
INTRODUCTION
Over-production of reactive oxygen species (ROS) shall cause oxidative damages to human osteoblasts. It is the key contributor of osteoporosis or even osteonecrosis [1–4]. Hydrogen peroxide (H2O2) is one primary ROS [5, 6]. H2O2 elevation causes profound oxidative stress, osteoblast dysfunction and apoptosis [7–10]. In vitro studies have been adding H2O2 to the cultured human osteoblasts to establish a cellular model of osteoporosis/osteonecrosis [7–10]. This model would help to understand the pathological mechanisms of ROS-induced osteoblast injuries, and to develop possible intervention strategies [11–16].
Nrf2 (NF-E2-related factor 2)-ARE (anti-oxidant response element) pathway is principally mediated by the ubiquitin proteasome system [17–22]. In the resting state, Nrf2 forms a complex with its repressor protein Keap1 (Kelch-like erythroid cell-derived protein with CNC homology [ECH]-associated protein 1) and ubiquitin E3 ligase cullin 3 (Cul3). This leads to Nrf2 protein degradation via ubiquitin-mediated proteolysis [17–22]. Conversely, Nrf2 activation will result in impairment of the Nrf2 ubiquitination and degradation [23–25]. That will allow Nrf2 stabilization, accumulation and translocation to the nucleus, where it transcriptionally activates targeted anti-oxidant genes [17–22].
MicroRNAs (miRs) bind to 3’-untranslated region (UTR) of targeted-mRNAs, thereby causing mRNA degradation and/or the translation inhibition [26, 27]. miRs could be a novel and promising strategy to activate Keap1-Nrf2 signaling [28, 29]. It has been shown that miR-7 targeted Keap1, leading to Nrf2 protein stabilization and subsequent heme oxygenase-1 (HO1) expression [30]. Similarly, miR-141 activated Nrf2 signaling via silencing Keap1 [31]. Meanwhile, miR-141-activated Nrf2 signaling also protected human retinal pigment epithelium cells and retinal ganglion cells from UV radiation [29]. Further, miR-200a expression resulted in Keap1 degradation, leading to Nrf2 nuclear translocation and expression of anti-oxidant gene NADPH quinone oxidoreductase 1 (NQO1) [32].
Here, we identified microRNA-455 (miR-455) as a putative Cul3-targeting miRNA. More importantly, forced-expression of miR-455 activated Nrf2 signaling possibly via silencing Cul3, which protected human osteoblasts from H2O2.
RESULTS
miR-455 expression silences Cul3, causing Nrf2 protein stabilization in human osteoblastic cells
First, the miRNA database TargetScan was consulted, and potential Cul3-targeting miRNA was searched. We discovered that miR-455 (“-3p.1”) putatively targets the 3-UTR of Cul3 mRNA at position 28-34 (Figure 1A). Thereafter, a miR-455-expressing vector (pSuper-GFP-puro) was constructed (See Method), which was introduced to hFOB1. 19 human osteoblastic cells. Via puromycin selection, two stable hFOB1. 19 cell lines with the construct, namely miR-455 Vec (1)/(2), were established. As shown in Figure 1B, miR-455 (-3p) expression level was significantly increased in the stable cells. Remarkably, miR-455 expression dramatically decreased Cul3 mRNA expression in hFOB1. 19 cells (Figure 1C). Moreover, Cul3 protein was also downregulated in miR-455-expressing cells (Figure 1D). Consequently, Nrf2 protein (Figure 1D), but not Nrf2 mRNA (Figure 1E), was upregulated, indicating Nrf2 protein stabilization. Notably, Keap1 protein (Figure 1D) and mRNA (Figure 1F) were unchanged after miR-455 expression. The microRNA-control (“miRC”) (Figure 1B), as expected, had no significant effect on expression of Nrf2, Keap1 nor Cul3 (Figure 1C-1F). These results suggest that expression of miR-455 targets and downregulates Cul3, causing Nrf2 protein stabilization.
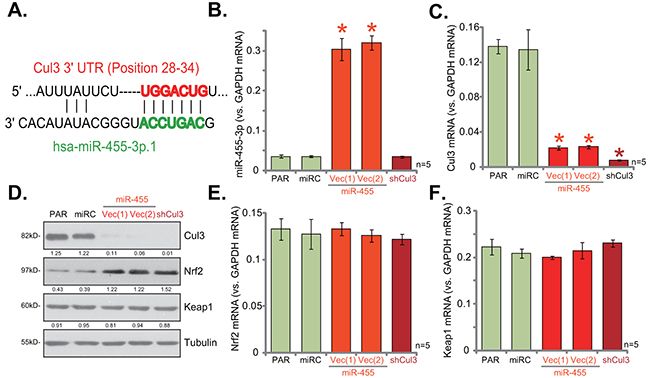
Figure 1: miR-455 expression silences Cul3, causing Nrf2 protein stabilization in human osteoblastic cells. miR-455 (3p) targets the 3-UTR of Cul3 mRNA at position 28-34 (A). Stable hFOB1. 19 osteoblastic cells (puromycin-selected), expressing miRNA-455 Vector [two lines, “Vec (1)/(2)”], microRNA-control (“miRC”) or the Cul3-shRNA (“shCul3”), as well as the parental control hFOB1. 19 cells (“PAR”) were subjected to qRT-PCR assay (B, C, E and F) and Western blotting assay (D) of listed miRNA and genes. Expression of listed proteins was quantified, and was normalized to loading control Tubulin (D). Data were shown as mean (n=5) ± standard deviation (SD). *p<0.05 vs. “PAR” cells. Experiments in this figure were repeated four times, and similar results were obtained.
