
INTRODUCTION
The therapy of B chronic lymphocytic leukemia (B-CLL) is rapidly evolving, as inhibitors of B-cell receptor (BCR) signaling have shown substantial activity in the absence of traditional immune-chemotherapy [1, 2]. Among the inhibitors of BCR, Ibrutinib was the first described inhibitor of the Bruton tyrosine kinase (BTK) that irreversibly inhibits the BTK kinase through covalent binding [3]. Recently, the FDA approval for Ibrutinib has been extended to CLL patients regardless of their treatment history (treatment-naïve and previously-treated patients) and Ibrutinib is now recommended by the National Comprehensive Cancer Network (NCCN) for first line treatment of frail CLL patients with significant comorbidities, as well as for high-risk CLL patients with 17p deletion [3, 4]. Although the vast majority of B-CLL patients treated with Ibrutinib shows evident clinical benefit [4-7], some B-CLL patients develop progressive disease after prolonged treatment. Such secondary resistance to Ibrutinib has been associated, in some cases, with acquired mutations in either BTK or in its downstream target PLCγ [8, 9]. Recently it has been reported that the TP53 mutation/17pdel combination, which represents a major determinant of resistance to immune-chemotherapy in B-CLL [10], might negatively interferes also with Ibrutinib efficacy [11, 12]. Regardless of the mutational status, the activation of NOTCH1 signaling, through interactions with its surface ligands, might render B-CLL cells more resistant to spontaneous and chemotherapy-induced apoptosis [13-15]. Indeed, the binding to NOTCH1 ligands, belonging to the Jagged or Delta-like ligand (DLL) families, triggers multiple proteolytic cleavages of the NOTCH1 protein, the last of which is operated by the γ-secretase enzyme, causing nuclear translocation of the intra-cellular domain of NOTCH1 (ICN) [16, 17].
Therefore, in order to start to elucidate the factors governing sensitivity/resistance to Ibrutinib, we sought to analyze: i) the clonal evolution of TP53 mutations in a pilot group of B-CLL patients undergoing Ibrutinib therapy in a 12 months follow-up; ii) the potential anti-leukemic activity of the combination of Ibrutinib with γ-secretase inhibitors (GSI) by in vitro assays performed using B-CLL primary cells.
RESULTS
In vivo evolution of the frequency of TP53mut clones in response to ibrutinib therapy in a small subset of B-CLL patients
For the present study, we analyzed a B-CLL population of 30 patients at different disease stage and characterized by different canonical clinical prognostic markers (CD38, IgHV status, chromosomal aberrations and TP53 mutations) (Table 1). Among the B-CLL population analyzed, all characterized for having unmutated BTK and PLCγ2, six patients underwent to Ibrutinib therapy and three of them carried TP53 functional mutations, in different genetic sites and at different clonal frequency (Table 1 and Figure 1). For these patients we could perform TP53 analysis at different time points after Ibrutinib therapy. As reported in Figure 1, the TP53-clonal frequency of Pt.#1 declined from 11% to 1% after 12 months of Ibrutinib therapy. Similar trend was observed also in Pt.#2 (from 73% to 50%) and in Pt.#4 (from 95% to 54%) after 12 months of Ibrutinib therapy (Figure 1). These findings reinforce the notion of the efficacy of Ibrutinib in B-CLL carrying TP53 mutations [18, 19], and provide the first evidence concerning the ability of Ibrutinib to target the TP53mut clones.
Table 1: Clinical and laboratory characteristics of B-CLL patients at the moment of in vitro treatment with ibrutinib
Pt. # |
Age (years)/ gender |
CD38+/ ZAP70+ |
IgHV status |
Cytogenetic abnormalities |
TP53 mutation (clone frequency) |
Therapy |
|---|---|---|---|---|---|---|
1 |
74/M |
pos/na |
unmut |
11qdel, 13qdel |
c.644G>A (8%) |
Ibrutinib |
2 |
70/M |
neg/neg |
unmut |
11qdel, 17pdel, 13qdel |
c.770T>C (60%) |
Ibrutinib |
3 |
76/M |
pos/na |
na |
na |
c.380C>T (39.6%) |
None |
4 |
63/F |
neg/neg |
unmut |
11qdel, 13qdel |
c.394A>C (90%) |
Ibrutinib |
5 |
58/M |
neg/neg |
unmut |
neg |
unmut |
None |
6 |
83/F |
pos/neg |
mut |
13qdel, Trisomy 12 |
unmut |
Steroid |
7 |
71/M |
neg/neg |
mut |
13qdel |
unmut |
None |
8 |
69/M |
neg/neg |
mut |
neg |
unmut |
None |
9 |
56/M |
pos/na |
na |
11qdel |
unmut |
Rituximab |
10 |
68/M |
neg/neg |
unmut |
11qdel |
unmut |
Ibrutinib |
11 |
58/F |
pos/pos |
unmut |
13qdel |
unmut |
None |
12 |
57/F |
pos/neg |
unmut |
neg |
unmut |
Chl |
13 |
59/M |
neg/pos |
