
INTRODUCTION
Oculocutaneous albinism (OCA) is a heterogeneous and autosomal recessive disorder with an estimated prevalence of 1/17,000 worldwide, and the carrier rate is approximately 1 in 70. OCA is characterized by a reduction or complete loss of pigment in the skin, hair, and eyes accompanied by photophobia, nystagmus, strabismus, and reduced visual acuity due to melanin biosynthesis deficiency [1, 2]. OCA is broadly classified as non-syndromic and syndromic OCA based on the presence of other symptoms such as immunodeficiency, bleeding diathesis, or neurological dysfunction [3, 4]. Non-syndromic OCA includes four types, OCA1-4, and the clinical diagnosis of OCA subtype is difficult because of its variable clinical phenotype. Emerging evidence shows that molecular and genetic analyses can provide accurate diagnosis and genetic counselling.
The prevalence of different OCA subtypes significantly differs in different ethnic populations. OCA1 and OCA2 are the two most frequent types of OCA, making up 50% and 30% of all OCA cases worldwide, respectively [1, 5]. OCA1 is caused by mutations of TYR. OCA2-4, which are somewhat milder, are caused by mutations in OCA2, TYRP1, and SLC45A2, respectively. OCA2 is mainly found in Africa, and the frequencies of OCA3 and OCA4 are approximately 3% and 17% worldwide, respectively [6–8].
OCA2 is characterized by yellow, brown, or golden hair at birth. To date, 154 OCA2 mutations have been identified, including 4 nonsense mutations, 92 missense mutations, 17 splicing mutations, 1 regulatory mutation, 20 small deletions, 7 small insertions, 2 small indels, 9 gross deletions, and 2 gross insertions/duplications. Wei et al. reported that 8 of 52 Chinese families including patients with OCA were identified with mutations in OCA2, accounting for 15.4% [9].
Genetic tests were carried out to provide an accurate genetic diagnosis and genetic counselling for a Chinese family with two patients affected by OCA characterized by yellow hair, milky skin, photophobia, nystagmus, and reduced visual acuity. Two compound heterozygous mutations (c.808-3C>G and c.2080-2A>G) in OCA2 were identified, which may result in pathogenic splice site mutation and may be responsible for some clinical manifestations of OCA.
RESULTS
Clinical phenotype
The pedigree chart and the clinical features of the male patient (proband) affected by OCA are shown in Figure 1. Both the patients have yellow eyebrows and hair, milky skin, and heterochromia iridis, accompanied with photophobia, impaired visual acuity, and nystagmus. On the other hand, unaffected family members present normal pigmentation.
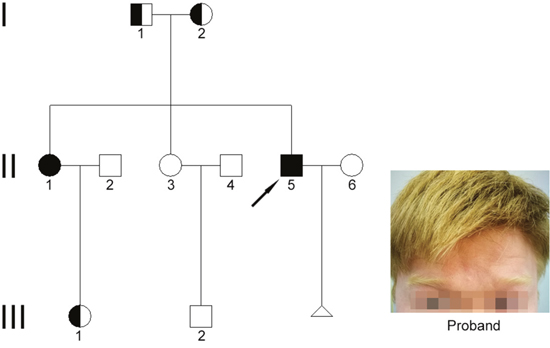
Figure 1: Clinical features and family pedigree. A simplified pedigree is shown. Arrow indicates the proband. Square symbols denote males, and the circles denote females. A filled square indicates affected male, and filled circle indicates affected female. Half-filled square and circle indicate carriers. Triangle denotes pregnancy. Image of the hair and eyebrow of the proband is shown.
Mutation identification and analysis of OCA2
No TYR or TYRP1 mutation was identified in the two patients. Compound heterozygous mutations in OCA2 (c.808-3C>G and c.2080-2A>G) were identified in the two patients (Figure 2 and Figure 3, Table 1). OCA2 c.808-3C>G has been previously reported in a Hispanic family with OCA, and this mutation is predicted to affect splicing [10]. OCA2 c.2080-2A>G mutation is novel, and it has not been found in 1000Genomes (http://www.internationalgenome.org/), Human Gene Mutation Database (HGMD, http://www.hgmd.cf.ac.uk/), or the Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org/) database.