If miR-455 expression targets Cul3 to induce Nrf2 accumulation, Cul3 knockdown should also stabilize Nrf2. Thus, the lentiviral Cul3-shRNA was utilized. Expression of Cul3-shRNA (“shCul3”) almost completely depleted Cul3 mRNA (Figure 1C) and protein (Figure 1D) in hFOB1. 19 cells. Similarly, Nrf2 protein was significantly increased (Figure 1D). Yet, Nrf2 mRNA (Figure 1E) and Keap1 mRNA (Figure 1F) expressions were not changed by the Cul3-shRNA. Thus, Cul3 silence causes Nrf2 stabilization in hFOB1. 19 cells.
Cul3 knockdown by expressing miR-455 or targeted-shRNA protects hFOB1. 19 cells from H2O2
The results above demonstrated that Cul3 depletion, by expressing miR-455 or targeted-shRNA, caused Nrf2 protein stabilization in hFOB1. 19 cells. Nrf2, once stabilized, shall translocate to nuclei and dictate transcription of anti-oxidant genes [18, 33]. Several ARE-dependent genes were then tested, including NADPH quinone oxidoreductase 1 (NQO1), heme oxygenase-1 (HO1) and glutamate cysteine ligase catalytic subunit (GCLC) [34, 35]. Results from quantitative real-time PCR (“qRT-PCR”) assay showed that, as compared to parental (“PAR”) hFOB1. 19 cells, mRNA expressions of NQO1, HO1 and GCLC were significantly increased in cells with miR-455 or Cul3-shRNA (Figure 2A). These results indicated Nrf2 signaling activation. Moreover, H2O2-induced ROS production was largely attenuated by either miR-455 expression or Cul3-shRNA in hFOB1. 19 cells (Figure 2B). Consequently, H2O2-induced hFOB1. 19 cell viability loss (MTT OD reduction, Figure 2C), cell death (Trypan blue increase, Figure 2D) and apoptosis (Histone DNA ELISA OD increase, Figure 2E) were also dramatically attenuated after expressing miR-455 or Cul3-shRNA. The microRNA-control (“miRC”) had no significant effect on ARE gene expression (Figure 2A) nor ROS production (Figure 2B). These results indicate that Cul3 knockdown by expressing miR-455 or targeted-shRNA protects hFOB1. 19 cells from H2O2.
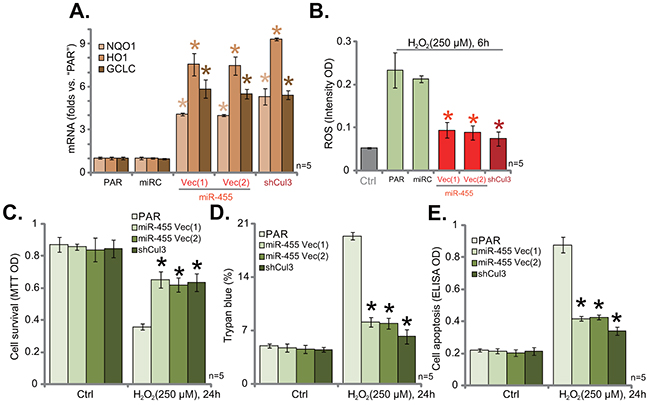
Figure 2: Cul3 knockdown by expressing miR-455 or targeted-shRNA protects hFOB1. 19 cells from H2O2. Stable hFOB1. 19 cells (puromycin-selected), expressing miRNA-455 Vector [two lines, “Vec (1)/(2)”], microRNA-control (“miRC”) or the Cul3-shRNA (“shCul3”), as well as the parental control hFOB1. 19 cells (“PAR”) were treated with/out H2O2 (250 μM, for applied time), relative NQO1, HO1 and GCLC mRNA expressions were tested by qRT-PCR assay (A). Cellular ROS intensity was tested by DCFH-DA fluorescent dye assay (B). Cell viability, cell death and apoptosis were tested by MTT assay (C), Trypan blue staining assay (D) and histone DNA ELISA assay (E), respectively. Data were shown as mean (n=5) ± standard deviation (SD). *p<0.05 vs. “PAR” cells. Experiments in this figure were repeated three times, and similar results were obtained.
miR-455 anti-sense induces Cul3 upregulation and Nrf2 degradation
To further confirm that miR-455 selectively targets Cul3, the miR-455 anti-sense (“Anti-miR-455”) was introduced to hFOB1. 19 cells. As shown in Figure 3A, Anti-miR-455 indeed depleted miR-455 in hFOB1. 19 cells. Consequently, Cul3 mRNA (Figure 3B) and protein (Figure 3C) expressions were upregulated. Nrf2 protein, on the other hand, was degradated (Figure 3C). Nrf2 mRNA, along with Keap1 mRNA, were yet unchanged (Figure 3D). Therefore, Anti-miR-455 depleted miR-455, causing Cul3 upregulation and Nrf2 degradation in hFOB1. 19 cells. These results further confirm that miR-455 selectively targets Cul3 in hFOB1. 19 cells.