unmut |
neg |
unmut |
FCL |
14 |
76/M |
neg/neg |
mut |
13qdel |
unmut |
R-Benda |
15 |
57/F |
neg/neg |
mut |
13qdel |
unmut |
None |
16 |
83/M |
pos/neg |
mut |
13qdel |
unmut |
None |
17 |
74/M |
neg/pos |
mut |
13qdel |
unmut |
None |
18 |
70/M |
neg/neg |
mut |
13qdel |
unmut |
None |
19 |
82/F |
neg/neg |
mut |
Trisomy 12 |
unmut |
None |
20 |
76/M |
pos/neg |
unmut |
13qdel |
unmut |
R-Benda |
21 |
75/M |
neg/neg |
mut |
neg |
unmut |
None |
22 |
71/F |
neg/neg |
mut |
13qdel |
unmut |
None |
23 |
84/F |
neg/neg |
mut |
13qdel |
unmut |
na |
24 |
77/F |
neg/neg |
mut |
Trisomy 12 |
unmut |
None |
25 |
85/M |
neg/na |
na |
neg |
unmut |
None |
26 |
36/F |
pos/na |
unmut |
neg |
unmut |
FCR |
27 |
65/M |
neg/pos |
mut |
neg |
unmut |
None |
28 |
47/M |
pos/na |
na |
Trisomy 12 |
unmut |
None |
29 |
66/M |
neg/neg |
unmut |
13qdel |
unmut |
Ibrutinib |
30 |
68/F |
pos/na |
na |
na |
unmut |
None |
Pt., patient; R-Benda, rituximab-bendamustine; Chl, chlorambucil; FCR, fludarabine-cyclophosphamide-rituximab; FCL, fludarabine-cyclophosphamide-lenalidomide; neg, negative; pos, positive; del, deletion; na, not available; mut, mutated; unmut, unmutated
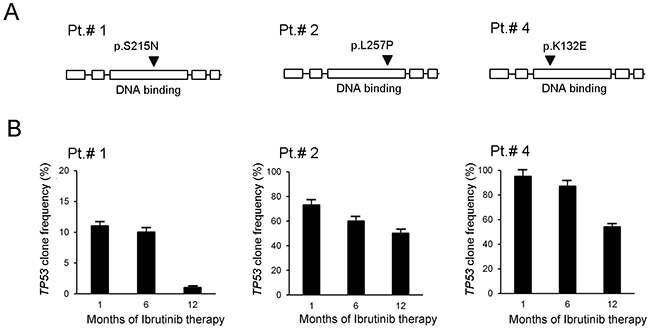
Figure 1: In vivo evolution of frequency of TP53mut clones in response to ibrutinib. Longitudinal follow-up of clone frequency relatively to TP53 analyzed by NGS on B-CLL primary cells of 3 patients treated in vivo with Ibrutinib therapy and in partial remission at the time of the 12th -month of follow-up. In (A), schematic representations of P53 protein domains (from left the boxes represent: transactivation domain, proline-rich domain, DNA-binding domain, tetramerization domain and regulatory domain) show the relative mutation site on the protein (arrowhead) characteristic of each patient. As shown, all mutations map in the DNA-binding domain that is responsible for DNA binding and target gene transactivation. In (B), results of the TP53mut clone frequency follow-up are reported for each patient as mean±SD of analyses performed in triplicates.
In vitro cytotoxic effect of ibrutinib+GSI combination in B-CLL cells
Cell cultures obtained from the same cohort of B-CLL patients (Table 1) were exposed in vitro to Ibrutinib, used at the concentration corresponding to the IC50 mean value determined in previous studies of our group in primary B-CLL cultures [11] and in line with other groups [20-23]. As shown in Figure 2A, in vitro treatment with Ibrutinib revealed a progressive reduction of cell viability coupled to the induction of apoptosis, with mean±SD (percentage of apoptotic cells over basal levels) of 18±12 and 32±15 at 24 and 48 hours of treatment, respectively. In particular, the in vitro response to Ibrutinib at 48 hours of treatment was comparable in B cell samples obtained from naïve B-CLL patients (mean±SD: 26±15) with respect to the patients under therapy with Ibrutinib and/or with chemo-immunotherapy (mean±SD: 35±13). Moreover, patient samples carrying TP53 mutations showed a susceptibility to Ibrutinib cytotoxicity comparable to unmutated patient samples. These in vitro data are therefore in line with the in vivo data illustrated above.
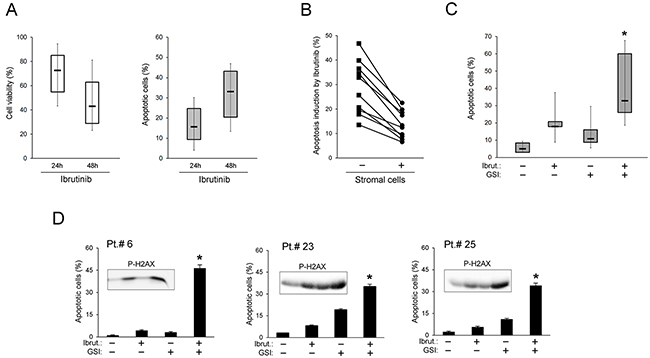
Figure 2: In vitro cytotoxic effect of Ibrutinib+GSI combination in primary B-CLL cell cultures. Patients’ derived B-CLL cells were exposed in vitro to Ibrutinib±GSI for 24/48 hours. In (A), cell viability, calculated as percentage with respect to the control untreated cultures (set to 100%), and apoptosis induction in response to Ibrutinib are shown. In (B), apoptosis induction by Ibrutinib was comparatively assessed in primary B-CLL cells either kept in suspension or co-cultured with stromal cells. In the graph each line connects results of single patients. In (C) and (D), cytotoxic effect of Ibrutinib±GSI, assessed in primary B-CLL cells co-cultured with stromal cells, was comparatively evaluated as induction of apoptosis. In C, results performed on B-CLL from 12-15 patients are shown. In D, results of cell cultures from representative patients are reported as mean±SD of three independent experiments and are shown together with P-H2AX levels analyzed by Western blotting (inserts). In A and C, horizontal bars are median, upper and lower edges of box are 75th and 25th percentiles, lines extending from box are 10th and 90th percentiles. Apoptosis induction was calculated as percentage of Annexin V/PI double positive cells. The asterisk indicates p<0.05 with respect to the single compound.
