
INTRODUCTION
Immune escape is one of the main reasons for the rapid progression of cancer and the poor efficacy of immunotherapy. The inactivation of natural killer (NK) cells contributes to the immune escape of tumor cells. NK cells serve as an antitumor defense through their direct cytotoxic and indirect immune-regulatory capacities. The NK cell activation is controlled by signals derived from the ligation of activating receptors. Natural killer group 2 member D (NKG2D) is one of the most prominent activating receptors of NK cells [1]. In humans, the NKG2D ligands (NKG2DLs) comprise eight surface glycoproteins, including the major histocompatibility complex class I chain-related proteins A and B (MICA/B) and the UL16-binding protein family. These NKG2DLs are considered to be promising targets by which the immunogenicity of cancer cells can be improved [2]. While NKG2DL expression is low or undetectable on normal cells, it can be induced during stress and malignant transformation. However, some malignantly transformed cells can still evade immune detection and elimination by reducing their surface levels of NKG2DLs by various mechanisms, such as by downregulating the expression of these proteins [3] and/or shedding them from the cell surface [4].
To this end, many researchers have aimed to induce the expression of NKG2DLs with agents such as decitabine, bortezomib, suberoylanilide hydroxamic acid, doxorubicin and melphalan [5-8]. However, most of these agents are antitumor drugs with inevitable side effects, so it would be meaningful to identify safer agents. The regulation of NKG2DLs by dietary compounds has been investigated to a lesser extent. Resveratrol, a naturally occurring dietary compound, has been suggested to enhance the immune response to certain cancers, including leukemia, lung cancer and hepatic cancer [9-11]. Of particular interest, resveratrol was reported to upregulate NKG2DLs by activating the ataxia–telangiectasia mutated kinase (ATM) in leukemia cells [10]. However, the mechanism whereby resveratrol upregulates NKG2DLs is not fully understood. On the other hand, it is known that the levels of some NKG2DLs are regulated by cancer-relevant microRNAs (miRNAs), such as miR-20a, miR-34a, miR-34c, miR-106b, miR-93, miR-373, and miR-520 [12-15].
In the current study, we hypothesized that resveratrol could upregulate MICA/B expression (and, in turn, cancer cell lysis by NK cells) by altering miRNA expression, and provided experimental validation for this hypothesis in human breast cancer. In examining the mechanism whereby resveratrol regulates MICA/B, we discovered a novel c-Myc/miR-17 pathway that governs MICA/B expression in breast cancer.
RESULTS
Resveratrol increases MICA and MICB expression in breast cancer cell lines and xenograft tumors
To determine whether resveratrol could increase the expression of MICA and MICB in breast cancer cells, we exposed BCap37, MDA-MB-231, Hs 578T and MCF-7 cells to 6.25 μM or 25 μM resveratrol for 48 h. The surface-protein and mRNA levels of MICA and MICB were detected by flow cytometry and qRT-PCR, respectively. As shown in Figure 1, resveratrol treatment increased the surface levels of MICA and MICB on breast cancer cells in a dose-dependent manner (Figure 1A-1E). In parallel, the mRNA levels of MICA and MICB were also upregulated (Figure 1F-1I). The upregulation was most obvious in the BCap37 and MDA-MB-231 cell lines. A 48-h cell viability assay revealed that, at a resveratrol concentration of 25 μM, the cell viability was 98.3±1.9% in MDA-MB-231 cells and 92.9±2.9% in BCap37 cells (Supplementary Figure 1A). In an apoptosis assay, 25 μM resveratrol induced apoptosis in less than 4% of cells (Supplementary Figure 1B). Thus, at the concentrations adopted in this study, the direct toxicity of resveratrol to the target cells was limited.
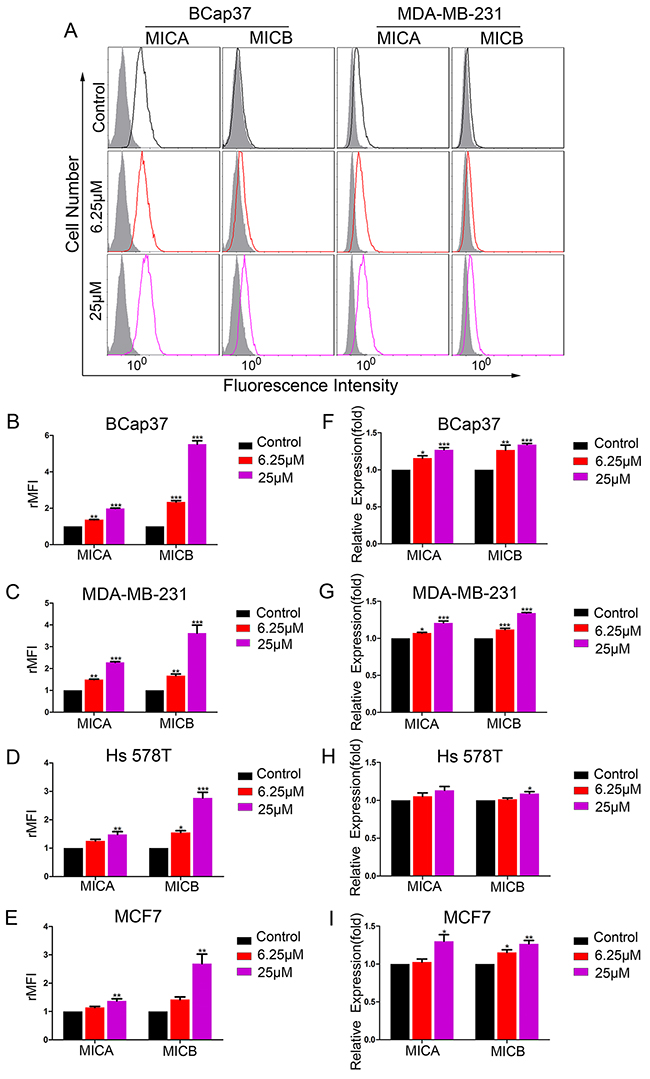
Figure 1: Resveratrol upregulates MICA and MICB expression in breast cancer cells in vitro. Breast cancer cell lines were treated with resveratrol (6.25 μM or 25 μM) or the control for 48 h. (A-E) The protein levels of MICA and MICB on BCap37, MDA-MB-231, Hs 578T and MCF-7 cells were detected by flow cytometry. (A) depicts representative results from (B) and (C). (F-I) The mRNA levels of MICA and MICB were detected by qRT-PCR, with HPRT1 as a reference. Error bars represent the SEM obtained from three independent experiments. *P<0.05, **P<0.01, ***P<0.001.
We further explored the effects of resveratrol in vivo. BCap37 cells were subcutaneously implanted into the right hind flanks of female BALB/c (nu/nu) mice to form xenograft tumors, and resveratrol was injected intraperitoneally at different doses (0, 25 or 100 mg/kg/d) for 28 days. In line with the results in vitro, resveratrol dose-dependently increased the expression of MICA/B in xenograft tumors (Figure 2). Moreover, at the endpoint of the study, all the mice were alive and in good condition, and their body weights did not differ significantly (Supplementary Figure 2A), indicating that 100-mg/kg/d resveratrol treatment for 28 days was safe for mice. We also found that resveratrol directly reduced tumor growth, as has been reported by other researchers (Supplementary Figure 2B, 2C) [16, 17]. These data indicate that resveratrol can upregulate MICA and MICB in breast cancer cells in vitro and in vivo.
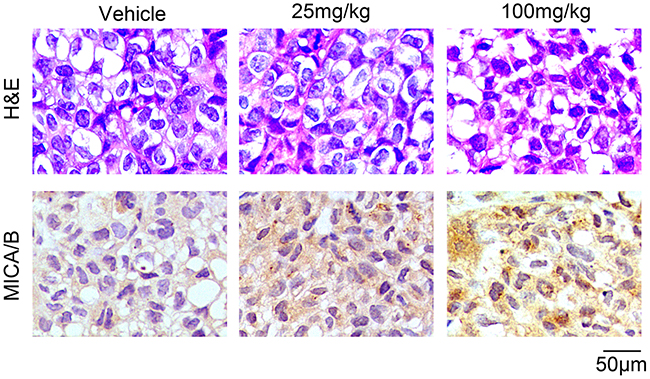
Figure 2: Resveratrol upregulates MICA/B expression in breast cancer xenograft tumors. Representative images from hematoxylin & eosin staining and an immunohistochemistry assay. BCap37 cells were subcutaneously implanted into the right hind flanks of female BALB/c (nu/nu) mice. Tumor tissues were collected after 28-day administration of resveratrol or vehicle. Paraffin sections of the tumor samples were prepared, and the cellular morphology was detected by hematoxylin & eosin staining (upper). MICA/B expression was examined with an immunohistochemistry assay (lower) (magnification ×400).