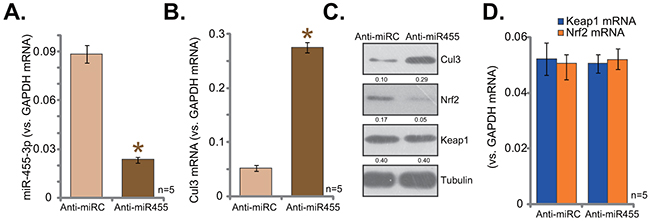
Figure 3: miR-455 anti-sense induces Cul3 upregulation and Nrf2 degradation. hFOB1. 19 cells were transfected with miR-455 anti-sense (“Anti-miR-455”, for 5 rounds) or miR anti-sense control (“Anti-miRC”), cells were then subjected to qRT-PCR assay (A, B, and D) and Western blotting assay (C) of listed miRNA and genes. Expression of listed proteins was quantified, and was normalized to loading control Tubulin (C). Data were shown as mean (n=5) ± standard deviation (SD). *p<0.05 vs. “Anti-miRC” cells. Experiments in this figure were repeated four times, and similar results were obtained.
Exogenous over-expression of Cul3 causes Nrf2 degradation
Based on the above results, we speculate that Cul3 over-expression should cause Nrf2 protein degradation. To test this hypothesis, the Cul3 expression vector was constructed, and was introduced to hFOB1. 19 cells. Via puromycin selection, two hFOB1. 19 cell lines with the Cul3 construct were established, namely “Cul3 Vec(1)/(2)”. As shown in Figure 4A, Cul3 mRNA level was significantly increased in the two stable lines. Western blotting assay results in Figure 4B confirmed the expression of exogenous Cul3 (Flag-tagged) and endogenous Cul3 in the two lines. Notably, exogenous over-expression of Cul3 indeed led to Nrf2 protein degradation in hFOB1. 19 cells. Nrf2 mRNA (Figure 4C) and Keap1 expression (Figure 4B and 4D) were unchanged with Cul3 over-expression.
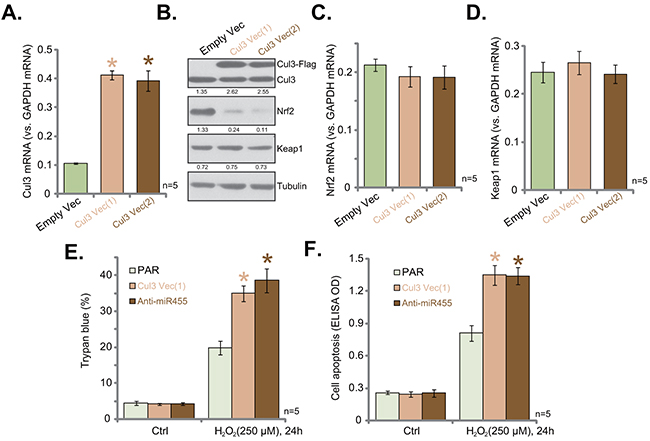
Figure 4: Exogenous over-expression of Cul3 causes Nrf2 degradation. Stable hFOB1. 19 cells, expressing Cul3-expressing Vector [two lines, “Vec (1)/(2)”] or empty vector (“Empty Vec”, pSuper-puro-Flag-GFP) were subjected to qRT-PCR assay (A, C and D) and Western blotting assay (B) of listed genes. Expression of listed proteins was quantified, and was normalized to loading control Tubulin (B). Stable hFOB1. 19 cells, expressing Cul3-expressing Vector [“Vec (1)”], or miR-455 anti-sense (“Anti-miR-455”) as well as the parental control cells (“PAR”) were treated with/out H2O2 (250 μM) for 24 hours. Cell death and apoptosis were tested by Trypan blue staining assay (E) and histone DNA ELISA assay (F), respectively. Data were shown as mean (n=5) ± standard deviation (SD). *p<0.05 vs. “Empty Vec” cells (A). *p<0.05 vs. “PAR” cells (E and F). Experiments in this figure were repeated five times, and similar results were obtained.
Cul3 upregulation and Nrf2 degradation were observed in hFOB1. 19 cells expressing Anti-miR-455 (Figure 3) and Cul3-expressing construct (Figure 4A-4D), we then tested H2O2 sensitivity in these cells. As compared to the parental control cells (“PAR”), H2O2 (250 μM)-induced cell death (Figure 4E) and apoptosis (Figure 4F) were dramatically exacerbated in cells expressing Anti-miR-455 or Cul3 vector. These results suggested that Cul3 upregulation induced Nrf2 degradation and facilitated H2O2-induced killing of hFOB1. 19 cells.
miR-455-induced Cul3 silence protects primary human osteoblasts from H2O2
The results above indicated that Cul3 silence could protect hFOB1. 19 cells from H2O2. Next, we tested this hypothesis in primary human cells. Primary-cultured human osteoblasts were constructed with miR-455-expressing vector [“Vec (1)”] or the Cul3-shRNA. Via puromycin selection, stable cells were established. miR-455 (-3p) level was only increased in cells expressing miR-455 vector, but not Cul3-shRNA (Figure 5A). Notably, Cul3 downregulation (Figure 5B and 5C) and Nrf2 protein stabilization (Figure 5C) were observed in cells expressing miR-455 or Cul3-shRNA. Consequently, upregulation of ARE-dependent genes, NQO1 and HO1, was achieved in above mentioned primary cultured human osteoblasts (Figure 5D). More importantly, H2O2 (250 μM)-induced cell death (Figure 5E) and apoptosis (Figure 5F) were largely inhibited in the primary cells expressing miR-455 or Cul3-shRNA. Thus, we imply that Cul3 silence, by expressing miR-455 or targeted-shRNA, protects primary human osteoblasts from H2O2.
