
INTRODUCTION
Hypopharyngeal cancer (HPC) mainly originated in the pyriform sinus, followed by sites not otherwise specified, and the posterior hypopharyngeal wall [1]. These tumors frequently present at an advanced stage, and display early submucosal spread, resulting in poor prognosis, among the worst of all head and neck subsites [2]. Surgery is difficult to achieve due to the multifocal disease and early lymphatic spread. Although combined surgical therapy and chemoradiotherapy have improved the survival for patients with HPC over the past 3 decades, the mortality rate in late-stage diagnosis of HPC is unsatisfactory [3]. Detection of HPC at an earlier stage would be beneficial to patients. Many molecules, such as p16, bcl-2, microRNAs, and cyclin-D1, have been evaluated as candidate biomarkers for HPC [4, 5], but none has been widely used in practice because of each belongs to signaling pathways of multiple known or unknown proteins [6]. Therefore, more effective biomarkers for early diagnosis of HPC are necessary. And understanding of the molecular mechanisms involved in HPC development, progression, and treatment response is also necessary.
Recently, next generation sequencing has become very useful tools for identifying gene alterations and novel biomarkers and therapeutic targets [7]. For instance, Sawada G et al performed whole-exome sequence analysis of tumor and nontumor esophageal tissues collected from 144 patients with esophageal squamous cell carcinoma, and found that many tumors contained mutations in genes that regulate the cell cycle (TP53, CCND1, CDKN2A), epigenetic processes and receptor-tyrosine kinase-phosphoinositide 3-kinase signaling pathways (PIK3CA, EGFR, ERBB2) [8]. Whole exome sequencing on cisplatin-resistant metastatic squamous cell carcinoma of head and neck tumors revealed that inactivation of REV3L may inform treatment options in patients of recurrent squamous cell carcinoma of head and neck tumors [9]. Thus, whole exome sequencing can provide comprehensive insights into the mutational signatures of cancers and identify markers for early diagnosis and potential therapeutic targets. However, the mutations in hypopharyngeal cancer remain largely unknown.
In this study, we performed whole-exome sequencing (WES) of 23 hypopharyngeal tumor and paired adjacent normal tissue and confirmed the expression of NSD1 and PRB4 in an independent cohort containing 88 hypopharyngeal tumor and 36 adjacent normal tissues. In addition, we further investigate the function of PRB4 in hypopharyngeal tumor and the underlying mechanisms.
RESULTS
Clinicopathologic features of the case for whole-exome sequencing
Twenty-four pared adjacent and tumor tissues from patients with hypopharyngeal carcinoma were collected to perform whole-exome sequencing. As the low quality of DNA extracted from No. 22 tissue, this tissue was excluded in analysis. The information of patients on demography, risk factors, clinical stage, and histopathological features were recorded. All the patients were male, age was between 41-80 years old, 64% (14/23) were exposed to tobacco and alcohol, 78.2% (18/23) presented at advanced stage III/IV, and nodal metastasis (Figure 1B).
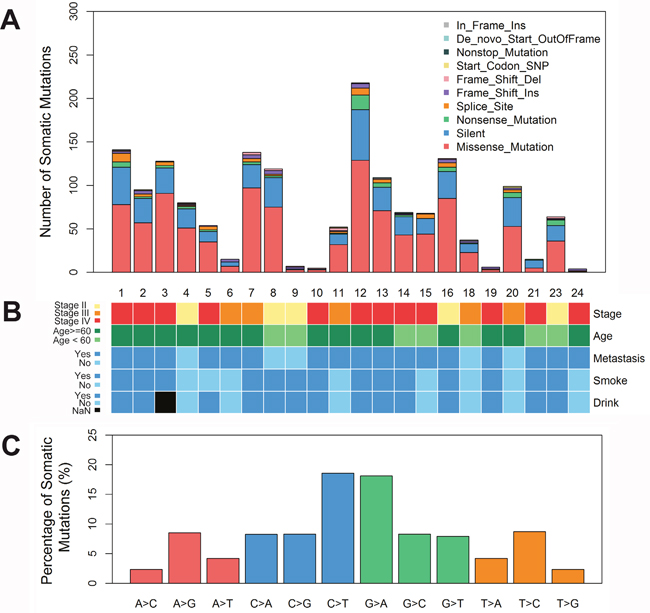
Figure 1: Somatic mutation of hypopharyngeal cancer tissues. (A) A summary of all kinds of somatic mutations in hypopharyngeal cancer tissues. Each bar represents individual mutations in each tissue. (B) The clinicopathological features of hypopharyngeal cancer patients. (C) The bars show percentage of somatic single nucleotide variations identified by whole exome sequencing in hypopharyngeal cancer tissues, compared to adjacent tissues. del, deletion; Ins, insert.
Whole-exome sequencing
Whole-exome sequencing of 23 hypopharyngeal carcinoma tumors and matched adjacent normal tissues from patients was performed. Approximately 3.32~4.82 Gb of cleaned sequencing data were obtained for each sample. About 99.02% of the sequencing reads were mapped to the human genome 19, with mean 57× (range 34–80×) sequencing depth. The somatic mutations mainly included missence mutation (50-70%), Silent mutation (20-30%), and nonsence mutation (1-5%) (Figure 1A). The most common type of mutation in the exomic region was C>T and G>A transition, followed by A>G and T>C transition (Figure 1C).
We searched for enrichment of somatically mutated and CNVs (amplication or deletion) genes with p<0.05 in the KEGG pathways including the Cell Cycle signaling, PI3K signaling, RAS signaling, Hippo signaling, and cell adhesion through GSEA algorithm combined with mutational analysis. PI3K signaling pathway was identified as the most significantly altered (FDR≤0.05). Fifteen somatic mutations (TP53, REC8, PRB4, EI24, NSD1, CDKN2A, KLK3, ALDH2, BICD1, CDK2AP1, PIK3CA, PEG3, CNGA4, SULF1, LATS1) and three CNVs genes (ATF1, CDKN2A, CDKN2B) were identified (Figure 2). Hence, these data shed light on the essential role of dysregulation of these critical pathways in tumorigenesis of hypopharyngeal carcinoma.
