
INTRODUCTION
The dithiocarbamate disulfiram (DSF) has been used for several decades as a deterrent in the treatment of alcohol addiction [1]. Recently, the role of DSF in the treatment of cancer has been recognised although its mechanism of action has not been established. DSF has been shown to inhibit proteasome activity [2], angiogenesis [3], P-glycoprotein (P-gp) [4] and aldehyde dehydrogenase [5, 6]. Furthermore, DSF generates oxidative stress through the inhibition of NF-κB activation [7], by inhibition of superoxide dismutase (SOD) [8] and as a result of its induction of an increase in the ratio of oxidised glutathione to its reduced form [9]. It has also been shown that copper is required for DSF cytotoxicity [10] and that DSF acts as a copper ionophore [11, 12]. Therefore, intracellular copper deposition could be cytotoxic through the generation of oxidative stress resulting from Fenton reactions [producing cupric ions and hydroxyl radicals] or through enzymatic inhibition by virtue of copper’s binding to peptidic bonds. Moreover, copper complexes have been shown to intercalate between the base pairs of DNA owing to their planar conformation [13-15]. These reports raise the possibility that copper confers upon DSF a favourable conformation for DNA intercalation, resulting in alteration to the structure of DNA and cell death. It has been suggested that DSF and its reduced form, diethyldithiocarbamate, inhibit DNA synthesis through inhibition of DNA polymerase and ribonucleotide reductase [16, 17]. Finally, topoisomerase I, an enzyme whose activity relieves torsional stress generated at the replication fork during DNA replication, has been shown to be inhibited by DSF via binding to critical thiol residues [18]. A similar mechanism of inhibition of DNA synthesis has been reported for the nucleotide analogue gemcitabine which interacts with ribonucleotide reductase via cysteine [19].
Therefore, the cytotoxicity of the disulfiram and copper complex (DSF:Cu) may result from the generation of oxidative stress, inhibition of DNA replication or modulation of the activity of other critical cell regulatory pathways. Despite the multiplicity of potential targets, DSF-induced side-effects are reversible upon cessation of treatment and have been safely managed clinically since 1948 [20, 21].
DSF and its dithiocarbamate derivatives have been shown to be radioprotectors as measured by increased survival in yeast [22], decreased clonogenic cell kill [23], a longer survival in mice [24] and by a reduction in lipid peroxidation and DNA damage [25]. In contrast, dithiocarbamate derivatives have been reported to be radiosensitisers as measured by increased tumour growth delay in mice [26], increased clonogenic cell kill [23, 27-28] and by increased erythrocyte heamolysis [29]. However, these studies employed inappropriately high, hence clinically irrelevant, concentrations of DSF in experimental conditions uncontrolled for the presence of copper. Recently, we have shown that clinically pertinent concentrations of DSF:Cu sensitised tumour cells grown as monolayers, spheroids or xenografts to γ-radiation and radiopharmaceutical treatments [10]. The mechanism of radiosensitisation remains unknown. We hypothesised that DSF:Cu sensitised cancer cells to ionising radiation treatment via the modulation of cell cycle progression.
RESULTS
Proteasome-independent radiosensitisation by DSF:Cu
DSF:Cu enhanced the clonogenic cell kill induced by ionising radiation in SK-N-BE(2c) (Figure 1A) and UVW cells (Figure 1B). The IC50 values following irradiation decreased 2.64-fold or 2.84-fold in the presence of 0.3 μM DSF:Cu in SK-N-BE(2c) or UVW cells, respectively. The interaction between DSF:Cu and ionising radiation, as evaluated by isobologram analysis, was synergistic in SK-N-BE(2c) cells (Figure 1C) and additive in UVW cells (Figure 1D). Crucially, we previously reported that Cu alone does not influence radiosensitivity [10].
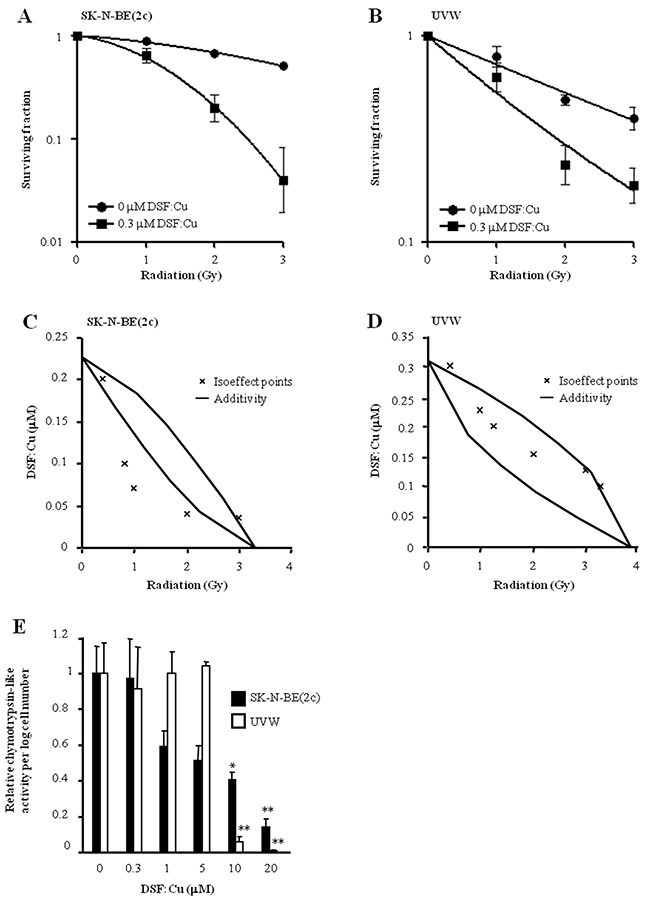
Figure 1: The effect of DSF:Cu on cellular radiosensitivity. SK-N-BE(2c) (A) and UVW cells (B) were exposed to 0.3 μM DSF:Cu for 24 h and irradiated at the start of exposure to DSF:Cu. Data are means ± SEM, n=3. The nature of the interaction between IR and DSF:Cu was evaluated by isobologram analysis using SK-N-BE(2c) (C) and UVW cells (D). (E) The effect of DSF:Cu on the chymotrypsin-like activity of the proteasome was evaluated. Data are means ± SEM, n=3. The independent t-test was used to compare the means of test groups with those of untreated control: *P<0.05; **P<0.01.
It has been shown that DSF:Cu inhibits proteasome activity [2, 30]. However, in SK-N-BE(2c) and UVW cells, proteasome activity was not inhibited following radiosensitising treatment consisting of 0.3 μM DSF:Cu (Figure 1E). This suggested that proteasome inhibition was not required for radiosensitisation by DSF:Cu. Together, these results confirmed the previously reported radiosensitising property of DSF:Cu [10], but indicated that a mechanism other than proteasome inhibition is responsible. The absence of a correlation between proteasome activity inhibition by DSF:Cu and cytotoxicity has also been suggested by others [31].
DSF:Cu inhibits DNA synthesis
Both SK-N-BE(2c) and UVW cells were arrested in early S phase by thymidine treatment. Their synchronous progression through S phase following release from thymidine block was indicated by the increase in propidium iodide intensity over 8 h (Figure 2A). DSF:Cu inhibited S phase progression (Figure 2A) and bromodeoxyuridine (BrdU) incorporation (Figure 2B). For instance, 8 h following release from the thymidine block, the proportion of G2 cells that incorporated BrdU was decreased 4.5-fold (P<0.001) in SK-N-BE(2c) cells and 1.6-fold in UVW cells by treatment with DSF:Cu (Figure 2B).
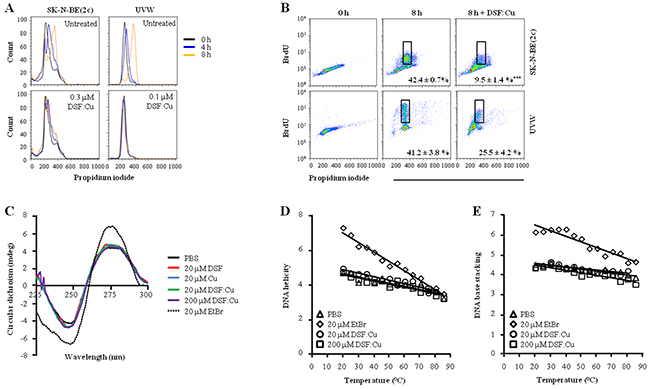
Figure 2: The effect of DSF:Cu on DNA synthesis. Following release from thymidine block, synchronised SK-N-BE(2c) and UVW cells were exposed for 8 h to 0.3 or 0.1 μM DSF:Cu, respectively. The effect of DSF:Cu on DNA synthesis was monitored by flow cytometric analysis of propidium iodide-stained (A) or BrdU-stained (B) synchronised cells. The data are representatives of triplicate determinations. The proportions of BrdU-positive cells in G2 are means ± SEM, n=3. Independent t-test: *** P<0.001. (C) The interaction between DSF:Cu and DNA was investigated by circular dichroism (CD) using treatment with ethidium bromide (EtBr) as a positive control. (D, E) The effect of DSF:Cu on the stability of the DNA molecule in response to heat treatment was investigated by CD. The absolute CD values at 245 nm (D, nadir of the negative wave) and at 275 nm (E, zenith of the positive wave) were plotted against temperature to measure change in helicity and base stacking, respectively.
