
INTRODUCTION
Ovarian cancer is the leading cause of death from gynecological malignancies in developed countries [1]. Despite their clinical importance, little is known about the early stages of development for this neoplasm, largely owing to the absence of adequate animal models, the clinical inaccessibility of human ovaries, and the unfortunate paucity of symptoms until higher stages. The most common form of ovarian cancer is epithelial ovarian cancer (EOC). EOCs are believed to originate from normal ovarian surface epithelium or from its derivatives, which include the crypts and inclusion cysts on the epithelial surface [2]. A series of recent evidence also suggest that ovarian high-grade serous carcinoma (HGSC) originates from the fimbrial portion of the fallopian tube, which has been termed “serous tubal intraepithelial carcinoma” (STIC) [3-6]. Understanding the molecular basis of EOC should significantly refine the diagnosis and management of these tumors and eventually lead to the development of more specific and more effective treatment modalities.
S100 proteins, a large subgroup of the EF-hand protein family, are small calcium-binding proteins that can function as both intracellular and extracellular signaling molecules [7]. They exert a broad range of functions by modulating their subcellular localization and by interacting with specific target proteins responsible for the regulation of inflammation, cell growth, cell motility, cell survival, and apoptosis [7-9]. Altered expression of a large number of S100 proteins has been reported in various human malignancies [10]. In addition, some S100 proteins, namely S100A2, S100A4, and S100P, have been implicated in tumor invasion and metastasis [11-13].
S100A14 has recently been identified as an S100 calcium-binding protein with unknown biological function [14]. The S100A14 gene was originally cloned and characterized in human lung cancer cell line, but was found to be differentially expressed in a variety of cell types. It was also reported to be upregulated in several tumor types, including ovarian, lung, breast, and uterine cancer, but downregulated in others, such as kidney, colon, rectal, and esophageal cancer [14]. S100A14 has also been shown to play vital roles in bladder tumorigenesis and tumor progression [15]. S100A14 can also regulate oral squamous cell carcinoma cell invasion by modulating the expression of matrix metalloproteinase (MMP) 1 and MMP9 [16]. However, the role of S100A14 in ovarian cancer development and its underlying molecular mechanisms remain unclear. We have previously identified S100A14 to be upregulated in EOC by microarray [17] and have confirmed this by real-time PCR. In this study, we further investigate the clinical significance and functional role of S100A14 in EOC.
RESULTS
High S100A14 expression correlates with poor prognosis in EOC
We previously observed that the transcription level of S100A14 was upregulated 7.85-fold in three EOC cell lines by microarray analysis [17]. To eliminate the possibility that S100A14 gene expression only occurred in newly established EOC cell lines in in vitro culture, we performed validation studies using PCR and immunohistochemistry (IHC) in various EOC cell lines and tissues. Reverse transcriptase-PCR (RT-PCR) and real-time PCR revealed thatS100A14 mRNA levels were abundantly expressed in ovarian cancer cell lines, except TOV112D, OVCA433, and YDOV-151, whereas S100A14 expression was almost undetectable in HOSE cell lines (Fig. 1A). Lysates from HEK293T cells transfected with pCDH/S100A14 plasmids were loaded as a positive control. Expression of S100A14 at the protein level was confirmed by immunoblot (Fig. 1B). S100A14 was highly expressed in SNU840, RMUG-S, and YDOV-139 cell lines, but could not be detected in HOSE cell lines (Fig. 1B). These observations suggest that S100A14 expression primarily occurs in fully transformed cells.
To determine whether S100A14 overexpression is linked to clinicopathological features of EOC, we performed IHC analysis of S100A14 in 104 ovarian tissue specimens. Most immunoreactivity was observed in the cytoplasm of malignant cells (Fig. 1C). Scoring results from IHC analyses are summarized in Supplementary Table 2. EOC tissues had higher S100A14 expression levels than in borderline, benign, or normal tissues (P < 0.001). Furthermore, S100A14 immunoreactivity significantly correlated with features also associated with poor prognosis, including tumor stage (P < 0.001), histologic subtype (P = 0.004), and tumor grade (P < 0.001). Specifically, advanced stage including recurrence, serous histologic subtype, and poor differentiation correspond to higher IHC scores (Fig. 1D, Supplementary Table 1). To further analyze S100A14 expression in ovarian cancer, we combined seven microarray data sets (GSE55510, GSE55512, GSE27651, GSE14001, GSE14407, GSE28724, and GSE51373) downloaded from the GEO database according to the literature [14]. Overall, 149 ovarian cancers from genome-wide gene expression data were available. As expected, we found that S100A14 mRNA expression was significantly higher in ovarian cancer (n = 149) than in normal ovarian surface epithelium (n = 29) (P < 0.001).
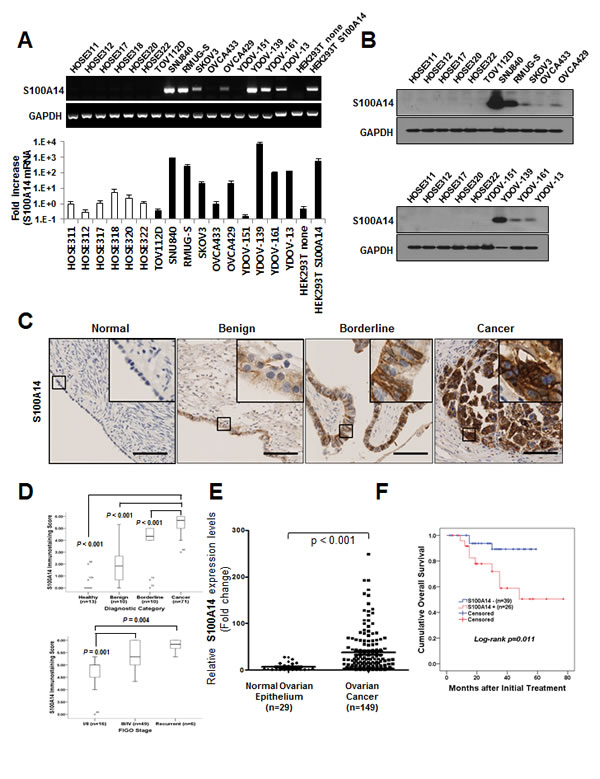
Figure 1: S100A14 is highly expressed in human ovarian cancer cells and tissue specimens, and its expression correlates with tumor stage and outcome of disease. A. S100A14 mRNA levels were assessed using RT-PCR (upper panel) and real-time PCR (lower panel) in human ovarian surface epithelial (HOSE) cells and ovarian cancer cells. Expression of GAPDH was included as an internal loading control. Kruskal-Wallis analysis of variance and post hoc Dunn method revealed statistically significant differences between EOC and HOSE cell lines (P = 0.019). Each value is expressed as a mean of triplicate samples. The reference cell line, HOSE 311, was considered to have a value of 1. B. S100A14 protein levels were analyzed using immunoblot. GAPDH was included as an internal loading control. C. Representative immunohistochemical staining for S100A14 in formalin-fixed, paraffin-embedded EOC tissues. Bars: 100 µm. D. IHC staining scores of S100A14 in EOC samples were significantly higher than those of healthy controls, benign ovarian tumors, and borderline ovarian tumors (P < 0.001). E. When we combined seven microarray data sets from GEO database, S100A14 mRNA expression was significantly increased in ovarian cancer (n = 149) when compared to normal ovarian surface epithelium (n = 29) (P < 0.001). F. Kaplan-Meier plots for patients with epithelial ovarian cancer were stratified according to S100A14 expression.