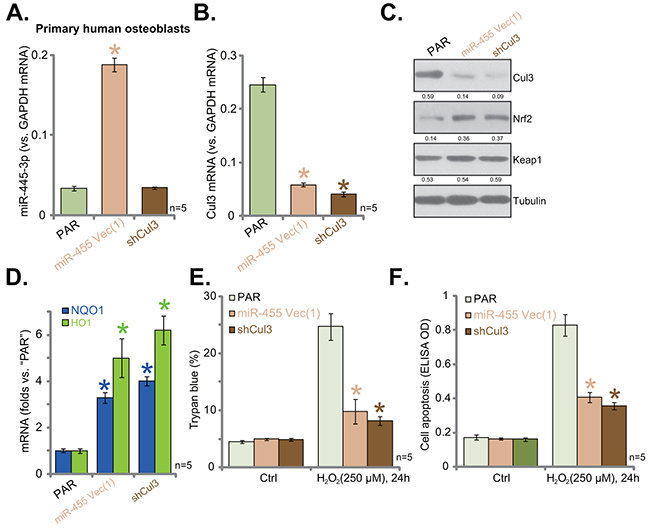
Figure 5: miR-455-induced Cul3 silence protects primary human osteoblasts from H2O2. Puromycin-selected stable primary human osteoblasts, expressing miRNA-455 Vector [“Vec (1)”] or the Cul3-shRNA (“shCul3”), as well as the parental control cells (“PAR”) were subjected to qRT-PCR assay (A, B and D) and Western blotting assay (C) of listed microRNA or genes. Expression of listed proteins was quantified, and was normalized to loading control Tubulin (C). Cells were also treated with/out H2O2 (250 μM, for 24 hours). Cell death and apoptosis were tested by Trypan blue staining assay (E) and histone DNA ELISA assay (F), respectively. Data were shown as mean (n=5) ± standard deviation (SD). *p<0.05 vs. “PAR” cells. Experiments in this figure were repeated twice, and similar results were obtained.
DISCUSSION
Nrf2 stabilization and activation induces ARE-dependent transcription of multiple antioxidant defense genes [34, 35], including NQO1, HO1, GCLC [21, 36–38]. Nrf2-ARE signaling has become an attractive target for prevention of human osteoblast injuries. Li et al., previously demonstrated that SC79, a novel Akt activator, protected osteoblasts from dexamethasone though activating Akt downstream Nrf2 signaling [39]. Meanwhile, icariside II-induced osteoblast cytoprotection requires Nrf2 activation [40]. Further, cytoprotection of chlorogenic acid against H2O2-induced oxidative stress in osteoblasts also relies on activation of Nrf2-HO-1 signaling [41]. Nrf2 signaling activation is primarily based on Nrf2 dissociation from its inactive repressor protein Keap1, and the subsequent translocation of Nrf2 to the cell nuclei [21, 36–38]. Many natural and synthetic chemicals have been shown to interfere Nrf2-Keap1 association, causing Nrf2 protein stabilization and activation [28, 29, 31, 42]. Other studies have been able to provoke Nrf2 activation via inhibition, silence, mutation or depletion of Keap1 [28, 29, 31, 42].
Cul3 is a member of the cullin-based ubiquitin ligase family, which is required for Nrf2 degradation [43]. Cul3 forms a complex with Hrt1 and BTB-domain containing proteins, which functions as an E3 ligase to bring Keap1 to ubiquitination and degradation [43]. On the other hand, Cul3 inhibition, silence or mutation will cause inhibition of Nrf2 degradation, and Nrf2 protein stabilization [44]. Recent studies have proposed that miRNA could be a novel and promising strategy to provoke Nrf2 signaling activation (mostly by targeting Keap1) [28, 29]. Very few have focused on miRNA-mediated targeting of Cul3.
Our results here demonstrated that miR-455 is a Cul3-targeting miR in human osteoblasts. Forced-expression of miR-455 in human osteoblasts led to Cul3 degradation, Nrf2 protein stabilization and subsequent transcription of ARE-dependent genes (NQO1, HO1 and GCLC). Remarkably, Cul3 silencing by miR-455 expression or targeted-shRNA protected human osteoblasts from H2O2. On the other hand, miR-455 depletion by miR-455 anti-sense led to Cul3 upregulation and Nrf2 protein degradation, which then exacerbated H2O2 damages in human osteoblasts. These results together indicate that miR-455 expression could be a novel strategy to provoke Nrf2-ARE signaling activation in human osteoblasts. It will also be interesting to test the in vivo function of miR-455 against oxidative-damaged human osteoblasts. Expressions of miR-455 and Cul3 in human osteoporosis and osteonecrosis tissues should also be tested in future studies.
CONCLUSIONS
Together, our results suggest that miR-455 activates Nrf2 signaling via silencing Cul3, and protects human osteoblasts from oxidative stress.
MATERIALS AND METHODS
Reagents
Puromycin was purchased from Sigma Aldrich (St. Louis, MO). All the antibodies were purchased from Cell Signaling Tech (Beverly, MA). Cell culture reagents were obtained from Gibco (Nantong, China).