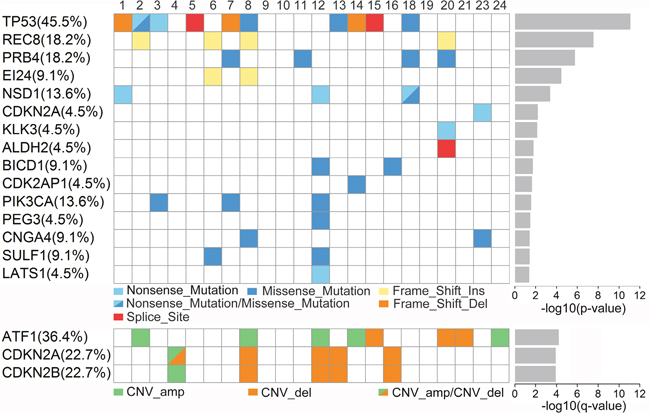
Figure 2: Mutations of genes in hypopharyngeal cancer tissues. Upper chart indicates number and type of mutations for each gene. Lower chart shows the copy number variations (CNVs) of ATF1, CDNK2A and CDNK2B. amp, amplification; del, deletion; Ins, insert.
As shown in Table 1, mutations resulted in down-regulation or up-regulation of genes expression. In order to explore the possible predicting biomarkers of hypopharyngeal carcinoma, we especially concerned with the amplication genes. Among them, the somatic mutation of PRB4 and NSD1 caused gene activation (Fold change > 5). Other activated genes included SYCP1 PDE4DIP, KIAA1383, DUSP19, hsa-mir-28, ATF1, EEA1 and SYCP2. And the top ten of downregulated genes included EPHA5, SERF1B, PCDHB9, CDKN2A, EFNB2, IL9R, hsa-mir-1302-9, LOC100129138, FOXD4L4, STX19 (Table 1).
Table 1: The top ten upregulated and downregulated genes in adjacent and tumor tissues
Upregulated genes |
cytoband |
Expression |
Fold change (tumor vs. adjacent) |
|
|---|---|---|---|---|
adjacent |
tumor |
|||
PRB4 |
12p13.2 |
376655 |
2069567 |
5.52 |
NSD1 |
5q35.3 |
45497 |
311837 |
6.91 |
SYCP1 |
1p13.2 |
121440 |
451095 |
3.73 |
PDE4DIP |
1q21.1 |
323511 |
974559 |
3.04 |
KIAA1383 |
1q42.2 |
116503 |
266925 |
2.30 |
DUSP19 |
2q33.1 |
227069 |
948717 |
4.24 |
hsa-mir-28 |
3q25.2 |
252910 |
729638 |
2.95 |
ATF1 |
12q13.12 |
193112 |
586193 |
3.02 |
EEA1 |
12q22 |
200547 |
723458 |
3.64 |
SYCP2 |
20q13.33 |
241605 |
996799 |
4.17 |
Downregulated genes |
|
|
|
|
EPHA5 |
4q13.1 |
592005 |
199657 |
0.34 |
SERF1B |
5q13.2 |
817299 |
169158 |
0.21 |
PCDHB9 |
5q31.3 |
958396 |
296799 |
0.31 |
CDKN2A |
9p21.3 |
788582 |
304950 |
0.39 |
EFNB2 |
13q33.3 |
996799 |
391073 |
0.39 |
IL9R |
Yq11.223 |
1076166 |
537999 |
0.50 |
hsa-mir-1302-9 |
9p24.3 |
816827 |
149433 |
0.18 |
LOC100129138 |
1p21.1 |
527704 |
131291 |
0.25 |
FOXD4L4 |
9p11.2 |
387398 |
131880 |
0.34 |
STX19 |
3p12.1 |
811353 |
311972 |
0.38 |
Upregulation of PRB4 and NSD1 is predictive of a poor outcome of patient with hypopharyngeal carcinoma
We focused on the role of PRB4 and NSD1 in hypopharyngeal carcinoma. An independent cohort containing 88 tumor tissues and 36 adjacent tissues was used to confirm their expressions using IHC staining. We found that PRB4 and NSD1 were dramatically increased in hypopharyngeal carcinoma tissues compared with adjacent tissues (Figure 3A). And the Kaplan-Meier survival cure showed that the patients with high PRB4 and NSD1 expression had shorter survival time than those with low PRB4 and NSD1 expression (Figure 3B). We also found that the expression of PRB4 was positively correlated with NSD1 expression (Figure 3C). In addition, PRB4 and NSD1 expression was associated with tumor size (p=0.010), metastasis (p=0.029) and clinical stage (p=0.005), but both their expressions were not associated with smoking and alcohol (Table 2). Moreover, we investigated the factors that could predict the prognosis of hypopharyngeal cancer patients by using Cox proportional hazard regression model for the univariate and multivariate analysis. Univariate analysis data indicated that the PRB4 level (P=0.013) and NSD1 level (P=0.010), as well as the tumor size (P=0.027), metastasis (P=0.006), and clinical stage (P=0.008) was significantly associated with the survival (Table 3). Moreover, as demonstrated in Table 4, the PRB4 level (P=0.027), NSD1 level (P=0.018), tumor size (P=0.030), metastasis (P=0.012) and clinical stage (P=0.021) were found to be independent factors for predicating the prognosis of hypopharyngeal cancer patients. Thus, the results suggest that PRB4 and NSD1 might contribute to the development of hypopharyngeal cancer.
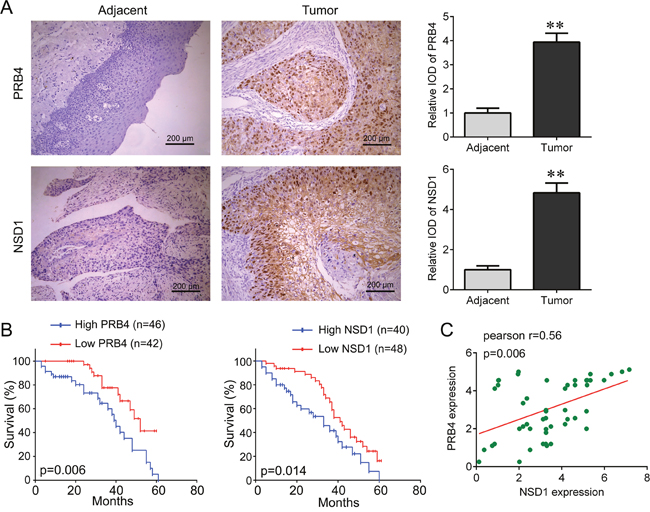
Figure 3: PRB4 and NSD1 are increased in human hypopharyngeal carcinoma tissues. (A) PRB4 and NSD1 were identified as positively expressed by immunohistochemical staining analysis and their expression was significantly higher in human hypopharyngeal carcinoma tissues (n=88) than in adjacent tissues (n=36). Scale bar, 200 μm. **p<0.01. (B) The relationship between PRB4 expression (left), NSD1 expression (right) and overall survival rate of hypopharyngeal carcinoma patients. The patients with high PRB4 or NSD1 expression have a shorter survival time than that with low PRB4 or NSD1 expression. (C) The correlation between PRB4 and NSD1 expression in human hypopharyngeal carcinoma tissues.