We next examined the relationship between S100A14 expression and outcome. Clinicopathological and outcome information was available for 65 EOC patients. Tumor samples in six cases were obtained at recurrence and excluded from the survival analysis. The follow-up period of EOC ranged from 5 to 77 months with a mean of 30.8 months. Kaplan-Meier plots demonstrated that patients with advanced stage (III/IV) and patients whose tumors were S100A14+ (IHC score of 6) displayed significantly worse overall survival (P = 0.029 and P = 0.011, respectively) (Fig. 1E, Supplementary Fig. 1A-1C). A Cox multivariate proportional hazards analysis showed that advanced stage (hazard ratio [HR] = 3.25, P = 0.038 and HR = 4.31, P = 0.024, respectively) and S100A14+ status (HR = 3.10, P = 0.017 and HR = 4.53, P = 0.029, respectively) were independent prognostic factors of disease-free and overall survival (Table 1).
Table 1: Univariate and multivariate analyses of the associations between prognostic variables and disease-free (DFS) and overall survival (OS) in EOC patients.
DFS hazard ratio (95% CI), P value |
OS hazard ratio (95% CI), P value |
|||
Univariate |
Multivariate |
Univariate |
Multivariate |
|
FIGO stage (III/IV) |
||||
Tumor grade (poor) |
NS |
NA |
||
Cell type (Serous) |
NS |
NA |
||
CA125+† |
NS |
NA |
NS |
NA |
S100A14+‡ |
||||
CI, confidence interval; NS, not significant; NA, not applicable, CA125+† (> 35 U ml−1), S100A14+‡ (IHC score of > 5).
Lentiviral-mediated overexpression and knockdown of S100A14 reveal that high S100A14 expression increases cell proliferation and colony formation in EOC cells
Given that the biological function of S100A14 in EOC is largely unknown, we set out to investigate the potential role of S100A14 in the development of a malignant phenotype in EOC cells by modulating intracellular S100A14 expression. We first established stable S100A14-overexpressed and -knocked down EOC cell lines by antibiotic selection of a pool of lentivirus-infected cells. For our S100A14 overexpression experiments, we used TOV112D and YDOV-151 cell lines, which have low levels of endogenous S100A14. Conversely, we used SNU840 and OVCA429 cell lines that have high levels of endogenous S100A14 for our knockdown experiments. Strong S100A14 immunoreactivity indicated excellent enrichment of S100A14 in TOV112D (S100A14 #6, #8) and YDOV-151 (S100A14 #6, #10) cells when compared to empty vector-transfected cells. Real-time PCR results were similar to those obtained from immunoblots, wherein higher S100A14 mRNA expression was shown in exogenous S100A14-expressing cells (Fig. 2A). In contrast, protein levels of S100A14 decreased by short-hairpin S100A14 RNAi in SNU840 (sh-S100A14 #2, #5) and OVCA429 (sh-S100A14 #2, #5) cells (Fig. 2B). Ultimately, the two most efficiently overexpressing or knocked down cell cultures for each cell type were selected to further investigate phenotypic changes arising from S100A14 overexpression or downregulation.
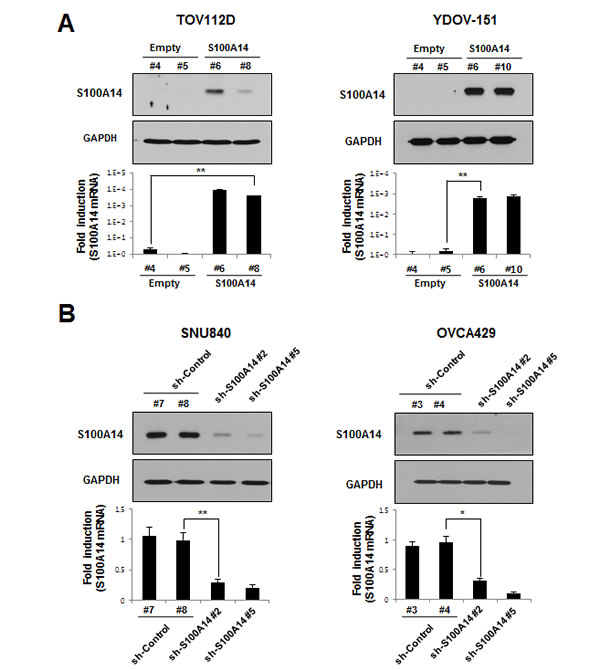
Figure 2: Ectopic S100A14 expression and knockdown in lentivirus-mediated stable cells. A and B. Whole cell lysates and total RNA pool were collected from TOV112D (A, left : transfected with empty clone #4, #5 or S100A14 clone #6, #8), YDOV-151 (A, right : transfected with empty clone #4, #5 or S100A14 clone #6, #10). SNU840 (B, left: transfected with empty clone #7, #8 or S100A14 shRNA sequence #2, #5), or OVCA429 (B, right: transfected with empty clone #3, #4 or S100A14 shRNA sequence #2, #5) stable cell lines. All shRNA sequences were different from each other. Expression of S100A14 protein was analyzed by immunoblot (upper panels), and mRNA level was measured by real-time PCR (lower panels). GAPDH was included as an internal loading control. An asterisk (*) indicates a p-value < 0.05, and a double asterisk (**), a p-value < 0.01.
We analyzed the effect of S100A14 overexpression in TOV112D and YDOV-151 using two assays to investigate short-term proliferative impact (WST-1 assay) and long-term proliferative ability (colony formation assay). In all S100A14-overexpressing cell lines, rates of cell growth were significantly higher than in empty vector-expressing controls (Fig. 3A). Similar results were also observed in colony formation assays (Fig. 3B). When S100A14 expression was repressed in SNU840 or OVCA429 cells, cell growth was not significantly altered by the WST-1 assay (Fig. 3C). However, knockdown of S100A14 significantly decreased colony formation in SNU840 and OVCA429 cells when compared to the shRNA-control group (Fig. 3D). We next performed a soft agar colony assay to assess S100A14’s role in tumorigenesis and found that the number of colonies formed from S100A14-transfected TOV112D cells was significantly higher than those resulting from empty vector-expressing controls (Fig. 3E).