Resveratrol increases the lysis of breast cancer cells by NK cells by promoting NKG2D/NKG2DL binding in vitro
We next examined whether resveratrol could increase the susceptibility of breast cancer cells to lysis by NK cells. Bcap37 cells were treated with 6.25 μM or 25 μM resveratrol for 48 h and then used in NK cytotoxicity assays. As shown in Figure 3A, the upregulation of MICA and MICB by resveratrol was functional, as it moderately induced the killing of target cells tested at various effector-to-target ratios (Figure 3A, left). Furthermore, resveratrol-enhanced cytotoxicity was caused by increasing NKG2D recognition, as pretreatment of NK cells with an anti-NKG2D monoclonal antibody (mAb) resulted in equivalent killing of all targets (Figure 3A, right). Thus, resveratrol upregulates MICA and MICB and promotes NKG2D recognition to induce the death of target cells by NK cells.
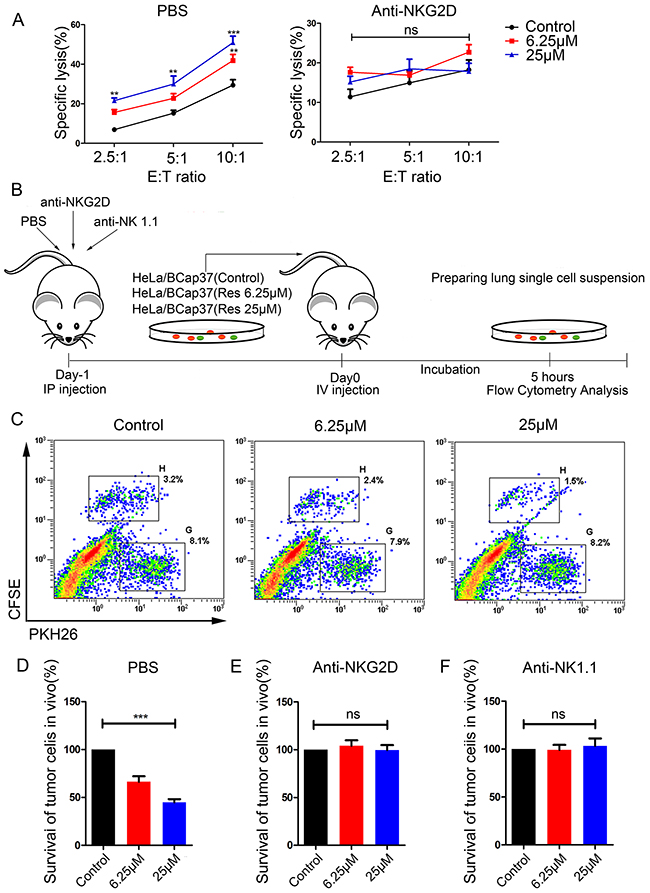
Figure 3: Resveratrol increases the susceptibility of breast cancer cells to cytotoxicity in vitro and in vivo. (A) BCap37 cells were exposed to different concentrations of resveratrol for 48 h. NK-92MI cells were pretreated with anti-NKG2D Abs (right) or PBS (left) for 1 h before the cytotoxicity assay. Cytotoxicity assays were performed with NK-92MI cells as effector cells at different effector-to-target ratios. (B) Schematic representation of the in vivo experimental procedures. (C-F) Mice were injected with PBS (C, D), anti-NKG2D (E) or anti-NK 1.1 (F) 24 h before being injected with a mixture of BCap37 and HeLa cells. Flow cytometry was used to analyze the ratios of BCap37 cells to HeLa cells in lung single-cell suspensions. (C) depicts representative results from (D). Error bars represent the SEM obtained from three independent experiments. **P<0.01, ***P<0.001. Abbreviations: IP, intraperitoneal; IV, intravenous.
Resveratrol contributes to immune clearance in vivo
To further explore the potential effects of resveratrol on breast cancer cell clearance in vivo, we performed experiments with C57BL/6 mice. The murine NKG2D receptor recognizes the human ligands and vice versa, though the human and mouse NKG2D ligands differ [18, 19]. A short-term in vivo assay (illustrated in Figure 3B) was adopted, and HeLa cells were used as an internal control.
In accordance with the results of our in vitro experiment, the survival rate of resveratrol-treated BCap37 cells injected into mice declined dose-dependently (Figure 3C, 3D). That is, as higher concentrations of resveratrol were used to treat the BCap37 cells, more BCap37 cells were cleared by the murine immune system and fewer carboxyfluorescein succinimidyl ester (CFSE)-labeled BCap37 cells were detected by flow cytometry. To explore whether the enhanced immune clearance was due to the binding between NKG2D and NKG2DLs, we intraperitoneally injected mice with anti-NKG2D or anti-NK1.1 mAbs to block the related receptors. The in vivo blocking of NKG2D activity with the anti-NKG2D mAb (Figure 3E) abolished the effects of resveratrol that had been observed in the phosphate-buffered saline (PBS) control group (Figure 3C, 3D). NK cells were the main effectors of the clearance, as the depletion of NK cells with the anti-NK1.1 mAb completely abolished the effects of resveratrol (Figure 3F). Thus, the upregulation of MICA and MICB by resveratrol promotes the immune recognition and clearance of tumor cells in vivo.
Resveratrol reduces the expression of miR-17
The miR-17-92 cluster, which is processed from the transcript of Chromosome 13 open reading frame 25 (C13orf25), consists of six microRNAs: miR-17, miR-18a, miR-19a, miR-20a, miR-19b, and miR-92a [7]. This cluster, known as oncomiR-1, is always overexpressed in tumors. We analyzed 56 pairs of breast tumor and peri-tumor specimens, and found that miR-17 expression was higher in breast tumor tissues than in non-tumorous tissues (Figure 4A). To determine whether resveratrol could suppress the expression of the miR-17-92 cluster, we examined the expression of this gene cluster in BCap37 and MDA-MB-231 cells pretreated with resveratrol. When breast cancer cells were treated with resveratrol (6.25 μM or 25 μM) for 48 h, the transcription of the miR-17-92 cluster declined dose-dependently (Figure 4B).
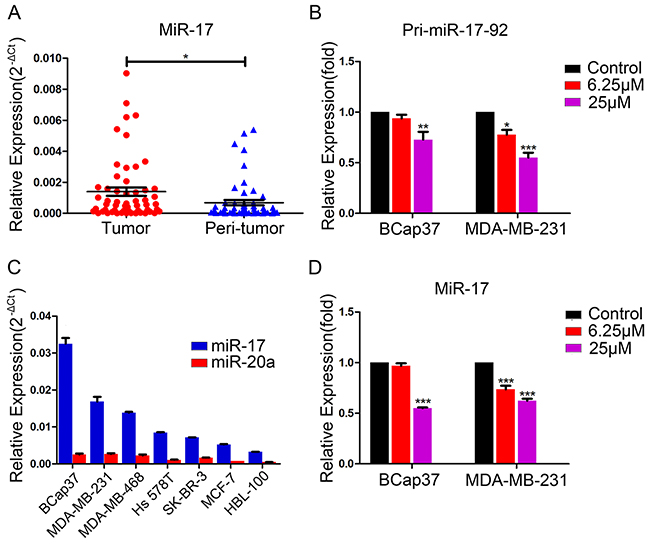
Figure 4: Endogenous expression of miR-17 in breast cancer cells and suppression of pri-miR-17-92 and miR-17 by resveratrol. (A) Endogenous miR-17 levels in breast tumor tissues and non-tumorous tissues from 56 patients were detected by qRT-PCR, with U6 as a reference. (B) BCap37 and MDA-MB-231 cells were pretreated with vehicle or different concentrations of resveratrol for 48 h. Transcription of the miR-17-92 cluster was detected by qRT-PCR, with HPRT1 as a reference. (C) The endogenous levels of miR-17 and miR-20a in six breast cancer cell lines and one normal breast cell line were assessed by qRT-PCR, with U6 as a reference. (D) BCap37 and MDA-MB-231 cells were treated as in (B). MiR-17 levels were assessed by qRT-PCR, with U6 as a reference. Data represent the mean ± SEM of three independent experiments. *P<0.05, **P<0.01, ***P<0.001.