For most patient samples, B-CLL cells were treated with Ibrutinib also in co-culture with stromal cells, mimicking the microenvironment of lymph node niches. As shown in Figure 2B, under co-culture conditions the response to Ibrutinib-cytotoxicity was reduced with respect to suspension B-CLL cultures, consistently with the protective role of B-CLL/stroma interactions against anti-leukemic drugs [24, 25]. On the other hand, the anti-leukemic cytotoxicity of Ibrutinib was enhanced by the combination with γ-secretase inhibitors (GSI, both PF-03084014 and L-685,458), as evaluated in terms of apoptosis and of P-H2AX levels (Figure 2C-2D). This effect was more evident in the B-CLL/stroma co-cultures than in suspension (Supplementary Figure 1) due to the lower toxicity of the treatment with the single drugs.
Down-modulation of NOTCH1 and c-MYC pathways by ibrutinib±GSI in B-CLL cells
Since attachment of B-CLL cells to the stroma is known to protect B-CLL cells through the activation of different pro-survival pathways, including the NOTCH1 pathway [24, 25], in the next experiments the levels of NOTCH1 activation were analyzed by Western blotting. As expected, a marked NOTCH1 activation was observed in the B-CLL cells upon stromal co-culture, as documented by higher levels of the NOTCH1 intracellular domain (ICN) with respect to the untreated suspension culture, which were reduced by treatment with GSI (Figure 3). Unexpectedly, in both the culture settings (suspension and co-culture) also exposure to Ibrutinib alone down-regulated NOTCH1 activation, and the effect was enhanced when used in combination with GSI (Figure 3 and Supplementary Figure 2).
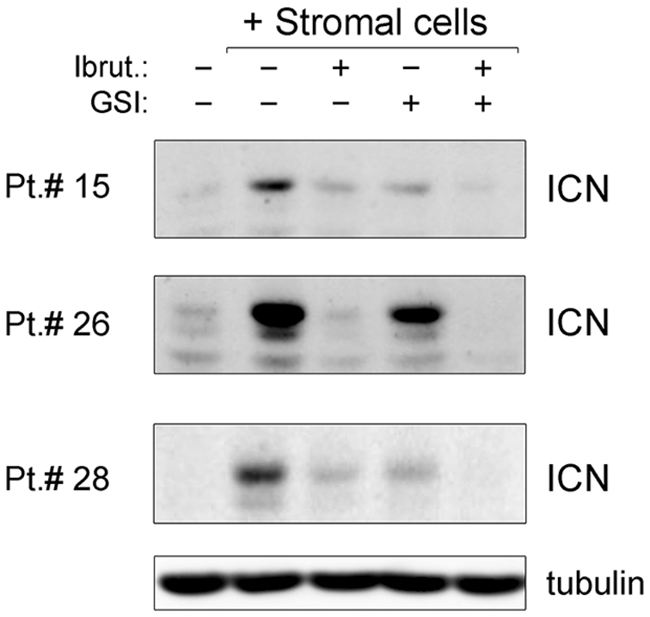
Figure 3: Down-modulation of NOTCH1 pathway by ibrutinib±GSI in primary B-CLL cell cultures. Patients’ derived B-CLL cells co-cultured with stromal cells were exposed to Ibrutinib±GSI for 24 hours or were cultured untreated in suspension as control. Western blotting analyses of cleaved intracellular NOTCH1 (ICN) are shown for representative primary B-CLL patients. For clarity, tubulin is shown as loading control for one patient.
In parallel, we have analyzed the oncogenic transcription factor c-MYC expression, which has recently been shown to be a target of both BTK [26, 27] and NOTCH1 [24, 25] pathways in different hematological malignancies, including B-CLL. Moreover, over-expression of c-MYC is involved in B cell transformation [28] and has been linked to potential Ibrutinib resistance [27]. As shown in Figure 4, under co-culture conditions a significant induction of c-MYC was documented in B-CLL samples both by protein and mRNA expression analysis. Of note, both GSI and, even more, Ibrutinib, either alone or in combination, counteracted the upregulation of c-MYC induced by the contact with stroma (Figure 4A-4B). Although the baseline c-MYC mRNA levels were significantly lower in B-CLL cells suspension, respect to the same samples co-cultured with stroma (Figure 4B), a significant down-modulation induced by the combination was observed also in this setting (Supplementary Figure 3).
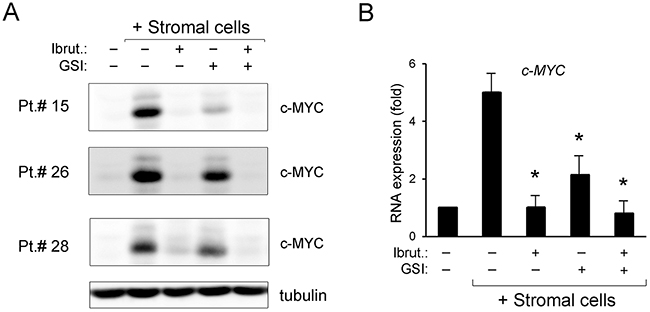
Figure 4: Down-regulation of c-MYC pathway by ibrutinib±GSI in primary B-CLL cell cultures. Patients’ derived B-CLL cells co-cultured with stromal cells were exposed to Ibrutinib±GSI for 24 hours or were grown untreated in suspension as control. In (A), Western blotting analyses of c-MYC protein levels are shown for representative primary B-CLL patients. For clarity, tubulin is shown as loading control for one patient. In (B), levels of c-MYC mRNA were analyzed by qRT-PCR and are expressed as fold of modulation with respect to the untreated B-CLL cultures grown in suspension set at 1. Results are reported as mean±SD of four independent experiments, performed in duplicate. The asterisk indicates p<0.05 with respect to untreated B-CLL cells co-cultured with stromal cells.