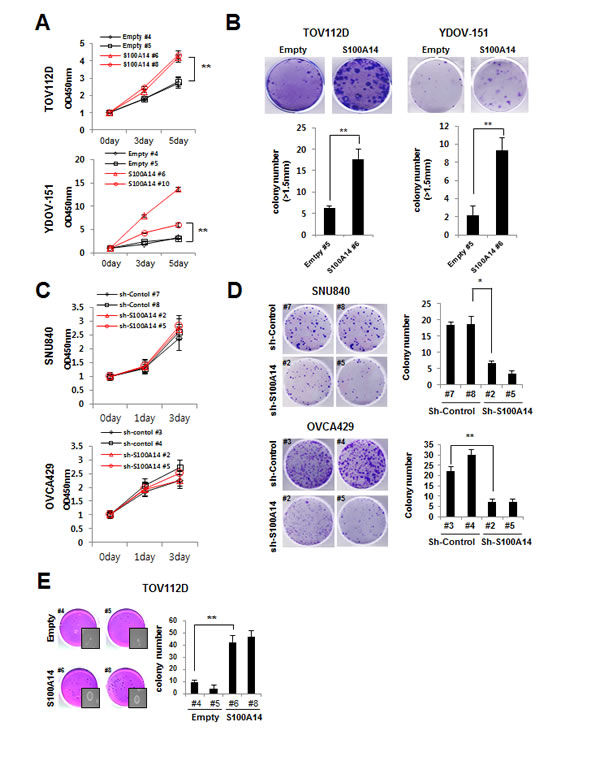
Figure 3: S100A14 increases cell proliferation and clonogenicity. A. Cell proliferation was determined by WST-1 assay. Cell proliferation curves for TOV112D and YDOV-151 cells at various indicated times. Error bars represent mean ± SD of triplicate experiments. B. A clonogenic assay was performed on TOV112D and YDOV-151 cells for 3 weeks. Left panel shows representative images; and right panel shows quantification of colonies. Error bars represent mean ± SD of triplicate experiments. C. Cell proliferation was determined by WST-1 assay. Cell proliferation curves for SNU840 and OVCA429 cells at various indicated times. Error bars represent mean ± SD of triplicate experiments. D. A clonogenic assay was performed on SNU840 and OVCA429 cells for 3 weeks. Left panel shows representative images; right panel shows quantification of colonies. Error bars represent mean ± SD of triplicate experiments. E. Soft-agar colony formation assay was performed using TOV112D cells. Left panel shows a representative picture; right panel shows quantification of colonies. Error bars represent mean ± SD of triplicate experiments. An asterisk (*) indicates a p-value < 0.05, and a double asterisk (**), a p-value < 0.01.
Lentiviral-mediated overexpression or knockdown of S100A14 reveals that high S100A14 expression increase cell motility and invasion in EOC cells
To investigate whether S100A14 plays a role in migration in EOC cells, we conducted a wound-healing assay for cell migration. Overexpression of S100A14 resulted in increased motility of TOV112D cells when compared to empty vector-expressing cells (Fig. 4A). In contrast, shRNA-mediated S100A14 knockdown inhibited cell motility in SNU840 cells (Fig. 4A). To evaluate whether S100A14 could also enhance invasion in EOC cells, a Matrigel invasion assay was performed. SNU840 and OVCA429, both of which have higher levels of endogenous S100A14, demonstrate significantly higher invasive potential than TOV112D and YDOV-151, which both express endogenous S100A14 at much lower levels (Fig. 4B). To further confirm the possible role of S100A14 in cell invasion, we evaluated the invasive potential in TOV112D, YDOV-151, and OVCA429. S100A14 overexpression in TOV112D (S100A14#8) and YDOV-151 (S100A14 #6, #10) resulted in increased invasion when compared to empty vector-expressing controls (Fig. 4C). In contrast, S100A14 knockdown in OVCA429 (sh-S100A14 #2, #5) cells led to a decrease in invasion capability (Fig. 4D). To explore the molecular mechanism behind S100A14’s promotion of cell migration and invasion, we characterized the expression of matrix metalloproteinase genes, MMP1, MMP2, and MMP9, by RT-PCR (Supplementary Fig. 2A). Interestingly, among the MMPs detected, expression of MMP1 and MMP9 was dramatically upregulated in S100A14-overexpressed TOV112D cells. Real-time PCR analysis further confirmed the dramatic increase of MMP1 and MMP9 expression (Supplementary Fig. 2B). However, no significant difference in p53 expression was observed between S100A14-overexpressed cells and empty vector-transfected cells (Supplementary Fig. 2C-D). Taken together, these results demonstrated that S100A14 plays an important role in cell motility and invasion.
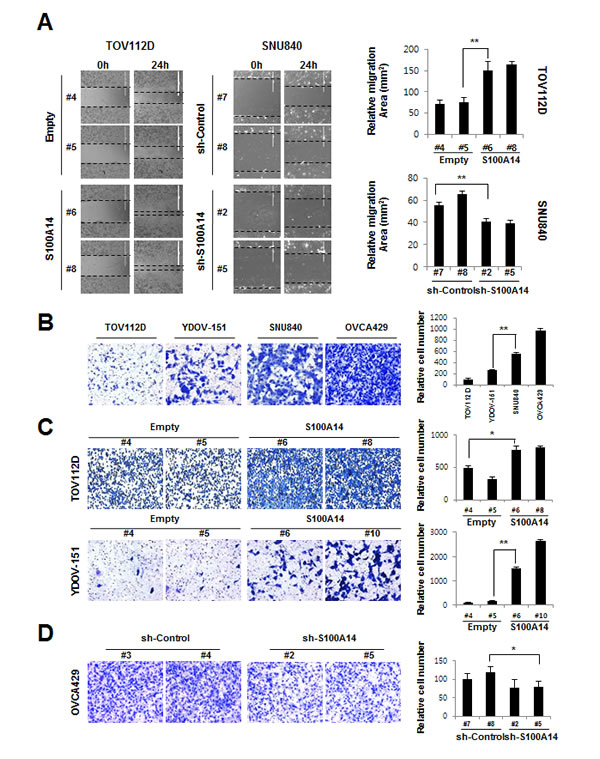
Figure 4: S100A14 promotes cell migration and invasion. A. Cell migration analyses of TOV112D and SNU 840 cells by wound healing assay. Left panel: Representative images of migrations assays. Right panel: Quantitative results of migration experiments. Results are presented as relative migration area. B-D. Cell invasion analysis of untreated EOC cells (B), TOV112D (C), YDOV-151 (C) and OVCA429 (D) cells using Matrigel invasion assay. Left panel: Representative figures of cell invasion. Right panel: Quantitative results of invasion experiments. Results are presented as relative number of invading cells. Cells were counted in 4 randomly selected fields. An asterisk (*) indicates a p-value < 0.05, and a double asterisk (**), a p-value < 0.01.
S100A14 overexpression in TOV112D cells increases tumor growth in nude mice xenografts
To explore whether S100A14 can affect tumor growth in vivo, we inoculated TOV112D (S100A14 #6, #8, Empty #4, #5) as xenografts into nude mice (Fig. 5A). Tumor volume and weight were measured. Mean tumor volumes (1998.5±397.0 and 1442.4±491.7 mm3, respectively) at day 49 in mice receiving S100A14-overexpressed TOV112D (S100A14 #6, #8) cells were significantly larger than those (114.8±97.8 and 86.3±64.7 mm3, respectively) in mice receiving empty vector-expressing cells (Empty #4, #5) (P < 0.001) (Fig. 5B-D). Tumor weight correlated with tumor volume, as determined by calipers (P < 0.001; r2 = 0.935) (Fig. 5D). These data indicated that S100A14 can promote tumor growth in vivo and further support our initial hypothesis that S100A14 plays a functional role in the malignant transformation of EOC.
