
Role of epigenetic regulators in breast cancer
Breast cancer is one of the most prevalent tumors and a leading cause of death in women worldwide [1]. Although early detection and adjuvant treatment have improved survival in early stage breast cancer, disseminated cancer is still an incurable disease where novel therapies have shown limited efficacy [2]. In addition, the fact that most breast cancer patients progress after a given period of treatment reinforce the necessity for the identification of novel therapeutics that can contribute to improve patient outcome.
Breast cancer has been classified in several different subtypes based on molecular and genomic findings [2]. Around 20% of tumors overexpress the transmembrane protein HER2 due to gene amplification [3, 4]. These tumors benefit from therapies against this protein, including monoclonal antibodies like trastuzumab or pertuzumab, or small tyrosine kinase inhibitors like lapatinib or neratinib [5, 6]. Recently targeting tumoral cells with an antibody drug conjugate (ADC) against HER2 has also shown efficacy in the metastatic setting [7]. Expression of hormone receptors (HR), including estrogen and progesterone receptors, is observed in 80% of tumors and strategies to block these receptors or decrease the presence of activating hormones have been used with successful results for a long time [8]. Novel agents designed to block the estrogen receptor have also reached the clinical setting [8, 9]. Finally, breast cancer has been classified by gene expression analyses in several subtypes that mimic the histological classification in some aspects. The HER2 enriched, the luminal and the basal-like subtype mimic tumors overexpressing HER2, HR positive and the triple negative subtype, respectively [8, 10].
Epigenetic regulators are promising therapeutic targets in hematologic malignancies and solid tumors [11]. For instance, deacetylase inhibitors have shown activity in the clinical setting in some hematological diseases [12]. Deregulation in chromatin modulators and transcription factors (TFs) are common findings in cancer [11, 13, 14]. The bromodomain and extra-terminal (BET) family of proteins is involved in the recruitment of proteins of the transcription machinery to regulate gene expression [13, 15]. To do so, they recognize acetylated-lysine residues in nucleosomal histones [13, 14]. An example is the recruitment of the positive transcription elongation factor (p-TEFb) [16]. There are around 46 bromodomain-containing proteins of which the BET family in mammals is composed of four members: BRD2, BRD3, BRD4 and the testis-specific BRDT. They contain two conserved N-terminal bromodomains (BD1 and BD2), which recognize and bind acetylated lysine residues on histone tails [17] (Figure 1). Inhibition of these proteins has shown antitumoral efficacy in a wide range of solid tumors including breast cancer [11, 18]. It should be mention, that the inhibition of each individual component can have a different effect that the inhibition of all members and sometimes one member can have an opposite function [19]. On the other hand, most available BET inhibitors can inhibit all members although there are currently under development inhibitors of specific members, like MZ1 that induces selective removal of BRD4 [20].
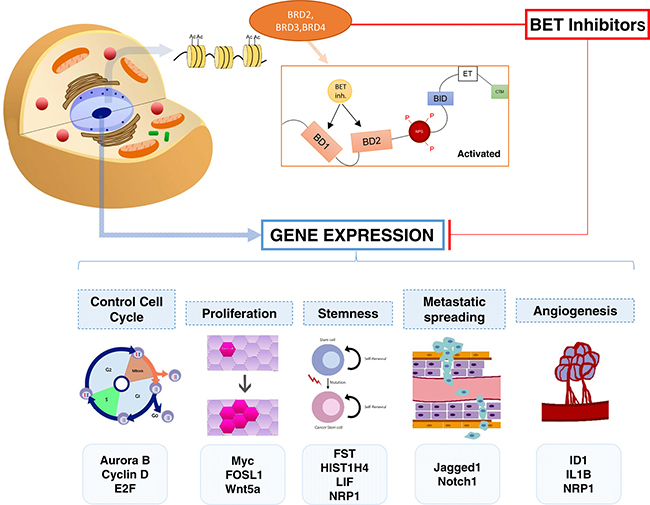
Figure 1: Representation of the mechanisms of action of BET inhibitors and the biological functions inhibited by these compounds. The acetylation of lysine residues plays a major role in the regulation of chromatin and the control of its transcription. The extra terminal bromodomains are the main readers of the acetylated lysines producing the transcription of genes related to multiple functions like cell cycle, proliferation, stemness, metastatic spreading and angiogenesis. BET inhibitors suppress the transcription of genes including Aurora B, Cyclin D, E2F, MYC, FOSL1 and Wnt5a, among others.