Next, using various databases, we tabulated the predicted binding scores of miR-17-92 cluster members to the MICA/B 3′-UTRs, and calculated the aggregate binding scores from these databases (Supplementary Tables 1, 2). MiR-17 and miR-20a received the highest scores, so we chose these two miRNAs for further experiments. We evaluated the expression of miR-17 and miR-20a in six different breast cancer cell lines (BCap37, MDA-MB-231, MDA-MB-468, Hs 578T, SK-BR-3, and MCF-7) and one normal breast cell line (HBL-100) by qRT-PCR. As shown in Figure 4C, these breast cancer and normal cells expressed miR-17 and miR-20a at different levels. The expression of miR-17 was higher than that of miR-20a in both breast cancer and normal cells, and was higher in BCap37 and MDA-MB-231 cells than in the other cell lines. Considering this, along with the fact that miR-17 and miR-20a belong to the same microRNA family and share the same seed sequence, we focused on miR-17 expression in BCap37 and MDA-MB-231 cells in the following experiments. When the expression of miR-17 was investigated in BCap37 and MDA-MB-231 cells treated with resveratrol at various concentrations for 48 h, resveratrol was found to downregulate miR-17 dose-dependently (Figure 4D). These data indicate that resveratrol suppresses the transcription of the miR-17-92 cluster and subsequently downregulates miR-17.
MiR-17 downregulates MICA and MICB expression by binding to their 3′-untranslated regions (3′-UTRs)
We next assessed the endogenous expression of miR-17 and MICA/B in seven breast cancer cell lines (BCap37, MDA-MB-231, MDA-MB-468, Hs 578T, SK-BR-3, BT-474 and MCF-7) and two normal breast cell lines (HBL-100 and Hs 578Bst). MiR-17 levels correlated negatively with MICA/B levels in all the tested cell lines (Figure 5A). To further explore the regulation of MICA and MICB expression by miR-17, we transiently transfected BCap37 and MDA-MB-231 cells with a miR-17 mimic, miR-17 inhibitor, or the corresponding negative control (NC). MICA and MICB were upregulated in miR-17 inhibitor-transfected BCap37 cells, and were downregulated in miR-17 mimic-transfected BCap37 cells (Figure 5B, 5C). Similar observations were made in MDA-MB-231 cells (Figure 5D).
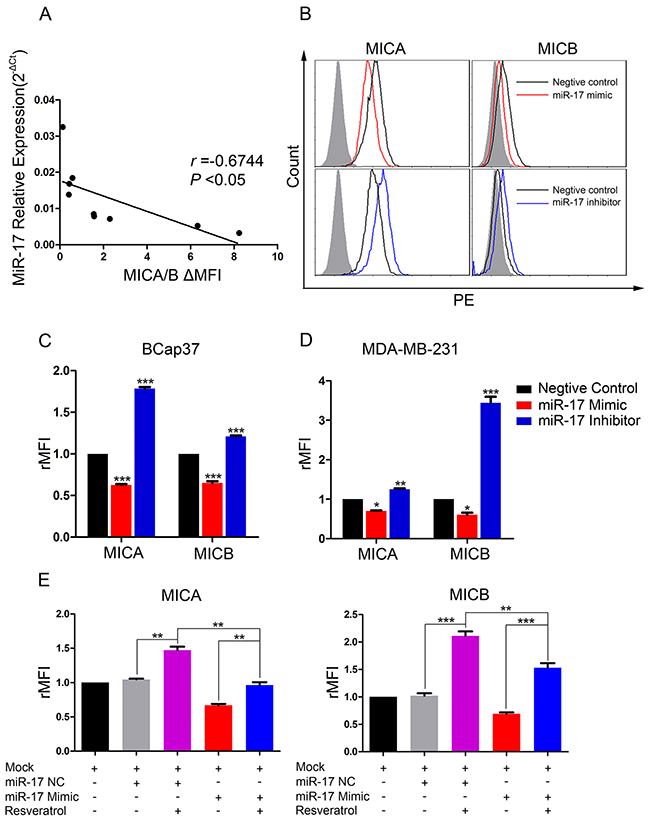
Figure 5: MiR-17 downregulates MICA and MICB expression, which can be blocked by resveratrol. (A) MiR-17 and MICA/B levels in seven breast cancer cell lines and two normal breast cell lines were assessed by qRT-PCR and flow cytometry, respectively. The correlation between the two was analyzed by linear regression. (B-D) BCap37 (B, C) and MDA-MB-231 (D) cells were transfected with a 50 nM miR-17 mimic, inhibitor, or the corresponding NC for 24 h. The surface levels of MICA and MICB were detected by flow cytometry 72 h after transfection. (B) depicts representative results from (C). (E) BCap37 cells transfected with a miR-17 mimic or NC for 24 h were treated with culture medium or 25 μM resveratrol for another 48 h. Then, the levels of MICA and MICB on cells were detected by flow cytometry. Error bars represent the SEM obtained from three independent experiments. *P<0.05, **P<0.01, ***P<0.001.
To evaluate whether resveratrol could prevent the downregulation of MICA and MICB by miR-17, we transiently transfected BCap37 cells with the miR-17 mimic or NC for 24 h and then treated them with 25 μM resveratrol for another 48 h. As shown in Figure 5E, the miR-17 mimic downregulated MICA and MICB, but resveratrol exposure remarkably blocked this downregulation.
To determine whether miR-17 directly targeted MICA and MICB, we performed luciferase reporter assays in BCap37 cells. Wild-type 3′-UTRs of MICA and MICB and mutated sequences (mut-MICA and mut-MICB) were individually cloned into reporter plasmids (psiCHECK-2) downstream of the Renilla luciferase gene (Figure 6A). BCap37 cells were transfected with a reporter plasmid only, or co-transfected with the reporter plasmid and either 50 nM miR-17 mimic or 50 nM NC. The luciferase activities of the reporters with wild-type MICA or MICB 3′-UTRs were significantly suppressed by miR-17 mimic transfection, while the activities of the reporters with mutant MICA or MICB 3′-UTRs did not change significantly (Figure 6B). These results demonstrate that miR-17 binds directly to MICA and MICB.
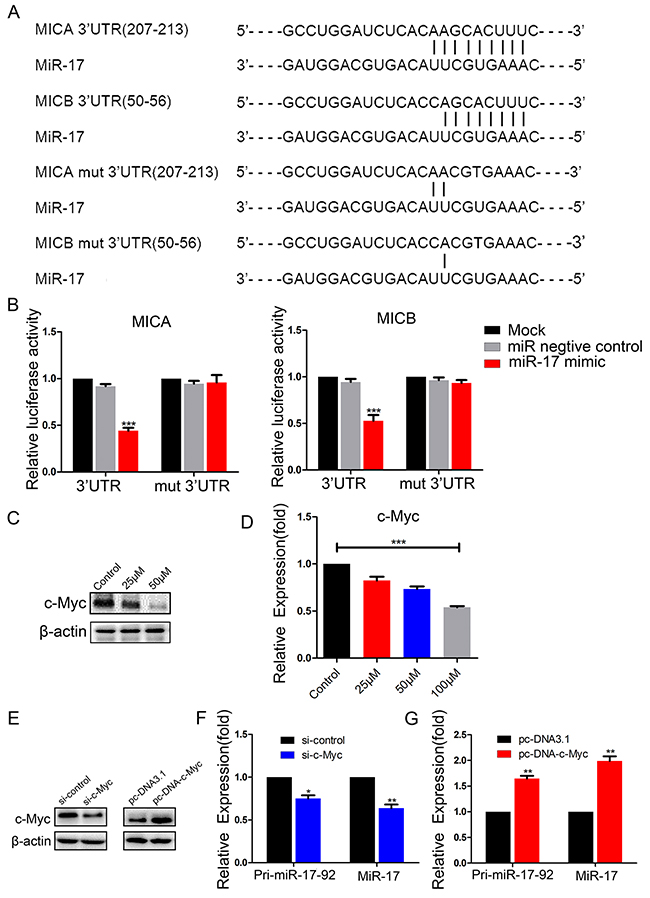
Figure 6: MiR-17 binds to the 3′-UTRs of MICA and MICB, and c-Myc promotes the expression of miR-17. (A) Schematic representation of predicted miR-17 binding sites in the 3′-UTRs of MICA/B, and 3′-UTR mutated alignment. (B) Luciferase reporter assay of BCap37 cells co-transfected with a miR-17 mimic, NC or mock and either wild-type or mutated luciferase plasmid. (C, D) BCap37 cells were treated with different concentrations of resveratrol for 48 h. The expression of c-Myc was detected by Western blotting (C) and qRT-PCR (D). (E) c-Myc protein levels in BCap37 cells transfected with control siRNA (si-control) or c-Myc siRNA (si-c-Myc) (left) and with pc-DNA3.1 or pc-DNA-c-Myc (right) were analyzed by Western blotting. β-actin is shown as a loading control. (F, G) Pri-miR-17-92 and miR-17 levels were evaluated in c-Myc-knockdown (F) or c-Myc-overexpressing (G) BCap37 cells by qRT-PCR. Error bars represent the SEM obtained from three independent experiments. *P<0.05, **P<0.01, ***P<0.001.