Table 2: Clinical association between PRB4 and NSD1 expression and clinicopathological variables in hypopharyngeal cancer patients
Variable |
PRB4 expression |
p |
NSD1 expression |
p |
||
|---|---|---|---|---|---|---|
Low (n=42) |
High (n=46) |
Low (n=48) |
High (n=40) |
|||
Age |
|
|
0.834 |
|
|
0.834 |
<60 |
20 |
23 |
|
24 |
19 |
|
≥60 |
22 |
23 |
|
24 |
21 |
|
Gender |
|
|
|
|
|
|
Male |
42 |
46 |
- |
48 |
40 |
- |
Female |
0 |
0 |
|
|
|
|
Tumor size |
|
|
0.018 |
|
|
0.010 |
<3cm |
25 |
15 |
|
28 |
12 |
|
≥3cm |
17 |
31 |
|
20 |
28 |
|
Metastasis |
|
|
0.005 |
|
|
0.029 |
No |
24 |
12 |
|
25 |
11 |
|
Yes |
18 |
34 |
|
23 |
29 |
|
Stage |
|
|
0.030 |
|
|
0.005 |
I-II |
23 |
14 |
|
27 |
10 |
|
III-IV |
19 |
32 |
|
21 |
30 |
|
Smoking |
|
|
0.598 |
|
|
0.602 |
No |
10 |
8 |
|
11 |
7 |
|
Yes |
32 |
38 |
|
37 |
33 |
|
Alcohol |
|
|
0.659 |
|
|
0.074 |
No |
14 |
18 |
|
13 |
19 |
|
Yes |
28 |
28 |
|
35 |
21 |
|
Metastasis, lymph node metastasis or distant metastasis; stage, clinical stage; p, chi-square test.
Table 3: Univariate analysis of prognostic factors of hypopharygeal cancer
Variable |
Hazard ratio |
p value |
|---|---|---|
Age (≥60/<60) |
1.23 |
0.671 |
Tumor size (≥3cm/<3cm) |
2.35 |
0.027 |
Metastasis (Yes/No) |
4.35 |
0.006 |
Clinical stage (III-IV/I-II) |
4.26 |
0.008 |
PRB4 expression (High/Low) |
3.02 |
0.013 |
NSD1 expression (High/Low) |
3.65 |
0.010 |
Table 4: Multivariate analysis of independent prognostic factors of hypopharygeal cancer
Variable |
Hazard ratio |
p value |
|---|---|---|
Tumor size |
2.53 |
0.030 |
Metastasis |
3.87 |
0.012 |
Clinical stage |
3.11 |
0.021 |
PRB4 expression |
2.63 |
0.027 |
NSD1 expression |
3.73 |
0.018 |
Knockdown of PRB4 by siRNA inhibits the cell proliferation and invasion of hypopharyngeal cancer cells
To investigate whether PRB4 affected the growth of Fadu and Tu686 cells, the cells were transfected with PRB4 siRNA. QPCR and western blot were used to confirm knockdown of PRB4 expression (Figure 4A, 4B). As shown in Figure 4C and 4D, PRB4 downregulation significantly impeded the cell viability and the ability of colony formation in Fadu and Tu686 cells. A corresponding effect on cell invasion was also observed in transwell assay, which showed a significant reduction of invasive cells in Fadu and Tu686 cells compared with the respective NC group (Figure 4E), and the expression of MMP2 was also significantly decreased by PRB4 downregulation (Figure 4B).
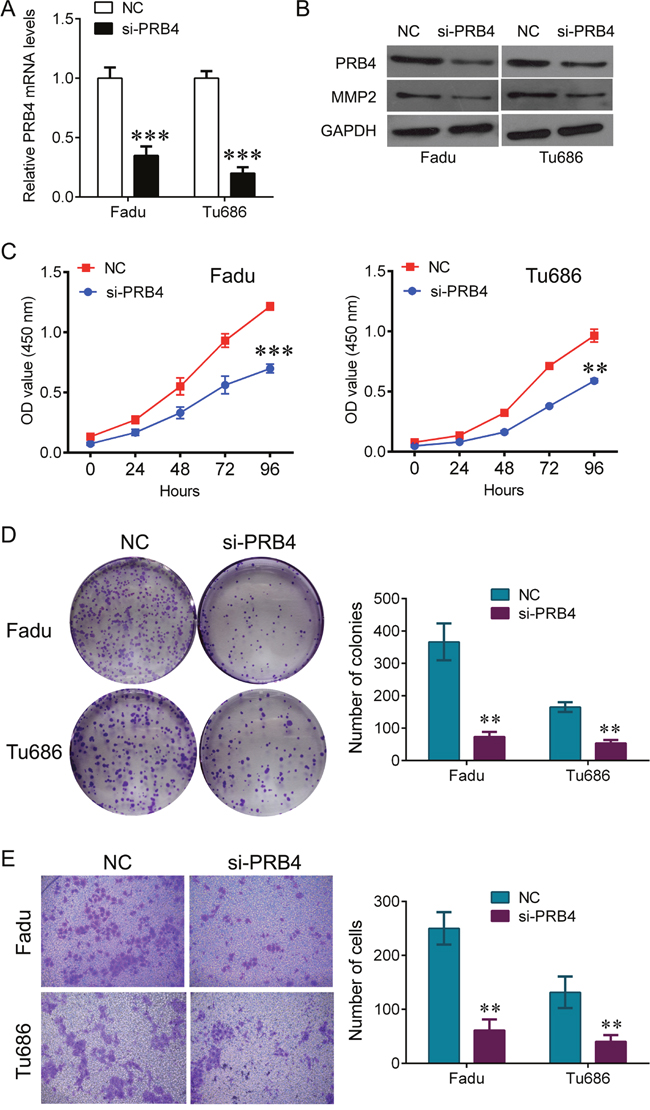
Figure 4: knockdown of PRB4 represses Fadu and Tu686 cells growth and invasion. (A) QPCR detected the expression of PRB4 in Fadu and Tu686 cells after si-PRB4 transfection. (B) Western blot detected the expression of PRB4 and MMP2 in Fadu and Tu686 cells after si-PRB4 transfection. (C) CCK-8 was used to measure the cells proliferation in Fadu and Tu686 cells after si-PRB4 transfection. (D) Colony formation assay was used to measure the ability of colony formation in Fadu and Tu686 cells after si-PRB4 transfection. (E) Transwell assay was used to measure the ability of invasion in Fadu and Tu686 cells after si-PRB4 transfection. Data are expressed as mean ± standard deviation. **P<0.01, ***P<0.001 vs. negative control.