Effect of ibrutinib±GSI on CXCR4/SDF-1α-mediated migration of B-CLL cells
CXCR4 has been identified as a key regulator in CLL-cell retention in bone marrow and lymphoid tissues and it has recently been shown that BTK inhibition impairs CXCR4 expression in B-CLL [29]. In addition, in the multiple myeloma setting a direct positive control of CXCR4 by NOTCH1 has been recently proposed [30]. On these bases, in the last series of experiments we have assessed the effect of Ibrutinib, alone or in combination with GSI, on CXCR4 expression in B-CLL primary cells. As shown in Figure 5A, the exposure to Ibrutinib, as well as to GSI, significantly decreased the levels of CXCR4. The functional relevance of the CXCR4 down-regulation induced by Ibrutinib, GSI and combination thereof, was assessed by migration assays (Figure 5B). As shown in Figure 5B, the leukemic cell migration in response to recombinant human SDF-1α (10 ng/ml) was significantly impaired by pretreatment with Ibrutinib and GSI used alone, and completely abrogated by the drug combination.
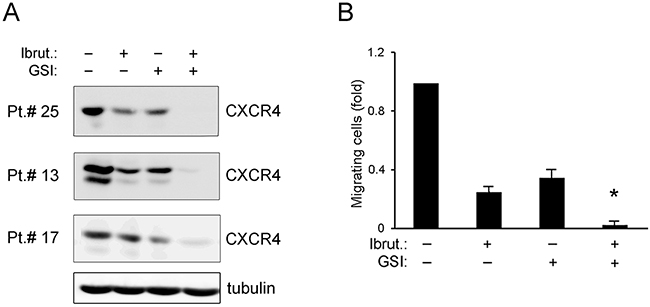
Figure 5: Effect of ibrutinib±GSI on CXCR4/SDF-1α-mediated migration of primary B-CLL cell cultures. Patients’ derived B-CLL cells were exposed in vitro to Ibrutinib±GSI for 24 hours. In (A), Western blotting analyses of CXCR4 total protein levels are shown for representative primary B-CLL cultures. For clarity, tubulin is shown as loading control for one patient. In (B), the number of migrated B-CLL cells towards SDF-1α is expressed as fold with respect to control untreated cultures set at 1. Results are reported as mean±SD of four independent experiments. The asterisk indicates p<0.05 with respect to single treatments.
DISCUSSION
Summarizing the major findings of our study, we found that B-CLL samples with TP53 mutations showed sensitivity to Ibrutinib treatment, as suggested by both in vivo and in vitro analyses. Indeed, in the pilot group of patients under treatment with Ibrutinib for 12 months we documented a progressive decrease of the percentages of TP53mut clones. Moreover, the ability of Ibrutinib to induce apoptosis in vitro in B-CLL patient cultures carrying TP53 mutations was similar to unmutated B-CLL samples.
The in vitro experiments performed by treating primary B-CLL cultures with Ibrutinib used in combination with GSI showed enhanced B-CLL cell apoptosis both in the absence or presence of stroma, a condition mimicking the B-CLL microenvironment. Although the concentrations of Ibrutinib used in our in vitro assays exceeded the values found in the plasma of patients treated in vivo with Ibrutinib [21], these doses were necessary to evaluate the anti-leukemic effect in different biological assays and comparatively among different B-CLL samples. In this respect, the results obtained in the co-culture setting are particularly relevant since it is well established that the stromal niche exerts a protective effect on CLL cells also attenuating their drug sensitivity [24]. Of note, at the molecular level, the in vitro treatment with Ibrutinib down-regulated: i) the pro-survival NOTCH1 pathway, which represents a major molecular target of GSI [31] and plays a relevant role in B-CLL leukemogenesis [32]; ii) the c-MYC expression, which is constitutively activated in situ in the lymph nodes of B-CLL patients [33], and has been proposed to confer resistance to Ibrutinib in B-NHL [27]; iii) CXCR4 expression, which plays a key role in the retention of B-CLL into the microenvironmental niches [34], coupled to reduction of migratory response of B-CLL cells to SDF-1α. The effects of Ibrutinib on the NOTCH1 and c-MYC pathways, as well as on CXCR4/SDF-1α were enhanced by the combination with GSI.
Overall, our findings are of particular relevance since in vivo the B-CLL cells in secondary lymphoid organs are in contact with several cell types and secreted molecules that together constitute the B-CLL microenvironment [35]. It has been demonstrated that leukemia cell-to-microenvironment interactions are weakened upon exposure to Ibrutinib, and a fraction of B-CLL cells egresses the lymphoid organs and relocates to the blood stream [36]. Thus, it is particularly noteworthy that Ibrutinib+GSI combination promoted enhanced cytotoxicity also on B-CLL cells co-cultured in the presence of stromal cells.
Another important issue is represented by the potential clinical relevance of our data with respect to the feasibility of using the Ibrutinib+GSI combination for the treatment of B-CLL patients. In this respect, it should be noticed that a number of GSI have been clinically evaluated as anti-cancer agents, including semagacestat (LY450139), RO4929097, avagacestat (BMS-708163), PF-03084014 and 3-[(1r, 4s)-4-(4-chloroph enylsulfonyl)-4-(2,5-difluorophenyl) cyclohexyl] propanoic acid (MK-0752) (data concerning these trials are available at clinicaltrials.gov). In spite of initial disappointing results [31], recent phase I clinical trials with the novel GSI PF-03084014, used for our in vitro assays, documented a complete response and several partial responses in advanced cancers [37]. Besides application of GSI as single agent, a wide number of combination studies incorporating GSI with established anti-cancer treatments have been evaluated and/or are currently undergoing evaluation for therapeutic efficacy (details are available from clinicaltrials.gov), including drugs with different molecular targets: capecitabine, a fluorouracil prodrug; bicalutamide, an androgen antagonist; letrozole, a nonsteroidal aromatase inhibitor; temozolomide, an alkylating agent; tamoxifen, an antiestrogen; erlotinib hydrochloride, an inhibitor of epidermal growth factor receptor tyrosine kinase; gemcitabine hydrochloride, an antimetabolite; vinblastine and docetaxel, microtubule targeting agents; cisplatin, a DNA targeting agent; and cediranib maleate, a vascular endothelial growth factor receptor-2 tyrosine kinase inhibitor. Moreover, of particular relevance for the aim of this study, promising results have been obtained in multiple myeloma, by employing GSI in combination with proteasome inhibitors [38], as well as in B-CLL by using GSI in association with conventional chemotherapy [39, 40]. Finally, it is also noteworthy that novel pan-NOTCH GSI have been recently described [41, 42].