Taken together, these findings suggest that miR-17 downregulates MICA and MICB by directly binding to their 3′-UTRs, while resveratrol can block this downregulation.
Resveratrol downregulates miR-17 expression by suppressing c-Myc
To determine how resveratrol downregulated miR-17, we focused on the transcription factor c-Myc, which has been reported to bind directly to the miR-17-92 cluster genomic locus in human lymphoma cells [20]. We previously found that there were c-Myc binding sites in the miR-17-92 cluster transcriptional initiation site in breast cancer cells, and that c-Myc promoted the transcription of this gene cluster (Wengong Si et al., revised). Here, we hypothesized that c-Myc was involved in the regulation of miR-17-92 cluster expression by resveratrol. To test this hypothesis, we investigated the effect of resveratrol on c-Myc expression in BCap37 cells. Following 48-h resveratrol treatment, c-Myc mRNA and protein levels decreased dose-dependently (Figure 6C, 6D).
To determine the effect of c-Myc on the transcription of the miR-17-92 cluster, we transfected BCap37 cells with c-Myc siRNA or overexpression plasmids to knock down or overexpress c-Myc (Figure 6E). Knockdown of c-Myc reduced the expression of pri-miR-17-92 and miR-17 (Figure 6F). On the other hand, overexpression of c-Myc significantly increased pri-miR-17-92 and miR-17 expression (Figure 6G). These observations suggest that resveratrol reduces miR-17 expression by suppressing c-Myc.
MiR-17 expression correlates inversely with MICA/B expression and overall survival in breast cancer patients
To investigate the significance of miR-17 in breast cancer patients, we analyzed the correlations of miR-17 expression with MICA/B expression, disease stage, overall survival and cancer recurrence. The levels of miR-17 and MICA/B were evaluated in 56 clinical breast cancer specimens. The median relative level of miR-17 or MICA/B was adopted as the threshold value, such that miRNA or mRNA levels above or below this level were considered as high or low expression, respectively. In line with other studies [21], the overall survival probability was poorer and the cancer recurrence rate was higher in patients with higher levels of miR-17 than in those with lower levels (Figure 7A, Supplementary Table 3). The expression of miR-17 correlated inversely with the expression of MICA and MICB in the 56 specimens (Figure 7B, 7C). These specimens were then divided into three groups according to the WHO staging system (stage I, II, and III). A trend for higher miR-17 expression was observed in late-stage patients (Supplementary Figure 3).
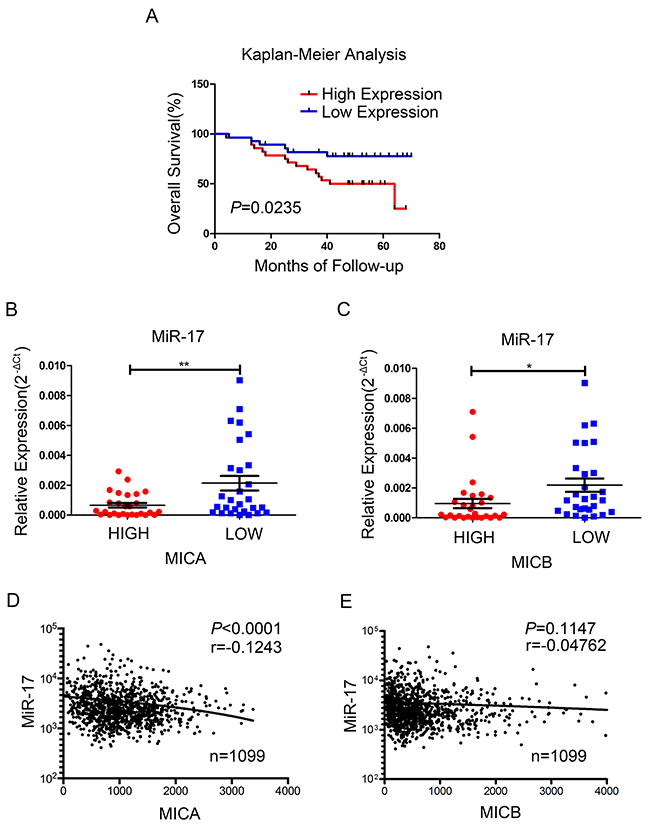
Figure 7: Association of miR-17 with the expression of MICA/B and survival in human breast cancer. (A-C) Data from 56 clinical breast cancer patients. (A) Kaplan-Meier overall survival curves comparing high and low miR-17 expression groups; P value was based on a log-rank test. (B, C) The expression of miR-17 was compared in high and low MICA (B) or MICB (C) expression groups. (D, E) The association of miR-17 expression with MICA (D) and MICB (E) expression in 1099 breast cancer samples from TCGA. Error bars in B and C represent the mean ± SEM. *P<0.05, **P<0.01.
Because of our small sample size, we further analyzed 1099 available breast cancer samples from The Cancer Genome Atlas (TCGA). In these samples, miR-17 expression correlated negatively with MICA expression, but did not correlate significantly with MICB expression (Figure 7D, 7E).
Thus, higher miR-17 levels correlate with a poorer overall survival probability, higher cancer recurrence rate and lower MICA/B level in breast cancer patients.
DISCUSSION
NKG2D-NKG2DL binding is important in the antitumor immune response, as it ensures innate immunity to tumor cells and co-stimulates T-cell subset activation [22, 23]. High levels of NKG2DLs are associated with the increased cytotoxic activity of effector cells [24]. However, tumor cells have developed various strategies to downregulate NKG2DLs, allowing them to avoid immune recognition and clearance. Thus, the NKG2DL system has attracted vivid interest as a potential target of treatments to improve antitumor immune responses [2].
Functional analyses in this study demonstrated that resveratrol increased the susceptibility of breast cancer cells to lysis by NK cells in vitro and in vivo by inducing MICA and MICB expression. Resveratrol is a natural polyphenolic flavonoid compound with multiple health-promoting effects, mainly against oxidative stress and inflammation. Resveratrol is produced by many commonly consumed plants, including grapes, apples, raspberries, blueberries, plums and peanuts [11]. Consecutive high-dose administration of resveratrol (up to 5.0 g daily for 29 days) was verified to be safe and well-tolerated in healthy volunteers [25, 26]. In our animal study, we found that consecutive administration of 100 mg/kg/d resveratrol for 28 days was safe in mice. Thus, resveratrol may be a safer candidate than other NKG2DL-inducing agents such as proteasome inhibitors, histone deacetylases and some chemotherapeutic drugs [27-29].
The regulation of MICA and MICB expression is a complex process that involves various transcriptional and post-transcriptional mechanisms. Numerous signals and pathways have been identified as regulators of NKG2DLs, including the ATM (ataxia-telangiectasia, mutated) and ATR (ATM and Rad3-related) pathways, the NF-κB pathway, heat shock factor-1, Sp-family transcription factors and the ERK signaling pathway [4, 6, 30-32]. Here, we have described a novel c-Myc/miR-17 pathway whereby resveratrol upregulates MICA and MICB.
MiR-17, which belongs to the oncogenic miR-17-92 cluster, is upregulated in a variety of cancers [33, 34]. We found that miR-17 expression was higher in breast tumor tissues than in peri-tumor tissues, consistent with previous reports. Thus far, miR-17 has been reported to promote tumorigenesis and progression by sustaining cell survival, proliferation, self-renewal, migration and angiogenesis. MiR-17 was found to stimulate cancer genesis and progression by suppressing P130 and PTEN [35, 36]. In addition, miR-17 was reported to impair cancer cell apoptosis by downregulating the pro-apoptotic gene Bim, and to impede cell cycle arrest by downregulating the cell cycle inhibitor p21 [33, 37-40]. Overexpression of miR-17 also improved the invasion and migration abilities of cancer cells [41-43]. MiR-17 may promote cancer progression by inducing angiogenesis in solid tumors [44]. Although miR-17 is known to drive tumorigenesis and progression by inhibiting certain tumor suppressors, there has been little evidence of its tumorigenicity from the immunological standpoint. MiR-20a, another component of the miR-17-92 cluster, was reported to bind to the mRNA of MICA and MICB [7, 45, 46]. Therefore, we postulated that miR-17 could promote tumor progression by inhibiting MICA/B expression, thus allowing tumor cells to escape from immune recognition.