Copper complexes have been shown to be cytotoxic via binding to DNA [13-15]. Moreover, it has been suggested that DSF:Cu is a planar molecule [32], raising the possibility that DSF:Cu impedes DNA replication via insertion between the base pairs of DNA. Accordingly, we tested whether DSF:Cu could interact with DNA using circular dichroism (CD). In contrast to the known DNA intercalator ethidium bromide (EtBr), DSF:Cu did not modify the helicity nor the base stacking of calf thymus-DNA, as indicated by the lack of modification of the negative and positive waves of the CD spectrum, respectively (Figure 2C). Furthermore, the stability of calf thymus-DNA was tested by thermal denaturation in the presence or in the absence of DSF:Cu in order to determine whether interaction between DSF:Cu and calf thymus-DNA could occur via a mechanism other than intercalation. In control conditions without DSF:Cu, both helicity (Figure 2D) and base stacking (Figure 2E) of the DNA helix were lost as temperature increased. In contrast to the consequence of EtBr treatment, the rate of loss of DNA conformation due to increasing temperature was unaffected by DSF:Cu (Figure 2D and 2E). These results suggested that interaction between DSF:Cu and DNA is unlikely and that an alternative mechanism is responsible for the inhibition of DNA synthesis by DSF:Cu. One possibility is a negative regulatory impact of DSF:Cu upon enzymes responsible for DNA synthesis.
DSF:Cu does not abrogate G2 arrest induced by ionising radiation
We next evaluated whether DSF:Cu affected cell cycle progression through the G2 phase of the cell cycle. Exposure to DSF:Cu for 12 h resulted in increased proportions of SK-N-BE(2c) and UVW cells in G2-M from 30.5% to 34.5% and from 23.0% to 33.9% (P<0.01), respectively (Table 1), indicating G2 arrest. This conclusion was supported by the observed decrease in the level of expression of total CDK1 following treatment of SK-N-BE(2c) (P<0.001) or UVW cells with DSF:Cu (Figure 3A, 3B). Decreased CDK1 expression has previously been shown to prevent entry into mitosis [33, 34].
Table 1: The effect of DSF:Cu and ionising radiation on cellular accumulation at the G2 checkpoint.
|
Control |
DSF:Cu |
5 Gy |
DSF:Cu + 5 Gy |
|---|---|---|---|---|
SK-N-BE(2c) |
30.5 ± 0.63 |
34.5 ± 1.63 |
53.7 ± 5.05* |
40.4 ± 0.51¶ |
UVW |
23.0 ± 0.83 |
33.9 ± 2.57** |
56.1 ± 1.40*** |
44.0 ± 0.50††¶¶ |
The percentages of unsynchronised SK-N-BE(2c) and UVW cells in G2-M, 12 h after irradiation and exposure to 0.3 μM DSF:Cu, were determined by FACS analysis of propidium-iodide-stained cells. Data are means ± SEM, n=3. One-way ANOVA with Bonferroni correction was used to compare the means of the test groups with those of the untreated control (*), with those of the irradiated group (†) or compared with those treated with DSF:Cu alone (¶). One symbol: P<0.05; two symbols: P<0.01; three symbols: P<0.001.
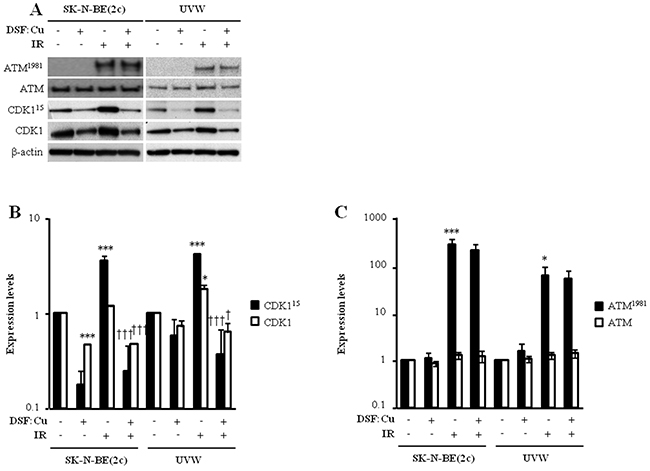
Figure 3: The effect of DSF:Cu on the phosphorylation of ATM and CDK1 induced by ionising radiation. (A) The expression levels of ATM and ATM1981 were determined by immunoblotting 1 h following irradiation whereas CDK1 and CDK115 expression levels were determined 12 h following irradiation. SK-N-BE(2c) and UVW cells were exposed to 0.3 μM DSF:Cu and irradiated with 5 Gy at the start of exposure to DSF:Cu. The immunoblots are representative examples of triplicate determinations. (B, C) Histogram data are fold changes in expression levels above background compared with untreated controls and normalised to the loading control β-actin. Data are means ± SEM, n=3. One-way ANOVA with Bonferroni correction was used to compare the means of the test groups with those of the control (*) or with those of the irradiated group (†). One symbol P<0.05, two symbols P<0.01; three symbols P<0.001.
One of the hallmarks of the cellular response to ionising radiation is the activation of the G2 checkpoint prior to entry into mitosis. To determine whether radiosensitisation by DSF:Cu was due to the inhibition of G2 checkpoint activation following irradiation, the distribution of unsynchronised SK-N-BE(2c) and UVW cells throughout the phases of the cell cycle was assessed 12 h after irradiation in the absence or in the presence of DSF:Cu. Irradiation alone resulted in the activation of the G2 checkpoint in unsynchronised SK-N-BE(2c) and UVW cells (Table 1), as indicated by increases in the G2-M populations from 30.5% to 53.7% (P<0.05) and from 23.0% to 56.1% (P<0.001), respectively. In comparison with irradiation alone, the G2-M populations, following irradiation in the presence of 0.3 μM DSF:Cu, were decreased from 53.7% to 40.4% in SK-N-BE(2c) cells and from 56.1% to 44.0% (P<0.01) in UVW cells (Table 1), indicating that DSF:Cu may inhibit irradiation-induced G2 arrest. The activation of G2 arrest was also evaluated by immunoblotting of ATM1981 and CDK115 (Figure 3). ATM1981 detects DNA damage in the form of double-strand breaks (DSB) and activates the G2 checkpoint as well as DSB repair [35]. CDK115, a downstream effector of ATM1981, prevents entry into mitosis to allow for the repair of DNA damage [35]. The G2 checkpoint was activated following irradiation, as indicated by an increase in expression level of ATM1981 (P<0.05) and CDK115 (P<0.001) in SK-N-BE(2c) and UVW cells (Figure 3B, 3C). In comparison with irradiation alone, expression of ATM1981 was unaltered by irradiation in the presence of DSF:Cu (Figure 3C), suggesting that DSF:Cu does not affect DSB detection following irradiation. In contrast, expression of CDK115 was reduced (P<0.001) following irradiation in the presence of DSF:Cu in comparison with irradiation alone (Figure 3B). However, the decrease in CDK115 expression is likely due to a decrease in total CDK1 expression.
We reasoned that if DSF:Cu suppressed the phosphorylation of CDK1 at tyrosine 15 as well as reducing the expression level of total CDK1, the levels of total CDK1 may be too low to trigger the mitotic process. Consequently, we anticipated that the apparent inhibition of the irradiation-induced G2 arrest may not be due to a bypass of G2 arrest but, instead, to the failure to complete S phase before entry into G2. Therefore, we studied cell cycle progression using synchronised populations of SK-N-BE(2c) and UVW cells. Synchronised cells were irradiated at the S-G2 border and immediately exposed to DSF:Cu. Cell cycle profiles were obtained 6 h after the commencement of irradiation and DSF:Cu treatments. At this timepoint, 30.5% of untreated SK-N-BE(2c) cells and 86.5% of untreated UVW cells had reached G1, indicating progress through G2 and mitosis (Table 2). In comparison, there were fewer SK-N-BE(2c) (17.2%, P<0.001) and UVW (4.6%, P<0.001) cells in G1 following DSF:Cu treatment alone (Table 2). The decrease in the proportion of cells in G1 following DSF:Cu treatment was accompanied by an increase in the proportion of SK-N-BE(2c) and UVW cells in G2, indicating failure to progress through mitosis (Table 2). These results corroborated the observation made in Table 1 that DSF:Cu causes arrest in G2.