Table 1: Mutation summary of this family member
|
OCA2 c.808-3C>G |
OCA2 c.2080-2A>G |
SLC45A2 c.814G>A |
SLC45A2 c.890C>T |
|---|---|---|---|---|
I-1 |
Het |
WT |
WT |
Het |
I-2 |
WT |
Het |
Het |
WT |
II-1 |
Het |
Het |
WT |
Het |
II-3 |
WT |
WT |
WT |
Het |
II-5 |
Het |
Het |
Het |
Het |
II-6 |
WT |
WT |
WT |
WT |
III-1 |
WT |
Het |
Het |
Het |
WT, wild type; Het, Heterozygous; Hom, Homozygous.
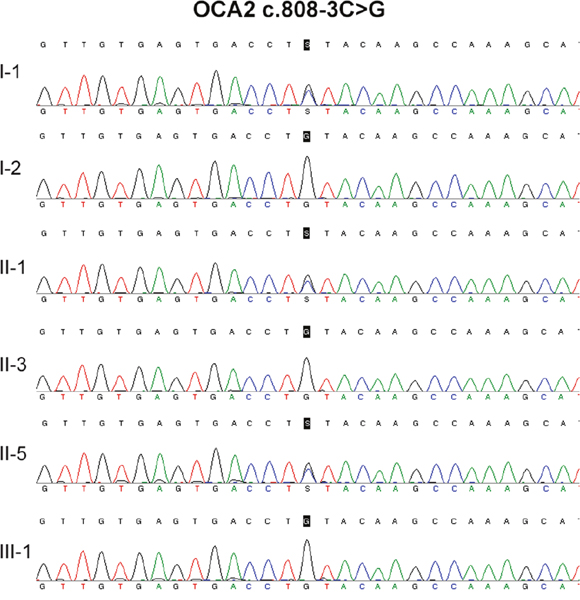
Figure 2: DNA sequencing result of OCA c.808-3C>G.
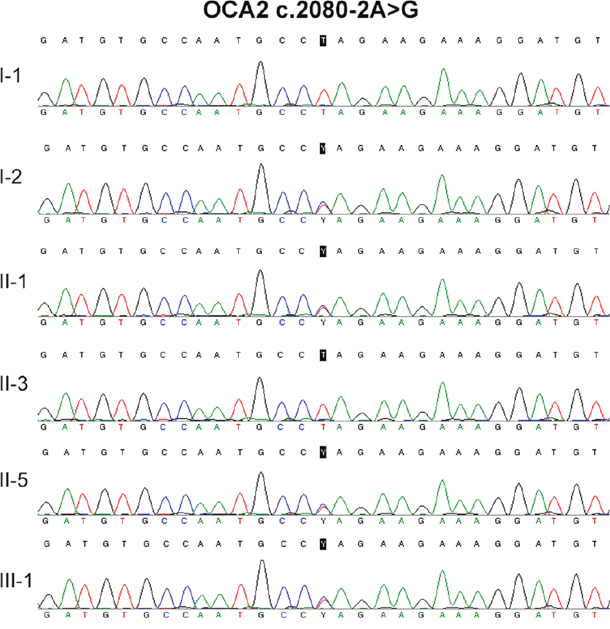
Figure 3: DNA sequencing result of OCA c.2080-2A>G.
Several in silico analysis programs were used to predict the effect of the compound heterozygous mutations in OCA2 (c.808-3C>G and c.2080-2A>G) on splicing. The GENIE program performs splice site score calculation. The results indicated that the mutants showed a remarkable decrease of the splice site score (Table 2). The NetGene2 program showed that both OCA2 c.808-3C>G and c.2080-2A>G mutations abolished a previously predicted splice site (Figure 4A). The NNSPLICE program showed that OCA c.808-3C>G mutation generated a novel splice site, and the two nucleotides (AG) from the authentic splice site were incorporated into the coding region, creating a frame shift mutation. The NNSPLICE program showed results similar to those obtained with NetGene2 for OCA2 c.2080-2A>G (Figure 4B). These data suggest that both OCA2 c.808-3C>G and c.2080-2A>G mutations may affect OCA2 mRNA splicing, and compound heterozygous mutations (c.808-3C>G and c.2080-2A>G) in OCA2 might be responsible for some clinical manifestations of OCA.