We discovered that resveratrol suppressed the transcription of the miR-17-92 cluster host gene and subsequently reduced the expression of miR-17. MICA and MICB expression increased in response to resveratrol treatment, an effect that correlated inversely with miR-17 expression. By performing the dual luciferase reporter assays, miR-17 was verified to reduce MICA and MICB expression by binding to their 3′-UTRs. These data indicated that resveratrol increased the susceptibility of breast cancer cells to lysis by NK cells through a miR17/MICA/MICB pathway.
However, the mechanism whereby resveratrol suppressed the expression of miR-17 was still unknown. Certain transcription factors, such as c-Myc, p-STAT3 and E2F, are known to transcriptionally activate the miR-17-92 cluster [7, 20, 47]. C-Myc, which is essential for cell growth, proliferation and tumorigenesis, is dysregulated in a wide range of malignancies, including breast cancer [48, 49]. Our study demonstrated that c-Myc was repressed in resveratrol-treated breast cancer cells. Knockdown or overexpression of c-Myc respectively reduced or induced the expression of miR-17-92, supporting previous findings that c-Myc could directly bind to the miR-17-92 cluster promoter [20]. Therefore, we deduced that resveratrol suppressed the expression of c-Myc and consequently inhibited the transcription of miR-17-92.
Our findings in breast cancer cells and animal models prompted us to investigate the clinical association of miR-17 with MICA/B and its prognostic impact on breast cancer patients. Overexpression of miR-17 has been associated with poor prognosis in certain malignancies [21, 36]. Accordingly, we found that the overall survival was shorter and the cancer recurrence rate was higher in breast cancer patients with higher levels of miR-17. In our analysis of 56 clinical breast cancer specimens and data from TCGA, it was obvious that miR-17 expression correlated inversely with MICA and MICB expression, potentially indicating that miR-17 inhibited MICA/B expression. Though the difference was not significant, patients with late-stage disease were more likely to express higher levels of miR-17 than those with early-stage disease. Thus, high expression of miR-17, which showed a negative correlation with MICA and MICB levels, might predict a poor prognosis in breast cancer patients.
In conclusion, resveratrol elevated MICA and MICB expression in breast cancer cells by suppressing the c-Myc/miR-17 pathway, thus increasing the susceptibility of breast cancer cells to lysis by NK cells. Therefore, resveratrol treatment could be a feasible way to improve the MICA and MICB mediated anticancer effects in breast cancer patients.
MATERIALS AND METHODS
Clinical samples
Paraffin-embedded sections of breast cancer tissues and adjacent non-tumorous breast tissues were derived from 56 patients who underwent surgical resection at the First Affiliated Hospital of Zhejiang University College of Medicine from 2010 to 2011. The respective adjacent non-tumorous breast tissue was used as a reference sample for each cancer tissue. Total RNA (including miRNA) was isolated with the RNeasy® FFPE Kit (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol, and was stored at −80°C for further study. Clinical parameters, including age, sex, pathological features and TMN stage, were obtained from patients’ medical records. Detailed sample information is listed in Supplementary Table 3. The study protocol was reviewed and approved by the Ethics Committee of Zhejiang University College of Medicine. Written informed consent was obtained from each patient.
Mice and cell lines
Male C57BL/6 mice (8-9 weeks old) were purchased from Vital River (Beijing, China). Female BALB/c (nu/nu) mice (6 weeks old) were purchased from the Shanghai Laboratory Animal Center (Shanghai, China). All animal experiments were approved by the Ethics Committee of Zhejiang University College of Medicine.
All cell lines were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China) and maintained according to the manufacturer’s recommendations. Experiments were initiated when the cells exhibited logarithmic growth.
Cell viability assay
Cell viability was determined by the microculture tetrazolium (MTT) assay. Cells were seeded into 96-well culture plates, incubated for one night, and treated with different concentrations of resveratrol or control medium for 48 h. Four hours prior to the endpoint, the MTT solution was added. The plates were incubated again for 4 h at 37°C in the dark. Then, the medium containing MTT in each well was replaced with 150 μL of dimethyl sulfoxide (DMSO, Sinopharm Chemical Regent Co., Shanghai, China) to dissolve the formazan crystals. The absorbances of the individual wells were determined at 570 nm with a microplate reader (Bio-Rad, Sunnyvale, CA, USA).
Apoptosis assay
Cells were seeded in six-well plates. Twelve hours later, the cells were treated with different concentrations of resveratrol or control medium for 48 h. Fluorescein Annexin V-fluorescein isothiocyanate (FITC) and propidium iodide (PI) double labeling was performed with an Annexin V-FITC/PI apoptosis detection kit (Beyotime, Haimen, China) in accordance with the manufacturer’s instructions. The percentage of apoptotic cells was determined by flow cytometry analysis on a BD FACSCalibur™ flow cytometry system (BD Biosciences, Franklin Lakes, NJ, USA).
Plasmids
For gene knockdown experiments, c-Myc siRNA (sense: AUGAUGUUUU UGAUGAAGGUC; anti-sense: CCUUCAUCAAAAACAUCAUCA) and negative control siRNA were obtained from GenePharma (Shanghai, China). For gene overexpression vector construction, the open reading frames and downstream 3′-UTR of c-Myc were cloned into the pcDNA3.1+ vector (Invitrogen) between the HindIII and EcoRI sites and driven by the cytomegalovirus promoter. For the luciferase reporter assay, the 3′-UTR fragment of MICA or MICB was amplified and cloned into the XhoI and NotI sites downstream of the SV40 promoter-driven Renilla luciferase cassette in the psiCHECK-2 plasmid (Promega, Madison, WI, USA). A Fast Mutagenesis kit (Vazyme Biotech, Nanjing, China) was used to mutate the miR-17 binding sites in the MICA or MICB 3′-UTR vectors according to the manufacturer’s instructions.
Transfection and drug treatments
BCap37 and MDA-MB-231 cells were seeded in 6-cm cell culture dishes at 1.6×105 and 1.2×105 cells/mL, respectively. The cells were transfected with a 50 nM miR-17 mimic, miR-17 inhibitor or the corresponding scrambled NC (Ribobio, Guangzhou, China) with lipofectamine 2000 (Invitrogen) per the manufacturer’s instructions. Twenty-four hours later, the medium was replaced and the cells were prepared for subsequent experiments.
Resveratrol was purchased from Selleck Chemicals (Houston, TX, USA) and stored at −20°C. It was dissolved in DMSO to a 109.5 mM stock. Cells were treated with 6.25, 25, 50 or 100 μM resveratrol for 48 h.
RNA extraction and quantitative real-time PCR (qRT-PCR) analysis of mRNA and miRNA
Total RNA was isolated from breast cancer cells with RNAiso Plus (TaKaRa, Kusatsu, Japan). The extracted RNA was synthesized into cDNA with a PrimeScriptTM RT reagent Kit (#RR037A, TaKaRa) to produce a template suitable for qRT-PCR. For mRNA reverse-transcriptase PCR (RT-PCR), random hexamer primers were used, and for miRNA RT-PCR, specific primers (Ribobio) were used.
qRT-PCR was used to detect the expression of MICA, MICB, pri-miR-17-92 and c-Myc, with hypoxanthine phosphoribosyltransferase 1 (HPRT1) as an internal normalized reference. The sequences of the PCR primers are listed in Supplementary Table 4 Mature miRNA expression was quantified by qRT-PCR with specific BulgeLoop miRNA qRT-PCR primers (Ribobio), with U6 small nuclear RNA as an internal normalized reference (Supplementary Table 5). qRT-PCR was performed in a LightCycler 480II system (Roche Diagnostics, Basel, Switzerland) with a SYBR Premix EX Tag kit (#RR420A, TaKaRa). Relative expression was calculated as 2-ΔCt or 2-ΔΔCt after normalization with the reference control.
Flow cytometry analysis
For cell-surface protein expression analysis, cells were harvested, washed with PBS, and incubated with antibodies for 25 min at 4°C in the dark. Data acquisition and analysis were carried out on a BD FACSCalibur™ flow cytometry system with the CellQuest software package (BD Biosciences). The following antibodies were purchased from R&D systems (R&D, Minneapolis, MN, USA): MICA-PE (FAB1300P-100), MICB-PE (FAB1599P-025), and mouse IgG2B PE-conjugated antibody (IC0041P). The increase in mean fluorescence intensity (ΔMFI) was calculated as: (MFI with specific mAb – MFI with isotype control) / MFI with isotype control. The relative MFI (rMFI) was employed to illustrate the difference between the ΔMFI of a specific treatment and that of the control, and was calculated as: ΔMFI of specific treatment / ΔMFI of control treatment [50].