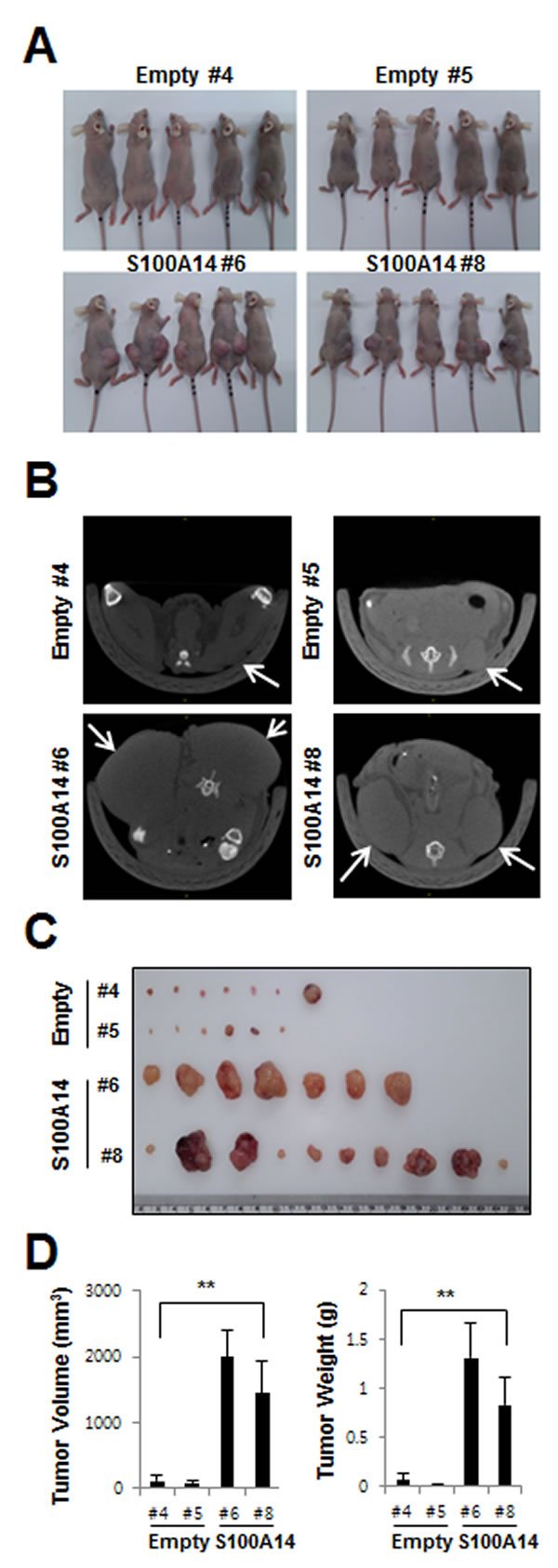
Figure 5: S100A14 overexpression increases in vivo xenograft tumor growth. A. For the xenograft, a total of 1.5x106 cells were injected subcutaneously into the left and right flank, respectively. B. Representative microCT images of mice seven weeks after injection. C. Gross images of tumor masses from representative mice from each group. D. S100A14 overexpression increased tumor volume and weight of xenografted TOV112D cells in BALB/c-nu mice. Mean tumor volume and weight for each group was calculated at seven weeks after injection. Error bars were calculated as standard error of means, and two-way ANOVA was used for statistical analyses (n = 5 mice/group). A double asterisk (**) indicates a p-value < 0.01.
S100A14 promotes a malignant phenotype in EOC cells through the PI3K/Akt signaling pathway
To determine the mechanism by which S100A14 promotes a malignant phenotype in ovarian cells, we assessed the status of important signaling cascades controlled by Akt and Erk in S100A14-overexpressed and S100A14-shRNA-knockdown cells. S100A14 overexpression in TOV112D (S100A14 #6, #8) and YDOV-151 (S100A14 #6, #10) cells led to increased pAkt expression (Fig. 6A), while S100A14 knockdown in SNU840 (sh-S100A14 #2, #5) and OVCA429 (sh-S100A14 #2, #5) cells led to decreased pAkt expression (Fig. 6B). There was no detectable change, however, in either pErk or Erk expression after S100A14 transduction. To confirm the relationship between S100A14 and pAkt, we prepared immunoblots in a series of non-transfected EOC cells. TOV112D and YDOV-151 cells, which lack endogenous S100A14 expression, showed lower levels of pAkt expression compared to cells with higher endogenous S100A14 expression (Fig. 6C). These data suggest that S100A14 could potentially modulate the activity of Akt.
To define the role of Akt in S100A14-modulated cell proliferation and invasion, we treated TOV112D (S100A14 #6, #8) cells with either a PI3K (Akt upstream kinase) inhibitor, wortmannin, or an Akt inhibitor, MK-2206, and evaluated their effects on cell proliferation and invasion. First, we confirmed the specificity of these inhibitors by immunoblot (Fig. 6D), wherein increased levels of pAkt in S100A14-overexpressed cells were significantly decreased by both wortmannin and MK-2206. Next, we observed that wortmannin and MK-2206 both significantly decreased S100A14-enhanced cell proliferation and invasion via colony formation assay and invasion assay (Fig. 6E and 6F). Taken together, these data demonstrate that S100A14 controls the invasive potential and proliferative ability of cells through regulation of the PI3K/Akt pathway.
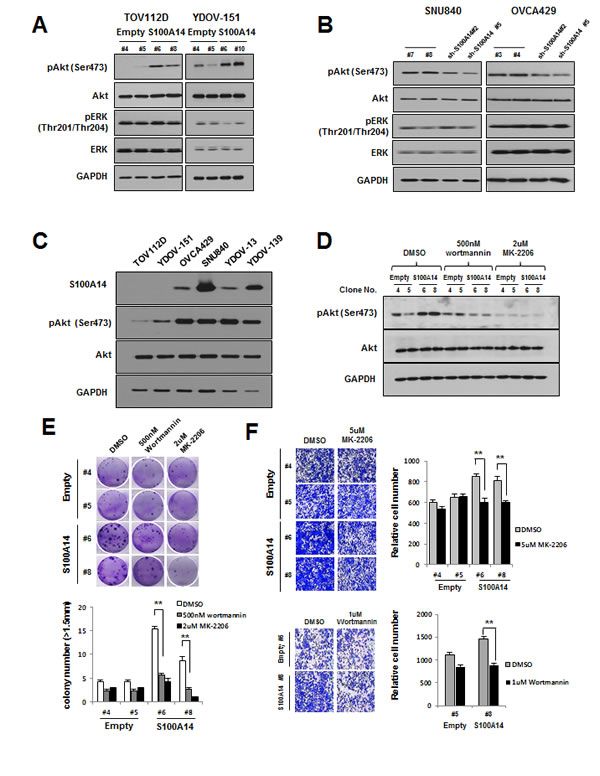
Figure 6: S100A14 controls oncogenic phenotypes of EOC cells through PI3K/Akt pathway. A and B. Expression of pAkt (ser478), Akt, pERK (thr201/204), and ERK was assessed by immunoblot in TOV112D and YDOV-151 cells (A) and SNU840 and OVCA429 cells (B). C. Protein levels of S100A14, pAkt (ser478), and Akt were determined by immunoblot in non-transfected EOC cells. GAPDH was included as an internal loading control. D. Immunoblot analysis of pAkt inhibition in TOV112D cells treated with wortmannin, MK-2206, or control (DMSO) for eight hours. E. A clonogenic assay was performed on TOV112D cells for three weeks in the presence of wortmannin, MK-2206, or control (DMSO). Left panel shows representative images, and right panel shows quantification of colonies. Error bars represent mean ± SD of triplicate experiments. F. Cell invasion analysis of TOV112D cells with wortmannin, MK-2206, or control (DMSO). All drugs were applied to the upper chamber. Left panel: Representative figures of cell invasion. Right panel: Quantitative results of invasion experiments. Results are presented as relative number of invading cells. Cells were counted in six randomly selected fields. A double asterisk (**) indicates a p-value < 0.01.