Culture of osteoblastic cell line
The hFOB1.19 human osteoblastic cell line [45, 46] was obtained from the Cell Bank of Shanghai Institute of Biological Science (Shanghai, China). Cells were maintained in α-modified essential medium (α-MEM) supplemented with 10% FBS, under 37°C in the presence of 5% CO2. Cells were fully differentiated as described [47].
Primary culture of human osteoblasts
The trabecular bone fragments from healthy donors were minced into small pieces, which were digested by incubation with 5 mg/mL collagenase D (Sigma) for 90 min at 37 °C with agitation. The resulting trabecular bone fragments were further digested with 0.5 mg/mL collagenase D overnight at 37 °C. Cells were then filtered through a 70-μm nylon mesh, and were placed onto the culture flasks with the described medium [48]. Medium was changed three times a week until reaching confluence, and were fully differentiated as described [47]. Primary human osteoblasts were used for further experiments stating at passage 3. The protocols of using human tissues and cells were approved by Ethics Board of Nantong University. Written-informed consent was obtained from each donor.
Cell viability assay
Human osteoblasts (5000 cells per each well) were initially seeded onto 96-well plates. Following the applied treatment, the MTT dye (20 μL/per well, 5 mg/mL, Sigma) was added to the supernatant for two hours. Afterwards, the optic density (OD) absorbance of MTT at 450 nm was measured by a microplate reader to reflect cell viability.
Cell death assay
Trypan blue staining assay was performed to test cell death after applied treatment. Cells excluding the dye were considered alive. Trypan blue positive cells were considered dead, and the ratio was recorded using an automatic cell counter.
Apoptosis quantification by ELISA assay
Thenucleosomal histone-bound DNA fragmentation is the characteristic marker of cell apoptosis, which was examined by the commercial available ELISA kit (Roche, Shanghai, China), using the anti-histone antibody and a secondary anti-DNA antibody. The ELISA OD at 450 nm was tested as the quantitative measurement of cell apoptosis.
Forced-expression of miR-455
The miR-455 precursor was purchased from RiboBio (Guangzhou, China), which was inserted to the pSuper-GFP-puro vector (Ambion, Shanghai, China) to establish the miR-455-expression vector. Human osteoblasts were transfected with the miR-455 construct or the scramble non-sense microRNA control (“miRC”, Genepharm, Shanghai, China) using the Lipofectamine 2000 reagent (Invitrogen). Stable cells were selected by puromycin (2.5 μg/mL, Sigma) for another 96 hours. Over 95% of stable cells were GFP positive. miR-455 (3p) expression was always verified by the qRT-PCR assay.
miR-455 anti-sense expression
The hFOB1. 19 osteoblasticcells were transfected with 20 nM of miR-455 anti-sense (“Anti-miR-455”, Ambion, Shanghai, China) by Lipofectamine 2000 (Invitrogen). After two days, cells were split and were transfected with Anti-miR-455 again. This process was repeated for five rounds for a total of 10 days. Expression of miR-455 in the stable cells was examined by qRT-PCR assay. The Ambion miRNA anti-sense negative control (“Anti-miRC”) was transfected to hFOB1. 19 cells as the control cells.
Western blotting assay
Equivalent amount of total cellular proteins (30 μg per lane) were extracted by a RIPA buffer (Biyuntian, Wuxi, China), and were separated by the 10% SDS gel, prior to transfer onto polyvinylidene difluoride (PVDF) membranes (Millipore, Shanghai, China). The blots were then blocked in 5 % (m/v) milk dissolved in Tris-buffered saline with 0.05 % (w/v) Tween-20 (TBS-T), and were probed with the designated primary and secondary antibodies. The protein signals were visualized under an enhanced chemiluminescence (ECL) system (Amersham Bioscience, Shanghai, China). β-Tubulin (“Tubulin”) was always tested as the loading control. The images were analyzed with Image J software.
Cul3-shRNA
The lentiviral Cul3-shRNA particles (with GFP-tag) were purchased from Santa Cruz Biotech (sc-35130-V, Nanjing, China). The lentiviral particles (20 μL/mL, per each well) were added to cultured human osteoblasts for 48 hours. Stable cells were again selected by puromycin (2.5 μg/mL, Sigma) for 96 hours. Over 98% of stable cells were GFP positive. Cul3 knockdown in the stable cells was verified by Western blotting assay and qRT-PCR assay. For the control cells, the lentiviral scramble control shRNA particles (Santa Cruz Biotech) were added.
Exogenous Cul3 over-expression
The full-length human Cul3 cDNA was synthesized by Genepharm (Shanghai, China), which was inserted to the pSuper-puro-GFP-Flag vector (Addgene, Shanghai, China). Lipofectamine 2000 was applied to transfect the Cul3 construct to human osteoblasts. Puromycin (2.5 μg/mL, Sigma) was added to select stable cells for 96 hours. Over 98% of cells were GFP positive. Expression of endogenous and exogenous (Flag-tagged) Cul3 in the stable cells was verified by Western blotting assay and qRT-PCR assay. The empty pSuper-puro-GFP-Flag vector was transfected to the control cells.
Quantitative RT-PCR
Trizol reagents (Invitrogen) were utilized to extract total cellular RNA, and the High Capacity cDNA Reverse Transcription Kit was applied to synthesize cDNA from 0.5 μg mRNA per treatment. Quantitative real-time PCR (“qRT-PCR”) assay was performed by the Power SYBR Green RT-PCR Reagents Kit using the ABI-7500 system [49]. We utilized 2ΔΔCt method to yield relative mRNA fold expression (as compare to GAPDH mRNA). mRNA primers for HO-1 and GCLC were described previously [50]. mRNA primers for Nrf2, Keap1, Cul3, NQO1 and GAPDH were described early [51]. miR-455 (-3p) expression was tested via the TaqMan microRNA assay [52] (Applied Biosystems, Shanghai, China), from 5 ng of total RNA [53].