Bioinformatic analysis
MiRNAs that bind to the 3′-UTRs of MICA and MICB were predicted with different bioinformatic algorithms from various databases, including TargetScan (http://www.targetscan.org/), miRanda (http://www.microrna.org/microrna/home.do), RNA22 (https://cm.jefferson.edu/rna22/), miRWalk (http://www.umm.uni-heidelberg.de/apps/zmf/mirwalk/), and Starbase (http://starbase.sysu.edu.cn/targetSite.php). If a miRNA was predicted to bind to the 3′-UTR of a certain gene, it received a score of “1”; otherwise, it received a score of “0”. The miRNAs with the highest aggregate scores were chosen for further experiments.
TCGA (https://cancergenome.nih.gov/) provides breast cancer patient mRNA and miRNA sequence results. All the available level-3 data (1099 breast cancer samples) with detailed miR-17 and MICA/B information were extracted from the data portal of TCGA and used as input for correlation analysis by the R package edgeR [51].
Luciferase reporter assay
BCap37 cells were cultured in 96-well plates at a density of 2×104 cells per well. The cells were co-transfected with a 50 nM miR-17 mimic or NC, and 50 ng of the MICA or MICB 3′-UTR wild-type or mutant psiCHECK-2 reporter plasmid (Promega). The transfection was performed with Lipofectamine 2000 according to the manufacturer’s instructions. After 48 h of transfection, a luciferase assay was performed with a Dual Luciferase Reporter Assay System Kit (Promega) as described in the manufacturer’s protocol. Luminometry readings were obtained with a Varioskan Flash Spectral Scanning Multimode Reader (Thermo Scientific, Waltham, MA, USA). Firefly luciferase activity was normalized to constitutive Renilla luciferase activity. Each transfectant was assayed in triplicate.
Cytotoxicity assay in vitro
After pretreatment with 6.25 μM or 25 μM resveratrol for 48 h, BCap37 cells were used as target cells and seeded into a round-bottom 96-well plate at a density of 1×104 cells per well. Cells from the NK cell line NK-92MI were pretreated with PBS or anti-NKG2D antibodies (50 mg/mL, Novus Biologicals, Littleton, CO, USA) for 1 h before the cytotoxicity assay, and then were added to the wells at effector-to-target ratios of 10:1, 5:1 and 2.5:1. After a 4-h co-incubation at 37°C in a humidified atmosphere of 5% CO2, the supernatants were harvested and analyzed by the CytoTox 96 Non-Radioactive Cytotoxicity Assay (Promega). The cytotoxicity of the effector cells against the target cells was assessed with the following equation: Cytotoxicity = (Experimental - Effector spontaneous - Target spontaneous) / (Target maximum - Target spontaneous) ×100%.
Lung clearance assay
Male C57BL/6 mice (8-9 weeks old) were divided into three groups and intraperitoneally injected with PBS, anti-mouse NKG2D monoclonal antibodies (300 μg per mouse, #191004, Novus Biologicals) or anti-NK1.1 antibodies (300 μg per mouse, #108712, Biolegend, San Diego, CA, USA), respectively. Twenty-four hours later, HeLa cells (which are not efficiently killed by mouse NK cells, and thus served as an internal control) were labeled with PKH26 (MINI26-1KT, Invitrogen), and various BCap37 cells (pretreated with 6.25 μM, 25 μM resveratrol or control for 48 h) were labeled with CFSE (C34554, Invitrogen). Stained cells (5×106 cells of each population) were mixed in 1 mL PBS, and 0.4 mL of this mixture was injected into the tail vein of each mouse. Five hours later, the lungs were harvested and single-cell suspensions were prepared for flow cytometry. The ratio of tested target cells to HeLa cells was calculated. In each group, the ratio of control-medium-pretreated BCap37 cells to HeLa cells was set as 100% [13, 52].
Immunohistochemistry in a xenograft model
BCap37 cells (2×106 cells in 0.2 mL PBS) were subcutaneously implanted into the right hind flanks of female BALB/c (nu/nu) mice. The length and width of the tumor were measured every three days with a caliper, and the tumor volume was calculated as (length × width × width × 0.5). When the tumor size was approximately 40 mm3, the mice were divided into three groups. Two groups were given resveratrol (dissolved in DMSO: polyethylene glycol 400: distilled and deionized water [1:1:3]) intraperitoneally at a daily dose of 25 or 100 mg/kg for four consecutive weeks, whereas the control group received the vehicle only [53]. Body weights and tumor volumes were recorded every week. After four weeks of treatment, the mice were sacrificed. The tumors were fixed in 4% paraformaldehyde (Sinopharm Chemical Reagent Co., Shanghai, China) and sectioned. The slides were stained with hematoxylin and eosin (Hematoxylin and Eosin Staining Kit, C0105, Beyotime Biotechnology, Shanghai, China) so that cell types and morphologic changes could be observed. Specific antibodies against MICA/B (#Ab54413, Abcam, Cambridge, MA, USA) were used for immunohistochemical staining. The slides were then incubated with a goat anti-rabbit IgG/horseradish peroxidase (HRP) complex (PV-6001, ZSGB-BIO, Beijing, China) for 45 minutes at room temperature. After being washed twice in PBS, the sections were incubated with HRP-conjugated streptavidin (DAB kit, ZLI-9019, ZSGB-BIO) for 30 minutes and then washed twice in PBS. Positive results were visualized with 3, 3′-diaminobenzidine. The results were analyzed by observation under a microscope, and images were acquired at ×400 magnification.
Western blot
After treatment with resveratrol or transfection with siRNA or expression vectors, BCap37 cells were harvested. Total proteins were extracted in RIPA lysis buffer (Beyotime Institute of Biotechnology, China) plus 1 mM phenylmethylsulfonyl fluoride and a 1x protease inhibitor cocktail (Sigma-Aldrich, Sigma, MO, USA). The protein content was detected with a bicinchoninic acid (BCA) kit (Thermo Fisher Scientific, MA, USA). Then, 40 ug lysate was run on a 10% polyacrylamide gel and transferred onto polyvinylidene fluoride membranes (Roche, Switzerland). The membranes were probed with specific antibodies against c-Myc (32072, Abcam) and β-Actin (A2228, Sigma-Aldrich). β-Actin was adopted as an internal control. Signals from HRP-conjugated secondary antibodies were detected with enhanced chemiluminescence reagents (Thermo Scientific, Darmstadt, Germany) according to the manufacturer’s instructions. Blots were scanned with a ChemiDoc Touch Imaging System (Bio-Rad, CA, USA). Experiments were repeated independently at least three times.
Statistical analysis
All data are presented as mean ± standard error of the mean (SEM) from at least three independent experiments. Statistical significance was assessed with Student’s t-test or one-way analysis of variance. Overall survival was defined as the time period between diagnosis and disease-related death. Overall survival was censured when the patient either was alive at the last follow-up or died from causes unrelated to the disease. Overall survival probability rates were calculated by the Kaplan–Meier method, and compared through log-rank test univariate analysis. Linear correlation and Pearson’s correlation were used to evaluate the correlation between two variants. Statistical analyses were performed with GraphPad Prism Ver. 5.01 (San Diego, CA, USA) in accordance with the guidelines for the software. Statistical significance was set at p < 0.05 and displayed as *** for p<0.001; ** for p<0.01 and * for p<0.05.
Abbreviations
MICA, Major histocompatibility complex class I chain-related protein A
MICB, Major histocompatibility complex class I chain-related protein B
NKG2D, Natural killer group 2 member D
NKG2DL, Natural killer group 2 member D ligand
3′-UTR, 3′-untranslated region
NC, negative control
OS, overall survival
TCGA, The Cancer Genome Atlas
Author contributions
JP and WF planned and guided all the experiments. JP and JS performed all the experiments. WS, DC and LX participated in the operation of some experiments. CD, MY and PF collected the specimens. JP analyzed the data and wrote the manuscript. WF obtained funding for this project. All authors reviewed and approved of the final submitted version.
ACKNOWLEDGMENTS
We thank Youfa Zhu, Yanwei Li, Li Liu, Yingying Huang and the staff members of the Core Facilities, Zhejiang University College of Medicine for providing technical support in the flow cytometry and immunohistochemistry experiments.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FINANCIAL SUPPORT
This work was supported by the National Natural Science Foundation of China (81372462, 81572987), the Department of Science and Technology of Zhejiang Province (2014C03012) and the Health and Family Planning Commission of Zhejiang Province (2012KYB045).