DISCUSSION
In addition to identifying novel diagnostic markers for ovarian cancer, further understanding of the molecular mechanisms underlying the progression and metastasis of ovarian cancer is needed to develop more effective therapeutic treatments. Among various genes and proteins previously identified to be expressed or modified in ovarian cancer[17], S100A14 attracted research interest due to its marked and consistent overexpression in all six EOC cell lines and its reported role in cell growth and survival in other cancers [18-20]. In the present study, we investigated the functional role and clinical significance of S100A14 expression in ovarian cancer using EOC cell lines and mouse xenograft models.
It has recently been proposed that ovarian cancer can be divided into two main categories, type I and type II tumors, based on their distinct pathogenesis[21]. Type I tumors include well-differentiated serous, mucinous, endometrioid, malignant Brenner, and clear cell tumors that generally exhibit slow, indolent growth with Ras and/or Raf mutations. In contrast, type II tumors include moderately or poorly differentiated serous, undifferentiated, and malignant mixed mesodermal tumors with p53 mutation. Type II tumors are highly aggressive and present at an advanced stage, where currently available therapies are seldom curative[22]. When we consider the fact that type II tumors are more aggressive than type I tumors and constitute approximately 75% of ovarian cancer [23], it is reasonable to focus on type II tumors when identifying potential biomarkers in the future. We therefore performed subgroup analyses of IHC results in type I and type II tumors to compare the molecular subtypes of ovarian cancer. S100A14 protein expression was greater in type II tumors (IHC score = 5.42, n = 52) when compared to type I tumors (IHC score = 4.85, n = 19) (P < 0.001). Although the difference was small in our study, subsequent analysis using more samples may demonstrate a significant difference in S100A14 expression between type I and type II tumors. Notably, S100A14+ status and advanced tumor stage were independent prognostic factors for both disease-free and overall survival on multivariate analysis (Table 1). Taken together, these results suggest that S100A14 may play an important role in the pathogenesis of EOC. However, it should be noted that our relatively small clinical sample size is a limitation of this study and that the small sample size and mixture of different histological subtypes may weaken the strength of our clinical analysis. We therefore plan to incorporate larger numbers of specific histological subgroups to generalize our findings in a future study.
Increasing evidence suggests a critical role for the S100 family in cell growth, invasion, and cancer metastasis. We therefore investigated the potential roles of S100A14 in EOC based on our prior analysis of clinical specimens. S100A14 overexpression resulted in significant increases in cell proliferation and clonogenicity. Conversely, S100A14 knockdown led to the suppression of these phenotypes (Fig. 3). Importantly, S100A14 expression positively correlated with cell migration and invasion in EOC cell in vitro cultures (Fig. 4) and in vivo xenograft-bearing mice (Fig. 5). Another limitation of our study should be noted, however. Only subcutaneous xenograft models were used to investigate EOC cell behavior in vivo. Cancer cells implanted subcutaneously allow for rapid and quantitative tumor formation, being more suitable for studies that require continuous measurement of the tumor. In contrast, intraperitoneal or orthotopic xenograft models are inherently difficult to quantitatively monitor tumor growth, but can provide a more relevant tumor microenvironment. Despite this limitation, our findings suggest that S100A14 is one of the critical proteins contributing to EOC carcinogenesis and progression.
Interestingly, there was a discrepancy in the level of S100A14 expression (Fig. 2A) with the gain of phenotype, such as cell growth (Fig. 3A), colony formation (Fig. 3E), migration (Fig. 4A), and invasion (Fig. 4C) in S100A14-transfected TOV112D and YDOV-151 stable cell lines. There may be many reasons for the observed discrepancy, such as the characteristics of the cell line itself (e.g., TOV112D originates from endometrioid EOC and YDOV-151 is from mucinous EOC), cell shape, and cellular localization of S100A14. It is well known that the function of protein could be controlled by cell shape or its subcellular localization [24,25]. Thus, it will be very useful to examine cell morphology and/or subcellular localization of S100A14 in TOV112D and YDOV-151 stable cell lines in future studies.
It is well known that some S100 proteins regulate cell invasion and metastasis by modulating expression and activity of MMPs [11,16,33]. In particular, Chen et al. reported that S100A14 promotes cell motility and invasion by regulating the expression and function of MMP2 in a p53-dependent manner in esophageal squamous cell carcinoma [25]. In the present study, MMP1 and MMP9 expression was enhanced in S100A14-overexpressed TOV112D cells (Supplementary Fig. 2A-2B). However, S100A14 overexpression did not increase the expression of p53 in TOV112D cells (Supplementary Fig. 2C-D). We believe that this is mainly because of TOV112D’s p53R175 mutant status (http://www.atcc.org/products/all/CRL-11731.aspx#characteristics). In contrast to our study, Sapkota et al. demonstrated that S100A14 overexpression was associated with suppression of MMP9 gelatinolytic activity in CaLH3 cells[16]. Thus, the function of S100A14 can be different depending on the anatomical origin of these malignant cells.
Finally, we investigated whether Akt or Erk signaling pathway is compromised upon S100A14 overexpression or knockdown. There was an obvious relationship between increased levels of phosphorylated Akt and overexpression of S100A14 in TOV112D and YDOV-151 cells (Fig. 6A). Conversely, S100A14 knockdown decreased phosphorylated Akt in SNU840 and OVCA429 cells (Fig. 6B). However, there was no detectable change in either expression or activation of Erk1/2 after S100A14 modulation (Fig. 6A-B). Inhibition of PI3K/Akt signaling with wortmannin or MK-2206 resulted in significant decreases in phosphorylated Akt (Fig. 6D), cell proliferation (Fig. 6E), and cell invasion (Fig. 6F) in TOV112D or YDOV-151 cells. Thus, S100A14 may contribute to the malignant phenotype of EOC cells through activated PI3K/Akt signaling.
In summary, we demonstrate that S100A14 expression was lowest in normal ovarian epithelium, slightly increased in benign and borderline ovarian tumors, and highest in EOC. Enhanced S100A14 expression correlated positively with clinicopathological parameters in both in vitro and in vivo ovarian cancer systems, lending support for the use of S100A14 to determine clinicopathological stage and/or prognosis in ovarian cancer. In addition, S100A14 could be a potential therapeutic target, as the present study demonstrated the mechanistic role of S100A14 in promoting cell motility and invasion through the PI3K/Akt pathway.