Table 2: The splice site score for the 3' site calculated by GENIE program
|
OCA2 c.808-3C>G |
OCA2 c.2080-2A>G |
|---|---|---|
Wild type score |
5.2 |
11.5 |
Mutant score |
-5.3 |
0.6 |

Figure 4: Splice site prediction. (A), NetGene2 was used for splice site prediction. (B), NNSPLICE was used for splice site prediction. OCA2 c.808-3C>G wild type (acctagaccgagcagtgccagatcccagatggtgtctcaggtgaaaagcctcaccataacttatgctttggcttgtaCaggtcactcacaactggacggtgt atttaaatccgaggagaagcgagcactcagtgatgagcaggacctttgaggtactgaccaggtgagttctcagtgagtgaggtgttggggcaggctct) and the mutant (acctagaccgagca gtgccagatcccagatggtgtctcaggtgaaaagcctcaccataacttatgctttggcttgtaGaggtcactcacaactggacggtgtatttaaatccgaggagaagcgagcact cagtgatgagcaggacctttgaggtactgaccaggtgagttctcagtgagtgaggtgttggggcaggctct) were used for sequence input. OCA2 c.2080-2A>G wild type (tcattttcaagactttttttttaaatcttgcatatattttcggttctaaactgattctcaccacacatcctttcttctAggcattggcacatctccactt
aatagaatatgttggagaacaaactgctttgctaataaaggtaaaataaatgctata atagaaggcactccagccactgttctttgattttgtgaaaaaa) and the mutant (tcattttcaagactttttttttaaatcttgcatatattttcggttctaaactg attctcaccacacatcctttcttctGggcattggcacatctccacttaatagaatatgttggagaacaaactgctttgctaataaaggtaaaataaatgctataatagaaggcactccagccactgttctttgattttgtg aaaaaa) were used for sequence input. The capital words indicate the mutation site.
In order to detect rearrangements (deletion/duplication) of TYR and OCA2, Multiplex Ligation-dependent Probe Amplification (MLPA) assay was used and the results showed that no significant exon rearrangement (deletion/duplication) of TYR and OCA2 occurred in the two patients (Figure 5).
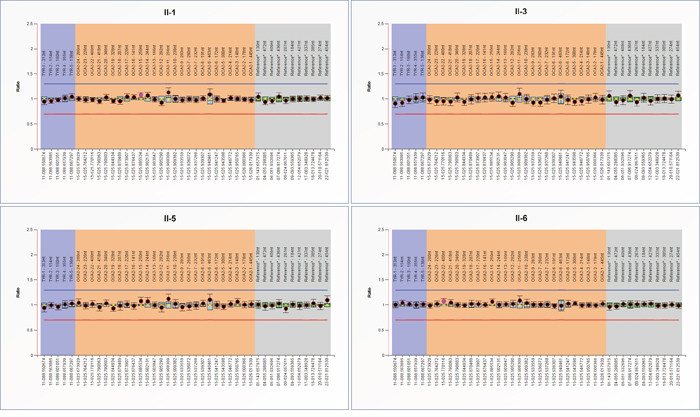
Figure 5: MLPA analysis of TYR and OCA2. The two patients, their unaffected sister, and the proband’s wife were analyzed for exon rearrangements (deletion/duplication) of TYR and OCA2.
Mutation identification and analysis of SLC45A2
Compound heterozygous mutations in SLC45A2 (c.814G>A and c.890C>T, which result in p.Glu272Lys and p.Thr297Ile, respectively) have been identified in both the male patient and a healthy unaffected girl in the family. These data suggest that compound heterozygous mutations in SLC45A2 (c.814G>A and c.890C>T) may not be associated with OCA in this family.
DISCUSSION
OCA2 accounts for 30% of all OCA cases worldwide with an estimated prevalence of 1/38,000–1/40,000. It is the most frequent form of OCA in the African population with a higher prevalence of 1/3,900–1/1,500 and in the African-American population, the prevalence is estimated to be 1/10,000 [11, 12]. OCA2 is characterized by variable hypopigmentation of the skin and hair, which may range from minimal to near normal, accompanied with ocular changes. Nystagmus is present before 3–4 months of age. Strabismus and visual inattention may occur in the first six months of age. Iris color ranges from blue to brown. Hair color may darken over time, although the hair color ranges from light yellow to light brown in newborns [13, 14].
