytotoxic T lymphocytes recognize the same HER2/neu-derived peptide. Proc Natl Acad Sci U S A. 1995; 92:432-436.
46. Chou PY, Fasman GD. Conformational parameters for amino acids in helical, beta-sheet, and random coil regions calculated from proteins. Biochemistry. 1974; 13:211-222.
47. Nicolau DV Jr, Paszek E, Fulga F, Nicolau DV. Mapping hydrophobicity on the protein molecular surface at atom-level resolution. PLoS One. 2014; 9:e114042.
48. Zhang CT. Prediction of protein structural classes. Crit Rev Biochem Mol Biol. 1995; 30:275-349.
49. Doytchinova IA, Flower DR. Toward the quantitative prediction of T-cell epitopes: coMFA and coMSIA studies of peptides with affinity for the class I MHC molecule HLA-A*0201. J Med Chem. 2001; 44:3572-3581.
50. del Guercio MF, Sidney J, Hermanson G, Perez C, Grey HM, Kubo RT, Sette A. Binding of a peptide antigen to multiple HLA alleles allows definition of an A2-like supertype. J Immunol. 1995; 154:685-693.
51. Tsai V, Southwood S, Sidney J, Sakaguchi K, Kawakami Y, Appella E, Sette A, Celis E. Identification of subdominant CTL epitopes of the GP100 melanoma-associated tumor antigen by primary in vitro immunization with peptide-pulsed dendritic cells. J Immunol. 1997; 158:1796-1802.
52. Vitiello A, Sette A, Yuan L, Farness P, Southwood S, Sidney J, Chesnut RW, Grey HM, Livingston B. Comparison of cytotoxic T lymphocyte responses induced by peptide or DNA immunization: implications on immunogenicity and immunodominance. Eur J Immunol. 1997; 27:671-678.
53. Kawakami Y, Eliyahu S, Jennings C, Sakaguchi K, Kang X, Southwood S, Robbins PF, Sette A, Appella E, Rosenberg SA. Recognition of multiple epitopes in the human melanoma antigen gp100 by tumor-infiltrating T lymphocytes associated with in vivo tumor regression. J Immunol. 1995; 154:3961-3968.
54. Sette A, Sidney J, del Guercio MF, Southwood S, Ruppert J, Dahlberg C, Grey HM, Kubo RT. Peptide binding to the most frequent HLA-A class I alleles measured by quantitative molecular binding assays. Mol Immunol. 1994; 31:813-822.
55. Sette A, Vitiello A, Reherman B, Fowler P, Nayersina R, Kast WM, Melief CJ, Oseroff C, Yuan L, Ruppert J, Sidney J, del Guercio MF, Southwood S, et al. The relationship between class I binding affinity and immunogenicity of potential cytotoxic T cell epitopes. J Immunol. 1994; 153:5586-5592.
56. Ruppert J, Sidney J, Celis E, Kubo RT, Grey HM, Sette A. Prominent role of secondary anchor residues in peptide binding to HLA-A2.1 molecules. Cell. 1993; 74:929-937.
57. Lin HH, Ray S, Tongchusak S, Reinherz EL, Brusic V. Evaluation of MHC class I peptide binding prediction servers: applications for vaccine research. BMC Immunol. 2008; 9:8.
58. Cramer RD, Patterson DE, Bunce JD. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J Am Chem Soc. 1988; 110:5959-5967.
59. Zhao X, Chen M, Huang B, Ji H, Yuan M. Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA) studies on α(1A)-adrenergic receptor antagonists based on pharmacophore molecular alignment. Int J Mol Sci. 2011; 12:7022-7037.
60. Klebe G, Abraham U. Comparative molecular similarity index analysis (CoMSIA) to study hydrogen-bonding properties and to score combinatorial libraries. J Comput Aided Mol Des. 1999; 13:1-10.
61. Klebe G, Abraham U, Mietzner T. Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J Med Chem. 1994; 37:4130-4146.
62. Chou KC, Shen HB. Recent progresses in protein subcellular location prediction. Anal Biochem. 2007; 370:1-16.
63. Shen HB. Cell-PLoc 2.0: an improved package of web-servers for predicting subcellular localization of proteins in various organisms. Nat Sci. 2010; 2:1090-1103.
64. Chou KC. Prediction of protein cellular attributes using pseudo amino acid composition. Proteins. (Erratum: ibid, 2001; 44: 60). 2001; 43:246-255.
65. Zhong WZ, Zhou SF. Molecular science for drug development and biomedicine. Int J Mol Sci. 2014; 15:20072-20078.
66. Chou KC. An unprecedented revolution in medicinal chemistry driven by the progress of biological science. Curr Top Med Chem. 2017; 17:2337-2358.
67. Chou KC. Pseudo amino acid composition and its applications in bioinformatics, proteomics and system biology. Curr Proteomics. 2009; 6: 262-274.
68. Chen W, Lin H. Pseudo nucleotide composition or PseKNC: an effective formulation for analyzing genomic sequences. Mol Biosyst. 2015; 11:2620-2634.
69. Liu B, Wu H. Pse-in-One 2.0: an improved package of web servers for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nat Sci. 2017; 9:67-91.