METHODS
Cell lines and reagents
Five EOC cell lines (YDOV-13, YDOV-151, YDOV-139, YDOV-161, and YDOV-13) and 6 human ovarian surface epithelial (HOSE) cells, which were established and characterized in our laboratory, were cultured as described previously [28,29]. SNU-840 was purchased from Korean Cell Line Bank (KCLB, Seoul, Korea). RMUG-S was purchased from Health Science Research Resources Bank (HSRRB, Osaka, Japan). SKOV3, TOV112D, OVCA429, and OVCA433 cell lines were purchased from American Type Culture Collection (ATCC, Manassas, VA). All purchased cell lines were maintained as recommended. AKT inhibitor MK-2206 was purchased from Sellect (Houston, TX), and PI3 kinase inhibitor wortmannin was purchased from Sigma Aldrich (St. Louis, MO). The stock solutions of 10 mM MK-2206 and 1 mM wortmannin were dissolved in dimethyl sulfoxide (DMSO).
Patients and tumor specimens
Tumor samples from 71 EOCs (54 serous, 6 mucinous, 4 endometrioid, 2 transitional cell, 2 mixed, 2 clear cell, and 1 Brenner tumor), 10 borderline ovarian tumors (6 serous and 4 mucinous), 10 mucinous cystadenomas, and 13 normal ovary specimens were included in this study and were provided by the Korea Gynecologic Cancer Bank through Bio & Medical Technology Development Program of the Ministry of Education, Science and Technology, Korea. Tumor staging was performed according to the International Federation of Gynecology and Obstetrics (FIGO) classification. All FIGO Stage I/II ovarian cancer patients underwent pelvic and para-aortic lymph node dissection according to the National Comprehensive Cancer Network (NCCN) clinical practice guidelines. Medical records were reviewed to collect data including age, surgical procedure, survival time, and survival status. Response to therapy was assessed according to Response Evaluation Criteria in Solid Tumors (RECIST; version 1.0) by spiral computed tomography [30]. Data for tumor grades and cell types were obtained by reviewing pathology reports. All biological samples were obtained with appropriate informed consent from participants according to institutional review board (IRB) guidelines.
Immunohistochemistry
Paraffin tissue sections were deparaffinized in two changes of xylene, rehydrated in graded ethanol, and treated for 30 min with 3% H2O2 solution in methanol to block endogenous peroxidase. After blocking in 10% goat serum in TBS for 30 minutes, sections were incubated with rabbit polyclonal anti-Human S100A14 antibody (Proteintech Group, Inc., Chicago, IL) diluted to 1:100 for 1 hr at room temperature, followed by detection using Dako LSAB+ (Dako, Glostrup, Denmark). The reaction product was developed with DAB (3,3’-diaminobenzidine) chromogen solution (Dako). Sections were counterstained with hematoxylin and mounted in Faramount aqueous mounting medium (Dako). Appropriate negative and positive controls were concurrently performed. Representative photomicrographs were recorded using a digital camera (Nikon, Tokyo, Japan). Negative controls were processed by omitting the primary antibody. Human pancreas tissue was used as a positive control for S100A14 immunoreactivity.
Staining for S100A14 was scored as positive when tumor or epithelial cells showed cytoplasmic immunoreactivity. S100A14 staining results were scored based on staining intensity (0 = negative, 1 = weak, 2 = moderate, 3 = strong) and the percentage of positive cells (0 = 0%, 1 = 1 – 25%, 2 = 26 – 50%, 3 = 51 – 100% positive cells), as described previously [31]. For the immunostaining score, the intensity and positivity scores were added, resulting in a value between 0 and 6. The overall score for each patient was further simplified by dichotomizing as either negative (overall score of ≤ 5) or positive (score of 6). Slides were scored in the absence of any clinical data, and the final immunostaining score was the average score of three expert pathologists.
Real-time PCR
Total RNA was extracted using an RNeasy Mini Kit (Qiagen, Valencia, CA). cDNA was generated using a SuperScript TM III First–Strand Synthesis System (Invitrogen, Carlsbad, CA). Real-time PCR was performed to quantify messenger RNA expressions using SYBR® Green PCR Master Mix (Applied Biosystems, Foster City, CA) and the ABI PRISM® 7300 real-time PCR system (Applied Biosystems). A housekeeping gene, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), was used to normalize the quantity of cDNA used in the PCR reaction. Each assay was performed in triplicate and expressed as the mean ± SD. The primers used for the PCR analysis were as follows: for S100A14 forward 5’-GTG TCG GTC AGC CAA CGC AGA-3’, S100A14 reverse 5’-TGC TGG GTG ACC AGG TCC CGT-3’, MMP1 forward 5’-CTG CTG CTG CTG TTC TGG GGT-3’, MMP1 reverse 5’-CCA CTG GGC CAC TAT TTC TCC GCT-3’, MMP2 forward 5’- GAT ACC CCT TTG ACG GTA AGG A -3’, MMP2 reverse 5’CCT TCT CCC AAG GTC CAT AGC -3’, MMP9 forward 5’- AGA CGG GTA TCC CTT CGA CG -3’, MMP9 reverse 5’- AAA CCG AGT TGG AAC CAC GAC -3’, GAPDH forward 5’- GAA GGT GAA GGT CGG AGT -3’, GAPDH reverse 5’- GAA GAT GGT GAT GGG ATT TC -3’. The primer for TP53 was purchased from Bioneer (Daejeon, Korea; Cat.# P188330). The comparative CT method was used to calculate relative quantification of gene expression as described previously [32].
Transfection and generation of stable cell lines
To generate pCDH/S100A14, cDNA encoding human S100A14 was amplified from pOTB7 S100A14 cDNA clone (MHS1011-60727, Open Biosystems) using the primer set 5’-TTC TAG AGC CAC CAT GGG ACA GTG TCG GTC AG-3’ (forward) and 5’-TTG CGG CCG CTC AGT GCC CCC GGA CAG-3’ (reverse). The amplified cDNA was cloned into Not I /Xba I restriction sites of the pCDH-Promoter-MCS-EF1 Lentivector (System Biosciences, Mountain View, CA). Stable cell lines expressing S100A14 in TOV112D and YDOV-151 were generated using the pPACKH1 Lentivector Packaging Kit (System Biosciences), and selection of S100A14-transduced cells was accomplished using 2.0 μg/mL puromycin (Invitrogen).
For the generation of sh-S100A14 stable cell lines, pLKO.1 S100A14 shRNA libraries were purchased from Open Biosystems (Waltham, MA). Of five lentiviral constructs tested, two with the best knockdown efficiency were used for the experiments presented here, using the human S100A14 sequence ATCACTGAATTCCTGAGCATC for shRNA #2 and TGGTGAAAGTTCTTGATGAGG for shRNA #5. The non-target shRNA control vector (SHC002) was purchased from Sigma (Sigma Aldrich). Stable cell lines expressing sh-S100A14 were generated using the viral packaging plasmids composed of pCMV delta and pMDG. Virus particles were collected after 48 hours and 72 hours post-transfection. SNU840 and OVCA 429 cells were transduced, and positively transduced cells were selected with puromycin (2 μg/mL).