Antitumor activity and mechanism of action of BET inhibitors in breast cancer
The efficacy of BET inhibitors was mainly explored in hematologic malignancies and pediatric cancers where well known transcription factors play a key role in the oncogenesis of the disease, as is the case of C-MYC in neuroblastomas or leukemia [18, 21]. Later on, the significant antitumor activity of these compounds in other solid tumors like prostate, non-small cell lung cancer or pancreatic cancer was evaluated and reported [22–24]. Indeed, the efficacy of these agents can be more relevant in biological situations like in cancer where chromatin is in a constitutively open situation available for gene transcription.
BET inhibitors have shown activity in different breast cancer subtypes including HER2 positive tumors, HR tumors and triple negative breast cancer (TNBC). For a comprehensive review of BET inhibitors in clinical development we suggest to read the article by Andrieu G et al. [25].
BRD4 activity is necessary for the proliferation of breast and endometrial tumors expressing estrogen receptors as it regulates ERα-induced gene expression by affecting elongation-associated phosphorylation of RNA polymerase II (RNAPII) and histone H2B monoubiquitination [26]. Indeed BET inhibition with JQ1 impaired E2-induced transcriptional activation [27].
In HER2 and HR positive tumors, BET inhibitors have contributed to overcome resistance to currently approved therapies like lapatinib in HER2 positive tumors, or tamoxifen and everolimus in HR positive cancers, among other therapies [28–30]. These data will be described in detailed in the next section of this review.
Given the fact that BET inhibitors can repress the expression of several transcription factors, the mechanism of action of these compounds is heterogeneous affecting several biological functions including: control of cell cycle, proliferation, stemness, metastatic spreading or angiogenesis, among others [31–34]. Of note doses used in some of the studies were extremely high leading to conclusions that have no relation with the biological mechanism of action [29]. Therefore this data cannot be considered as a good example of how BET inhibitors suppress certain transcription factors and cause arrest of the cell cycle, unless the goal is to generate chemoresistance clones.
Several studies have described the function of Bromodomains (BRD) and particularly BRD2 and BRD4, in cell cycle regulation [35]. Indeed, experience from our group and others shows that treatment with BET inhibitors affects proliferation by inducing cell cycle arrest at G1 with an increase of p27 [33, 34, 36]. Inhibition of proliferation is likely mediated by a reduction of genes involved in cell division [28] and particularly of those associated with the mitotic process like Aurora kinases A and B, Polo-like kinases (PLKs) or TTK protein kinases [33, 36].
Inhibitors of BET proteins reduce cell migration and invasion, and recent data suggests that this mechanism is mediated by inhibition of the Jagged1/Notch1 signaling pathway through modulation of BRD4 [31]. In addition, BET inhibition reduces angiogenesis in xenografted tumors by targeting the hypoxic response [32]. BET inhibition also produce morphologic changes inducing a stem cell phenotype [31, 34, 37]. Interestingly the mechanism of action of BET inhibitors in TNBC is not mainly mediated by the inhibition of MYC or by affecting known pathways linked with this disease like the JAK/STAT pathway [34, 36]. By contrast, other transcription factors affected by the compound have been described like LM04, DEPDC or FOXM1 [33].