In this context, we have demonstrated for the first time that the GSI enhanced the cytotoxicity induced by Ibrutinb in B-CLL both in the absence and presence of stromal cells, by down-modulating the NOTCH1 and the c-MYC pathways. In addition, the Ibrutinib+GSI combination down-regulated the level of CXCR4 in B-CLL and, subsequently, migratory ability towards SDF-1α. Considering that combination therapies with Ibrutinib are advancing into the clinic, our data provide a rationale for the use of novel Ibrutinib-based combinations with GSI to overcome stroma/NOTCH1-mediated drug resistance.
MATERIALS AND METHODS
B-CLL patient samples collection
Peripheral blood samples were collected in heparin coated tubes, following informed consent, in accordance with the Declaration of Helsinki and in agreement with institutional guidelines (University-Hospital of Ferrara), from a cohort of 30 B-CLL patients. The clinical, laboratory and cytogenetic data (CD38 and ZAP70 surface expression, FISH and IgHV status) of each patient were abstracted from clinical and laboratory records. TP53, BTK and PLCγ2 mutations were analyzed by NGS as previously described [43]. To identify pathogenic variations, mutations that did not affect the protein coding regions (intronic, 3’ and 5’ UTR variations, silent exonic mutations and polymorphisms) were filtered out.
All patients have been without prior therapy at least for three weeks before peripheral blood collection. Peripheral blood mononuclear cells (PBMC) were isolated from B-CLL patient’s by gradient centrifugation with lymphocyte cell separation medium (Cedarlane Laboratories, Hornby, ON). T lymphocytes, NK lymphocytes, granulocytes and monocytes were negatively depleted from B-CLL PBMC with immunomagnetic microbeads (MACS microbeads, Miltenyi Biotech, Auburn, CA), with a purity >95% of resulting CD19+ population. Freshly isolated primary cells were cultured in RPMI-1640 medium containing 10% FBS, L-glutamine and penicillin/streptomycin (Gibco, Grand Island, NY), or conserved within Hematopathology collection of our institution (Department of Morphology, Surgery and Experimental Medicine and LTTA Centre, University of Ferrara). For co-culture experiments, primary B-CLL cells were added to a sub-confluent monolayer of stromal cells and cultured in complete RPMI-1640 medium (Gibco) at 37°C in a humidified atmosphere containing 5% CO2.
Culture treatments and assessment of cell viability and apoptosis
For in vitro treatments of cell cultures, Ibrutinib (PCI-32765; Selleckchem, Houston, TX) was used either alone or in combination with GSI (PF-03084014 or L-685,458; Sigma-Aldrich, St. Louis, MO). B-CLL cells were seeded, either alone or in co-culture with stromal cells, at a density of 2x106 cells/mL. Optimal concentrations for the compounds (1-10 μM each) were determined in dose-response assays. At different time points after treatment, cell viability was examined by Trypan blue dye exclusion and MTT (3-(4,5-dimethilthiazol-2yl)-2,5-diphenyl tetrazolium bromide) colorimetric assay (Roche Diagnostics Corporation, Indianapolis, IN) for data confirmation, as previously described [44]. The amount of apoptosis was quantified by Annexin V-FITC/propidium iodide (PI) staining (Beckman Coulter Inc., Brea, CA) using a FACSCalibur flow cytometer (BD Biosciences, San Josè, CA). To avoid non-specific fluorescence from dead cells, live cells were gated tightly using forward and side scatter, as described [45].
RNA and protein analyses
Total RNA was extracted from cells using the QIAGEN miRNeasy Mini kit (QIAGEN, Hilden, Germany), accordingly to the supplier’s instructions. Genomic DNA was removed with RNase-Free DNase set. For each sample, total RNA (300 ng) was transcribed into cDNA and amplified using the Express One-Step Superscript qRT-PCR Kit, universal (Thermo Fisher Scientific, Rockford, IL). Analysis of human c-MYC expression was carried out with validated TaqMan Gene Expression Assays specific PCR primers sets (Thermo Fisher Scientific). All samples were run in duplicate using the real time thermal analyzer Applied Biosystems 7500 Fast Real-Time PCR System (Thermo Fisher Scientific). Expression values were normalized to the housekeeping gene POLR2A amplified in the same sample.
For Western blotting analysis, cells were lysed as previously described [46]. Protein determination was performed by BCA Protein Assay (Thermo Scientific). Equal amounts of proteins for each sample were migrated in SDS-polyacrylamide gels and blotted onto nitrocellulose filters. The following Abs were used: anti cleaved NOTCH1 (Val1744) (D3B8), anti CXCR4 (D4Z7W), anti c-MYC (D3N8F) and anti phospho-histone H2A.X (Ser139), all from Cell Signaling (Danvers, MA); anti tubulin, from Sigma-Aldrich. After incubation with anti-mouse or anti-rabbit IgG horseradish peroxidase-conjugated secondary antibodies (Sigma-Aldrich), specific reactions were revealed with the ECL Lightning detection kit (Perkin Elmer, Waltham, MA). Images acquisition was performed using the ImageQuant™ LAS 4000 biomolecular imager (GE Healthcare, Buckinghamshire, UK).