NSD1 epigenetically activates PRB4 expression and PI3K signaling
Due to the positive correlation between NSD1 and PRB4, we further investigated whether NSD1, a histone transmethylase, could regulate the expression of PRB4. We found that knockdown of NSD1 could significantly decrease the mRNA and protein levels of PRB4, whereas overexpression of NSD1 increased the expression of PRB4 (Figure 5). Interestingly, knockdown of NSD1 significantly inactivated PI3K signaling evaluated by decreased PI3K and phosphorylated Akt expression and increased Bad expression. Overexpression of NSD1 exhibited the opposite effects on PI3K signaling (Figure 5). We further investigated the mechanism by which NSD1 regulates PRB4. We performed MSP to examine whether NSD1 could regulate the methylation status of PRB4 promoter. And we found that NSD1 overexpression hypermethylated PRB4 promoter in Fadu and Tu686 cells (Figure 6). ChIP assay was performed to evaluate whether NSD1 could bind to PRB4 promoter. The results showed that NSD could bind to the promoter regions of PRB4 (Figure 7A). In addition, the promoter region (2000 bp) of PRB4 was inserted into a PGL3 luciferase reporter vector, and dual-Luciferase reporter analysis showed that NSD1 could bind to this region and activate luciferase (Figure 7B). Moreover, we also found that overexpression of NSD1 could reduce the binding of H3K27me2 on PRB4 promoter, while increase the binding of H3K36me2 on PRB4 promoter (Figure 7C). However, knockdown of NSD1 reversed these bindings in Fadu and Tu686 cells (Figure 7D). These results indicate that PRB4 upregulation in hypopharyngeal cancer may be epigenetically activated by NSD1.
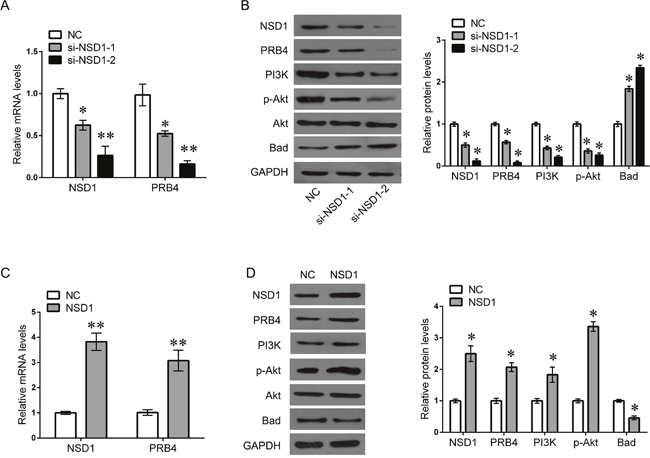
Figure 5: NSD1 activates PRB4 and PI3K signaling. (A) QPCR detected the expression of PRB4 and NSD1 in Fadu cells after si-NSD1 transfection. (B) Fadu cells were transfected with si-NSD1 for 48 h. And then the expression of NSD1, PRB4, PI3K, Akt, p-Akt and Bad protein were measured by western blot, and quantification. (C) QPCR detected the expression of PRB4 and NSD1 in Fadu cells after NSD1 transfection. (D) Fadu cells were transfected with NSD1 for 48 h. And then the expression of NSD1, PRB4, PI3K, Akt, p-Akt and Bad protein were measured by western blot, and quantification. Data are expressed as mean ± standard deviation. *P<0.05, **P<0.01 vs. negative control.
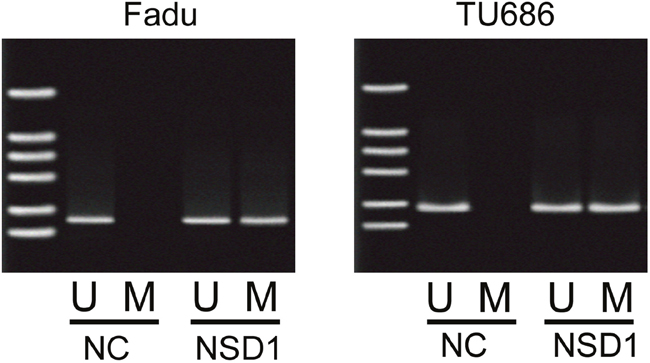
Figure 6: The methylation levels of promoter of PRB4 in Fadu and Tu686 cells. MSP assay was performed to determine the methylation levels of promoter of PRB4 after overexpressing NSD1. U, unmethylation; M, methylation.
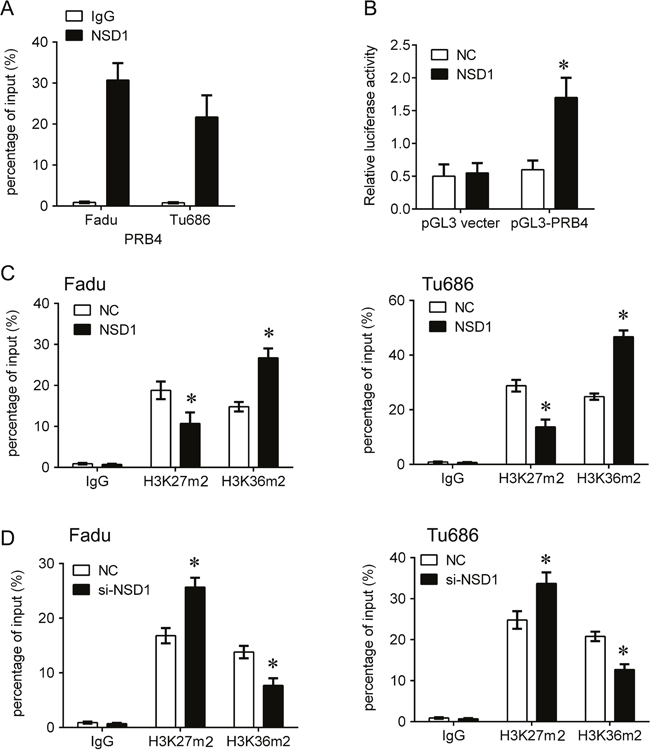
Figure 7: NSD1 epigenetically activates PRB4 by recruiting H3K36m2. (A) ChIP-qPCR analysis of NSD1 occupancy in the PRB4 promoter regions in the Fadu and Tu686 cells. IgG was used as a negative control. (B) Luciferase reporter analysis of luciferase activity in the Fadu cells cotransfected with pGL3-PRB4 and NSD1 lentivirus or an empty lentivirus. (C, D) ChIP-qPCR analysis of H3K27m2 and H3K36m2 occupancy in the PRB4 promoters in the Fadu and Tu686 cells after transfection with NSD1 lentivirus (C) or si-NSD1 (D). IgG was used as a negative control. *P < 0.05 vs. negative control.