Finally, several mechanisms have been reported in relation to resistance to BET inhibitors. The activation of casein kinase II (CK2) has been associated with resistance to BET inhibitors and agents targeting this kinase have shown to overcome such resistance [23]. Similarly, the decrease of the PP2A phosphatase that opposes the CK2 function has been linked with resistance to BET inhibitors [24]. In addition, inhibition of BCL-X and BCL-2, revert the resistance to BET inhibitors [34]. Confirming these findings, novel BET inhibitors with a more potent mechanism of action have shown to be synergistic with agents that induce apoptosis [38]. Of note this has also been observed in other tumors beyond breast cancer, like in T-cell acute lymphoblastic leukemia [39]. Figure 1 summarizes the mechanism of action of BET inhibitors.
BET inhibitors can overcome resistance to breast cancer therapies
If resistance to a given therapy is mediated by upregulation of specific transcription factors, BET inhibitors can overcome resistance to this specific therapy by reducing their expression. For instance, resistance to everolimus in estrogen receptor positive tumors was mediated by the expression of MYC, and reverted by treatment with BET inhibitors that downregulated MYC expression [29]. Other studies have shown activity of BET inhibitors in HR positive tumors that are resistant to tamoxifen or increasing the effect of the ER degrader fulvestrant [28]. In addition, JQ1 increases the efficacy of everolimus in MCF7 xenograted models [29].
Similarly BET inhibitors can overcome the resistance to PI3K inhibitors [40]. In addition, OTX015, a novel inhibitor, increases the activity of everolimus in some triple negative breast cancer cells [41].
In HER2 positive breast cancer, BET inhibitors have been shown to overcome resistance to lapatinib [30]. Resistance to the anti-HER2 small tyrosine kinase inhibitor lapatinib was mediated by reprograming of the kinome and activation of ErbB2/ErbB3 receptors [30]. BET inhibition reduced the transcription of some of the genes involved in this pathway like ERBB3, IGF1R, DDR1 or MET, among others [30].
Potential combinations, biomarker discovery and future clinical development
Preclinical data from our group showed that BET inhibitors synergized with chemotherapies used for the treatment of breast cancer including antimitotic agents like taxanes or vinorelbine and DNA damaging compounds like cisplatin and carboplatin [33]. In addition, the combination produced a remarkable induction of apoptosis compared to treatment with each agent given alone [33]. BET inhibitors synergized with agents targeting key kinases involved in the mitotic spindle formation like polo-like kinase 1 [37]. These data opened options for the future clinical development of BET inhibitors in triple negative breast cancer in combination with chemotherapies or PLK1 inhibitors [37].
Similarly BET inhibitors have shown a synergistic interaction with inducers of apoptosis, like BCL-X and BCL-2 inhibitors or with CK2 inhibitors, through the control or relevant phosphatases [34, 38].
Combination of BET inhibitors with immunotherapies like checkpoint inhibitors is an attractive approach. Indeed some articles have suggested that PD-L1 expression is suppressed by treatment with JQ1, and this could be the bases to explore this regimen in several tumors [42].
Also a potent inhibitory effect has been observed with the combination of BET inhibitors and HDAC inhibitors in triple negative and estrogen receptor positive breast cancer cells, mainly through an induction of the ubiquitin-specific protease (USP17) [43]. And finally, the BRD family of proteins has been involved in several mechanisms associated with resistance to cell signaling inhibitors by avoiding the activation of complementary signaling pathways [44]. For instance, inhibition of BRD4 overcomes resistance to MEK inhibitors in triple negative breast cancer [45].
Although not yet explored in breast cancer, in other tumor types like in leukemia the presence of stem cells is associated with resistance to BET inhibitors through an increase of the Wnt/β-catenin pathway [46]. In addition, activation of the AMPK pathway is associated with JQ1-induced autophagy in resistant cells. AMPK promoted JQ1-induced apoptosis in acute myeloide Leukemia stem cells [47].
Table 1 shows the list of BET inhibitors in clinical development. Of note two of them OTX015 and MK-8628 are under evaluation in triple negative breast cancer. Indeed OTX015 has shown a good tolerability profile, with mild side effects like hematologic toxicities, diarrhoea or fatigue, in recently published phase I studies in lymphoma and acute leukemia [48, 49].