Table 2: The effect of DSF:Cu and ionising radiation on the progression of synchronised cells through G2.
|
Control |
DSF:Cu |
5 Gy |
DSF:Cu + 5 Gy |
|---|---|---|---|---|
SK-N-BE(2c) |
|
|
|
|
G1 |
30.5 ± 0.70 |
17.2 ± 0.27*** |
16.0 ± 0.79*** |
17.2 ± 0.12*** |
S |
18.8 ± 0.31 |
28.4 ± 0.48*** |
17.0 ± 1.10 |
28.3 ± 0.76*** |
G2 |
33.7 ± 0.40 |
39.2 ± 0.62 |
42.4 ± 3.15* |
37.3 ± 1.09 |
UVW |
|
|
|
|
G1 |
86.5 ± 0.18 |
4.6 ± 0.70*** |
13.2 ± 1.49*** |
5.1 ± 0.12*** |
S |
2.4 ± 0.08 |
1.2 ± 0.06 |
1.6 ± 0.06 |
1.8 ± 0.53 |
G2 |
9.6 ± 0.33 |
93.8 ± 0.79*** |
84.3 ± 1.62*** |
92.8 ± 1.65*** |
The percentages of synchronised SK-N-BE(2c) and UVW cells in G1, S and G2 were determined by FACS analysis of propidium-iodide-stained cells 6 h after irradiation at the S-G2 border and treatment with 0.3 μM DSF:Cu. Data are means ± SEM, n=3. One-way ANOVA with Bonferroni correction was used to compare the means of the test groups with those of the untreated control (*). One symbol: P<0.05; two symbols: P<0.01; three symbols: P<0.001.
Irradiation at the S-G2 border increased the proportion of SK-N-BE(2c) (P<0.05) and UVW (P<0.001) cells in G2 (Table 2). The increase in the proportion of cells in G2 following irradiation was accompanied by a decrease in the proportion of both SK-N-BE(2c) (P<0.001) and UVW (P<0.001) cells in G1. There was no inhibition of irradiation-induced cellular accumulation in G2 by DSF:Cu, indicated by similar proportions of cells in G2 and G1 in the irradiated group and in the group irradiated in the presence of DSF:Cu (Table 2). These observations indicated the failure of synchronised cells to bypass the irradiation-induced G2 arrest in the presence of DSF:Cu.
Taken together, these results indicated the ability of DSF:Cu treatment to cause accumulation of cells in G2 and to inhibit DNA synthesis. The apparent prevention of the irradiation-induced accumulation of unsynchronised cells in G2 by DSF:Cu (Table 1) is likely due to a cytostatic effect of DSF:Cu on cell cycle progression through S phase.
Enhancement of cellular radiosensitivity by combination of DSF:Cu with the nucleotide analogue gemcitabine
Our results indicate that DSF:Cu is a radiosensitiser and that it inhibits DNA replication. However, the radiosensitivity of cells in S phase relies on a moving replication fork for the creation of lethal DSB in DNA [36]. Therefore, the radiosensitivity of S phase cells was evaluated in the presence of DSF:Cu. A clonogenic cell kill of 55% was achieved by irradiating unsynchronised SK-N-BE(2c) cells with 3 Gy whereas 2 Gy was sufficient to achieve the same kill in SK-N-BE(2c) cells synchronised in early S phase (Figure 4A). Similarly, 40% kill of UVW clonogens was achieved by irradiating unsynchronised UVW cells with 3 Gy whereas 1 Gy was sufficient to achieve the same kill of UVW cells synchronised in early S phase (Figure 4B). These observations indicated that cells in early S phase are more sensitive to irradiation than unsynchronised populations. DSF:Cu radiosensitised SK-N-BE(2c) cells in S phase to a lesser degree than unsynchronised cells (P<0.01) whereas DSF:Cu failed to radiosensitise UVW cells in S phase (P<0.001) (Figure 4). These data indicated the resistance of cells in S phase to combination treatment consisting of ionising radiation and DSF:Cu.
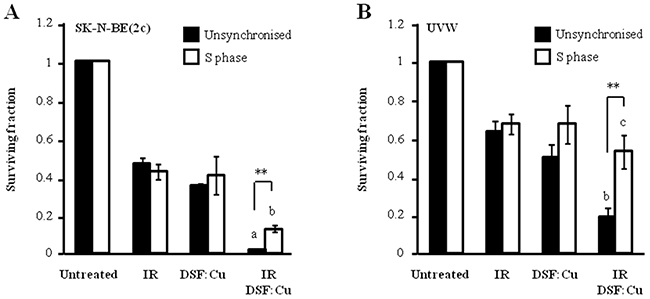
Figure 4: The effect of DSF:Cu on cellular radiosensitivity in S phase. (A) Unsynchronised SK-N-BE(2c) cells were irradiated with 3 Gy in the absence or the presence of 0.15 μM DSF:Cu. Alternatively, following synchronisation, SK-N-BE(2c) cells were irradiated at the start of S phase with 2 Gy in the absence or the presence of 0.15 μM DSF:Cu. Data are means ± SEM, n=3. (B) Unsynchronised UVW cells were irradiated with 3 Gy in the absence or the presence of 0.1 μM DSF:Cu. Alternatively, following synchronisation, UVW cells were irradiated at the start of S phase with 1 Gy in the absence or the presence of 0.1 μM DSF:Cu. Data are means ± SEM, n=3. One-way ANOVA with Bonferroni correction was used to compare the means of the combination treatment with those treated with either single agent (a, P<0.001; b, P<0.01, c, non-significant). The independent t-test was used to compare the means of the combination treatment in unsynchronised cells versus those of S phase cells (**P<0.01).
Gemcitabine is a nucleotide analogue which kills cancer cells specifically in S phase. DSF:Cu has been shown to enhance gemcitabine efficacy through the inhibition of NF-κB [37], the generation of oxidative stress [38] or the eradication of aldehyde dehydrogenase-positive cancer cells [5, 6, 39]. Since S phase cells showed resistance to treatment consisting of ionising radiation and DSF:Cu, we hypothesised that gemcitabine treatment could increase its efficacy by targeting S phase cells. In view of the fact that gemcitabine toxicity to S phase cells depends on its incorporation into DNA and that DSF:Cu inhibits DNA synthesis, we hypothesised that modulation by gemcitabine of the efficacy of the treatment consisting of ionising radiation and DSF:Cu may be schedule-dependent. Preliminary investigations indicated that Cu does not influence the sensitivity of SK-N-BE(2c) nor SH-SY5Y cells to gemcitabine treatment (Supplementary Figure 1).
Exposure of SK-N-BE(2c) or UVW cells to gemcitabine for 24 h inhibited DNA synthesis, indicated by a significant decrease in the proportion of cells incorporating BrdU (P<0.001) (Figure 5A and 5C). However, in SK-N-BE(2c) cells, BrdU positivity significantly increased from 1.4 ± 0.2 to 62.4 ± 1.6 % (P<0.001) 8 h after removal of gemcitabine from the medium, indicating the reversible nature of the inhibition of DNA synthesis in SK-N-BE(2c) cells by gemcitabine (Figure 5A). In UVW cells, although BrdU positivity significantly increased from 2.4 ± 1.2 to 14.0 ± 0.1 % (P<0.05) 8 h following removal of gemcitabine from the medium, there was no increase in BrdU-positive cells from 4 to 8 h, indicating that DNA synthesis did not restart within 8 h of removal of gemcitabine (Figure 5C). Pre-treatment of unsynchronised SK-N-BE(2c) and UVW cells with gemcitabine for 24 h before exposure to DSF:Cu for 8 h (schedule 1) resulted in increased radiation DEF values in comparison with treatment with gemcitabine or DSF:Cu alone (Figure 5B, 5D, Table 3). The nature of the interaction between DSF:Cu, gemcitabine and irradiation was evaluated by combination index (CI) analysis. Synergism was indicated by CI values less than 1 at toxicity levels greater than 0.5 in SK-N-BE(2c) cells and greater than 0.25 in UVW cells (Table 4). These results indicated that re-initiation of DNA synthesis following removal of gemcitabine from the culture medium in SK-N-BE(2c) cells, but not in UVW cells, did not affect the efficacy of drug administration according to schedule 1.
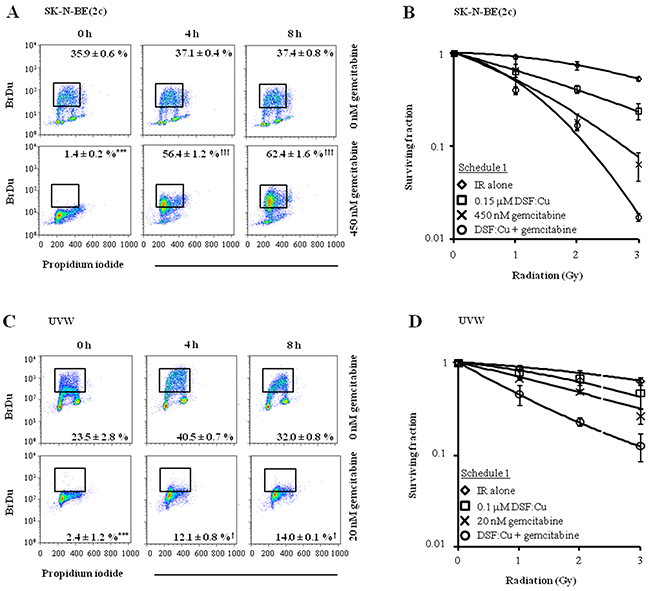
Figure 5: The effect of drug administration according to schedule 1 on clonogenic survival of SK-N-BE(2c) and UVW cells. (A, C) Unsynchronised SK-N-BE(2c) and UVW cells were exposed, for 24 h, to 450 nM or 20 nM gemcitabine, respectively. These concentrations of gemcitabine corresponded to their IC50 values. The ability of SK-N-BE(2c) and UVW cells to incorporate BrdU into the DNA was measured by flow cytometry 0, 4 and 8 h following removal of gemcitabine from the culture medium. The data are representative examples of triplicate determinations. The proportions of BrdU-positive cells are means ± SEM, n=3. One-way ANOVA with Bonferroni correction was used to compare the means of the test groups with those of the control (*) or with those of group treated with gemcitabine at the 0 h time-point (†). One symbol P<0.05, 3 symbols P<0.001. (B, D) Schedule 1 consisted of pre-treatment of cells with gemcitabine for 24 h immediately followed by treatment with DSF:Cu for 8 h. The cells were irradiated at the commencement of exposure to DSF:Cu. Data are means ± SEM, n=3.