DISCUSSION
In order to better understand the mutations in hypopharyngeal carcinoma, WES was performed in 23 paired tumor and adjacent normal tissues. Our study identifies novel candidate driver genes associated with hypopharyngeal carcinoma. These genes are different from earlier exome-sequencing studies on head and neck squamous cell carcinoma with lower mutation frequency and higher C>T transition predominantly occurred at CpG sites [7, 12], while are similar with esophageal squamous cell carcinoma, which may be due to the same histological origin in esophagus and hypopharyngeal carcinoma and share the same risk factors (alcohol and gastroesophageal reflux).
Moreover, we identified several copy number variants (CNVs) genes were associated with hypopharyngeal carcinoma, one with amplification (ATF1) and two with deletions (CDKN2A, CDKN2B). CDKN2A and CDKN2B encode protein p16INK4a and p15INK4b respectively, which belong to a family of cyclin-dependent kinases (CDKs) inhibitor proteins that can inactivate CDKs [13]. Both of proteins can specially bind to CDK4/CDK6 and subsequently induce an allosteric conformational change to inhibit the formation of the CDK4/6 complex and cyclin D, leading to G1 phase cell cycle arrest [14]. They therefore potentially act as tumor suppressors, and their inactivation corresponds to human carcinogenesis. The loss of CDKN2A and CDKN2B was observed in various cancers through deletion, inactivating mutations, epigenetic silencing or post-translational modification [15, 16]. In this study, CDKN2A was found down-regulated expression (61.0%) and point mutation (4.5%), suggesting CDKN2A and CDKN2B deficiency and inactivation may contribute to hypopharyngeal carcinoma progression.
In addition, we identified 15 SMGs associated with hypopharyngeal carcinoma. Among these genes, TP53 and PIK3CA are well-known cancer-associated genes. TP53 is known as the ‘‘gatekeeper’’ gene in the cellular defense against genotoxic damage and functions as a critical tumor suppressor. We found that nearly half of hypopharyngeal carcinoma (45.5%) has TP53 mutations, supporting that the loss of function of TP53 is a key initiating or tumor promoting event in hypopharyngeal carcinoma. PIK3CA gene, an important molecule in the PI3K (RAS/PI3K/AKT) pathway, is the main genetic driving force of this pathway in human cancers. Mutations of PIK3CA were found in 13.6% of hypopharyngeal carcinoma, and the E545K and H1047R mutant sites, two of hotspots mutations of PIK3CA, have been demonstrated that they can increase PIK3CA kinase activity and promote cellular transformation [17, 18]. These findings indicate aberrantly PIK3CA activation is another fundamental mechanism in hypopharynx tumorigenesis.
We also discovered nine new genes (PRB4, NSD1, REC8, ZNF772, ZNF69, EI24, CYFIP2, NEFH, KRTAP4-5) associated with hypopharyngeal carcinoma. Proline-rich proteins are the most conserved oral salivary proteins among mammals. The high polymorphism of proline-rich proteins gives an important contribution to the high heterogeneity, possibly involved in different biological activities [19]. Mutations of proline rich protein BstNI subfamily (PRB) 4 and 1 were found in primary gastric cancer and matched peritoneal metastatic cancer tissues [20]. However, the functions of PRB4 and NSD1 (nuclear receptor binding SET domain protein 1) are not clear in hypopharyngeal carcinoma. Their expression was significantly upregulated in hypopharyngeal carcinoma estimated by WES, which was confirmed in an independent cohort using IHC. There was a positive relationship between PRB4 and NSD1. We knocked down PRB4 expression hypopharyngeal carcinoma cells, and found that downregulation of PRB4 could inhibit cell growth, colony formation and cell invasion, and we speculate that the mutations in this gene enhanced it oncogenic effects. NSD1 is a SET-domain histone methyltransferase that methylates lysine 36 of histone 3 (H3K36) [21], which is associated with chromosomal instability by methylation [22]. Initially, NSD1 mutations were found to lead to the Sotos syndrome. More recently, many studies show that the NSD1 were overexpressed, amplified or somatically mutated in multiple types of cancer, suggesting their critical role in cancer [23, 24]. Its dysfunction results in epigenetic aberrations which play a fundamental role in oncogenesis [25]. Notably, we here demonstrate that NSD1 positively regulates PRB4 expression and PI3K signaling. Further mechanism investigation reveals that NSD could bind to the promoter regions of PRB4 and activate promoter activity by reducing the binding of H3K27me2 and increasing the binding of H3K36me2 on PRB4 promoter. These results indicate that PRB4 upregulation in hypopharyngeal cancer may be epigenetically activated by NSD1. Therefore, PRB4 and NSD1 mutations might contribute to hypopharyngeal carcinoma tumorigenesis.
In summary, we pinpoint the predominant underlying mutational processes in hypopharyngeal carcinoma by WES, highlighting the substantial genetic alterations contributing to hypopharyngeal carcinoma tumorigenesis. We also indentify a novel epigenetically regulatory between PRB4 and NSD1 that contribute to hypopharyngeal carcinoma tumorigenesis. They may become potential prognostic biomarkers and therapeutic target for hypopharyngeal carcinoma treatment.
MATERIALS AND METHODS
Samples and clinical data
This study was approved by the Xiangya Ethics Committee of the Central South University. Written informed consent was obtained from individual patients who participated in the study. All human frozen and paraffin-embedded tissues as well as clinical information were obtained from The Xiangya Hospital of Central South University. Twenty-four tumor tissues and matched adjacent tissues obtained from patients with hypopharyngeal carcinoma from January 2011 to December 2015 were used for whole exome sequencing (WES) analysis. After resection, samples were immediately preserved in liquid nitrogen. The tissues were verified using routine H&E staining by pathologists. The cancer tissues containing more than 90% squamous cells and their paired adjacent tissues containing more than 95% normal cells were included for whole-exome sequencing. One pair of samples was excluded due to low quality, and 23 pairs of tumor-adjacent frozen biopsies were applied for WES. Another independent cohort containing 88 tumor tissues and 36 normal adjacent tissues was used for verification of candidate genes by immunohistochemistry (IHC) analysis.