Migration assays
Migration assays were performed in trans-well plates (Corning Costar, Cambridge, MA) 6.5 mm in diameter, with 5-μm pore filters, as described [47]. Briefly, CD19+ cells derived from B-CLL patients either left untreated or pretreated with Ibrutinib and GSI, alone or in combination, were added to the upper chamber, while SDF-1α (10 ng/ml, R&D Systems, Minneapolis, MN) was added to the lower chamber as chemoattractant. After 3 hours of incubation at 37°C in 5% CO2, the upper side of the filters was removed and cells migrated to the lower chamber were counted using a FACSCalibur flow cytometer (BD Biosciences). Each experiment was done in duplicate.
Statistical analysis
Statistical analysis data were calculated as median or mean±SD. Box plots were used to show the median and interquartile values for each group of data. The results were evaluated by using an ANOVA with subsequent comparisons by a Student’s t-test and with the MannWhitney rank-sum test. Statistical significance was defined as p<0.05.
Abbreviations
BCR: B-cell receptor; B-NHL: B-cell Non-Hodgkin lymphoma; BTK: Bruton’s tyrosine kinase; CD: cluster of differentiation; Chl: chlorambucil; CLL: chronic lymphocytic leukemia; CXCR4: C-X-C chemokine receptor type 4; FCL: fludarabine-cyclophosphamide-lenalidomide; FCR: fludarabine-cyclophosphamide-rituximab; FDA: Food and Drug Administration; FISH: fluorescent in situ hybridization; H2AX: Histone Family Member X; ICN: intra-cellular domain of NOTCH1; IgHV: immunoglobulin heavy-chain variable region; GSI: γ-secretase inhibitors; MTT: 3-(4,5-dimethilthiazol-2yl)-2,5-diphenyl tetrazolium bromide; NCCN: National Comprehensive Cancer Network; NGS: Next Generation Sequencing; PBMC: Peripheral blood mononuclear cells; PLCγ: phospholipase C-gamma; R-Benda, rituximab-bendamustine; SDF-1alpha: stromal cell-derived factor 1α; ZAP70: 70 kDa zeta-associated protein.
Author contributions
PS and GZ analyzed the data, wrote and revised the manuscript; RV organized, designed the study and performed experiments, acquired and analyzed the data and wrote the manuscript; ER, EM, EA, VT, SG performed experiments, analyzed the data, revised and approved the manuscript.; GMR provided CLL samples and the clinical data, revised and approved the manuscript.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
Grant by MIUR-FIRB Project (RBAP11Z4Z9_002 to G.Z.) and fellowship from Italian Association for Cancer Research (AIRC; 18055 to S.G.).
REFERENCES
1. Stevenson FK, Krysov S, Davies AJ, Steele AJ, Packham G. B-cell receptor signaling in chronic lymphocytic leukemia. Blood. 2011; 118: 4313-4320.
2. Spaargaren M, de RooijMF, Kater AP, Eldering E. BTK inhibitors in chronic lymphocytic leukemia: a glimpse to the future. Oncogene. 2015; 34: 2426-2436.
3. Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, Li S, Pan Z, Thamm DH, Miller RA, Buggy JJ. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci U S A. 2010; 107: 13075-13080.
4. Burger JA, Tedeschi A, Barr PM, Robak T, Owen C, Ghia P, Bairey O, Hillmen P, Bartlett NL, Li J, Simpson D, Grosicki S, Devereux S, et al. Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. N Engl J Med. 2015; 373: 2425-2437.
5. Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, Grant B, Sharman JP, Coleman M, Wierda WG, Jones JA, Zhao W, Heerema NA, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013; 369: 32-42.
6. O’Brien S, Furman RR, Coutre SE, Sharman JP, Burger JA, Blum KA, Grant B, Richards DA, Coleman M, Wierda WG, Jones JA, Zhao W, Heerema NA, et al. Ibrutinib as initial therapy for elderly patients with chronic lymphocytic leukaemia or small lymphocytic lymphoma: an open-label, multicentre, phase 1b/2 trial. Lancet Oncol. 2014; 15: 48-58.
7. Byrd JC, Furman RR, Coutre SE, Burger JA, Blum KA, Coleman M, Wierda WG, Jones JA, Zhao W, Heerema NA, Johnson AJ, Shaw Y, Bilotti E, et al. Three-year follow-up of treatment-naive and previously treated patients with CLL and SLL receiving single-agent ibrutinib. Blood. 2015; 125: 2497-2506.
8. Woyach JA, Furman RR, Liu TM, Ozer HG, Zapatka M, Ruppert AS, Xue L, Li DH, Steggerda SM, Versele M, Dave SS, Zhang J, Yilmaz AS, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014; 370:2286-2294.
9. Furman RR, Cheng S, Lu P, Setty M, Perez AR, Guo A, Racchumi J, Xu G, Wu H, Ma J, Steggerda SM, Coleman M, Leslie C, et al. Ibrutinib resistance in chronic lymphocytic leukemia. N Engl J Med. 2014; 370: 2352-2354.
10. Secchiero P, di Iasio MG, Gonelli A, Zauli G. The MDM2 inhibitor Nutlins as an innovative therapeutic tool for the treatment of haematological malignancies. Curr Pharm Des. 2008; 14: 2100-2110.
11. Voltan R, Rimondi E, Melloni E, Rigolin GM, Casciano F, Arcidiacono MV, Celeghini C, Cuneo A, Zauli G, Secchiero P. Ibrutinib synergizes with MDM-2 inhibitors in promoting cytotoxicity in B chronic lymphocytic leukemia. Oncotarget. 2016; 7: 70623-70638. https://doi.org/10.18632/oncotarget.12139.
12. Amin NA, Balasubramanian S, Saiya-Cork K, Shedden K, Hu N, Malek SN. Cell-intrinsic determinants of Ibrutinib-induced apoptosis in chronic lymphocytic leukemia. Clin Cancer Res. 2017; 23: 1049-1059.
13. Rosati E, Sabatini R, Rampino G, Tabilio A, Di Ianni M, Fettucciari K, Bartoli A, Coaccioli S, Screpanti I, Marconi P. Constitutively activated Notch signaling is involved in survival and apoptosis resistance of B-CLL cells. Blood. 2009; 113: 856-865.