OCA2 is caused by mutations in OCA2, a human homologue of mouse pink-eye dilution gene located on chromosome 15q11.2-q12 containing 24 exons (23 coding domains). The encoded protein, known as the P protein, is an integral membrane protein composed of 838 amino acid residues that consists of 12 transmembrane spanning regions and is involved in tyrosine transport, which is a precursor to melanin synthesis and pigmentation in the skin, hair, and eye [15, 16]. P protein is involved in the regulation of the pH of melanosomes or served as a melanosomal tyrosine transporter [17]. The most common mutation in OCA2 is a 2.7-kb deletion, which removes exon 7 and results in a frame shift mutation in the first luminal loop of OCA2 protein, producing a truncated and non-functional protein. This mutation is detected in Africans, sub-Saharan African heritage, African-Americans, and Haitian, suggesting a founder effect [18, 19]. Rooryck et al. stated that rearrangements of OCA2 might be present in more than 20% of patients with OCA2 [20]. P protein may disturb the pigmentation characteristics by altering the melanosomal tyrosine or tyrosinase function due to OCA2 mutations, but melanocytes of patients with OCA2 still produce small amounts of melanin. OCA2 is caused by homozygous or compound heterozygous OCA2 mutation, and recessive compound heterozygous mutation indicated that the mutant alleles of both copy are at different locations on the same gene. Patients in the compound heterozygous state may present with a less severe phenotype compared with those presenting with the homozygous form [13]. In the HGMD database, 154 mutations were included, and missense mutations account for 60%, while splicing mutations account for 11%.
No TYR or TYRP1 mutation was identified in the two patients. Two splice site mutations in OCA2 (c.808-3C>G and c.2080-2A>G) have been identified in both patients, while the healthy family members presented with only one of the two mutations. OCA2 c.808-3C>G was first identified in a Hispanic family with OCA, and this is the first report in Chinese population. OCA2 c.2080-2A>G has not been reported in any ethnic population yet. GENIE, NNSPLICE, and NetGene2 programs have been used to predict the effect of the two splice site mutations on OCA2 mRNA splicing. All three programs showed that both OCA2 c.808-3C>G and c.2080-2A>G mutations may affect OCA2 mRNA splicing by abolishing previous splice sites or generating a new splice site. The expression of OCA2 is very low in leukocytes, and we failed to amplify OCA2 mRNA. In vitro experiments may further confirm these effects.
To exclude the large deletion or duplication of exons in TYR and OCA2, MLPA was also performed to detect exon rearrangements, and the results showed that no deletion or duplication of exons was found in the patients.
In the current study, compound heterozygous mutations in SLC45A2 (c.814G>A and c.890C>T) were identified in both a male patient and an unaffected girl in the family. Therefore, compound heterozygous mutations in SLC45A2 may not be the causative mutation for OCA in this family.
In conclusion, this study expands the mutation spectrum of OCA. Compound heterozygous mutations (c.808-3C>G and c.2080-2A>G) in OCA2 might be responsible for some clinical manifestations of OCA.
MATERIALS AND METHODS
Subjects and clinical evaluation
This study was approved by the Ethics Committee of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology. All procedures were carried out in accordance with the approved guidelines. One patient was a 25-year-old male, and another patient was a 37-year-old female. Both of them presented the same clinical ophthalmologic characteristics, including heterochromia iridis, milky skin, yellow hair, photophobia, nystagmus, and reduced visual acuity. Family history and pedigree chart were drawn to evaluate the inheritance model. Written informed consent was obtained from all participants and authorization to publish personal photographs was obtained from the male patient only. However, he only allowed us to publish photographs of his hair and eyebrows, but not of his eyes.
Strategy for mutational screening
Mutational screening of TYR was prioritized for patients with OCA. OCA2, SLC45A2, and TYRP1 were sequentially screened for mutations when no mutation was found in TYR.