Whole-exome sequencing
Genomic DNA samples were extracted from tumor and adjacent tissues obtained from patients with hypopharyngeal carcinoma using TIANamp Genomic DNA Kits (Qiagen, Valencia, CA, USA). Qubit® 2.0 Fluorometer (Life technologies) was used to evaluate DNA quality and quantity. Two micrograms of genomic DNA from each sample was used for generating the sequencing library using the Agilent SureSelect Library Prep Kit according to the manufacturer’s protocol. Exome regions were captured and enriched with the SeqCap EZ Human Exome Library v3.0 (Nimblegen). Captured DNAs were subjected to high-throughput sequencing using an Illumina HiSeq 2000/2500 platform to generate 101 bp paired-end reads.
Variant calling and prioritization
Base calling was performed using the Real-Time Analysis software (Illumina). The whole-exome sequencing data were analyzed with the following criterion of quality control:1) reads contaminated by adapter sequences together with their mate pair reads were removed; 2) reads with more than 50% of low-quality bases(Q<5) were removed; and 3) reads with a high N rate (>0.1) and their mate pair reads were removed. BWA (Burrows-Wheeler Aligner) was used to align clean reads against to the human reference assembly hg19. SAM tools were used for conversion of SAM files to BAM files. Reads with multiple mapping loci in the genome and reads with more than 3 mismatches, with more than 1 gap, or with a gap more than 20 bases long were removed. More than one file from a patient was merged for further genomic analysis. The duplicated reads derived from PCR amplification were marked using Picard tools (http://broadinstitute.github.io/picard/). Local realignments and base quality recalibrations were performed using Genome Analysis Toolkit (GATK).
Statistical inferences on nature of genomic alteration
The Mutect algorithm and VarScan algorithm were applied to identify somatic single-nucleotide variants (SNV) in targeted regions. All these SNVs were annotated with Oncotator (http://www.broadinstitute.org/cancer/cga/oncotator/). SNVs were filtered according the following conditions:
1) SNV was detected by more than one software; 2) Filter SNV: alternative allele depth >=5 and total depth >= 30 in cancer and total depth >= 30 in normal, 1000 genome was freq<=0.05; AndVarScan was used to identify somatic Indels with the default parameters and subjected to filter with the following criterion of somatic indels: Varscan Somatic and Total Detph>= 50 in Normal and Tumor Sample.
Copy number variation
To identify CNAs (copy number alterations), VarScan was used to compare read depths between tumors and matched adjacent samples for contiguous regions of coverage. And then a circular binary segmentation (CBS) algorithm was used to delineate segments using DNA copy package, pick out significant change-points and merge these regions with mergeSegment.pl.
Immunohistochemistry (IHC) and staining evaluation
IHC staining was performed as previously described [10, 11]. Tissues sections were deparaffinized in xylene and rehydrated in alcohol, and then pretreated with citrate buffer (10 mmol/l, pH 6.0) for 20 min at 100°C in a microwave oven; 3% hydrogen peroxide was used to block endogenous peroxidase activity for 15 min at room temperature. After that, nonspecific binding sites were blocked by 10% normal goat serum for 30 min at 37°C. The sections were incubated with antibody (rabbit polyclonal anti-PRB4, 1:200 dilution, rabbit monoclonal anti-NSD1 1:200 dilution, Abcam) overnight at 4°C. The sections incubated with PBS instead of the primary antibody were used as negative controls. Sections were incubated with biotinylated goat anti-rabbit IgG (1:1000 dilution, Zhongshan Chemical) for 20 min at 37°C after rinsing with PBS. Finally, tissue sections were incubated with 3', 3'-diami-nobenzidine (DAB; Maixin, Fuzhou), then counterstained with Harris modified hematoxylin (Zhongshan Chemical). For evaluating the expression of PRB4 and NSD1 in tissues, the integral optical density (IOD) was obtained by ImageJ (National Institutes of Health, USA). To compare the expression of PRB4 and NSD1 between adjacent and tumor tissues, the IOD of PRB4 and NSD1 expression was normalized to the average score of them in normal tissues. The IOD of PRB4 and NSD1 higher than their mean IOD in adjacent tissues was identified as high expression, otherwise as low expression.
Cell lines
Two kinds of hypopharyngeal carcinoma cell lines, Fadu and Tu686 cells, were obtained from Cell Bank of Chinese Academy of Sciences (Shanghai, China). All cells were grown in DMEM/F-12 (Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Gibco) at 37°C in 5% CO2. Knockdown of PRB4 and NSD1 in Fadu and Tu686 cells was achieved by transfection with lentivirus containing PRB4 siRNA or NSD1 siRNA (si-PRB4, si-NSD1-1, siNSD1-2, Genepharma, Shanghai, China) using Lipofectamine 2000 (Invitrogen, CA, USA). Overexpression of NSD1 was achieved by using lentivirus containing NSD1 expressed plasmid (GeneCopoeia, Guangzhou, China) using Lipofectamine 2000 (Invitrogen, CA, USA). The cells transfected with empty lentivirus were used as negative control. Cells were plated in 6-well clusters or 96-well plates and transfected for 24 h or 48 h. Transfected cells were used in further assays or protein extraction.
RNA extraction and SYBR green quantitative PCR analysis
Total RNA was extracted from cells using Trizol reagent (Invitrogen, CA, USA). The expression of PRB4 and NSD1 was measured by SYBR green qPCR assay (Takara, Dalian, China) according to manufacturers’ instructions. Expression of β-actin was used as an endogenous control. The primers were used as followings: PRB4, sense, CCAGTCCCAAGGAAAGCCAC, anti-sense, CCTTGTTCCAATG TCACGGC; NSD1, sense, GGATGGATCAGACCTGTGAACT, anti-sense, TCTGG ATCATCCGAAAGGGCTG; β-actin, sense, AGGGGCCGGACTCGTCATACT, anti-sense, GGCGGCACCACCATGTACCCT. QPCR was performed at the condition: 95.0 °C for 3 min, and 39 circles of 95.0 °C for 10s and 60 °C for 30 s. Data were processed using 2-ΔΔCT method.
Western blotting
Cultured or transfected cells were lysed in RIPA buffer with 1% PMSF. Western blot was performed on 10% SDS-PAGE using Mini-PROTEAN® Tetra Cell Systems (Bio-Rad). Proteins were transferred onto polyvinylidine difluoride (PVDF) membranes (Immobilon, Millipore). Membranes were incubated with anti-PRB4 rabbit polyclonal antibody (Abcam), anti-NSD1 rabbit monoclonal antibody (Abcam) or PI3K rabbit monoclonal antibody (Cell Signaling) or Akt mouse monoclonal antibody (Cell Signaling) or phospho-Akt rabbit monoclonal antibody (Cell Signaling) or Bad Rabbit monoclonal antibody (Cell Signaling) at 1:1000 dilution, or GAPDH specific antibody (Sigma-Aldrich) at 1:5000 dilution at 4°C overnight. Signals were visualized using ECL Substrates (Millipore, MA, USA).