Table 3: Radiation dose enhancement values at the 90% toxicity level.
|
DSF:Cu |
Gemcitabine |
DSF:Cu + gemcitabine |
|---|---|---|---|
SK-N-BE(2c) |
|
|
|
Schedule 1 |
1.59 ± 0.34 |
2.48 ± 0.28 |
2.97 ± 0.43 |
Schedule 2 |
1.64 ± 0.45 |
1.42 ± 0.09 |
2.59 ± 0.49 |
Schedule 3 |
2.39 ± 0.49 |
2.29 ± 0.19 |
4.50 ± 0.83 |
UVW |
|
|
|
Schedule 1 |
1.45 ± 0.35 |
2.17 ± 0.24 |
3.35 ± 0.35 |
Schedule 2 |
1.32 ± 0.26 |
1.41 ± 0.09 |
2.06 ± 0.45 |
Schedule 3 |
0.88 ± 0.19 |
1.15 ± 0.49 |
0.94 ± 0.05 |
The linear-quadratic equation was used to calculate the radiation dose enhancement factor at the 90% toxicity level following treatment with DSF:Cu, gemcitabine and ionising radiation according to administration schedules 1, 2 and 3. For SK-N-BE(2c) cells, the DSF:Cu and gemcitabine concentrations were 0.15 μM and 450 nM, respectively. For UVW cells, the DSF:Cu and gemcitabine concentrations were 0.1 μM and 20 nM, respectively. Data are means ± SEM, n=3.
Table 4: Combination indices describing the nature of the interaction between gemcitabine, DSF:Cu and ionising radiation
SK-N-BE(2c) |
|||
|---|---|---|---|
Affected fraction |
Schedule 1 |
Schedule 2 |
Schedule 3 |
0.25 |
2.67 |
2.49 |
2.54 |
0.5 |
1.37 |
1.33 |
1.31 |
0.75 |
0.82 |
0.81 |
0.81 |
0.95 |
0.42 |
0.42 |
0.46 |
UVW |
|||
Affected fraction |
Schedule 1 |
Schedule 2 |
Schedule 3 |
0.25 |
0.96 |
1.81 |
2.25 |
0.5 |
0.82 |
1.10 |
3.36 |
0.75 |
0.73 |
0.79 |
5.21 |
0.95 |
0.65 |
0.62 |
11.7 |
SH-SY5Y |
|||
Affected fraction |
Schedule 1 |
Schedule 2 |
Schedule 3 |
0.25 |
2.87 |
4.65 |
3.08 |
0.5 |
1.55 |
2.91 |
2.63 |
0.75 |
1.11 |
2.17 |
2.86 |
0.95 |
0.91 |
1.96 |
4.71 |
T98G |
|||
Affected fraction |
Schedule 1 |
Schedule 2 |
Schedule 3 |
0.25 |
3.55 |
5.53 |
3.21 |
0.5 |
1.69 |
2.13 |
2.80 |
0.75 |
1.13 |
1.36 |
3.96 |
0.95 |
0.92 |
1.04 |
11.3 |
The clonogenic assay was used to construct radiation survival curves. SK-N-BE(2c): ionising radiation, 1-2-3 Gy, 0.15 μM DSF:Cu, 450 nM gemcitabine; UVW: ionising radiation, 1-2-3 Gy, 0.1 μM DSF:Cu, 20 nM gemcitabine; SH-SY5Y: ionising radiation, 0.5-1-1.5 Gy, 0.05 μM DSF:Cu, 40 nM gemcitabine; T98G: ionising radiation, 1-2-3 Gy, 1 μM DSF:Cu, 50 nM gemcitabine. Combination index analysis was carried out to evaluate the nature of the interaction between ionising radiation, DSF:Cu and gemcitabine following administration schedules 1, 2 and 3.
Next, the efficacy of alternative schedules of drug administration was evaluated. Pre-treatment of unsynchronised SK-N-BE(2c) and UVW cells for 8 h with DSF:Cu followed by exposure to gemcitabine for 24 h (schedule 2) resulted in increased radiation DEF values in comparison with treatment with gemcitabine or DSF:Cu alone (Figure 6A, 6C, Table 3). Synergism was indicated by CI values less than 1 at toxicity levels greater than 0.5 in SK-N-BE(2c) and UVW cells (Table 4). The simultaneous treatment with gemcitabine, DSF:Cu and ionising radiation (schedule 3) resulted in increased radiation DEF values in comparison with treatment with gemcitabine or DSF:Cu alone in SK-N-BE(2c) cells (Figure 6B, Table 3), but not in UVW cells (Figure 6D, Table 3). Accordingly, synergism was observed only in SK-N-BE(2c) cells as indicated by CI values less than 1 at toxicity levels greater than 0.5 (Table 4).
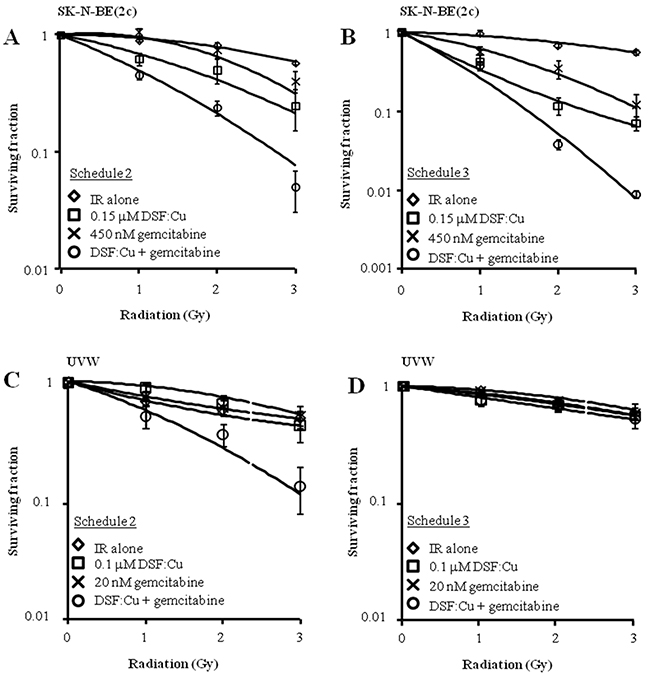
Figure 6: The effect of drug administration according to schedules 2 and 3 on clonogenic survival of SK-N-BE(2c) and UVW cells. (A, C) Schedule 2 consisted of pre-treatment of cells with DSF:Cu for 8 h, with irradiation at the commencement of exposure to DSF:Cu, immediately followed by treatment with gemcitabine for 24 h. Data are means ± SEM, n=3. (B, D) Schedule 3 consisted of simultaneous treatment of cells with gemcitabine for 24 h and DSF:Cu for 8 h, with irradiation at the commencement of drug treatment. Data are means ± SEM, n=3.
The nature of the interaction between ionising radiation, DSF:Cu and gemcitabine was further evaluated in the neuroblastoma cell line SH-SY5Y and the glioma cell line T98G. In agreement with the observations made using UVW cells, the simultaneous treatment with gemcitabine, DSF:Cu and ionising radiation (schedule 3) resulted in an antagonistic interaction in SH-SY5Y and T98G, as indicated by CI values greater than 1 (Table 4). For both alternate schedules 2 and 3, there was a trend towards an additive interaction between gemcitabine, DSF:Cu and ionising radiation in SH-SY5Y and T98G at toxicity levels greater than 0.75 (Table 4). However, pre-treatment of SH-SY5Y with DSF:Cu and ionising radiation prior gemcitabine treatment (schedule 2) did not result in an additive interaction despite CI values decreasing towards additivity with increasing toxicity levels (Table 4).