REFERENCES
1. Chitadze G, Lettau M, Luecke S, Wang T, Janssen O, Furst D, Mytilineos J, Wesch D, Oberg HH, Held-Feindt J, Kabelitz D. NKG2D- and T-cell receptor-dependent lysis of malignant glioma cell lines by human gammadelta T cells: Modulation by temozolomide and A disintegrin and metalloproteases 10 and 17 inhibitors. Oncoimmunology. 2016; 5: e1093276. https://doi.org/10.1080/2162402X.2015.1093276.
2. Ullrich E, Koch J, Cerwenka A, Steinle A. New prospects on the NKG2D/NKG2DL system for oncology. Oncoimmunology. 2013; 2: e26097. https://doi.org/10.4161/onci.26097.
3. Cheng M, Chen Y, Xiao W, Sun R, Tian Z. NK cell-based immunotherapy for malignant diseases. Cell Mol Immunol. 2013; 10: 230-52. https://doi.org/10.1038/cmi.2013.10.
4. Raulet DH, Gasser S, Gowen BG, Deng W, Jung H. Regulation of ligands for the NKG2D activating receptor. Annu Rev Immunol. 2013; 31: 413-41. https://doi.org/10.1146/annurev-immunol-032712-095951.
5. Tang KF, He CX, Zeng GL, Wu J, Song GB, Shi YS, Zhang WG, Huang AL, Steinle A, Ren H. Induction of MHC class I-related chain B (MICB) by 5-aza-2′-deoxycytidine. Biochem Biophys Res Commun. 2008; 370: 578-83. https://doi.org/10.1016/j.bbrc.2008.03.131.
6. Butler JE, Moore MB, Presnell SR, Chan HW, Chalupny NJ, Lutz CT. Proteasome regulation of ULBP1 transcription. J Immunol. 2009; 182: 6600-9. https://doi.org/10.4049/jimmunol.0801214.
7. Yang H, Lan P, Hou Z, Guan Y, Zhang J, Xu W, Tian Z, Zhang C. Histone deacetylase inhibitor SAHA epigenetically regulates miR-17-92 cluster and MCM7 to upregulate MICA expression in hepatoma. Br J Cancer. 2015; 112: 112-21. https://doi.org/10.1038/bjc.2014.547.
8. Soriani A, Iannitto ML, Ricci B, Fionda C, Malgarini G, Morrone S, Peruzzi G, Ricciardi MR, Petrucci MT, Cippitelli M, Santoni A. Reactive oxygen species- and DNA damage response-dependent NK cell activating ligand upregulation occurs at transcriptional levels and requires the transcriptional factor E2F1. J Immunol. 2014; 193: 950-60. https://doi.org/10.4049/jimmunol.1400271.
9. Lu CC, Chen JK. Resveratrol enhances perforin expression and NK cell cytotoxicity through NKG2D-dependent pathways. J Cell Physiol. 2010; 223: 343-51. https://doi.org/10.1002/jcp.22043.
10. Luis Espinoza J, Takami A, Trung LQ, Nakao S. Ataxia-telangiectasia mutated kinase-mediated upregulation of NKG2D ligands on leukemia cells by resveratrol results in enhanced natural killer cell susceptibility. Cancer Sci. 2013; 104: 657-62. https://doi.org/10.1111/cas.12141.
11. Szliszka E, Krol W. The role of dietary polyphenols in tumor necrosis factor-related apoptosis inducing ligand (TRAIL)-induced apoptosis for cancer chemoprevention. Eur J Cancer Prev. 2011; 20: 63-9. https://doi.org/10.1097/CEJ.0b013e32833ecc48.
12. Stern-Ginossar N, Mandelboim O. An integrated view of the regulation of NKG2D ligands. Immunology. 2009; 128: 1-6. https://doi.org/10.1111/j.1365-2567.2009.03147.x.
13. Tsukerman P, Stern-Ginossar N, Gur C, Glasner A, Nachmani D, Bauman Y, Yamin R, Vitenshtein A, Stanietsky N, Bar-Mag T, Lankry D, Mandelboim O. MiR-10b downregulates the stress-induced cell surface molecule MICB, a critical ligand for cancer cell recognition by natural killer cells. Cancer Res. 2012; 72: 5463-72. https://doi.org/10.1158/0008-5472.CAN-11-2671.
14. Heinemann A, Zhao F, Pechlivanis S, Eberle J, Steinle A, Diederichs S, Schadendorf D, Paschen A. Tumor suppressive microRNAs miR-34a/c control cancer cell expression of ULBP2, a stress-induced ligand of the natural killer cell receptor NKG2D. Cancer Res. 2012; 72: 460-71. https://doi.org/10.1158/0008-5472.CAN-11-1977.
15. Eissmann P, Evans JH, Mehrabi M, Rose EL, Nedvetzki S, Davis DM. Multiple mechanisms downstream of TLR-4 stimulation allow expression of NKG2D ligands to facilitate macrophage/NK cell crosstalk. J Immunol. 2010; 184: 6901-9. https://doi.org/10.4049/jimmunol.0903985.
16. Deus CM, Serafim TL, Magalhaes-Novais S, Vilaca A, Moreira AC, Sardao VA, Cardoso SM, Oliveira PJ. Sirtuin 1-dependent resveratrol cytotoxicity and pro-differentiation activity on breast cancer cells. Arch Toxicol. 2016. https://doi.org/10.1007/s00204-016-1784-x.
17. Medina-Aguilar R, Marchat LA, Arechaga Ocampo E, Gariglio P, Garcia Mena J, Villegas Sepulveda N, Martinez Castillo M, Lopez-Camarillo C. Resveratrol inhibits cell cycle progression by targeting Aurora kinase A and Polo-like kinase 1 in breast cancer cells. Oncol Rep. 2016; 35: 3696-704. https://doi.org/10.3892/or.2016.4728.
18. Lenac T, Arapovic J, Traven L, Krmpotic A, Jonjic S. Murine cytomegalovirus regulation of NKG2D ligands. Med Microbiol Immunol. 2008; 197: 159-66. https://doi.org/10.1007/s00430-008-0080-7.
19. Stern-Ginossar N, Gur C, Biton M, Horwitz E, Elboim M, Stanietsky N, Mandelboim M, Mandelboim O. Human microRNAs regulate stress-induced immune responses mediated by the receptor NKG2D. Nat Immunol. 2008; 9: 1065-73. https://doi.org/10.1038/ni.1642.
20. O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005; 435: 839-43. https://doi.org/10.1038/nature03677.
21. Robaina MC, Faccion RS, Mazzoccoli L, Rezende LM, Queiroga E, Bacchi CE, Thomas-Tikhonenko A, Klumb CE. miR-17-92 cluster components analysis in Burkitt lymphoma: overexpression of miR-17 is associated with poor prognosis. Ann Hematol. 2016; 95: 881-91. https://doi.org/10.1007/s00277-016-2653-7.
22. Benitez AC, Dai Z, Mann HH, Reeves RS, Margineantu DH, Gooley TA, Groh V, Spies T. Expression, signaling proficiency, and stimulatory function of the NKG2D lymphocyte receptor in human cancer cells. Proc Natl Acad Sci U S A. 2011; 108: 4081-6. https://doi.org/10.1073/pnas.1018603108.
23. Rincon-Orozco B, Kunzmann V, Wrobel P, Kabelitz D, Steinle A, Herrmann T. Activation of V gamma 9V delta 2 T cells by NKG2D. J Immunol. 2005; 175: 2144-51:
24. Leung WH, Vong QP, Lin W, Janke L, Chen T, Leung W. Modulation of NKG2D ligand expression and metastasis in tumors by spironolactone via RXRgamma activation. J Exp Med. 2013; 210: 2675-92. https://doi.org/10.1084/jem.20122292.
25. Brown VA, Patel KR, Viskaduraki M, Crowell JA, Perloff M, Booth TD, Vasilinin G, Sen A, Schinas AM, Piccirilli G, Brown K, Steward WP, Gescher AJ, et al. Repeat dose study of the cancer chemopreventive agent resveratrol in healthy volunteers: safety, pharmacokinetics, and effect on the insulin-like growth factor axis. Cancer Res. 2010; 70: 9003-11. https://doi.org/10.1158/0008-5472.CAN-10-2364.
26. Cottart CH, Nivet-Antoine V, Beaudeux JL. Review of recent data on the metabolism, biological effects, and toxicity of resveratrol in humans. Mol Nutr Food Res. 2014; 58: 7-21. https://doi.org/10.1002/mnfr.201200589.