Immunoblotting
Whole cell extraction was conducted using PRO-PRE Protein Extraction Solution (Intron Biotechnology, Seongnam, Korea). Equal amounts (20 µg) of each sample were separated on 8-15% SDS-PAGE and transferred to nitrocellulose membranes. The membranes were blocked with 5% nonfat dry milk in TBST (50 mM Tris, 150 mM NaCl, 0.1% Tween-20, pH 7.5) for 1 hr at room temperature, washed with TBST, and subsequently incubated with primary antibodies: anti-S100A14 (Proteintech, Chicago, IL), anti-phospho-Akt (ser473), anti-Akt, anti-phospho-Erk1/2, anti-Erk1/2 (Cell Signaling, Danvers, MA), anti-TP53 (Santa Cruz Biotechnology, Santa Cruz, CA) and anti-GAPDH (Santa Cruz Biotechnology). Primary antibodies against each protein were detected by secondary antibodies conjugated with horseradish peroxidase (GE Healthcare, Munich, Germany). Specific bands for each protein were detected on AGFA X-ray film (Agfa Health Care, Mortsel, Belgium) using the SuperSignal Chemiluminescence kit (Thermo Scientific, Rockford, IL).
Cell growth and clonogenicity
Cell proliferation was measured by WST-1 assay (DaeilLab, Seoul, Korea). In brief, cells were seeded at 3×103 viable cells/well onto 96-well microtiter plates in a final volume of 100 μL/well. Cells were incubated with WST-1 at 37oC for 2 hours and optical density (OD) values at 450 nm were recorded at days 0, 3, and 5 using a 96-well microplate reader (Bio-Rad Laboratories, Inc., Hercules, CA). The experiment was performed in triplicate.
In order to examine clonogenicity, cells were seeded into 60-mm dishes (0.02-0.1×104 cells/well) and cultured for 3 weeks. Colonies formed in each well were fixed with H2O containing 10% methanol and 10% acetic acid, stained with 0.5% crystal violet, and then counted visually. A three-dimensional clonogenic growth assay was performed using a soft agar culture system. Briefly, cells were mixed in 0.3% agar containing 10% FBS and layered on top of the base layer of 0.6% agar in a 60-mm dish (3×103 cells/dish). After a four-week incubation in a humidified CO2 incubator, colonies were counted visually after staining with 0.5% crystal violet. Each cell group was cultured in triplicate, and colonies greater than 1.5 mm in diameter were scored visually.
Cell migration and invasion assays
Cell migration was assessed by monolayer wound healing assay [33]. Briefly, cells were seeded into 12-well tissue culture dishes and allowed to grow to 90% confluency in complete medium. Cell monolayers were wounded with a plastic pipette tip (1 mm), and washed four times with complete medium to remove cell debris. After 24 hours of culture, phase contrast images were captured of the gap in the monolayers (EVOS®FL Cell Imaging System, Life Technologies, Carlsbad, CA). A cell invasion assay was performed in an invasion chamber (Neuro Probe 48-Well Micro Chemotaxis Chamber, Neuro Probe, Inc., Gaithersburg, MD) according to the manufacturer’s instruction. In brief, 5×104 cells were resuspended in serum-free medium (56 μL) and plated onto the upper chamber coated with Matrigel (BD Transduction Lab, San Jose, CA). The lower chamber was filled with medium containing 0.1-10% FBS (27 μL). After 24 hours of incubation, cells that had migrated through the membrane were stained with the Differential Quik Stain Kit (Triangle Biomedical Sciences, Inc., Durham, NC). Invading cells in six randomly selected fields were counted using Microscope Axio Imager.M2 (Carl Zeiss, Thornwood, NY, Magnification x200). Each experiment was repeated three times.
Animal studies
All animal procedures were performed under a protocol approved by the Institutional Animal Care and Use Committee of Gangnam Severance Hospital. A mouse ovarian cancer xenograft model was established using female BALB/C nude mice aged 4-5 weeks (OrientBio Inc., Sungnam, Gyunggi, Korea). TOV112D cells (1.5×106 cells/inoculation) were resuspended in 100 μL of a Matrigel (BD Biosciences, QC) and PBS mixture (1:1) and inoculated subcutaneously in both left and right flanks of each mouse. Seven weeks after inoculation, tumor-bearing mice were subjected to microCT imaging (NFR Polaris-G90 Micro CT scanner, Nano Focus Ray, Jeonju, Chunbuk, Korea). Briefly, mice were anesthetized using isoflurane/O2 (1.5-5% v/v) gas and scanned with the following parameter settings: 360 scan angle, X-ray voltage 65 kV, X-ray current 115 μA, X-ray spot size 9 μm, binning 1, and exposure time 40 ms. After microCT imaging, tumors were harvested from each mouse, weighted, and fixed in 10% neutral buffered formalin for histological assays. Tumor volumes were calculated using the following formula: tumor volume (mm3) = (L x W2)/2, where L is the length and W is width [34]. Data is represented as means ± standard errors.
Statistical analysis
Normality of distribution was assessed using the Shapiro-Wilk test. Because the distribution was not normal, univariate comparisons for quantitative variables between normal and cancerous specimens were made using non-parametric statistics (Kruskal–Wallis and Mann–Whitney U) where appropriate. Results with two-tailed p-values less than 0.05 were considered statistically significant. Statistical analyses were performed using SPSS software version 18.0 (SPSS Inc., Chicago, IL).
ACKNOWLEDGEMENTs
The authors thank Prof. Yong Il Kwon, Prof. Jong-Hyeok Kim, Prof. Nam Hoon Cho, and Prof. Jae Yong Cho for their assistance on technical aspects of this study and editing.
GRANT SUPPORT
This work was supported in part by grants from the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education, Science and Technology (2010-0011153, 2011-0010286, and 2011-0007146), the Korea Healthcare Technology R&D Project, Ministry for Health & Welfare Affairs, Republic of Korea (A121387), and faculty research grants from Yonsei University College of Medicine for 2013 (6-2013-0106).
CONFLICT OF INTEREST STATEMENT
The authors declare that there are no conflicts of interest.
REFERENCES
1. Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin. 2007; 57(1): 43-66.
2. Auersperg N, Wong AS, Choi KC, Kang SK, Leung PC. Ovarian surface epithelium: biology, endocrinology, and pathology. Endocr Rev. 2001; 22(2): 255-288.
3. Kuhn E, Kurman RJ, Vang R, Sehdev AS, Han G, Soslow R, Wang TL, Shih Ie M. TP53 mutations in serous tubal intraepithelial carcinoma and concurrent pelvic high-grade serous carcinoma--evidence supporting the clonal relationship of the two lesions. J Pathol. 2012; 226(3): 421-426.
4. Finch A, Shaw P, Rosen B, Murphy J, Narod SA, Colgan TJ. Clinical and pathologic findings of prophylactic salpingo-oophorectomies in 159 BRCA1 and BRCA2 carriers. Gynecol Oncol. 2006; 100(1): 58-64.
5. Piek JM, Kenemans P, Verheijen RH. Intraperitoneal serous adenocarcinoma: a critical appraisal of three hypotheses on its cause. Am J Obstet Gynecol. 2004; 191(3): 718-732.