14. Secchiero P, Melloni E, di Iasio MG, Tiribelli M, Rimondi E, Corallini F, Gattei V, Zauli G. Nutlin-3 up-regulates the expression of Notch1 in both myeloid and lymphoid leukemic cells, as part of a negative feedback antiapoptotic mechanism. Blood. 2009; 113: 4300-4308.
15. Arruga F, Gizdic B, Serra S, Vaisitti T, Ciardullo C, Coscia M, Laurenti L, D’Arena G, Jaksic O, Inghirami G, Rossi D, Gaidano G, Deaglio S. Functional impact of NOTCH1 mutations in chronic lymphocytic leukemia. Leukemia. 2014; 28: 1060-1070.
16. Guruharsha KG, Kankel MW, Artavanis-Tsakonas S. The Notch signalling system: recent insights into the complexity of a conserved pathway. Nat Rev Genet. 2012; 13: 654-666.
17. Ntziachristos P, Lim JS, Sage J, Aifantis I. From fly wings to targeted cancer therapies: a centennial for Notch signaling. Cancer Cell. 2014; 25: 318-334.
18. Farooqui MZ, Valdez J, Martyr S, Aue G, Saba N, Niemann CU, Herman SE, Tian X, Marti G, Soto S, Hughes TE, Jones J, Lipsky A, et al. Ibrutinib for previously untreated and relapsed or refractory chronic lymphocytic leukaemia with TP53 aberrations: a phase 2, single-arm trial. Lancet Oncol. 2015; 16: 169-176.
19. Lee HJ, Gallardo M, Ma H, Zhang X, Larsson CA, Mejia A, Hornbaker MJ, Qi Y, Su X, Pageon LR, Quintas-Cardama A, Post SM. p53-independent ibrutinib responses in an Eμ-TCL1 mouse model demonstrates efficacy in high-risk CLL. Blood Cancer J. 2016; 6: e434.
20. Vasyutina E, Boucas JM, Bloehdorn J, Aszyk C, Crispatzu G, Stiefelhagen M, Breuer A, Mayer P, Lengerke C, Döhner H, Beutner D, Rosenwald A, Stilgenbauer S, et al. The regulatory interaction of EVI1 with the TCL1A oncogene impacts cell survival and clinical outcome in CLL. Leukemia. 2015; 10: 2003-2014.
21. Da Roit F, Engelberts PJ, Taylor RP, Breij EC, Gritti G, Rambaldi A, Introna M, Parren PW, Beurskens FJ, Golay J. Ibrutinib interferes with the cell-mediated anti-tumor activities of therapeutic CD20 antibodies: implications for combination therapy. Haematologica. 2015; 100: 77-86.
22. Yeomans A, Lemm E, Wilmore S, Cavell BE, Valle-Argos B, Krysov S, Hidalgo MS, Leonard E, Willis AE, Forconi F, Stevenson FK, Steele AJ, Coldwell MJ, et al. PEITC-mediated inhibition of mRNA translation is associated with both inhibition of mTORC1 and increased eIF2α phosphorylation in established cell lines and primary human leukemia cells. Oncotarget. 2016; 7: 74807-74819. https://doi.org/10.18632/oncotarget.11655.
23. Yeomans A, Thirdborough SM, Valle-Argos B, Linley A, Krysov S, Hidalgo MS, Leonard E, Ishfaq M, Wagner SD, Willis AE, Steele AJ, Stevenson FK, Forconi F, Coldwell MJ, Packhamet al. Engagement of the B-cell receptor of chronic lymphocytic leukemia cells drives global and MYC-specific mRNA translation. Blood. 2016; 127: 449-457.
24. Nwabo Kamdje AH, Bassi G, Pacelli L, Malpeli G, Amati E, Nichele I, Pizzolo G, Krampera M. Role of stromal cell-mediated Notch signaling in CLL resistance to chemotherapy. Blood Cancer J. 2012; 2: e73.
25. Jitschin R, Braun M, Qorraj M, Saul D, Le Blanc K, Zenz T, Mougiakakos D. Stromal cell-mediated glycolytic switch in CLL cells involves Notch-c-Myc signaling. Blood. 2015; 125: 3432-3436.
26. Yeomans A, Thirdborough SM, Valle-Argos B, Linley A, Krysov S, Hidalgo MS, Leonard E, Ishfaq M, Wagner SD, Willis AE, Steele AJ, Stevenson FK, Forconi F, et al. Engagement of the B-cell receptor of chronic lymphocytic leukemia cells drives global and MYC-specific mRNA translation. Blood. 2016; 127: 449-457.
27. Moyo TK, Wilson CS, Moore DJ, Eischen CM. Myc enhances B-cell receptor signaling in precancerous B cells and confers resistance to Btk inhibition. Oncogene. 2017.
28. Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RD, Brinster RL. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985; 318: 533-538.
29. Chen SS, Chang BY, Chang S, Tong T, Ham S, Sherry B, Burger JA, Rai KR, Chiorazzi N. BTK inhibition results in impaired CXCR4 chemokine receptor surface expression, signaling and function in chronic lymphocytic leukemia. Leukemia. 2016; 30: 833-843.
30. Mirandola L, Apicella L, Colombo M, Yu Y, Berta DG, Platonova N, Lazzari E, Lancellotti M, Bulfamante G, Cobos E, Chiriva-Internati M, Chiaramonte R. Anti-Notch treatment prevents multiple myeloma cells localization to the bone marrow via the chemokine system CXCR4/SDF-1. Leukemia. 2013; 27: 1558-1566.
31. Kolb EA, Gorlick R, Keir ST, Maris JM, Lock R, Carol H, Kurmasheva RT, Reynolds CP, Kang MH, Wu J, Houghton PJ, Smith MA. Initial testing (stage 1) by the pediatric preclinical testing program of RO4929097, a γ-secretase inhibitor targeting notch signaling. Pediatr Blood Cancer. 2012; 58: 815-818.