Table 1: BET inhibitors in clinical development including indication and clinical stage
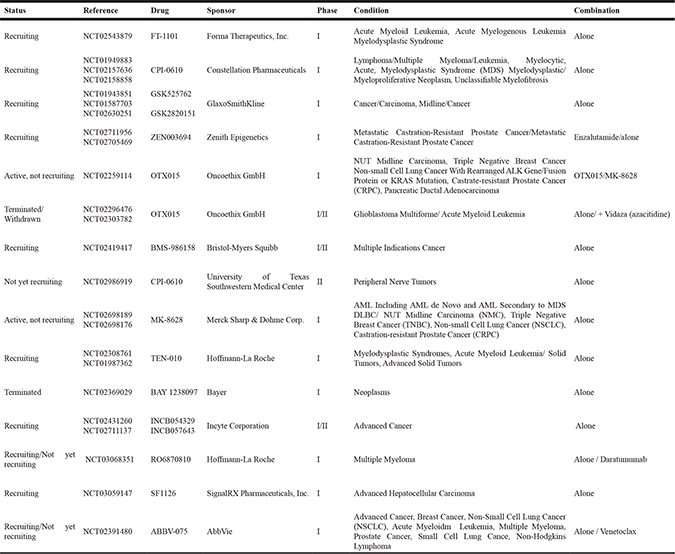
When observing the clinical development of these compounds, it is interesting to see that there are multiple indications and not clear biological rationale for the development of BET inhibitors in some of them, what suggests that this family of compounds are developed as a tolerated but nonselective molecule.
Strategies to augment the efficacy of these compounds include the designing of more potent agents or the identification of potential biomarkers of response. To increase the efficacy an approach is the development of bivalent BET inhibitors that are 100-fold more potent than monovalent antagonist [50]. To identify biomarkers of efficacy, a recent paper suggests potential genes like HEXIM1, SERPINI1, ZCCHC24 and ZMYND8 that could predict activity in a broader range of tumors [51]. However, these data needs to be confirmed in the clinical setting.
CONCLUSIONS
In summary, BET inhibitors have demonstrated preclinical activity in TNBC alone or in combination, and in some specific breast cancer scenarios mainly associated with resistance to existing therapies. Their effect goes beyond the regulation of oncogenes as they can act on a variety of cellular programs. The identification of patients whose tumors may benefit from these therapies together with studies to optimize the antitumoral properties of these compounds, represent important future challenges.
CONFLICTS OF INTEREST
No conflicts of interest to declare.
FUNDING
Instituto de Salud Carlos III (PI13/01444), ACEPAIN; Diputación de Albacete and CRIS Cancer Foundation (to AO). BAE (Beca Ampliación de Estudios) to AO for his stay at Yale University, CT, USA. Ministry of Economy and Competitiveness of Spain (BFU2015-71371-R), the Instituto de Salud Carlos III through the Spanish Cancer Centers Network Program (RD12/0036/0003) and CIBERONC, the scientific foundation of the AECC and the CRIS Foundation (to AP). The work carried out in our laboratories receive support from the European Community through the regional development funding program (FEDER).
REFERENCES
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016; 66:7–30. https://doi.org/10.3322/caac.21332.
2. Ocana A, Pandiella A. Targeting oncogenic vulnerabilities in triple negative breast cancer: biological bases and ongoing clinical studies. Oncotarget. 2017; 8:22218–34. https://doi.org/10.18632/oncotarget.14731.
3. Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987; 235:177–82. https://doi.org/10.1126/science.3798106.
4. Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, Levin WJ, Stuart SG, Udove J, Ullrich A. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989; 244:707–12. https://doi.org/10.1126/science.2470152.
5. Esparís-Ogando A, Montero JC, Arribas J, Ocaña A, Pandiella A. Targeting the EGF/HER Ligand-Receptor System in Cancer. Curr Pharm Des. 2016; 22:5887–98. https://doi.org/10.2174/1381612822666160715132233.