Since others have shown that the enhancement of gemcitabine cytotoxicity by DSF:Cu may be due to the generation of oxidative stress, we hypothesised that the difference in sensitivity to schedule 3 of drug administration between SK-N-BE(2c) and UVW cells may be due to their relative sensitivity to oxidative stress. It has also been shown that DSF induces the formation of reactive oxygen species (ROS) [5, 40]. In these studies, the generation of ROS following exposure of cells to DSF:Cu was prevented by treatment with the ROS scavenger N-acetyl cysteine (NAC) [5, 40]. Therefore we tested the sensitivity of SK-N-BE(2c) and UVW cells to schedule 3 in the absence and in the presence of the ROS scavenger NAC. However, since DSF interacts with thiols, the putative increased survival resulting from the presence of NAC may be due to direct interaction between NAC and DSF:Cu and not to the scavenging of ROS. Therefore, we also tested our hypothesis using the antioxidant Tiron whose mechanism of action is independent of thiol interaction [41]. We observed that the toxicity to SK-N-BE(2c) cells of schedule 3 treatment was reduced 10-fold (P<0.01) by treatment with 1 mM NAC or Tiron (Figure 7A). In contrast, NAC or Tiron failed to enhance the survival of UVW cells subjected to combination treatment using schedule 3 (Figure 7B). The enhanced kill of SK-N-BE(2c) cells, but not of UVW cells, induced by schedule 3 treatment may be due to a greater oxidative stress generated in SK-N-BE(2c) cells. In turn, the heightened sensitivity of SK-N-BE(2c) cells to oxidative stress may explain the synergistic interaction observed between ionising radiation and DSF:Cu in comparison with the additive interaction observed in UVW cells (Figure 1B, 1D). Alternatively, UVW cells may express higher levels of antioxidants or repair DNA more efficiently.
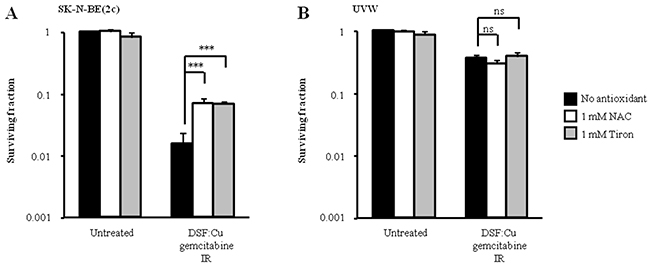
Figure 7: The effect of antioxidants on clonogenic survival following treatment administration schedule 3. (A) SK-N-BE(2c) cells were irradiated with 3 Gy and simultaneously treated with 0.15 μM DSF:Cu and 450 nM gemcitabine in the presence of 1 mM N-acetyl-cysteine (NAC) or 1 mM Tiron. (B) UVW cells were irradiated with 3 Gy and simultaneously treated with 0.1 μM DSF:Cu and 20 nM gemcitabine in the presence of 1 mM NAC or 1 mM Tiron. Data are means ± SEM, n=3. The independent t-test was used to compare the means of the groups treated in the absence of NAC or Tiron versus those treated in the presence of NAC or Tiron. ***P<0.001; ns, non-significant.
DISCUSSION
In summary, we report that DSF:Cu treatment sensitised cancer cells to ionising radiation via a mechanism independent of proteasome activity. The effects of DSF:Cu on cell cycle progression were manifest by inhibition of DNA synthesis, CDK1 depletion and blockade of entry into mitosis. Finally, the failure to radiosensitise cells in S phase by DSF:Cu was overcome by gemcitabine treatment in a cell-dependent and schedule-dependent manner. The cell cycle-specific effects of DSF:Cu and the strategy to enhance radiosensitisation by DSF:Cu are summarised in Figure 8.
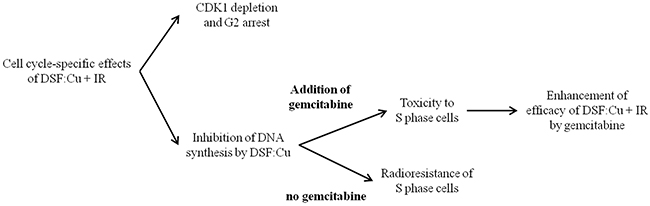
Figure 8: The summary of the strategy employed to enhance radiosensitisation by DSF:Cu.
Following exposure to stress, such as radiation, NF-κB is activated by phopsphorylation-induced proteasomal degradation of its inhibitor IkB [42]. NF-κB activation has been associated with radioresistance [43], notably via induction of anti-apoptotic and antioxidant gene expression [44, 45]. Furthermore, DSF has been shown to inhibit the proteasome [2] and NF-κB activation [7]. We have previously shown that specific proteasome inhibition, using bortezomib, is a potent radiosensitising strategy [46, 47]. However, in the present study, radiosensitisation by DSF:Cu could not be assigned to proteasome inhibition. Since NF-κB activation requires proteasomal activity, our data suggest that NF-κB inhibition is not the mechanism by which DSF:Cu sensitises cancer cells to ionising radiation.
We report that treatment with DSF:Cu reduced CDK1 expression levels, caused a G2 arrest and the failure to enter mitosis. CDC25 phosphatase is responsible for the dephosphorylation and activation of CDK1, which leads to progression through mitosis. CDC25 phosphatase contains a cysteine residue (C473) involved in its catalytic domain [48]. Given the ability of DSF to form disulfide bonds, a possible scenario is the formation of a mixed disulfide between DSF and C473 of CDC25. The resulting sustained phosphorylated state of CDK1, in conjunction with reduced levels of CDK1, prevents progression through mitosis and promotes accumulation of cells in G2. CDK1 is also an effector of S phase checkpoint activation following treatment with DNA damaging agents such as cisplatin and ionising radiation [34]. It follows that CDK1 depletion by DSF:Cu cannot explain inhibition of DNA synthesis. Interestingly, many DNA replication enzymes contain iron-sulfur clusters or thiol residues susceptible to mixed disulfide formation with DSF [49, 50]. Furthermore, the thiol-containing small molecules glutathione, glutaredoxin and thioredoxin are required to supply electrons to ribonucleotide reductase in order to convert ribonucleotides into deoxyribonucleotides for DNA synthesis [51]. It has also been reported that non-protein thiol levels increase during S phase [52]. Therefore, by virtue of DSF’s ability to form mixed disulfides with cellular components [53], the interaction of DSF:Cu with thiol-containing molecules may explain its inhibitory action on DNA synthesis.
It has been reported that CDK1 depletion leads to sensitisation of S phase cells to DNA damaging agents such as cisplatin and ionising radiation [34]. However, our observation that DSF:Cu depleted CDK1 levels but failed to sensitise S phase cells to irradiation precludes CDK1 depletion as the mechanism for DSF:Cu-induced radiosensitisation. Instead, the lack of sensitisation of S phase cells to ionising radiation by DSF:Cu was attributed to the inhibition of DNA replication. This hypothesis is consistent with reports suggesting that the mechanism of radiation-induced cell kill in S phase is dependent on a progressing replication fork, which, upon collision with radiation-induced DNA single strand breaks (SSB), generates lethal DSB. For instance, it has been shown that the collision of the replication fork with SSB created by treatment with topotecan generates lethal DSB which synergises with ionising radiation [54]. Similarly, radiosensitisation by olaparib, an inhibitor of SSB repair, was enhanced in populations synchronised in S phase and abrogated by concomitant exposure to the DNA polymerase α inhibitor aphidicolin [36].
Gemcitabine is a known radiosensitiser through the following hypothetical mechanisms. As a prodrug, the nucleoside analogue gemcitabine requires phosphorylation by deoxycytidine kinase (dCK) for its incorporation into DNA. It has been reported that ionising radiation increases dCK expression and activity in a dose- and time-dependent manner [55]. Secondly, it has been suggested that gemcitabine is a radiosensitiser through the inhibition of ribonucleotide reductase-mediated supply of dATP for DNA repair [56] and through the accumulation of cells in early S phase, a radiosensitive phase of the cell cycle [57]. However, it has been shown that glutathione confers upon cancer cells resistance to gemcitabine [58]. Therefore, increased oxidative stress resulting from the interaction between DSF:Cu and glutathione [53] could sensitise cancer cells to gemcitabine treatment. In preclinical studies, DSF, and its derivative pyrrolidine dithiocarbamate, have been successfully used to enhance the efficacy of gemcitabine, notably through NF-κB inhibition and generation of oxidative stress [5, 37, 38]. Alternatively, DSF:Cu has been shown to inhibit ALDH activity [5, 39, 40], the formation of tumorspheres in vitro [5] and the growth of ALDH-positive xenografts in vivo [6], indicating the toxicity of DSF:Cu to cancer stem cells. We cannot speculate whether enhancement of toxicity by the combination of DSF:Cu, ionising radiation and gemcitabine is mediated by ALDH inhibition and the depletion of cancer stem cells because, in our study, we cultured our cells in conditions not optimised for cancer stem cell enrichment. We complement these studies by suggesting an alternative mechanism by which gemcitabine enhances DSF:Cu toxicity, namely the targeting, by gemcitabine, of S phase cells which are resistant to radiosensitisation by DSF:Cu.
Clonogenic cell kill achieved by the combined treatment consisting of DSF:Cu and ionising radiation was enhanced by pre-administration or subsequent administration of gemcitabine in SK-N-BE(2c), UVW, SH-SY5Y and T98G cells. However, the simultaneous treatment with gemcitabine, DSF:Cu and ionising radiation resulted in enhanced clonogenic cell kill in SK-N-BE(2c) cells, but not in UVW, SH-SY5Y and T98G cells. Gemcitabine toxicity to S phase cells depends on its incorporation into DNA and DSF:Cu inhibits DNA synthesis. This may explain the failure of gemcitabine to enhance the efficacy of the combination treatment consisting of ionising radiation and DSF:Cu when administered simultaneously. Furthermore, it has been suggested that gemcitabine exerts cytotoxic actions in S phase by virtue of binding to critical thiols of ribonucleotide reductase [19]. Since DSF:Cu also interacts with thiols [18, 53], the competition for these binding sites may hinder enhancement of treatment efficacy.