DNA extraction and mutational analysis
DNA extraction and PCR-based Sanger sequencing were performed as previously described [21]. Briefly, each 50-μL PCR mixture contained 100 ng of genomic DNA, 2 μL of 10 μM forward and reverse primers (with a final concentration of 400 nM), and 25 μL of 2× Taq PCR MasterMix (Takara, Dalian, China). PCR was carried out in Veriti thermocycler (Applied Biosystems, Foster City, CA, USA) using the following protocol: 95°C for 3 min; 35 cycles of denaturation at 95°C for 30 s, annealing at 55°C for 30 s, and extension at 72°C for 45 s; and a final extension at 72°C for 7 min. The amplified products were purified with a cycle-pure kit (Axygen, Wujiang, China) and sequenced using an ABI 3500 Dx sequencer (Applied Biosystems). In order to detect exon rearrangements (deletion/duplication) of TYR and OCA2 and to increase the mutation rate, MLPA assay Kit (P325-OCA2) from MRC-Holland (Amsterdam, Netherlands) was used and the procedure was performed according to manufacturer’s instructions. The mutation was named according to the recommendation of sequence variants by Human Genomic Variation Society (HGVS: http://www.hgvs.org/). The interpretation of sequence variants was made according to the recommendation of the American College of Medical Genetics (ACMG) and Genomics and the Association for Molecular Pathology (AMP) [22]. All primer sequences are listed in Table 3.
Table 3: Primer sequences used in this work
Primer name |
Sequence |
PCR product |
|---|---|---|
TYR CD1 AF |
GCT GGA GGT GGG AGT GGT ATT |
459bp |
TYR CD1 AR |
GTC CCC AAA AGC CAA ACT TG |
|
TYR CD1 BF |
AAT GCA CCA CTT GGG CCT C |
536bp |
TYR CD1 BR |
TCC CGC CAG TCC CAA TAT G |
|
TYR CD1 CF |
CAA CAC CCA TGT TTA ACG ACA |
475bp |
TYR CD1 CR |
CAT TGA GAG TTC TTA ACA GGG C |
|
TYR CD2 F |
GAT TTC TCA GAA CAT ATC CCT G |
526bp |
TYR CD2 R |
AGC TAG GGT CAT TGT CGA TAT |
|
TYR CD3 F |
AGA GTC TCA ATA CGG AAT GAA TT |
519bp |
TYR CD3 R |
GTA TCC TGC CTA ATC CAC CTT |
|
TYR CD4 F |
CTG TTT CCA ATT TAG TTT TAT AC |
790bp |
TYR CD4 R |
TAC AAA ATG GCC TAT GTT AAG C |
|
TYR CD5 F |
TGT CTA CTC CAA AGG ACT GT |
924bp |
TYR CD5 R |
GGC ACT TAG CTG GAT GTG TT |
|
TYR CD4 Sequencing F |
CTC CAG ATT TTA ATA TAT GCC |
348bp |
TYR CD4 Sequencing R |
GTG TTA TCT CAA AAT AAA TTG G |
|
TYR CD5 Sequencing F |
GAT GGT GAT CGT AAC AAT GG |
311bp |
TYR CD5 Sequencing R |
TTT GGC CCT ACT CTA TTG CC |
|
OCA2 CD1 F |
CGA AGA AGC AAC CTT CCT ATT GTA C |
490bp |
OCA2 CD1 R |
CTA AGC CAG GAA AGT GAT CTA ATG C |
|
OCA2 CD2 F |
ATT CTT GAA TCT AGC ACC TGA GTG C |
306bp |
OCA2 CD2 R |
TGT CAA GGA TCT GGC AGA GGT TA |
|
OCA2 CD3 F |
ACC CAT TCC CAC CAG TAT GAG AGT |
456bp |
OCA2 CD3 R |
CAA AAC TCA TCC TCT TCT TCA CGC |
|
OCA2 CD4 F |
TGA GAT GGA AGT TAC TCA AGG CTG |
285bp |
OCA2 CD4 R |