6. Piek JM, van Diest PJ, Zweemer RP, Jansen JW, Poort-Keesom RJ, Menko FH, Gille JJ, Jongsma AP, Pals G, Kenemans P, Verheijen RH. Dysplastic changes in prophylactically removed Fallopian tubes of women predisposed to developing ovarian cancer. J Pathol. 2001; 195(4): 451-456.
7. Donato R. S100: a multigenic family of calcium-modulated proteins of the EF-hand type with intracellular and extracellular functional roles. Int J Biochem Cell Biol. 2001; 33(7): 637-668.
8. Donato R. Intracellular and extracellular roles of S100 proteins. Microsc Res Tech. 2003; 60(6): 540-551.
9. Marenholz I, Heizmann CW, Fritz G. S100 proteins in mouse and man: from evolution to function and pathology (including an update of the nomenclature). Biochem Biophys Res Commun. 2004; 322(4): 1111-1122.
10. Salama I, Malone PS, Mihaimeed F, Jones JL. A review of the S100 proteins in cancer. Eur J Surg Oncol. 2008; 34(4): 357-364.
11. Saleem M, Kweon MH, Johnson JJ, Adhami VM, Elcheva I, Khan N, Bin Hafeez B, Bhat KM, Sarfaraz S, Reagan-Shaw S, Spiegelman VS, Setaluri V, Mukhtar H. S100A4 accelerates tumorigenesis and invasion of human prostate cancer through the transcriptional regulation of matrix metalloproteinase 9. Proc Natl Acad Sci U S A. 2006; 103(40): 14825-14830.
12. Whiteman HJ, Weeks ME, Dowen SE, Barry S, Timms JF, Lemoine NR, Crnogorac-Jurcevic T. The role of S100P in the invasion of pancreatic cancer cells is mediated through cytoskeletal changes and regulation of cathepsin D. Cancer Res. 2007; 67(18): 8633-8642.
13. Bulk E, Sargin B, Krug U, Hascher A, Jun Y, Knop M, Kerkhoff C, Gerke V, Liersch R, Mesters RM, Hotfilder M, Marra A, Koschmieder S, Dugas M, Berdel WE, Serve H, Muller-Tidow C. S100A2 induces metastasis in non-small cell lung cancer. Clin Cancer Res. 2009; 15(1): 22-29.
14. Pietas A, Schluns K, Marenholz I, Schafer BW, Heizmann CW, Petersen I. Molecular cloning and characterization of the human S100A14 gene encoding a novel member of the S100 family. Genomics. 2002; 79(4): 513-522.
15. Yao R, Lopez-Beltran A, Maclennan GT, Montironi R, Eble JN, Cheng L. Expression of S100 protein family members in the pathogenesis of bladder tumors. Anticancer Res. 2007; 27(5A): 3051-3058.
16. Sapkota D, Bruland O, Costea DE, Haugen H, Vasstrand EN, Ibrahim SO. S100A14 regulates the invasive potential of oral squamous cell carcinoma derived cell-lines in vitro by modulating expression of matrix metalloproteinases, MMP1 and MMP9. Eur J Cancer. 2011; 47(4): 600-610.
17. Cho H, Lee YS, Kim J, Chung JY, Kim JH. Overexpression of glucose transporter-1 (GLUT-1) predicts poor prognosis in epithelial ovarian cancer. Cancer Invest. 2013; 31(9): 607-615.
18. Chen H, Ma J, Sunkel B, Luo A, Ding F, Li Y, He H, Zhang S, Xu C, Jin Q, Wang Q, Liu Z. S100A14 is a novel modulator of terminal differentiation of esophageal squamous cell carcinoma. Mol Cancer Res. 2013; doi:10.1158/1541-7786.mcr-13-0317.
19. Sapkota D, Costea DE, Ibrahim SO, Johannessen AC, Bruland O. S100A14 Interacts with S100A16 and Regulates Its Expression in Human Cancer Cells. PLoS One. 2013; 8(9): e76058.
20. Xu C, Chen H, Wang X, Gao J, Che Y, Li Y, Ding F, Luo A, Zhang S, Liu Z. S100A14,a member of EF-hand Calcium-Binding Proteins, is overexpressed in breast cancer and acts as a modulator of HER2 signaling. J Biol Chem. 2013; doi:10.1074/jbc.M113.469718.
21. Shih Ie M, Kurman RJ. Ovarian tumorigenesis: a proposed model based on morphological and molecular genetic analysis. Am J Pathol. 2004; 164(5): 1511-1518.
22. Kurman RJ, Visvanathan K, Roden R, Wu TC, Shih Ie M. Early detection and treatment of ovarian cancer: shifting from early stage to minimal volume of disease based on a new model of carcinogenesis. Am J Obstet Gynecol. 2008; 198(4): 351-356.
23. Kurman RJ, Shih Ie M. The origin and pathogenesis of epithelial ovarian cancer: a proposed unifying theory. Am J Surg Pathol. 2010; 34(3): 433-443.
24. Yu CS, Chen YC, Lu CH, Hwang JK. Prediction of protein subcellular localization. Proteins. 2006; 64(3): 643-651.
25. Huang S, Ingber DE. Shape-dependent control of cell growth, differentiation, and apoptosis: switching between attractors in cell regulatory networks. Exp Cell Res. 2000; 261(1): 91-103.
26. Moon A, Yong HY, Song JI, Cukovic D, Salagrama S, Kaplan D, Putt D, Kim H, Dombkowski A, Kim HR. Global gene expression profiling unveils S100A8/A9 as candidate markers in H-ras-mediated human breast epithelial cell invasion. Mol Cancer Res. 2008; 6(10): 1544-1553.
27. Chen H, Yuan Y, Zhang C, Luo A, Ding F, Ma J, Yang S, Tian Y, Tong T, Zhan Q, Liu Z. Involvement of S100A14 protein in cell invasion by affecting expression and function of matrix metalloproteinase (MMP)-2 via p53-dependent transcriptional regulation. J Biol Chem. 2012; 287(21): 17109-17119.
28. Cho H, Kang ES, Hong SW, Oh YJ, Choi SM, Kim SW, Kim SH, Kim YT, Lee KS, Choi YK, Kim JH. Genomic and proteomic characterization of YDOV-157, a newly established human epithelial ovarian cancer cell line. Mol Cell Biochem. 2008; 319(1-2): 189-201.
29. Cho H, Lim BJ, Kang ES, Choi JS, Kim JH. Molecular characterization of a new ovarian cancer cell line, YDOV-151, established from mucinous cystadenocarcinoma. Tohoku J Exp Med. 2009; 218(2): 129-139.
30. Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000; 92(3): 205-216.
31. Shibusa T, Shijubo N, Abe S. Tumor angiogenesis and vascular endothelial growth factor expression in stage I lung adenocarcinoma. Clin Cancer Res. 1998; 4(6): 1483-1487.
32. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001; 25(4): 402-408.
33. Valster A, Tran NL, Nakada M, Berens ME, Chan AY, Symons M. Cell migration and invasion assays. Methods. 2005; 37(2): 208-215.
34. Goodwin SC, Bonilla SC, Sacks D, Reed RA, Spies JB, Landow WJ, Worthington-Kirsch RL. Reporting standards for uterine artery embolization for the treatment of uterine leiomyomata. J Vasc Interv Radiol. 2003; 14(9 Pt 2): S467-476.