Our observation of modulation of cell cycle progression by DSF:Cu suggested that enhancement of the efficacy of DSF:Cu combined with ionising radiation may be achieved through the application of cytotoxic drugs acting preferentially on cells in S phase. Indeed, decreased clonogenic survival was obtained by the inclusion of the nucleoside analogue gemcitabine with DSF:Cu and ionising radiation treatments. However, the radiosensitising mechanism of DSF:Cu deserves further study. In particular, critical thiols in effectors of DNA repair, cell cycle checkpoint and other pathways linked to the cellular response to ionising radiation are possible targets for DSF:Cu-induced radiosensitisation. Recently, DSF:Cu treatment has been associated with decreased expression of genes involved in DNA repair [31]. The knowledge of the radiosensitising mechanism of DSF:Cu could be used to refine combination treatments and to define cancer patient sub-groups most likely to benefit from DSF:Cu and ionising radiation therapy.
It is noteworthy that the repurposing of well-established drugs such as DSF to cancer treatment has received recent interest [59]. In comparison to newly designed drugs, the advantages of DSF:Cu include cheaper and shorter clinical development due to prior knowledge of its favourable toxicological, pharmacokinetic and pharmacodynamic profiles. The clinical experience of DSF and gemcitabine treatments should facilitate the introduction of their combined use in clinical trials of radiotherapy.
MATERIALS AND METHODS
Cell culture
The neuroblastoma-derived cell lines SK-N-BE(2c) and SH-SY5Y and the glioma-derived cell line T98G were purchased from the American Type Culture Collection. SK-N-BE(2c) cells were maintained in Dulbecco-modified Eagle medium supplemented with 15% (v/v) foetal bovine serum (FBS, Autogen Bioclear, Wiltshire, UK), 2 mM L-glutamine and 1% (v/v) non-essential amino acids at 37°C in a 5% CO2 atmosphere. SH-SY5Y cells were maintained in Dulbecco-modified Eagle medium supplemented with 10% (v/v) foetal bovine serum FBS and 2 mM L-glutamine at 37°C in a 5% CO2 atmosphere. The glioma-derived cell lines UVW [60] and T98G were maintained in minimum essential medium supplemented with 10% (v/v) FBS and 2 mM L-glutamine at 37°C in a 5% CO2 atmosphere. Unless otherwise indicated, all cell culture reagents were obtained from Thermo Scientific (Paisley, UK).
Drug treatment and irradiation procedure
Gemcitabine, DSF, copper chloride, NAC and Tiron were purchased from Sigma-Aldrich (St Louis, MO, USA). Gemcitabine and DSF were dissolved in DMSO. The cells were exposed to a maximum concentration of 0.1 % (v/v) DMSO. Copper chloride was dissolved in distilled water. NAC and Tiron were dissolved in culture medium. DSF:Cu treatment consisted of equimolar concentrations of DSF and copper in serum-free culture medium in order to control for copper concentration [10]. Cells were irradiated using an RS225 irradiator (Xstrahl, Surrey, UK) at a dose-rate of 1.64 Gy/min. For combination treatment consisting of gemcitabine, DSF:Cu and ionising radiation, three schedules of treatment administration were assessed (Supplementary Figure 2). Schedule 1 consisted of pre-treatment of cells with gemcitabine for 24 h immediately followed by treatment with DSF:Cu for 8 h. The cells were irradiated at the commencement of exposure to DSF:Cu. Schedule 2 consisted of pre-treatment of cells with DSF:Cu for 8 h, with irradiation at the commencement of exposure to DSF:Cu, immediately followed by treatment with gemcitabine for 24 h. Schedule 3 consisted of simultaneous treatment of cells with gemcitabine for 24 h and DSF:Cu for 8 h, with irradiation at the commencement of drug treatment.
Clonogenic assay
After treatment, the medium was removed and the cells were washed twice with phosphate-buffered saline (PBS), suspended by treatment with trypsin and counted using a haemocytometer. For every treatment, 600 SK-N-BE(2c), 250 UVW, 1,000 SH-SY5Y and 700 T98G cells were seeded in triplicate in 5.3 cm diameter dishes (Thermo Scientific, Paisley, UK) and incubated at 37°C in 5% CO2. When the colonies comprised more than 50 cells, medium was removed and the colonies were fixed in 50% methanol (v/v) in PBS and stained with 1% (v/v) crystal violet in PBS before counting.
Isobologram analysis
Isobologram analysis at the 50% level of effect was performed in order to determine the nature of the interaction between DSF:Cu and ionising radiation treatment according to the method of Steel and Peckham [61]. Modes IIa and IIb were chosen to define the envelope of additivity because it accounts for non-linearity of the dose-effect curves for both DSF:Cu and ionising radiation. For mode IIa, a series of ionising radiation doses that produces a 0, 10, 20, 30, 40 and 50% reduction of clonogenicity was calculated using the median effect principle, as described previously [62]. This series of effects is then used as the initial level of cytotoxicity from which the increment of the concentration of DSF:Cu that produces a total level of toxicity of 50% was calculated. For mode IIb, a series of DSF:Cu concentrations that produces a 0, 10, 20, 30, 40 and 50% reduction of clonogenicity was calculated. This series of effects is then used as the initial level of cytotoxicity from which the increment of the ionising radiation dose that produces a total level of toxicity of 50% was calculated.
The coordinates of the isoeffect points were determined using the ionising radiation survival curves obtained in the absence and in the presence of various concentrations of DSF:Cu. Firstly, the surviving fractions were plotted against the dose of ionising radiation in the absence and in the presence of 0, 0.1, 0.2 and 0.3 μM concentrations of DSF:Cu. The doses of ionising radiation producing a 50% reduction in clonogenicity (IC50 value) in the presence of 0, 0.1, 0.2 and 0.3 μM of DSF:Cu were calculated using the median effect principle. The pairs consisting of the IC50 value for ionising radiation in the presence of a selected concentration of DSF:Cu were the coordinates of the isoeffect points at the 50% level of cytotoxicity. Secondly, the analogous procedure is performed using survival curves obtained in response to DSF:Cu treatment in the presence of 0, 1, 2 and 3 Gy ionising radiation.
If the isoeffect points lay to the left of the envelope of additivity, the response was supra-additive. If the isoeffect points lay within the envelope of additivity, the response was additive. If the isoeffect points lay to the right of the envelope of additivity, the response was infra-additive.
Combination index analysis
When experimental design did not include treatment with incremental doses of each component, the nature of the interaction between the components of a combination treatment was assessed by the calculation of the combination index. The procedure has been described previously [62]. Briefly, the median effect equation was used to calculate the doses of each component of the combination treatment corresponding to a given effect. The dose reduction index was then calculated for each agent of the combination by dividing the dose within the combination by the dose as a single agent that induced the same kill. The combination index (CI) was calculated as the sum of the dose reduction indices of each component of the combination.
Chymotrypsin-like activity assay
Chymotrypsin-like activity of the proteasome was measured using Proteasome-Glo™ Chymotrypsin-Like, Trypsin-Like and Caspase-Like Cell-based Assay kit (Promega, US). Exponentially growing cells were treated in 96-well plates with DSF:Cu for 8 h at 37°C and 5% CO2. The cells were then washed with PBS and resuspended in 100 μl of PBS and exposed to the manufacturer’s buffer containing luciferase and the proteasome substrate Suc-LLVY-luciferin. The luminescent signal, which was a measure of proteasomal activity, was measured using a Lmax luminometer (Molecule Devices, Sunnyvale, CA).
Cell cycle synchronisation
SK-N-BE(2c) cells were exposed twice to 20 mM thymidine for 24 h separated by incubation in thymidine-free culture medium for 24 h. UVW cells were exposed twice to 2 mM thymidine for 15 h separated by incubation in thymidine-free culture medium for 9 h. At the end of the synchronisation procedure, cells were reversibly arrested in early S phase.
Cell cycle analysis
Cells were exposed to 10 μM bromodeoxyuridine (BrdU; BD Biosciences, Oxford, UK) for 90 min after which cellular monolayers were trypsinized, washed twice with PBS and fixed overnight in 70% (v/v) ethanol in distilled H2O at −20°C. Ethanol was removed by centrifugation and resuspension of the pellet in PBS. Cells were incubated in 2 M hydrochloric acid (HCl) for 20 minutes and then washed with PBS and then with PBS buffer containing 0.5% (v/v) Tween-20 (PBST). The cells were resuspended in PBS buffer containing 0.05% (v/v) Triton X-100 and BrdU antibody (BD Biosciences, Oxford, UK) for 20 min at room temperature. Excess antibody was removed by centrifugation and resuspension in PBST. The cells were then resuspended in 200 μl of PBS containing 20 μg/ml of propidium iodide (Sigma-Aldrich, St Louis, MO, USA) and 200 μg/ml of RNase A (Qiagen Ltd., West Sussex, UK) for at least 10 min before flow cytometry using a FACScan analyzer (BD Biosciences, Oxford, UK). Data were analyzed using the FlowJo software (BD Biosciences, Oxford, UK).