6. Chan A, Delaloge S, Holmes FA, Moy B, Iwata H, Harvey VJ, Robert NJ, Silovski T, Gokmen E, von Minckwitz G, Ejlertsen B, Chia SK, Mansi J, et al, and ExteNET Study Group. Neratinib after trastuzumab-based adjuvant therapy in patients with HER2-positive breast cancer (ExteNET): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2016; 17:367–77. https://doi.org/10.1016/S1470-2045(15)00551-3.
7. Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, Pegram M, Oh DY, Diéras V, Guardino E, Fang L, Lu MW, Olsen S, Blackwell K, EMILIA Study Group. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012; 367:1783–91. https://doi.org/10.1056/NEJMoa1209124.
8. Zelnak AB, O’Regan RM. Optimizing Endocrine Therapy for Breast Cancer. J Natl Compr Canc Netw. 2015; 13:e56–64. https://doi.org/10.6004/jnccn.2015.0125.
9. Robertson JF, Bondarenko IM, Trishkina E, Dvorkin M, Panasci L, Manikhas A, Shparyk Y, Cardona-Huerta S, Cheung KL, Philco-Salas MJ, Ruiz-Borrego M, Shao Z, Noguchi S, et al. Fulvestrant 500 mg versus anastrozole 1 mg for hormone receptor-positive advanced breast cancer (FALCON): an international, randomised, double-blind, phase 3 trial. Lancet. 2016; 388:2997–3005. https://doi.org/10.1016/S0140-6736(16)32389-3.
10. Sorlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, Deng S, Johnsen H, Pesich R, Geisler S, Demeter J, Perou CM, Lønning PE, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci USA. 2003; 100:8418–23. https://doi.org/10.1073/pnas.0932692100.
11. Kumar R, Li DQ, Müller S, Knapp S. Epigenomic regulation of oncogenesis by chromatin remodeling. Oncogene. 2016; 35:4423–36. https://doi.org/10.1038/onc.2015.513.
12. San-Miguel JF, Hungria VT, Yoon SS, Beksac M, Dimopoulos MA, Elghandour A, Jedrzejczak WW, Günther A, Nakorn TN, Siritanaratkul N, Corradini P, Chuncharunee S, Lee JJ, et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. 2014; 15:1195–206. https://doi.org/10.1016/S1470-2045(14)70440-1.
13. Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov. 2014; 13:337–56. https://doi.org/10.1038/nrd4286.
14. Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, et al. Selective inhibition of BET bromodomains. Nature. 2010; 468:1067–73. https://doi.org/10.1038/nature09504.
15. Wu SY, Chiang CM. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J Biol Chem. 2007; 282:13141–45. https://doi.org/10.1074/jbc.R700001200.
16. Yang Z, Yik JH, Chen R, He N, Jang MK, Ozato K, Zhou Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell. 2005; 19:535–45. https://doi.org/10.1016/j.molcel.2005.06.029.
17. Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, Zhou MM. Structure and ligand of a histone acetyltransferase bromodomain. Nature. 1999; 399:491–96. https://doi.org/10.1038/20974.
18. Sahai V, Redig AJ, Collier KA, Eckerdt FD, Munshi HG. Targeting BET bromodomain proteins in solid tumors. Oncotarget. 2016; 7:53997–4009. https://doi.org/10.18632/oncotarget.9804.
19. Deeney JT, Belkina AC, Shirihai OS, Corkey BE, Denis GV. BET Bromodomain Proteins Brd2, Brd3 and Brd4 Selectively Regulate Metabolic Pathways in the Pancreatic β-Cell. PLoS One. 2016; 11:e0151329. https://doi.org/10.1371/journal.pone.0151329.
20. Zengerle M, Chan KH, Ciulli A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem Biol. 2015; 10:1770–77. https://doi.org/10.1021/acschembio.5b00216.
21. Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, Taylor MJ, Johns C, Chicas A, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011; 478:524–28. https://doi.org/10.1038/nature10334.
22. Garcia PL, Miller AL, Kreitzburg KM, Council LN, Gamblin TL, Christein JD, Heslin MJ, Arnoletti JP, Richardson JH, Chen D, Hanna CA, Cramer SL, Yang ES, et al. The BET bromodomain inhibitor JQ1 suppresses growth of pancreatic ductal adenocarcinoma in patient-derived xenograft models. Oncogene. 2016; 35:833–45. https://doi.org/10.1038/onc.2015.126.
23. Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, Escara-Wilke J, Wilder-Romans K, Dhanireddy S, Engelke C, Iyer MK, Jing X, Wu YM, et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature. 2014; 510:278–82. https://doi.org/10.1038/nature13229.
24. Shimamura T, Chen Z, Soucheray M, Carretero J, Kikuchi E, Tchaicha JH, Gao Y, Cheng KA, Cohoon TJ, Qi J, Akbay E, Kimmelman AC, Kung AL, et al. Efficacy of BET bromodomain inhibition in Kras-mutant non-small cell lung cancer. Clin Cancer Res. 2013; 19:6183–92. https://doi.org/10.1158/1078-0432.CCR-12-3904.
25. Andrieu G, Belkina AC, Denis GV. Clinical trials for BET inhibitors run ahead of the science. Drug Discov Today Technol. 2016; 19:45–50. https://doi.org/10.1016/j.ddtec.2016.06.004.
26. Nagarajan S, Hossan T, Alawi M, Najafova Z, Indenbirken D, Bedi U, Taipaleenmäki H, Ben-Batalla I, Scheller M, Loges S, Knapp S, Hesse E, Chiang CM, et al. Bromodomain protein BRD4 is required for estrogen receptor-dependent enhancer activation and gene transcription. Cell Reports. 2014; 8:460–69. https://doi.org/10.1016/j.celrep.2014.06.016.
27. Sengupta S, Biarnes MC, Clarke R, Jordan VC. Inhibition of BET proteins impairs estrogen-mediated growth and transcription in breast cancers by pausing RNA polymerase advancement. Breast Cancer Res Treat. 2015; 150:265–78. https://doi.org/10.1007/s10549-015-3319-1.
28. Feng Q, Zhang Z, Shea MJ, Creighton CJ, Coarfa C, Hilsenbeck SG, Lanz R, He B, Wang L, Fu X, Nardone A, Song Y, Bradner J, et al. An epigenomic approach to therapy for tamoxifen-resistant breast cancer. Cell Res. 2014; 24:809–19. https://doi.org/10.1038/cr.2014.71.
29. Bihani T, Ezell SA, Ladd B, Grosskurth SE, Mazzola AM, Pietras M, Reimer C, Zinda M, Fawell S, D’Cruz CM. Resistance to everolimus driven by epigenetic regulation of MYC in ER+ breast cancers. Oncotarget. 2015; 6:2407–20. https://doi.org/10.18632/oncotarget.2964.
30. Stuhlmiller TJ, Miller SM, Zawistowski JS, Nakamura K, Beltran AS, Duncan JS, Angus SP, Collins KA, Granger DA, Reuther RA, Graves LM, Gomez SM, Kuan PF, et al. Inhibition of Lapatinib-Induced Kinome Reprogramming in ERBB2-Positive Breast Cancer by Targeting BET Family Bromodomains. Cell Reports. 2015; 11:390–404. https://doi.org/10.1016/j.celrep.2015.03.037.
31. Andrieu G, Tran AH, Strissel KJ, Denis GV. BRD4 Regulates Breast Cancer Dissemination through Jagged1/Notch1 Signaling. Cancer Res. 2016; 76:6555–67. https://doi.org/10.1158/0008-5472.CAN-16-0559.