AGA CAG TCA GAG AAT CAG GCG AAG |
|
OCA2 CD5 F |
AGT AGC CCC ATC ATC ACA TCT GTT |
298bp |
OCA2 CD5 R |
AAA TTC GAG TGG TAA TGG CCT GT |
|
OCA2 CD6 F |
TTC TTC ACA CAC TGT CAG AGG AGG |
382bp |
OCA2 CD6 R |
GAA TTG ACT AAG AAT GGT GTC CTC G |
|
OCA2 CD7 F |
AAC AAA TAC CTA GAC CGA GCA GTG |
242bp |
OCA2 CD7 R |
TAT AGG TCA GAC TCC TTT AAA CGC A |
|
OCA2 CD8 F |
GCT GTG AGA TTG GGC GTT GG |
461bp |
OCA2 CD8 R |
GCA AAT ATT CCT GTA TGG TTC CCT T |
|
OCA2 CD9 F |
GCC TGA AAC ATC AAG ACC CAT |
460bp |
OCA2 CD9 R |
CCT TTC CTC CAC CAC GAT G |
|
OCA2 CD10 F |
CAG CGA TAT AAT CCA ACT TCA AAG G |
355bp |
OCA2 CD10 R |
GCA CTA ACA CTT CTC AGT CAA GCC |
|
OCA2 CD11 F |
TGT AAG GGA TCA TGC TGA TGT CG |
387bp |
OCA2 CD11 R |
CAC AAC GAT TCA ACC TGA GTA CCC |
|
OCA2 CD12 F |
AAT GTT AGT TTG GCT CCC TGT TCT T |
330bp |
OCA2 CD12 R |
TCA TGC ACC TGA GAA TGG AAC C |
|
OCA2 CD13 F |
ACT CTG GAA AGG AAT GTA ACT CTC G |
491bp |
OCA2 CD13 R |
CTT GAG ATG CCC AGT AGC ACT TAC |
|
OCA2 CD14 F |
ATC CAC CCA CCT CGG AAA GT |
329bp |
OCA2 CD14 R |
AGC ATC CAG CAA CCC ATC AA |
|
OCA2 CD15 F |
GTC TCG AGT GTG TGT CTG CTC TGT C |
425bp |
OCA2 CD15 R |
TGC AGA GCT CAG TGA GGG TTA GAT A |
|
OCA2 CD16 F |
ACA CTC CTT TCA TCA TTC AGG TCA T |
423bp |
OCA2 CD16 R |
AAC CTC AAC GTC TTG TGT ATA ACC A |
|
OCA2 CD17 F |
CTG TCG TGA TTC CAG TTG CGT AG |
489bp |
OCA2 CD17 R |
CAG TGC CCA CTC TAT ATT CCT CCT C |
|
OCA2 CD18 F |
GAG GTA CAA GAA CAT AGG CAT GAA T |
552bp |
OCA2 CD18 R |
AAA TCT CTC AGT GGC TAA GGT AAA G |
|
OCA2 CD19 F |
TCT GGG CCT ACC TTA TGT TCA CG |
324bp |
OCA2 CD19 R |
CAT CTC TGG GCT GCA CAG GAT AG |
|
OCA2 CD20 F |
CTA TGT CTG CCT TGG TCT CGT GAT |
379bp |
OCA2 CD20 R |
CTC TGC TCA CTT TCG TCC TCT ACA C |
|
OCA2 CD21 F |
GGT TTC TTT CCA CAA ATC TTA TGC T |
341bp |
OCA2 CD21 R |
CAT CCA GAC TCT CCT TCA TTT GCT |
|
OCA2 CD22 F |
CAA ATC AAA GCC TGT GAG ATG ATC T |
326bp |
OCA2 CD22 R |
CTC CCC TAC ACC ACA GTC TCT CTA C |
|
OCA2 CD23 F |
GAT GAA CAA ACA GAG GCT CCA |
477bp |
OCA2 CD23 R |
TAG CAT CTC CAG GGT AAG CAC |
|
SLC45A2 CD1 F |
CTG ACC ATC TCT GTT GGT TGC TC |
594bp |
SLC45A2 CD1 R |
CTA GGA AAG GTC AAA CAC ATG AAC A |
|
SLC45A2 CD2 F |
GGA AGA TGA TTT TAT GGC AAG AAG T |
357bp |
SLC45A2 CD2 R |
CGT GTA GAG ACA CTG GAT GGC TT |
|
SLC45A2 CD3 F |
CCC ACT GAA GGG GAG TGT CTA TG |
518bp |
SLC45A2 CD3 R |
CCA TGA AAC TCT TCT CGT CAA ACA G |
|
SLC45A2 CD4 F |
ACA CTT TGT GTG ATG GCT GAC TGA C |
358bp |
SLC45A2 CD4 R |
ACT GTG CCA ATC TTA GAG GAT AGC C |
|
SLC45A2 CD5 F |
GAC ATT TGC TCC CCA GAG GT |
451bp |
SLC45A2 CD5 R |
ACC CAC TGA TTC CAA GAG CAA A |
|
SLC45A2 CD6 F |
CCA CAG ATA AGG GGA TTC TTT TGT T |
449bp |
SLC45A2 CD6 R |
TTC CAG CTC TGC TCT ACA CAT TGC |
|
SLC45A2 CD7 F |
ATC CAC GAA GCC AAA GGT A |
459bp |
SLC45A2 CD7 R |
GAA ATC ACA ATA GTG GGC GT |
|