70. Liu B, Liu F, Wang X, Chen J. Pse-in-One: a web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res. 2015; 43:W65-W71.
71. Zhang CT. An eigenvalue-eigenvector approach to predicting protein folding types. J Protein Chem. 1995; 14:309-326.
72. Golub GH, van der Vorst HA. Eigenvalue computation in the 20th century. J Comput Appl Math. 2000; 123:35-65.
73. Diekmann O, Heesterbeek JA, Metz JA. On the definition and the computation of the basic reproduction ratio R0 in models for infectious diseases in heterogeneous populations. J Math Biol. 1990; 28:365-382.
74. Du QS, Huang RB, Wei YT, Du LQ. Multiple field three dimensional quantitative structure-activity relationship (MF-3D-QSAR). J Comput Chem. 2008; 29:211-219.
75. Du QS, Huang RB, Wei YT, Pang ZW. Fragment-based quantitative structure-activity relationship (FB-QSAR) for fragment-based drug design. J Comput Chem. 2009; 30:295-304.
76. Shen HB. Recent advances in developing web-servers for predicting protein attributes. Nat Sci. 2009; 1:63-92.
77. Jia J, Liu Z, Xiao X, Liu B. iCar-PseCp: identify carbonylation sites in proteins by Monto Carlo sampling and incorporating sequence coupled effects into general PseAAC. Oncotarget. 2016; 7:34558-34570. https://doi.org/10.18632/oncotarget.9148.
78. Qiu WR, Sun BQ, Xu ZC. iHyd-PseCp: identify hydroxyproline and hydroxylysine in proteins by incorporating sequence-coupled effects into general PseAAC. Oncotarget. 2016; 7:44310-44321. https://doi.org/10.18632/oncotarget.10027.
79. Zhang CJ, Tang H, Li WC, Lin H. iOri-Human: identify human origin of replication by incorporating dinucleotide physicochemical properties into pseudo nucleotide composition. Oncotarget. 2016; 7:69783-69793. https://doi.org/10.18632/oncotarget.11975.
80. Chen W, Feng P, Yang H, Ding H. iRNA-AI: identifying the adenosine to inosine editing sites in RNA sequences. Oncotarget. 2017; 8:4208-4217. https://doi.org/10.18632/oncotarget.13758.
81. Liu B, Wu H, Zhang D, Wang X. Pse-Analysis: a python package for DNA/RNA and protein/peptide sequence analysis based on pseudo components and kernel methods. Oncotarget. 2017; 8:13338-13343. https://doi.org/10.18632/oncotarget.14524.
82. Chen W, Ding H, Feng P. iACP: a sequence-based tool for identifying anticancer peptides. Oncotarget. 2016; 7:16895-16909. https://doi.org/10.18632/oncotarget.7815.
83. Qiu WR, Xiao X, Xu ZH. iPhos-PseEn: identifying phosphorylation sites in proteins by fusing different pseudo components into an ensemble classifier. Oncotarget. 2016; 7:51270-51283. https://doi.org/10.18632/oncotarget.9987.
84. Xiao X, Ye HX, Liu Z, Jia JH. iROS-gPseKNC: predicting replication origin sites in DNA by incorporating dinucleotide position-specific propensity into general pseudo nucleotide composition. Oncotarget. 2016; 7:34180-34189. https://doi.org/10.18632/oncotarget.9057.
85. Liu LM, Xu Y. iPGK-PseAAC: identify lysine phosphoglycerylation sites in proteins by incorporating four different tiers of amino acid pairwise coupling information into the general PseAAC. Med Chem. 2017. https://doi.org/10.2174/1573406413666170515120507.
86. Jia J, Zhang L, Liu Z. pSumo-CD: predicting sumoylation sites in proteins with covariance discriminant algorithm by incorporating sequence-coupled effects into general PseAAC. Bioinformatics. 2016; 32:3133-3141.
87. Liu B, Long R. iDHS-EL: identifying DNase I hypersensitive sites by fusing three different modes of pseudo nucleotide composition into an en-semble learning framework. Bioinformatics. 2016; 32:2411-2418.
88. Qiu WR, Sun BQ, Xiao X. iPTM-mLys: identifying multiple lysine PTM sites and their different types. Bioinformatics. 2016; 32:3116-3123.
89. Xu Y, Li C. iPreny-PseAAC: identify C-terminal cysteine prenylation sites in proteins by incorporating two tiers of sequence couplings into PseAAC. Med Chem. 2017. https://doi.org/10.2174/1573406413666170419150052.
90. Qiu WR, Jiang SY, Sun BQ. iRNA-2methyl: identify RNA 2’-O-methylation sites by incorporating sequence-coupled effects into general PseKNC and ensemble classifier. Med Chem. 2017. https://doi.org/10.2174/1573406413666170623082245.
91. Cheng X, Xiao X, Chou KC. pLoc-mPlant: predict subcellular localization of multi-location plant proteins via incorporating the optimal GO information into general PseAAC. Mol Biosyst. 2017. https://doi.org/10.1039/c7mb00267J.
92. Chou KC. Impacts of bioinformatics to medicinal chemistry. Med Chem. 2015; 11:218-234.