32. da Motta LL, Ledaki I, Purshouse K, Haider S, De Bastiani MA, Baban D, Morotti M, Steers G, Wigfield S, Bridges E, Li JL, Knapp S, Ebner D, et al. The BET inhibitor JQ1 selectively impairs tumour response to hypoxia and downregulates CA9 and angiogenesis in triple negative breast cancer. Oncogene. 2017; 36:122–32. https://doi.org/10.1038/onc.2016.184.
33. Pérez-Peña J, Serrano-Heras G, Montero JC, Corrales-Sánchez V, Pandiella A, Ocaña A. In Silico Analysis Guides Selection of BET Inhibitors for Triple-Negative Breast Cancer Treatment. Mol Cancer Ther. 2016; 15:1823–33. https://doi.org/10.1158/1535-7163.MCT-16-0004.
34. Shu S, Lin CY, He HH, Witwicki RM, Tabassum DP, Roberts JM, Janiszewska M, Huh SJ, Liang Y, Ryan J, Doherty E, Mohammed H, Guo H, et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature. 2016; 529:413–17. https://doi.org/10.1038/nature16508.
35. Maruyama T, Farina A, Dey A, Cheong J, Bermudez VP, Tamura T, Sciortino S, Shuman J, Hurwitz J, Ozato K. A Mammalian bromodomain protein, brd4, interacts with replication factor C and inhibits progression to S phase. Mol Cell Biol. 2002; 22:6509–20. https://doi.org/10.1128/MCB.22.18.6509-6520.2002.
36. Sahni JM, Gayle SS, Bonk KL, Vite LC, Yori JL, Webb B, Ramos EK, Seachrist DD, Landis MD, Chang JC, Bradner JE, Keri RA. Bromodomain and Extraterminal Protein Inhibition Blocks Growth of Triple-negative Breast Cancers through the Suppression of Aurora Kinases. J Biol Chem. 2016; 291:23756–68. https://doi.org/10.1074/jbc.M116.738666.
37. Nieto-Jiménez C, Alcaraz-Sanabria A, Pérez-Peña J, Corrales-Sánchez V, Serrano-Heras G, Galán-Moya EM, Serrano-Oviedo L, Montero JC, Burgos M, Llopis J, Pandiella A, Ocaña A. Targeting basal-like breast tumors with bromodomain and extraterminal domain (BET) and polo-like kinase inhibitors. Oncotarget. 2017; 8:19478–90. 10.18632/oncotarget.14465.
38. Bai L, Zhou B, Yang CY, Ji J, McEachern D, Przybranowski S, Jiang H, Hu J, Xu F, Zhao Y, Liu L, Fernandez-Salas E, Xu J, et al. Targeted degradation of BET proteins in triple-negative breast cancer. Cancer Res. 2017; 77:2476–87. https://doi.org/10.1158/0008-5472.CAN-16-2622.
39. Peirs S, Frismantas V, Matthijssens F, Van Loocke W, Pieters T, Vandamme N, Lintermans B, Dobay MP, Berx G, Poppe B, Goossens S, Bornhauser BC, Bourquin JP, Van Vlierberghe P. Targeting BET proteins improves the therapeutic efficacy of BCL-2 inhibition in T-cell acute lymphoblastic leukemia. Leukemia. 2017; Epub ahead of print. https://doi.org/10.1038/leu.2017.10.
40. Stratikopoulos EE, Dendy M, Szabolcs M, Khaykin AJ, Lefebvre C, Zhou MM, Parsons R. Kinase and BET Inhibitors Together Clamp Inhibition of PI3K Signaling and Overcome Resistance to Therapy. Cancer Cell. 2015; 27:837–51. https://doi.org/10.1016/j.ccell.2015.05.006.
41. Vázquez R, Riveiro ME, Astorgues-Xerri L, Odore E, Rezai K, Erba E, Panini N, Rinaldi A, Kwee I, Beltrame L, Bekradda M, Cvitkovic E, Bertoni F, et al. The bromodomain inhibitor OTX015 (MK-8628) exerts anti-tumor activity in triple-negative breast cancer models as single agent and in combination with everolimus. Oncotarget. 2017; 8:7598–613. 10.18632/oncotarget.13814.