TYRP1 CD1 F |
TTG AAA GTG GTT TGG GAA GG |
742bp |
TYRP1 CD1 R |
AGC TTC AAC TCC AAC CCT TTA C |
|
TYRP1 CD2 F |
AAT CAT GCA GTA AAT TGG AGA G |
687bp |
TYRP1 CD2 R |
GAA ATG CCA AAG ACA GGT TAG |
|
TYRP1 CD3 F |
CCC TCA GAC ACC GTT GAT ATA CT |
499bp |
TYRP1 CD3 R |
GGT GTT TAA TGA ATG CCT GGT AC |
|
TYRP1 CD4 F |
AAT GGG ACA TGG TAA CTT AGA |
570bp |
TYRP1 CD4 R |
GAT TTC CAA GGG CTT CAC |
|
TYRP1 CD5 F |
GTC ATC AAC CAT AGG TAC AGA G |
586bp |
TYRP1 CD5 R |
GAG AGA TGA TTT GGT TAG TCC |
|
TYRP1 CD6 F |
TAA TTT CTC ATC CTG CTG TAG TG |
470bp |
TYRP1 CD6 R |
ATC AAA TTC CTT CCC TTA TCC |
|
TYRP1 CD7 F |
CTA TCC CAA TAG GGT CCA CTC |
558bp |
TYRP1 CD7 R |
TTC TGA AAG GGT CTT CCC AG |
|
In silico analysis
To predict the effect of the missense alterations, the following programs were used: InterVar (http://wintervar.wglab.org/), MutationTaster (http://www.mutationtaster.org/), and Ensembl database (http://asia.ensembl.org/index.html) [23, 24]. To predict the splice site, the following programs were used: GENIE program (http://rulai.cshl.edu/new_alt_exon_db2/HTML/score.html), NNSPLICE (http://www.fruitfly.org/seq_tools/splice.html), and NetGene2 (http://www.cbs.dtu.dk/services/NetGene2/) [25–28].
ACKNOWLEDGMENTS
This work was partially supported by the National Natural Science Foundation of China (No. 81500925).
CONFLICTS OF INTEREST
The authors do not have any conflicts of interest.
REFERENCES
1. Kamaraj B, Purohit R. Mutational analysis of oculocutaneous albinism: a compact review. BioMed research international. 2014; 2014:905472.
2. Gronskov K, Ek J, Brondum-Nielsen K. Oculocutaneous albinism. Orphanet journal of rare diseases. 2007; 2:43.
3. El-Chemaly S, Young LR. Hermansky-Pudlak Syndrome. Clinics in chest medicine. 2016; 37:505-511.
4. Introne WJ, Westbroek W, Golas GA, Adams D. Chediak-Higashi Syndrome. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Ledbetter N, Mefford HC, Smith RJH, Stephens K, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2017.
5. Rooryck C, Morice-Picard F, Elcioglu NH, Lacombe D, Taieb A, Arveiler B. Molecular diagnosis of oculocutaneous albinism: new mutations in the OCA1-4 genes and practical aspects. Pigment cell & melanoma research. 2008; 21:583-587.
6. Manga P, Kerr R, Ramsay M, Kromberg JG. Biology and genetics of oculocutaneous albinism and vitiligo - common pigmentation disorders in southern Africa. South African medical journal. 2013; 103:984-988.
7. Suzuki T, Tomita Y. Recent advances in genetic analyses of oculocutaneous albinism types 2 and 4. Journal of dermatological science. 2008; 51:1-9.
8. Zhang KH, Li Z, Lei J, Pang T, Xu B, Jiang WY, Li HY. Oculocutaneous albinism type 3 (OCA3): analysis of two novel mutations in TYRP1 gene in two Chinese patients. Cell biochemistry and biophysics. 2011; 61:523-529.