ROS assay
As described previously [16, 40, 54–56], we utilized the dichloro-dihydro-fluorescein diacetate (DCFH-DA) fluorescent dye (Invitrogen) assay to determine the intracellular ROS intensity. Briefly, after the applied treatment, DCFH-DA dye (5.0 μg/mL) was added to cells, followed by three-founds wash in warm PBS. Afterwards, the DCFH-DA fluorescence OD, reflecting the relative ROS intensity, was examined by a Fluorescence/Multi-Detection Microplate Reader (Synergy 2, BioTek).
Statistics
Data were expressed as the mean ± SD [45, 57]. Comparisons between groups were performed via one-way ANOVA and then Student-Newman-Keuls test (SPSS 18.0). The p values < 0.05 were considered statistically significant.
Author contributions
All authors carried out the experiments, participated in the design of the study and performed the statistical analysis, participated in its design and coordination and helped to draft the manuscript.
ACKNOWLEDGMENTS
This work was supported by a grant from the National Natural Science Foundation of China (No. 81300955), three grants from the Scientific Research Program of Nantong (No. MS22015047, MS12016009, wq2015011), and also a grant from the Ke-Jiao-Qiang-Wei-Qing-Nian-Yi-Xue-Zhong-Dian-Ren-Cai Financial Assistance of Jiangsu Province (No. QNRC2016412).
CONFLICTS OF INTEREST
The authors have no conflict of interests.
REFERENCES
1. Souttou B, Raulais D, Vigny M. Pleiotrophin induces angiogenesis: involvement of the phosphoinositide-3 kinase but not the nitric oxide synthase pathways. J Cell Physiol. 2001; 187:59-64.
2. Himburg HA, Muramoto GG, Daher P, Meadows SK, Russell JL, Doan P, Chi JT, Salter AB, Lento WE, Reya T, Chao NJ, Chute JP. Pleiotrophin regulates the expansion and regeneration of hematopoietic stem cells. Nat Med. 2010; 16:475-482.
3. Baek KH, Oh KW, Lee WY, Lee SS, Kim MK, Kwon HS, Rhee EJ, Han JH, Song KH, Cha BY, Lee KW, Kang MI. Association of oxidative stress with postmenopausal osteoporosis and the effects of hydrogen peroxide on osteoclast formation in human bone marrow cell cultures. Calcif Tissue Int. 2010; 87:226-235.
4. Tare RS, Oreffo RO, Sato K, Rauvala H, Clarke NM, Roach HI. Effects of targeted overexpression of pleiotrophin on postnatal bone development. Biochem Biophys Res Commun. 2002; 298:324-332.
5. Herbst RS. Review of epidermal growth factor receptor biology. Int J Radiat Oncol Biol Phys. 2004; 59:21-26.
6. Talasila KM, Soentgerath A, Euskirchen P, Rosland GV, Wang J, Huszthy PC, Prestegarden L, Skaftnesmo KO, Sakariassen PO, Eskilsson E, Stieber D, Keunen O, Brekka N, et al. EGFR wild-type amplification and activation promote invasion and development of glioblastoma independent of angiogenesis. Acta Neuropathol. 2013; 125:683-698.
7. Liang D, Xiang L, Yang M, Zhang X, Guo B, Chen Y, Yang L, Cao J. ZnT7 can protect MC3T3-E1 cells from oxidative stress-induced apoptosis via PI3K/Akt and MAPK/ERK signaling pathways. Cell Signal. 2013; 25:1126-1135.
8. Koh HK. Cutaneous melanoma. N Engl J Med. 1991; 325:171-182.
9. Rigel DS, Friedman RJ, Kopf AW. The incidence of malignant melanoma in the United States: issues as we approach the 21st century. J Am Acad Dermatol. 1996; 34:839-847.
10. Salopek TG, Marghoob AA, Slade JM, Rao B, Rigel DS, Kopf AW, Bart RS. An estimate of the incidence of malignant melanoma in the United States. Based on a survey of members of the American Academy of Dermatology. Dermatol Surg. 1995; 21:301-305.
11. Fatokun AA, Stone TW, Smith RA. Hydrogen peroxide-induced oxidative stress in MC3T3-E1 cells: the effects of glutamate and protection by purines. Bone. 2006; 39:542-551.
12. Xu ZS, Wang XY, Xiao DM, Hu LF, Lu M, Wu ZY, Bian JS. Hydrogen sulfide protects MC3T3-E1 osteoblastic cells against H2O2-induced oxidative damage-implications for the treatment of osteoporosis. Free Radic Biol Med. 2011; 50:1314-1323.
13. She C, Zhu LQ, Zhen YF, Wang XD, Dong QR. Activation of AMPK protects against hydrogen peroxide-induced osteoblast apoptosis through autophagy induction and NADPH maintenance: new implications for osteonecrosis treatment? Cell Signal. 2014; 26:1-8.
14. Yang L, Wu Z, Yin G, Liu H, Guan X, Zhao X, Wang J, Zhu J. Stem cell factor (SCF) protects osteoblasts from oxidative stress through activating c-Kit-Akt signaling. Biochem Biophys Res Commun. 2014; 455:256-261.