27. Armeanu S, Krusch M, Baltz KM, Weiss TS, Smirnow I, Steinle A, Lauer UM, Bitzer M, Salih HR. Direct and natural killer cell-mediated antitumor effects of low-dose bortezomib in hepatocellular carcinoma. Clin Cancer Res. 2008; 14: 3520-8. https://doi.org/10.1158/1078-0432.CCR-07-4744.
28. Min D, Lv XB, Wang X, Zhang B, Meng W, Yu F, Hu H. Downregulation of miR-302c and miR-520c by 1,25(OH)2D3 treatment enhances the susceptibility of tumour cells to natural killer cell-mediated cytotoxicity. Br J Cancer. 2013; 109: 723-30. https://doi.org/10.1038/bjc.2013.337.
29. Zhang C, Wang Y, Zhou Z, Zhang J, Tian Z. Sodium butyrate upregulates expression of NKG2D ligand MICA/B in HeLa and HepG2 cell lines and increases their susceptibility to NK lysis. Cancer immunology, immunotherapy. 2009; 58: 1275-85. https://doi.org/10.1007/s00262-008-0645-8.
30. Wu X, Tao Y, Hou J, Meng X, Shi J. Valproic acid upregulates NKG2D ligand expression through an ERK-dependent mechanism and potentially enhances NK cell-mediated lysis of myeloma. Neoplasia. 2012; 14: 1178-89. https://doi.org/10.1593/neo.12123.
31. Venkataraman GM, Suciu D, Groh V, Boss JM, Spies T. Promoter region architecture and transcriptional regulation of the genes for the MHC class I-related chain A and B ligands of NKG2D. J Immunol. 2007; 178: 961-9. https://doi.org/10.4049/jimmunol.178.2.96.
32. Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature. 2005; 436: 1186-90. https://doi.org/10.1038/nature03884.
33. Wang M, Gu H, Qian H, Zhu W, Zhao C, Zhang X, Tao Y, Zhang L, Xu W. miR-17-5p/20a are important markers for gastric cancer and murine double minute 2 participates in their functional regulation. Eur J Cancer. 2013; 49: 2010-21. https://doi.org/10.1016/j.ejca.2012.12.017.
34. Takakura S, Mitsutake N, Nakashima M, Namba H, Saenko VA, Rogounovitch TI, Nakazawa Y, Hayashi T, Ohtsuru A, Yamashita S. Oncogenic role of miR-17-92 cluster in anaplastic thyroid cancer cells. Cancer Sci. 2008; 99: 1147-54. https://doi.org/10.1111/j.1349-7006.2008.00800.x.
35. Ma Y, Zhang P, Wang F, Zhang H, Yang Y, Shi C, Xia Y, Peng J, Liu W, Yang Z, Qin H. Elevated oncofoetal miR-17-5p expression regulates colorectal cancer progression by repressing its target gene P130. Nat Commun. 2012; 3: 1291. https://doi.org/10.1038/ncomms2276.
36. Gu J, Wang D, Zhang J, Zhu Y, Li Y, Chen H, Shi M, Wang X, Shen B, Deng X, Zhan Q, Wei G, Peng C. GFRalpha2 prompts cell growth and chemoresistance through down-regulating tumor suppressor gene PTEN via Mir-17-5p in pancreatic cancer. Cancer Lett. 2016; 380: 434-41. https://doi.org/10.1016/j.canlet.2016.06.016.
37. Matsubara H, Takeuchi T, Nishikawa E, Yanagisawa K, Hayashita Y, Ebi H, Yamada H, Suzuki M, Nagino M, Nimura Y, Osada H, Takahashi T. Apoptosis induction by antisense oligonucleotides against miR-17-5p and miR-20a in lung cancers overexpressing miR-17-92. Oncogene. 2007; 26: 6099-105. https://doi.org/10.1038/sj.onc.1210425.
38. Petrocca F, Vecchione A, Croce CM. Emerging role of miR-106b-25/miR-17-92 clusters in the control of transforming growth factor beta signaling. Cancer Res. 2008; 68: 8191-4. https://doi.org/10.1158/0008-5472.CAN-08-1768.
39. Molitoris JK, McColl KS, Distelhorst CW. Glucocorticoid-mediated repression of the oncogenic microRNA cluster miR-17~92 contributes to the induction of Bim and initiation of apoptosis. Mol Endocrinol. 2011; 25: 409-20. https://doi.org/10.1210/me.2010-0402.
40. Weng H, Huang H, Dong B, Zhao P, Zhou H, Qu L. Inhibition of miR-17 and miR-20a by oridonin triggers apoptosis and reverses chemoresistance by derepressing BIM-S. Cancer Res. 2014; 74: 4409-19. https://doi.org/10.1158/0008-5472.CAN-13-1748.
41. Li H, Bian C, Liao L, Li J, Zhao RC. miR-17-5p promotes human breast cancer cell migration and invasion through suppression of HBP1. Breast Cancer Res Treat. 2011; 126: 565-75. https://doi.org/10.1007/s10549-010-0954-4.
42. Qu Y, Zhang H, Duan J, Liu R, Deng T, Bai M, Huang D, Li H, Ning T, Zhang L, Wang X, Ge S, Zhou L, et al. MiR-17-5p regulates cell proliferation and migration by targeting transforming growth factor-beta receptor 2 in gastric cancer. Oncotarget. 2016; 7: 33286-96. https://doi.org/10.18632/oncotarget.8946.
43. Zhang J, Xiao Z, Lai D, Sun J, He C, Chu Z, Ye H, Chen S, Wang J. miR-21, miR-17 and miR-19a induced by phosphatase of regenerating liver-3 promote the proliferation and metastasis of colon cancer. Br J Cancer. 2012; 107: 352-9. https://doi.org/10.1038/bjc.2012.251.
44. Dews M, Homayouni A, Yu D, Murphy D, Sevignani C, Wentzel E, Furth EE, Lee WM, Enders GH, Mendell JT, Thomas-Tikhonenko A. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat Genet. 2006; 38: 1060-5. https://doi.org/10.1038/ng1855.
45. Xie J, Liu M, Li Y, Nie Y, Mi Q, Zhao S. Ovarian tumor-associated microRNA-20a decreases natural killer cell cytotoxicity by downregulating MICA/B expression. Cell Mol Immunol. 2014; 11: 495-502. https://doi.org/10.1038/cmi.2014.30.
46. Shen J, Pan J, Du C, Si W, Yao M, Xu L, Zheng H, Xu M, Chen D, Wang S, Fu P, Fan W. Silencing NKG2D ligand-targeting miRNAs enhances natural killer cell-mediated cytotoxicity in breast cancer. Cell Death Dis. 2017; 8: e2740. https://doi.org/10.1038/cddis.2017.158.
47. Petrocca F, Visone R, Onelli MR, Shah MH, Nicoloso MS, de Martino I, Iliopoulos D, Pilozzi E, Liu CG, Negrini M, Cavazzini L, Volinia S, Alder H, et al. E2F1-regulated microRNAs impair TGFbeta-dependent cell-cycle arrest and apoptosis in gastric cancer. Cancer Cell. 2008; 13: 272-86. https://doi.org/10.1016/j.ccr.2008.02.013.
48. Dang CV. MYC on the path to cancer. Cell. 2012; 149: 22-35. https://doi.org/10.1016/j.cell.2012.03.003.
49. Yuan Y, Anbalagan D, Lee LH, Samy RP, Shanmugam MK, Kumar AP, Sethi G, Lobie PE, Lim LH. ANXA1 inhibits miRNA-196a in a negative feedback loop through NF-kB and c-Myc to reduce breast cancer proliferation. Oncotarget. 2016; 7: 27007-20. https://doi.org/10.18632/oncotarget.8875.
50. Boissel N, Rea D, Tieng V, Dulphy N, Brun M, Cayuela JM, Rousselot P, Tamouza R, Le Bouteiller P, Mahon FX, Steinle A, Charron D, Dombret H, et al. BCR/ABL oncogene directly controls MHC class I chain-related molecule A expression in chronic myelogenous leukemia. J Immunol. 2006; 176: 5108-16.
51. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26: 139-40. https://doi.org/10.1093/bioinformatics/btp616.
52. Halfteck GG, Elboim M, Gur C, Achdout H, Ghadially H, Mandelboim O. Enhanced in vivo growth of lymphoma tumors in the absence of the NK-activating receptor NKp46/NCR1. J Immunol. 2009; 182: 2221-30. https://doi.org/10.4049/jimmunol.0801878.
53. Tan L, Wang W, He G, Kuick RD, Gossner G, Kueck AS, Wahl H, Opipari AW, Liu JR. Resveratrol inhibits ovarian tumor growth in an in vivo mouse model. Cancer. 2016; 122: 722-9. https://doi.org/10.1002/cncr.29793.