9. Wei A, Yang X, Lian S, Li W. Implementation of an optimized strategy for genetic testing of the Chinese patients with oculocutaneous albinism. Journal of dermatological science. 2011; 62:124-127.
10. Chiang PW, Fulton AB, Spector E, Hisama FM. Synergistic interaction of the OCA2 and OCA3 genes in a family. American journal of medical genetics Part A. 2008; 146A:2427-2430.
11. Kromberg JG, Bothwell J, Kidson SH, Manga P, Kerr R, Jenkins T. Types of Albinism in the Black Southern Africa Population. East African medical journal. 2012; 89:20-27.
12. Hong ES, Zeeb H, Repacholi MH. Albinism in Africa as a public health issue. BMC public health. 2006; 6:212.
13. Wang Y, Wang Z, Chen M, Fan N, Yang J, Liu L, Wang Y, Liu X. Mutational Analysis of the TYR and OCA2 Genes in Four Chinese Families with Oculocutaneous Albinism. PloS one. 2015; 10:e0125651.
14. Urtatiz O, Sanabria D, Lattig MC. Oculocutaneous albinism (OCA) in Colombia: first molecular screening of the TYR and OCA2 genes in South America. Journal of dermatological science. 2014; 76:260-262.
15. Oetting WS, Garrett SS, Brott M, King RA. P gene mutations associated with oculocutaneous albinism type II (OCA2). Human mutation. 2005; 25:323.
16. Lee ST, Nicholls RD, Bundey S, Laxova R, Musarella M, Spritz RA. Mutations of the P gene in oculocutaneous albinism, ocular albinism, and Prader-Willi syndrome plus albinism. The New England journal of medicine. 1994; 330:529-534.
17. Brilliant MH. The mouse p (pink-eyed dilution) and human P genes, oculocutaneous albinism type 2 (OCA2), and melanosomal pH. Pigment cell research. 2001; 14:86-93.
18. Durham-Pierre D, Gardner JM, Nakatsu Y, King RA, Francke U, Ching A, Aquaron R, del Marmol V, Brilliant MH. African origin of an intragenic deletion of the human P gene in tyrosinase positive oculocutaneous albinism. Nature genetics. 1994; 7:176-179.
19. Stevens G, Ramsay M, Jenkins T. Oculocutaneous albinism (OCA2) in sub-Saharan Africa: distribution of the common 2.7-kb P gene deletion mutation. Human genetics. 1997; 99:523-527.
20. Rooryck C, Morice-Picard F, Lasseaux E, Cailley D, Dollfus H, Defoort-Dhellemme S, Duban-Bedu B, de Ravel TJ, Taieb A, Lacombe D, Arveiler B. High resolution mapping of OCA2 intragenic rearrangements and identification of a founder effect associated with a deletion in Polish albino patients. Human genetics. 2011; 129:199-208.
21. Wang X, Zhu Y, Shen N, Peng J, Wang C, Liu H, Lu Y. Mutation analysis of a Chinese family with oculocutaneous albinism. Oncotarget. 2016; 7:84981-84988. https://doi.org/10.18632/oncotarget.13109.
22. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in medicine. 2015; 17:405-424.
23. Li Q, Wang K. InterVar: Clinical Interpretation of Genetic Variants by the 2015 ACMG-AMP Guidelines. American journal of human genetics. 2017; 100:267-280.
24. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nature methods. 2014; 11:361-362.
25. Smith PJ, Zhang C, Wang J, Chew SL, Zhang MQ, Krainer AR. An increased specificity score matrix for the prediction of SF2/ASF-specific exonic splicing enhancers. Human molecular genetics. 2006; 15:2490-2508.
26. Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie. Journal of computational biology. 1997; 4:311-323.
27. Hebsgaard SM, Korning PG, Tolstrup N, Engelbrecht J, Rouze P, Brunak S. Splice site prediction in Arabidopsis thaliana pre-mRNA by combining local and global sequence information. Nucleic acids research. 1996; 24:3439-3452.
28. Brunak S, Engelbrecht J, Knudsen S. Prediction of human mRNA donor and acceptor sites from the DNA sequence. Journal of molecular biology. 1991; 220:49-65.