Immunoblotting
Whole cellular protein extracts were resolved in reducing and denaturing conditions by sodium dodecyl sulphate polyacrylamide gel electrophoresis for the determination of expression levels of ATM, ATM phosphorylated at serine 1981 (ATM1981), CDK1 and phosphorylated CDK1 at tyrosine 15 (CDK115). Proteins were transferred onto polyvinylidene fluoride Immobilon-P® membranes (Millipore, UK). Membranes were blocked with 7.5% (w/v) milk (CDK1, ATM, ATM1981 and β-actin) or bovine serum albumin (CDK115), then incubated overnight at 4°C with primary antibodies against CDK1 (1:1,000; mouse monoclonal, Millipore, UK), CDK115 (1:50,000; rabbit polyclonal, Millipore, UK), ATM (1:1,000; mouse monoclonal, Abcam, UK), ATM1981 (1:1,000; mouse monoclonal, Novus Biologicals, UK) and loading controls β-actin (1:50,000; mouse monoclonal, Abcam, UK). Membranes were then washed and incubated with horseradish peroxidase-conjugated secondary anti-mouse antibodies (1:2,000; Cell Signalling, UK) or anti-rabbit antibody (1:2,000; Santa-Cruz, Heidelberg, Germany) to enable detection using enhanced chemolumiscence (Thermo Scientific, UK). The densitometric analysis was carried out using ImageJ software (National Institute of Health, USA). The intensity of the band above background was recorded, normalised to that of β-actin and expressed as a fold-change in comparison with untreated controls.
Circular dichroism
Calf thymus DNA (Sigma-Aldrich, MO, USA) was denatured at 90°C for 5 min and allowed to cool and anneal at room temperature for 40 min. PBS, ethidium bromide (EtBr; Sigma-Aldrich, MO, USA) or DSF:Cu were added to 0.05 mg/ml calf thymus-DNA in 1 ml PBS prior circular dichroism (CD) spectra acquisition using a Jasco-600 spectropolarimeter (Japan Spectroscopic Co., Tokyo, Japan). CD spectra (225 to 300 nm) were collected using a quartz cell with a path length of 0.5 cm. Alternatively, CD spectra (225 to 300 nm) were collected from 85 to 20°C at 5°C increments.
Author contributions
Tesson M: study design, data acquisition, analysis and interpretation, statistical analysis, drafting and editing of the manuscript. Anselmi G: data acquisition. Bell C: data acquisition. Mairs RJ: data interpretation, drafting and editing of the manuscript.
ACKNOWLEDGMENTS
We would like to thank Dr Sharon Kelly, University of Glasgow, for assistance in circular dichroism studies.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This work was supported by the joined Action Medical Research and Neuroblastoma UK grant GN2196.
REFERENCES
1. Hald J, Jacobsen E. A Drug sensitising the organism to ethyl alcohol. Lancet. 1948; 255:1001-1004.
2. Chen D, Cui QZC, Yang HJ, Dou QP. Disulfiram, a clinically used anti-alcoholism drug and copper-binding agent, induces apoptotic cell death in breast cancer cultures and xenografts via inhibition of the proteasome activity. Cancer Res. 2006; 66:10425-10433.
3. Shiah SG, Kao YR, Wu FYH, Wu CW. Inhibition of invasion and angiogenesis by zinc-chelating agent disulfiram. Mol Pharmacol. 2003; 64:1076-1084.
4. Loo TW, Bartlett MC, Clarke DM. Disulfiram metabolites permanently inactivate the human multidrug resistance P-glycoprotein. Mol Pharm. 2004; 1:426-433.
5. Liu P, Brown S, Goktug T, Channathodiyil P, Kannappan V, Hugnot JP, Guichet PO, Bian X, Armesilla AL, Darling JL, Wang W. Cytotoxic effect of disulfiram/copper on human glioblastoma cell lines and ALDH-positive cancer-stem-like cells. Br J Cancer. 2012; 107:1488-1497.
6. Liu X, Wang L, Cui W, Yuan X, Lin L, Cao Q, Wang N, Li Y, Guo W, Zhang X, Wu C, Yang J. Targeting ALDH1A1 by disulfiram/copper complex inhibits non-small cell lung cancer recurrence driven by ALDH-positive cancer stem cells. Oncotarget. 2016; 7:58516-58530. https://doi.org/10.18632/oncotarget.11305.
7. Wang W, McLeod HL, Cassidy J. Disulfiram-mediated inhibition of NF-kappaB activity enhances cytotoxicity of 5-fluorouracil in human colorectal cancer cell lines. Int J Cancer. 2003; 104:504-511.
8. Heikkila RE, Cabbat FS, Cohen G. In vivo inhibition of superoxide-dismutase in mice by diethyldithiocarbamate. J Biol Chem. 1976; 251:2182-2185.
9. Cen D, Gonzalez RI, Buckmeier JA, Kahlon RS, Tohidian NB, Meyskens FL Jr. Disulfiram induces apoptosis in human melanoma cells: a redox-related process. Mol Cancer Ther. 2002; 1:197-204.
10. Rae C, Tesson M, Babich JW, Boyd M, Sorensen A, Mairs RJ. The role of copper in disulfiram-induced toxicity and radiosensitization of cancer cells. J Nucl Med. 2013; 54:953-960.
11. Nagai M, Vo NH, Shin Ogawa L, Chimmanamada D, Inoue T, Chu J, Beaudette-Zlatanova BC, Lu R, Blackman RK, Barsoum J, Koya K, Wada Y. The oncology drug elesclomol selectively transports copper to the mitochondria to induce oxidative stress in cancer cells. Free Radic Biol Med. 2012; 52:2142-2150.
12. Tardito S, Bassanetti I, Bignardi C, Elviri L, Tegoni M, Mucchino C, Bussolati O, Franchi-Gazzola R, Marchio L. Copper binding agents acting as copper ionophores lead to caspase inhibition and paraptotic cell death in human cancer cells. J Am Chem Soc. 2011; 133:6235-42.
13. Li M, Kong LL, Gou Y, Yang F, Liang H. DNA binding, cytotoxicity and apoptosis induction activity of a mixed-ligand copper(II) complex with taurine Schiff base and imidazole. Spectrochim Acta A Mol Biomol Spectrosc. 2014; 128:686-693.
14. Lu J, Sun Q, Li JL, Jiang L, Gu W, Liu X, Tian JL, Yan SP. Two water-soluble copper(II) complexes: synthesis, characterization, DNA cleavage, protein binding activities and in vitro anticancer activity studies. J Inorg Biochem. 2014; 137:46-56.
15. Rajalakshmi S, Weyhermuller T, Dinesh M, Nair BU. Copper(II) complexes of terpyridine derivatives: a footstep towards development of antiproliferative agent for breast cancer. J Inorg Biochem. 2012; 117:48-59.
16. Levinson W, Mikelens P, Oppermann H, Jackson J. Effect of antabuse (disulfiram) on Rous sarcoma virus and on eukaryotic cells. Biochim Biophys Acta. 1978; 519:65-75.
17. Spath A, Tempel K. Diethyldithiocarbamate inhibits scheduled and unscheduled DNA synthesis of rat thymocytes in vitro and in vivo - Dose-effect relationships and mechanisms of action. Chem Biol Interact. 1987; 64:151-166.
18. Yakisich JS, Siden A, Eneroth P, Cruz M. Disulfiram is a potent in vitro inhibitor of DNA topoisomerases. Biochem Biophys Res Commun. 2001; 289:586-590.
19. Chen Z, Zhou J, Zhang Y, Bepler G. Modulation of the ribonucleotide reductase M1-gemcitabine interaction in vivo by N-ethylmaleimide. Biochem Biophys Res Commun. 2011; 413:383-388.
20. Dupuy O, Flocard F, Vial C, Rode G, Charles N, Boisson D, Flechaire A. Disulfuram toxicity - report of 3 cases. Rev Med Interne. 1995; 16:67-72.
21. Filosto M, Tentorio M, Broglio L, Buzio S, Lazzarini C, Pasolini MP, Cotelli MS, Scarpelli M, Mancopperso M, Choub A, Padovani A. Disulfiram neuropathy: Two cases of distal axonopathy. Clin Toxicol (Phila). 2008; 46:314-316.
22. Nemavarkar P, Chourasia BK, Pasupathy K. Evaluation of radioprotective action of compounds using Saccharomyces cerevisiae. J Environ Pathol Toxicol Oncol. 2004; 23:145-151.
23. Evans RG, Engel C, Wheatley C, Nielsen J. Modification of the sensitivity and repair of potentially lethal damage by diethyldithiocarbamate during and following exposure of plateau-phase cultures of mammalian cells to radiation and cis-diamminedichloroplatinum(II). Cancer Res. 1982; 42:3074-3078.
24. Vanbekkum DW. The protective action of dithiocarbamates against the lethal effects of X-irradiation in mice. Acta Physiol Pharmacol Neerl. 1956; 4:508-523.