Cell viability assay
Cell viability was detected using 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5- (2,4-disulfophenyl)-2H-tetrazolium, monosodium salt (CCK-8 Kit; Beyotime biotech Co. Ltd., Hangzhou, China). CCK-8 solution (10 μl) was added to each well of 96-well plates with the same amount of culture fluid. CCK-8 solution without cells was used as blank control. Optical density was determined at the time points on a microtiter plate reader at 450 nm.
Invasion assay
After indicate treatments, equal cell numbers of the Fadu (1×106 cells/chamber) or Tu686 (0.5×106 cells/chamber) cell lines for each condition were placed in deprivation media in the upper chamber (8.0 μ m pores, Corning, Inc) coated with Matrigel (BD Biosciences). Deprivation media was placed in the lower chamber. Cells were allowed to invade for 48 h. The cells remaining in the upper chamber were removed from the upper chamber by a cotton swab. After fixation with 4 % formalin and staining with Differential Quik Stain (Polysciences, Inc), invaded cells were acquired on an inverted Olympus IX81 microscope (Olympus, Tokyo, Japan). To evaluate the invasive ability, the invaded cells were counted.
Colony formation assay
Following a 48 hour treatment with PRB4 siRNA, cells were seeded at 300 cells per well in 6-well plates containing complete DMEM/F-12 and incubated at 37°C and 5% CO 2 for 15 days. On day 15, cells were fixed with 4% polyformaldehyde for 15 min and stained with 1% crystal violet. The experiments were performed in triplicate, and the numbers of colonies containing more than 50 cells were counted.
Measurement of PRB4 promoter methylation status by methylation specific PCR (MSP)
Genomic DNA was extracted using the Qiagen FFPE DNA Kit (Qiagen, CA, USA). Genomic DNA (1 μg per sample) was modified with bisulfite using the EZ DNA Methylation-Gold Kit (Zymo, Orange County, CA, USA) according to the manufacturer's instructions. Methylation-specific PCR (MSP) was performed on bisulfate-treated DNA. The primers used were un-methylated PRB4 forward, TAATATGATATTGTATGGTTTTTGT, and reverse, AACTACCCCACAACCTACTCAAA; and methylated PRB4 forward: TGGTAATATGATATCGTATGGTTTTC, and reverse, AAACTACCCCACAACCTACTCG. The annealing temperature was 67.5 °C for methylated-PCR and 65°C for un-methylated-PCR, with 35 cycles used for each.
Luciferase reporter assay
The PRB4 promoter region (2000 bp) was synthesized and inserted into a pGL3-basic vector (Promega, Madison, WI, USA). The successful constructs were verified by DNA sequencing. The Dual-Luciferase Assay Kit was used to assess luciferase activities, following manufacturer’s protocol. The cells were plated in 96-well clusters, then cotransfected with 100 ng pGL3-basic vector or pGL3-PRB4, together with NSD1 or negative control. At 48 h after transfection, luciferase activity was detected using a dual-luciferase reporter assay system (Promega, Madison, WI) and normalized to Renilla activity.
RNA immunoprecipitation (RIP)
RIP assay was used to determine whether PRB4 interacts with or binds to RNA-binding proteins NSD1 in the human hypopharyngeal carcinoma cells. The EZMagna RIP kit (Millipore, Billerica, MA, USA) was used to conduct the RIP experiment, following manufacturer’s protocol. The Fadu and Tu686 cells were lysed using complete RIP lysis buffer; then, the extract was incubated with magnetic beads conjugated with NSD1antibodies or control IgG (Millipore) for 8 h at 4 °C. Next, the beads were washed with washing buffer and incubated with proteinase K at 55 °C for 30 min to remove the proteins. Finally, purified RNA was reverse-transcribed into cDNA and subjected to qPCR analysis to determine the presence of PRB4 using specific primers.
Chromatin immunoprecipitation (ChIP) assay
The EZ-Magna ChIP kit (EMD Millipore) was used to conduct the ChIP assays in accordance with manufacturer’s protocol, the Fadu and Tu686 cells were fixed with 4% paraformaldehyde and incubated with glycine for 10 min to generate DNA–protein cross-links. Then, the cells were lysed with Cell Lysis Buffer and Nuclear Lysis Buffer and sonicated to generate chromatin fragments. Next, the lysates were immunoprecipitated with Magnetic Protein A Beads conjugated with H3K27me2 (Millipore), or H3K36me2-specific antibodies (Millipore), or IgG as a control. Finally, the precipitated DNA was analyzed by qRT-PCR.
Statistical analysis
The SPSS Statistics 17.0 package was used to analyze data. All data from 3 independent experiments were expressed as mean ± SD. Chi-square test was used to analyze the association between the level of PRB4 and NSD1 and clinicopathological parameters of hypopharyngeal cancer. Student’s t test was used for statistical analysis in two groups, and data from more than two groups were analyzed by one-way ANOVA. Overall survival (OS) rate was determined by the Kaplan-Meier curve with log-rank test. Cox proportional hazard regression model was used to estimate the independent predicators for the prognosis of hypopharyngeal cancer patients. p< 0.05 was statistically significant.
Author contributions
Ping Wu, Honglong Wu, Yaoyun Tang, Shi Luo, Xing Fang, Chubo Xie, Jian He, Xi Chen, Dongfang Li Huanming and Yang Jian Wang performed clinical sample collection and in vitro experiments, and wrote this paper.
Suping Zhao, Xiaofeng Wang and Jiajia Xu performed in vivo experiments.
Ping Wu and Yaoyun Tang contributed to study design, data analysis, and paper modification.
CONFLICTS OF INTEREST
No conflicts of interest exits in this study.
FUNDING
This work was supported by The National Natural Science Foundation of China (no. 81302355), and Xiangya Famous Doctors Foundation.
REFERENCES
1. Newman JR, Connolly TM, Illing EA, Kilgore ML, Locher JL, Carroll WR. Survival trends in hypopharyngeal cancer: a population-based review. Laryngoscope. 2015; 125:624-629.
2. Hall SF, Griffiths R. Did the addition of concomitant chemotherapy to radiotherapy improve outcomes in hypopharyngeal cancer? A population-based study. Curr Oncol. 2016; 23:266-272.