15. Zhu Y, Zhou J, Ao R, Yu B. A-769662 protects osteoblasts from hydrogen dioxide-induced apoptosis through activating of AMP-activated protein kinase (AMPK). Int J Mol Sci. 2014; 15:11190-11203.
16. Liu W, Mao L, Ji F, Chen F, Hao Y, Liu G. Targeted activation of AMPK by GSK621 ameliorates H2O2-induced damages in osteoblasts. Oncotarget. 2017; 8:10543-10552. doi: 10.18632/oncotarget.14454.
17. Vriend J, Reiter RJ. The Keap1-Nrf2-antioxidant response element pathway: a review of its regulation by melatonin and the proteasome. Mol Cell Endocrinol. 2015; 401:213-220.
18. Suzuki T, Yamamoto M. Molecular basis of the Keap1-Nrf2 system. Free Radic Biol Med. 2015; 88:93-100.
19. Ma Q, He X. Molecular basis of electrophilic and oxidative defense: promises and perils of Nrf2. Pharmacol Rev. 2012; 64:1055-1081.
20. Itoh K, Mimura J, Yamamoto M. Discovery of the negative regulator of Nrf2, Keap1: a historical overview. Antioxid Redox Signal. 2010; 13:1665-1678.
21. Li W, Kong AN. Molecular mechanisms of Nrf2-mediated antioxidant response. Mol Carcinog. 2009; 48:91-104.
22. Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007; 47:89-116.
23. Jang HJ, Hong EM, Kim M, Kim JH, Jang J, Park SW, Byun HW, Koh DH, Choi MH, Kae SH, Lee J. Simvastatin induces heme oxygenase-1 via NF-E2-related factor 2 (Nrf2) activation through ERK and PI3K/Akt pathway in colon cancer. Oncotarget. 2016; 7:46219-46229. doi: 10.18632/oncotarget.10078.
24. Chen Z, Zhang Y, Ma L, Ni Y, Zhao H. Nrf2 plays a pivotal role in protection against burn trauma-induced intestinal injury and death. Oncotarget. 2016; 7:19272-19283. doi: 10.18632/oncotarget.8189.
25. Ryoo IG, Choi BH, Kwak MK. Activation of NRF2 by p62 and proteasome reduction in sphere-forming breast carcinoma cells. Oncotarget. 2015; 6:8167-8184. doi: 10.18632/oncotarget.3047.
26. Pasquinelli AE. MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nat Rev Genet. 2012; 13:271-282.
27. Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet. 2015; 16:421-433.
28. Krajka-Kuzniak V, Paluszczak J, Baer-Dubowska W. The Nrf2-ARE signaling pathway: an update on its regulation and possible role in cancer prevention and treatment. Pharmacol Rep. 2017; 69:393-402.
29. Cheng LB, Li KR, Yi N, Li XM, Wang F, Xue B, Pan YS, Yao J, Jiang Q, Wu ZF. miRNA-141 attenuates UV-induced oxidative stress via activating Keap1-Nrf2 signaling in human retinal pigment epithelium cells and retinal ganglion cells. Oncotarget. 2017; 8:13186-13194. doi: 10.18632/oncotarget.14489.
30. Kabaria S, Choi DC, Chaudhuri AD, Jain MR, Li H, Junn E. MicroRNA-7 activates Nrf2 pathway by targeting Keap1 expression. Free Radic Biol Med. 2015; 89:548-556.
31. Shi L, Wu L, Chen Z, Yang J, Chen X, Yu F, Zheng F, Lin X. MiR-141 activates Nrf2-dependent antioxidant pathway via down-regulating the expression of Keap1 conferring the resistance of hepatocellular carcinoma cells to 5-fluorouracil. Cell Physiol Biochem. 2015; 35:2333-2348.
32. Eades G, Yang M, Yao Y, Zhang Y, Zhou Q. miR-200a regulates Nrf2 activation by targeting Keap1 mRNA in breast cancer cells. J Biol Chem. 2011; 286:40725-40733.
33. Zhang Q, Pi J, Woods CG, Andersen ME. A systems biology perspective on Nrf2-mediated antioxidant response. Toxicol Appl Pharmacol. 2010; 244:84-97.
34. Ji C, Huang JW, Xu QY, Zhang J, Lin MT, Tu Y, He L, Bi ZG, Cheng B. Gremlin inhibits UV-induced skin cell damages via activating VEGFR2-Nrf2 signaling. Oncotarget. 2016; 7:84748-84757. doi: 10.18632/oncotarget.12454.
35. Gong YQ, Huang W, Li KR, Liu YY, Cao GF, Cao C, Jiang Q. SC79 protects retinal pigment epithelium cells from UV radiation via activating Akt-Nrf2 signaling. Oncotarget. 2016; 7:60123-60132. doi: 10.18632/oncotarget.11164.
36. Bruns DR, Drake JC, Biela LM, Peelor FF 3rd, Miller BF, Hamilton KL. Nrf2 signaling and the slowed aging phenotype: evidence from long-lived models. Oxid Med Cell Longev. 2015; 2015:732596.
37. Nguyen T, Yang CS, Pickett CB. The pathways and molecular mechanisms regulating Nrf2 activation in response to chemical stress. Free Radic Biol Med. 2004; 37:433-441.
38. Itoh K, Tong KI, Yamamoto M. Molecular mechanism activating Nrf2-Keap1 pathway in regulation of adaptive response to electrophiles. Free Radic Biol Med. 2004; 36:1208-1213.