25. Gandhi NM, Gopalaswamy UV, Nair CC. Radiation protection by disulfiram: protection of membrane and DNA in vitro and in vivo against gamma-radiation. J Radiat Res. 2003; 44:255-259.
26. Kent CR, Blekkenhorst GH. in vivo radiosensitization by diethyldithiocarbamate. Radiat Res. 1988; 116:539-546.
27. Lin PS, Kwock L, Butterfield CE. Diethyldithiocarbamate enhancement of radiation and hyperthermic effects on Chinese hamster cells in vitro. Radiat Res. 1979; 77:501-511.
28. Westman G, Marklund SL. Diethyldithiocarbamate, a superoxide-dismutase inhibitor, decreases the radioresistance of Chinese hamster Cells. Radiat Res. 1980; 83:303-311.
29. Stone D, Lin PS, Kwock L. Radiosensitization of human erythrocytes by diethyldithiocarbamate. Int J Radiat Biol Relat Stud Phys Chem Med. 1978; 33:393-396.
30. Li L, Yang H, Chen D, Cui C, Dou QP. Disulfiram promotes the conversion of carcinogenic cadmium to a proteasome inhibitor with pro-apoptotic activity in human cancer cells. Toxicol Appl Pharmacol. 2008; 229:206-214.
31. Lun X, Wells JC, Grinshtein N, King JC, Hao X, Dang NH, Wang X, Aman A, Uehling D, Datti A, Wrana JL, Easaw JC, Luchman A, et al. Disulfiram when combined with copper enhances the therapeutic effects of temozolomide for the treatment of glioblastoma. Clinical Cancer Res. 2016; 22:3860-3875.
32. Burkitt MJ, Bishop HS, Milne L, Tsang SY, Provan GJ, Nobel CS, Orrenius S, Slater AF. Dithiocarbamate toxicity toward thymocytes involves their copper-catalyzed conversion to thiuram disulfides, which oxidize glutathione in a redox cycle without the release of reactive oxygen species. Arch Biochem Biophys. 1998; 353:73-84.
33. Vassilev LT, Tovar C, Chen S, Knezevic D, Zhao X, Sun H, Heimbrook DC,Chen L. Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc Natl Acad Sci U S A. 2006; 103:10660-10665.
34. Johnson N, Cai D, Kennedy RD, Pathania S, Arora M, Li YC, D’Andrea AD, Parvin JD, Shapiro GI. Cdk1 participates in BRCA1-dependent S phase checkpoint control in response to DNA damage. Mol Cell. 2009; 35:327-339.
35. Langerak P, Russell P. Regulatory networks integrating cell cycle control with DNA damage checkpoints and double-strand break repair. Philos Trans R Soc Lond B Biol Sci. 2011; 366:3562-71.
36. Dungey FA, Loser DA, Chalmers AJ. Replication-dependent radiosensitization of human glioma cells by inhibition of poly(ADP-ribose) polymerase: mechanisms and therapeutic potential. Int J Radiat Oncol Biol Phys. 2008; 72:1188-1197.
37. Guo X, Xu B, Pandey S, Goessl E, Brown J, Armesilla AL, Darling JL, Wang W. Disulfiram/copper complex inhibiting NFkappaB activity and potentiating cytotoxic effect of gemcitabine on colon and breast cancer cell lines. Cancer Lett. 2010; 290:104-113.
38. Dalla Pozza E, Donadelli M, Costanzo C, Zaniboni T, Dando I, Franchini M, Arpicco S, Scarpa A, Palmieri M. Gemcitabine response in pancreatic adenocarcinoma cells is synergistically enhanced by dithiocarbamate derivatives. Free Radic Biol Med. 2011; 50:926-933.
39. Kim SK, Kim H, Lee DH, Kim TS, Kim T, Chung C, Koh GY, Kim H, Lim DS. Reversing the intractable nature of pancreatic cancer by selectively targeting ALDH-high, therapy-resistant cancer cells. PLoS One. 2013; 8:e78130.
40. Yip NC, Fombon IS, Liu P, Brown S, Kannappan V, Armesilla AL, Xu B, Cassidy J, Darling JL, Wang W. Disulfiram modulated ROS-MAPK and NFκB pathways and targeted breast cancer cells with cancer stem cell-like properties. Br J Cancer. 2011; 104:1564-74.
41. Taiwo FA. Mechanism of tiron as scavenger of superoxide ions and free electrons. Spectroscopy. 2008; 22:491-498.
42. Russo SM, Tepper JE, Baldwin AS, Liu R, Adams J, Elliott P, Coppersack JC. Enhancement of radiosensitivity by proteasome inhibition: implications for a role of NF-kappa B. Int J Radiat Oncol Biol Phys. 2001; 50:183-193.
43. Honda N, Yagi K, Ding GR, Miyakoshi J. Radiosensitization by overexpression of the nonphosphorylation form of I kappa B-alpha in human glioma cells. J Radiat Res. 2002; 43:283-292.
44. Gordon GJ, Mani M, Mukhopadhyay L, Dong L, Yeap BY, Sugarbaker DJ, Bueno R. Inhibitor of apoptosis proteins are regulated by tumour necrosis factor-alpha in malignant pleural mesothelioma. J Pathol. 2007; 211:439-446.
45. Jang JH, Surh YJ. Bcl-2 protects against Abeta(25-35)-induced oxidative PC12 cell death by potentiation of antioxidant capacity. Biochem Biophys Res Commun. 2004; 320:880-886.
46. Rae C, Tesson M, Babich JW, Boyd M, Mairs RJ. Radiosensitization of noradrenaline transporter-expressing tumour cells by proteasome inhibitors and the role of reactive oxygen species. EJNMMI Res. 2013; 3:73.
47. Tesson M, Rae C, Nixon C, Babich JW, Mairs RJ. Preliminary evaluation of prostate-targeted radiotherapy using (131) I-MIP-1095 in combination with radiosensitising chemotherapeutic drugs. J Pharm Pharmacol. 2016; 68:912-21.
48. Sohn J, Kristjansdottir K, Safi A, Parker B, Kiburz B, Rudolph J. Remote hot spots mediate protein substrate recognition for the Cdc25 phosphatase. Proc Natl Acad Sci U S A. 2004; 101:16437-41.
49. Fuss JO, Tsai CL, Ishida JP, Tainer JA. Emerging critical roles of Fe-S clusters in DNA replication and repair. Biochim Biophys Acta. 2015; 1853:1253-1271.
50. Sengupta R, Holmgren A. Thioredoxin and glutaredoxin-mediated redox regulation of ribonucleotide reductase. World J Biol Chem. 2014; 5:68-74.
51. Pallardo FV, Markovic J, Garcia JL, Vina J. Role of nuclear glutathione as a key regulator of cell proliferation. Mol Aspects Med. 2009; 30:77-85.
52. Mauro F, Grasso A, Tolmach LJ. Variations in sulfhydryl, disulfide, and protein content during synchronous and asynchronous growth of HeLa cells. Biophys J. 1969; 9:1377-97.
53. Nagendra SN, Shetty KT, Subhash MN, Guru SC. Role of glutathione reductase system in disulfiram conversion to diethyldithiocarbamate. Life Sci. 1991; 49:23-28.
54. McCluskey AG, Boyd M, Ross SC, Cosimo E, Clark AM, Angerson WJ, Gaze MN, Mairs RJ. [I-131]meta-ldobenzylguanidine and topotecan combination treatment of tumors expressing the noradrenaline transporter. Clin Cancer Res. 2005; 11:7929-7937.
55. Wei S, Ageron-Blanc A, Petridis F, Beaumatin J, Bonnet S, Luccioni C. Radiation-induced changes in nucleotide metabolism of two colon cancer cell lines with different radiosensitivities. Int J Radiat Biol. 1999; 75:1005-1013.
56. Pauwels B, Korst AE, Lardon F, Vermorken JB. Combined modality therapy of gemcitabine and radiation. Oncologist. 2005; 10:34-51.
57. Sinclair WK. Cyclic x-ray responses in mammalian cells in vitro. Radiat Res. 1968; 33:620-643.
58. Lo M, Ling V, Wang YZ, Gout PW. The xc- cystine/glutamate antiporter: a mediator of pancreatic cancer growth with a role in drug resistance. Br J Cancer. 2008; 99:464-472.
59. Cvek B. Nonprofit drugs as the salvation of the world’s healthcare systems: the case of Antabuse (disulfiram). Drug Discov Today. 2012; 17:409-12.
60. Neshasteh-Riz A, Angerson WJ, Reeves JR, Smith G, Rampling R, Mairs RJ. Incorporation of iododeoxyuridine in multicellular glioma spheroids: implications for DNA-targeted radiotherapy using Auger electron emitters. Br J Cancer. 1997; 75:493-499.
61. Steel GG, Peckham MJ. Exploitable mechanisms in combined radiotherapy-chemotherapy - Concept of additivity. Int J Radiat Oncol Biol Phys. 1979; 5:85-91.
62. Chou TC, Talalay P. Quantitative-analysis of dose-effect relationships - The combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984; 22:27-55.