3. Chan JY, Wei WI. Current management strategy of hypopharyngeal carcinoma. Auris Nasus Larynx. 2013; 40:2-6.
4. Lu W, Feng L, Zhang Y, Ma Y, Li P, Wang Y, Du Y, Chen X, Wu S, Zhao G, Lou W. miR-15a induces cell apoptosis by targeting BCL2L2 and BCL2 in HPV-positive hypopharyngeal squamous cell carcinoma. Oncol Rep. 2016; 36:2169-2176.
5. Wu P, Quan H, Kang J, He J, Luo S, Xie C, Xu J, Tang Y, Zhao S. Downregulation of calcium binding protein S100A9 inhibits hypopharyngeal cancer cell proliferation and invasion ability through inactivation of NFkappaB signaling. Oncol Res. 2017.
6. Cao W, Wu W, Yan M, Tian F, Ma C, Zhang Q, Li X, Han P, Liu Z, Gu J, Biddle FG. Multiple region whole-exome sequencing reveals dramatically evolving intratumor genomic heterogeneity in esophageal squamous cell carcinoma. Oncogenesis. 2015; 4:e175.
7. Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, Kryukov GV, Lawrence MS, Sougnez C, McKenna A, Shefler E, Ramos AH, Stojanov P, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011; 333:1157-1160.
8. Sawada G, Niida A, Uchi R, Hirata H, Shimamura T, Suzuki Y, Shiraishi Y, Chiba K, Imoto S, Takahashi Y, Iwaya T, Sudo T, Hayashi T, et al. Genomic landscape of esophageal squamous cell carcinoma in a Japanese population. Gastroenterology. 2016; 150:1171-1182.
9. Huang KK, Jang KW, Kim S, Kim HS, Kim SM, Kwon HJ, Kim HR, Yun HJ, Ahn MJ, Park KU, Ramnarayanan K, McPherson JR, Zhang S, et al. Exome sequencing reveals recurrent REV3L mutations in cisplatin-resistant squamous cell carcinoma of head and neck. Sci Rep. 2016; 6:19552.
10. Luo Y, Wang X, Wang H, Xu Y, Wen Q, Fan S, Zhao R, Jiang S, Yang J, Liu Y, Li X, Xiong W, Ma J, et al. High bak expression is associated with a favorable prognosis in breast cancer and sensitizes breast cancer cells to paclitaxel. PLoS One. 2015; 10:e138955.
11. Luo Y, Wang X, Niu W, Wang H, Wen Q, Fan S, Zhao R, Li Z, Xiong W, Peng S, Zeng Z, Li X, Li G, et al. Elevated microRNA-125b levels predict a worse prognosis in HER2-positive breast cancer patients. Oncol Lett. 2017; 13:867-874.
12. Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, Fakhry C, Xie TX, Zhang J, Wang J, Zhang N, El-Naggar AK, Jasser SA, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in Notch1. Science. 2011; 333:1154-1157.
13. Nabel EG. CDKs and CKIs: molecular targets for tissue remodelling. Nat Rev Drug Discov. 2002; 1:587-598.
14. Singh SK, Banerjee S, Acosta EP, Lillard JW, Singh R. Resveratrol induces cell cycle arrest and apoptosis with docetaxel in prostate cancer cells via a p53/p21WAF1/CIP1 and p27KIP1 pathway. Oncotarget. 2017; 8:17216-17228. https://doi.org/10.18632/oncotarget.15303.
15. Chung CH, Guthrie VB, Masica DL, Tokheim C, Kang H, Richmon J, Agrawal N, Fakhry C, Quon H, Subramaniam RM, Zuo Z, Seiwert T, Chalmers ZR, et al. Genomic alterations in head and neck squamous cell carcinoma determined by cancer gene-targeted sequencing. Ann Oncol. 2015; 26:1216-1223.
16. McNeal AS, Liu K, Nakhate V, Natale CA, Duperret EK, Capell BC, Dentchev T, Berger SL, Herlyn M, Seykora JT, Ridky TW. CDKN2B loss promotes progression from benign melanocytic nevus to melanoma. Cancer Discov. 2015; 5:1072-1085.
17. Samuels Y, Diaz LJ, Schmidt-Kittler O, Cummins JM, Delong L, Cheong I, Rago C, Huso DL, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell. 2005; 7:561-573.
18. Davoli T, Mengwasser KE, Duan J, Chen T, Christensen C, Wooten EC, Anselmo AN, Li MZ, Wong KK, Kahle KT, Elledge SJ. Functional genomics reveals that tumors with activating phosphoinositide 3-kinase mutations are dependent on accelerated protein turnover. Genes Dev. 2016; 30:2684-2695.
19. Manconi B, Castagnola M, Cabras T, Olianas A, Vitali A, Desiderio C, Sanna MT, Messana I. The intriguing heterogeneity of human salivary proline-rich proteins: Short title: Salivary proline-rich protein species. J Proteomics. 2016; 134:47-56.
20. Zhang J, Huang JY, Chen YN, Yuan F, Zhang H, Yan FH, Wang MJ, Wang G, Su M, Lu G, Huang Y, Dai H, Ji J, et al. Whole genome and transcriptome sequencing of matched primary and peritoneal metastatic gastric carcinoma. Sci Rep. 2015; 5:13750.
21. Graham SE, Tweedy SE, Carlson HA. Dynamic behavior of the post-SET loop region of NSD1: Implications for histone binding and drug development. Protein Sci. 2016; 25:1021-1029.
22. Lee ST, Wiemels JL. Genome-wide CpG island methylation and intergenic demethylation propensities vary among different tumor sites. Nucleic Acids Res. 2016; 44:1105-1117.
23. Papillon-Cavanagh S, Lu C, Gayden T, Mikael LG, Bechet D, Karamboulas C, Ailles L, Karamchandani J, Marchione DM, Garcia BA, Weinreb I, Goldstein D, Lewis PW, et al. Impaired H3K36 methylation defines a subset of head and neck squamous cell carcinomas. Nat Genet. 2017; 49:180-185.
24. Lavallee VP, Lemieux S, Boucher G, Gendron P, Boivin I, Girard S, Hebert J, Sauvageau G. Identification of MYC mutations in acute myeloid leukemias with NUP98-NSD1 translocations. Leukemia. 2016; 30:1621-1624.
25. Vougiouklakis T, Hamamoto R, Nakamura Y, Saloura V. The NSD family of protein methyltransferases in human cancer. Epigenomics-UK. 2015; 7:863-874.