42. Zhu H, Bengsch F, Svoronos N, Rutkowski MR, Bitler BG, Allegrezza MJ, Yokoyama Y, Kossenkov AV, Bradner JE, Conejo-Garcia JR, Zhang R. BET Bromodomain Inhibition Promotes Anti-tumor Immunity by Suppressing PD-L1 Expression. Cell Reports. 2016; 16:2829–37. https://doi.org/10.1016/j.celrep.2016.08.032.
43. Borbely G, Haldosen LA, Dahlman-Wright K, Zhao C. Induction of USP17 by combining BET and HDAC inhibitors in breast cancer cells. Oncotarget. 2015; 6:33623–35. https://doi.org/10.18632/oncotarget.5601.
44. Stottrup C, Tsang T, Chin YR. Upregulation of AKT3 Confers Resistance to the AKT Inhibitor MK2206 in Breast Cancer. Mol Cancer Ther. 2016; 15:1964–74. https://doi.org/10.1158/1535-7163.MCT-15-0748.
45. Zawistowski JS, Bevill SM, Goulet DR, Stuhlmiller TJ, Beltran AS, Olivares-Quintero JF, Singh D, Sciaky N, Parker JS, Rashid NU, Chen X, Duncan JS, Whittle MC, et al. Enhancer Remodeling during Adaptive Bypass to MEK Inhibition Is Attenuated by Pharmacologic Targeting of the P-TEFb Complex. Cancer Discov. 2017; 7:302–21. https://doi.org/10.1158/2159-8290.CD-16-0653.
46. Fong CY, Gilan O, Lam EY, Rubin AF, Ftouni S, Tyler D, Stanley K, Sinha D, Yeh P, Morison J, Giotopoulos G, Lugo D, Jeffrey P, et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature. 2015; 525:538–42. https://doi.org/10.1038/nature14888.
47. Jang JE, Eom JI, Jeung HK, Cheong JW, Lee JY, Kim JS, Min YH. AMPK-ULK1-mediated autophagy confers resistance to BET inhibitor JQ1 in acute myeloid leukemia stem cells. Clin Cancer Res. 2017; 23:2781–94. https://doi.org/10.1158/1078-0432.CCR-16-1903.
48. Amorim S, Stathis A, Gleeson M, Iyengar S, Magarotto V, Leleu X, Morschhauser F, Karlin L, Broussais F, Rezai K, Herait P, Kahatt C, Lokiec F, et al. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: a dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 2016; 3:e196–204. https://doi.org/10.1016/S2352-3026(16)00021-1.
49. Berthon C, Raffoux E, Thomas X, Vey N, Gomez-Roca C, Yee K, Taussig DC, Rezai K, Roumier C, Herait P, Kahatt C, Quesnel B, Michallet M, et al. Bromodomain inhibitor OTX015 in patients with acute leukaemia: a dose-escalation, phase 1 study. Lancet Haematol. 2016; 3:e186–95. https://doi.org/10.1016/S2352-3026(15)00247-1.
50. Tanaka M, Roberts JM, Seo HS, Souza A, Paulk J, Scott TG, DeAngelo SL, Dhe-Paganon S, Bradner JE. Design and characterization of bivalent BET inhibitors. Nat Chem Biol. 2016; 12:1089–96. https://doi.org/10.1038/nchembio.2209.
51. Lin X, Huang X, Uziel T, Hessler P, Albert DH, Roberts-Rapp LA, McDaniel KF, Kati WM, Shen Y. HEXIM1 as a Robust Pharmacodynamic Marker for Monitoring Target Engagement of BET Family Bromodomain Inhibitors in Tumors and Surrogate Tissues. Mol Cancer Ther. 2017; 16:388–96. https://doi.org/10.1158/1535-7163.MCT-16-0475.