39. Li ST, Chen NN, Qiao YB, Zhu WL, Ruan JW, Zhou XZ. SC79 rescues osteoblasts from dexamethasone though activating Akt-Nrf2 signaling. Biochem Biophys Res Commun. 2016; 479:54-60.
40. Liu W, Mao L, Ji F, Chen F, Wang S, Xie Y. Icariside II activates EGFR-Akt-Nrf2 signaling and protects osteoblasts from dexamethasone. Oncotarget. 2017; 8:2594-2603. doi: 10.18632/oncotarget.13732.
41. Han D, Chen W, Gu X, Shan R, Zou J, Liu G, Shahid M, Gao J, Han B. Cytoprotective effect of chlorogenic acid against hydrogen peroxide-induced oxidative stress in MC3T3-E1 cells through PI3K/Akt-mediated Nrf2/HO-1 signaling pathway. Oncotarget. 2017; 8:14680-14692. doi: 10.18632/oncotarget.14747.
42. Yang JJ, Tao H, Hu W, Liu LP, Shi KH, Deng ZY, Li J. MicroRNA-200a controls Nrf2 activation by target Keap1 in hepatic stellate cell proliferation and fibrosis. Cell Signal. 2014; 26:2381-2389.
43. Furukawa M, Xiong Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol Cell Biol. 2005; 25:162-171.
44. Pintard L, Willems A, Peter M. Cullin-based ubiquitin ligases: Cul3-BTB complexes join the family. EMBO J. 2004; 23:1681-1687.
45. Fan JB, Ruan JW, Liu W, Zhu LQ, Zhu XH, Yi H, Cui SY, Zhao JN, Cui ZM. miR-135b expression downregulates Ppm1e to activate AMPK signaling and protect osteoblastic cells from dexamethasone. Oncotarget. 2016; 7:70613-70622. doi: 10.18632/oncotarget.12138.
46. Ding H, Wang T, Xu D, Cha B, Liu J, Li Y. Dexamethasone-induced apoptosis of osteocytic and osteoblastic cells is mediated by TAK1 activation. Biochem Biophys Res Commun. 2015; 460:157-163.
47. Sato AY, Tu X, McAndrews KA, Plotkin LI, Bellido T. Prevention of glucocorticoid induced-apoptosis of osteoblasts and osteocytes by protecting against endoplasmic reticulum (ER) stress in vitro and in vivo in female mice. Bone. 2015; 73:60-68.
48. van der Meijden K, van Essen HW, Bloemers FW, Schulten EA, Lips P, Bravenboer N. Regulation of CYP27B1 mRNA expression in primary human osteoblasts. Calcif Tissue Int. 2016; 99:164-173.
49. Silverstein AM, Barrow CA, Davis AJ, Mumby MC. Actions of PP2A on the MAP kinase pathway and apoptosis are mediated by distinct regulatory subunits. Proc Natl Acad Sci U S A. 2002; 99:4221-4226.
50. Chapple SJ, Siow RC, Mann GE. Crosstalk between Nrf2 and the proteasome: therapeutic potential of Nrf2 inducers in vascular disease and aging. Int J Biochem Cell Biol. 2012; 44:1315-1320.
51. Loignon M, Miao W, Hu L, Bier A, Bismar TA, Scrivens PJ, Mann K, Basik M, Bouchard A, Fiset PO, Batist Z, Batist G. Cul3 overexpression depletes Nrf2 in breast cancer and is associated with sensitivity to carcinogens, to oxidative stress, and to chemotherapy. Mol Cancer Ther. 2009; 8:2432-2440.
52. Zhen YF, Li ST, Zhu YR, Wang XD, Zhou XZ, Zhu LQ. Identification of DNA-PKcs as a primary resistance factor of salinomycin in osteosarcoma cells. Oncotarget. 2016; 7:79417-79427. doi: 10.18632/oncotarget.12712.
53. Chen W, Chen L, Zhang Z, Meng F, Huang G, Sheng P, Liao W. MicroRNA-455-3p modulates cartilage development and degeneration through modification of histone H3 acetylation. Biochim Biophys Acta. 2016; 1863:2881-2891.
54. Ji F, Mao L, Liu Y, Cao X, Xie Y, Wang S, Fei H. K6PC-5, a novel sphingosine kinase 1 (SphK1) activator, alleviates dexamethasone-induced damages to osteoblasts through activating SphK1-Akt signaling. Biochem Biophys Res Commun. 2015; 458:568-575.
55. Guo S, Xie Y, Fan JB, Ji F, Wang S, Fei H. α-Melanocyte stimulating hormone attenuates dexamethasone-induced osteoblast damages through activating melanocortin receptor 4-SphK1 signaling. Biochem Biophys Res Commun. 2016; 469:281-287.
56. Guo S, Mao L, Ji F, Wang S, Xie Y, Fei H, Wang XD. Activating AMP-activated protein kinase by an alpha1 selective activator compound 13 attenuates dexamethasone-induced osteoblast cell death. Biochem Biophys Res Commun. 2016; 471:545-552.
57. Fan JB, Liu W, Zhu XH, Yi H, Cui SY, Zhao JN, Cui ZM. microRNA-25 targets PKCzeta and protects osteoblastic cells from dexamethasone via activating AMPK signaling. Oncotarget. 2017; 8:3226-3236. doi: 10.18632/oncotarget.13698.