32. Arruga F, Gizdic B, Serra S, Vaisitti T, Ciardullo C, Coscia M, Laurenti L, D’Arena G, Jaksic O, Inghirami G, Rossi D, Gaidano G, Deaglio S. Functional impact of NOTCH1 mutations in chronic lymphocytic leukemia. Leukemia. 2014; 28:1060-1070.
33. Herishanu Y, Pérez-Galán P, Liu D, Biancotto A, Pittaluga S, Vire B, Gibellini F, Njuguna N, Lee E, Stennett L, Raghavachari N, Liu P, McCoy JP, et al. The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood. 2011; 117: 563-574.
34. Pavlasova G, Borsky M, Seda V, Cerna K, Osickova J, Doubek M, Mayer J, Calogero R, Trbusek M, Pospisilova S, Davids MS, Kipps TJ, Brown JR, et al. Ibrutinib inhibits CD20 upregulation on CLL B cells mediated by the CXCR4/SDF-1 axis. Blood. 2016; 128: 1609-1613.
35. Herman SE, Mustafa RZ, Jones J, Wong DH, Farooqui M, Wiestner A. Treatment with ibrutinib inhibits BTK- and VLA-4-dependent adhesion of chronic lymphocytic leukemia cells in vivo. Clin Cancer Res. 2015; 21: 4642-4651.
36. Wodarz D, Garg N, Komarova NL, Benjamini O, Keating MJ, Wierda WG, Kantarjian H, James D, O’Brien S, Burger JA. Kinetics of CLL cells in tissues and blood during therapy with the BTK inhibitor ibrutinib. Blood. 2014; 123: 4132-4135.
37. Messersmith WA, Shapiro GI, Cleary JM, Jimeno A, Dasari A, Huang B, Shaik MN, Cesari R, Zheng X, Reynolds JM, English PA, McLachlan KR, Kern KA, et al. A Phase I, dose-finding study in patients with advanced solid malignancies of the oral γ-secretase inhibitor PF-03084014. Clin Cancer Res. 2015; 21: 60-67.
38. Chen F, Pisklakova A, Li M, Baz R, Sullivan DM, Nefedova Y. Gamma-secretase inhibitor enhances the cytotoxic effect of bortezomib in multiple myeloma. Cell Oncol (Dordr). 2011; 34: 545-451.
39. Rosati E, Sabatini R, De Falco F, Del Papa B, Falzetti F, Di Ianni M, Cavalli L, Fettucciari K, Bartoli A, Screpanti I, Marconi P. Gamma-Secretase inhibitor I induces apoptosis in chronic lymphocytic leukemia cells by proteasome inhibition, endoplasmic reticulum stress increase and Notch down-regulation. Int J Cancer. 2013; 132: 1940-1953.
40. López-Guerra M, Xargay-Torrent S, Rosich L, Montraveta A, Roldán J, Matas-Céspedes A, Villamor N, Aymerich M, López-Otín C, Pérez-Galán P, Roué G, Campo E, Colomer D. The γ-secretase inhibitor PF-03084014 combined with fludarabine antagonizes migration, invasion and angiogenesis in NOTCH1-mutated CLL cells. Leukemia. 2015; 29: 96-106.
41. Gavai AV, Quesnelle C, Norris D, Han WC, Gill P, Shan W, Balog A, Chen K, Tebben A, Rampulla R, Wu DR, Zhang Y, Mathur A, et al. Discovery of clinical candidate BMS-906024: a potent pan-Notch inhibitor for the treatment of leukemia and solid tumors. ACS Med Chem Lett. 2015; 6: 523-527.
42. Shan W, Balog A, Quesnelle C, Gill P, Han WC, Norris D, Mandal S, Thiruvenkadam R, Gona KB, Thiyagarajan K, Kandula S, McGlinchey K, Menard K, et al. BMS-871: a novel orally active pan-Notch inhibitor as an anticancer agent. Bioorg Med Chem Lett. 2015; 25: 1905-1909.
43. Athanasakis E, Melloni E, Rigolin GM, Agnoletto C, Voltan R, Vozzi D, Piscianz E, Segat L, Dal Monego S, Cuneo A, Secchiero P, Zauli G. The p53 transcriptional pathway is preserved in ATMmutated and NOTCH1mutated chronic lymphocytic leukemias. Oncotarget. 2014; 5: 12635-12645. https://doi.org/10.18632/oncotarget.2211.
44. Zauli G, Re MC, Furlini G, Giovannini M, La Placa M. Human immunodeficiency virus type 1 envelope glycoprotein gp120-mediated killing of human haematopoietic progenitors (CD34+ cells). J Gen Virol. 1992; 73: 417-421.
45. Zauli G, Bassini A, Vitale M, Gibellini D, Celeghini C, Caramelli E, Pierpaoli S, Guidotti L, Capitani S. Thrombopoietin enhances the αIIbβ3-dependent adhesion of megakaryocytic cells to fibrinogen or fibronectin through PI 3 kinase. Blood. 1997; 89: 883-895.
46. Voltan R, Rimondi E, Melloni E, Gilli P, Bertolasi V, Casciano F, Rigolin GM, Zauli G, Secchiero P. Metformin combined with sodium dichloroacetate promotes B leukemic cell death by suppressing anti-apoptotic protein Mcl-1. Oncotarget. 2016; 7: 18965-18977. https://doi.org/10.18632/oncotarget.7879.
47. Secchiero P, Melloni E, Corallini F, Beltrami AP, Alviano F, Milani D, D’Aurizio F, di Iasio MG, Cesselli D, Bagnara GP, Zauli G. Tumor necrosis factor-related apoptosis-inducing ligand promotes migration of human bone marrow multipotent stromal cells. Stem Cells. 2008; 26: 2955-2963.