
INTRODUCTION
Mixed-lineage kinase domain-like (MLKL) protein is a pseudokinase and downstream target of Receptor Interaction Protein kinase 3 (RIP3). RIP1/3 represent central players in necrosis [1–4], which is a caspase-independent programmed cell death [1, 5, 6]. Compared with apoptosis, the underlying mechanisms of necrosis remain poorly understood. Recent data show that MLKL forms complexes with RIP1 and RIP3 to induce necroptosis, a recently characterized type of programmed cell death [7]; moreover, MLKL knockdown prevents TNF-induced necrotic cell death, highlighting a critical role in the necrosis pathway [6–8].
In order to study necrotic signaling pathways, drug screens have been used to identify necrosis inhibitors like necrostatin-1 and Necrosulfonamide (NSA) [2, 7]. Necrostatin-1 is a RIP1 inhibitor that blocks TNF-induced necrosis and reduces infarct size after cerebral ischemia injury [1, 2]. NSA blocks MLKL by binding its N-terminal CC region, with similar results as MLKL knockdown by siRNA [7]. A recent study indicated that low MLKL expression is associated with poor prognosis in ovarian cancer patients [9]. This hints a role for MLKL in necrotic cell death in tumors and other diseases. Specifically, MLKL’s function in ischemic brain injury is unknown. In this study, we examined MLKL expression after cerebral ischemia reperfusion injury using a middle cerebral artery occlusion (MCAO) mouse model. Our results suggested that MLKL is a potential therapeutic target for ischemic stroke treatment.
RESULTS
MLKL expression showed time-dependent changes after I/R injury
Previous studies have indicated that MLKL plays a critical role in the process of RIP1/RIP3 complex-induced necrosis in vitro [7]. However, it is unknown whether MLKL expression is changed after cerebral ischemia injury. Therefore, ischemic tissues at different reperfusion time points (from 3 h to 7 d) were collected for western blot analysis. Compared with sham group, MLKL expression was significantly increased at 12 h after reperfusion, reached the peak at 48 h, and gradually decreased at 72 h and 7 d as shown in Figure 1. The results demonstrated that ischemic brain injury increased MLKL expression at the early stage of cerebral ischemia.
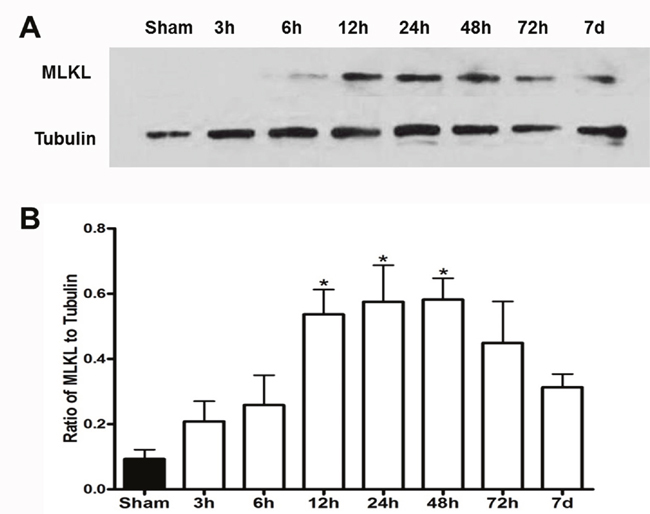
Figure 1: MLKL expression showed time-dependent changes after cerebral I/R injury. (A) Representative bands of MLKL at 3, 6, 12, 24, 48, 72 h and 7 d of reperfusion after 30 min of ischemia. β-tubulin used as a loading control. (B) Quantitative analysis of MLKL protein was performed. *P<0.05, versus sham group, n=4.
MLKL expression was reduced by NSA after cerebral I/R injury
NSA is a specific MLKL inhibitor [7], it is unclear whether NSA affects MLKL expression after cerebral I/R injury. To address this question, we treated mice with NSA (i.c.v.) 30 min before MCAO. Immunofluorescent staining results showed that MLKL expression significantly increased in the I/R group 48 h after reperfusion compared with both sham group and NSA alone group (Figure 2A). In addition, MLKL significantly decreased after NSA treatment compared with I/R group (Figure 2A). Consistent with staining results, Western blot analysis showed that NSA treatment markedly reduced MLKL expression during ischemia injury (P<0.05, Figure 2B, Figure 2C).
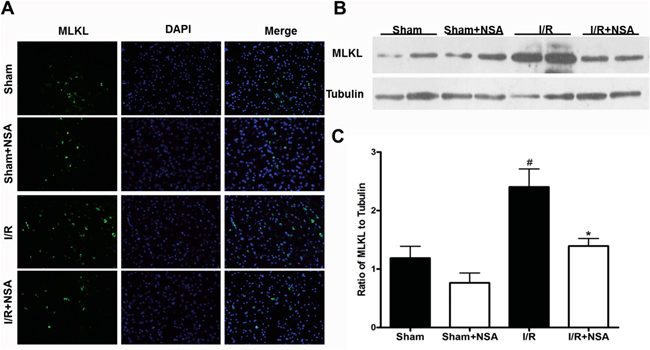
Figure 2: NSA treatment decreased MLKL expression after I/R injury. After 30 min of ischemia and 48 h of reperfusion, ischemic brain tissues were subjected to western blot analysis and brain sections were used for immunostaining. Mice were treated with NSA (1 μmol/kg) or vehicle 30 min before ischemia. (A) The representative photographs show MLKL levels in ischemic brain tissues in four groups. (B) The expression of MLKL was determined by Western blot in four groups. (C) Quantitative analysis of MLKL was performed in four groups. Bars represent mean ± SEM of 4-5 brains. #, P<0.05 versus sham group. *, P<0.05 versus I/R group.
NSA reduced infarct volume after cerebral ischemic injury
After neurological evaluations, brain tissue was sliced and TTC staining was performed. We found clearly large infarct volume after ischemic injury. Consistent with neurological deficit scores, NSA treatment reduced infarct volume compared with I/R group (Figure 3A). This suggested that NSA had protective effects during brain injury. To evaluate the protective efficiency, mice were treated with different doses of NSA (0.1, 0.5, 1 and 2 μmol/kg, i.c.v.). Our results showed that NSA offered protection in a dose-dependent manner (P<0.05, Figure 3B). To further examine clinical significance, we treated mice with NSA at 4 h and 6 h after reperfusion. We found NSA (1 μmol/kg) significantly reduced infarct volume at 4 h post-treatment (P<0.05, Figure 3C). However, no neuroprotection on infarct volume was seen at 6 h after reperfusion (P>0.05, Figure 3B and 3C). This suggests that NSA had a therapeutic window after ischemic injury.
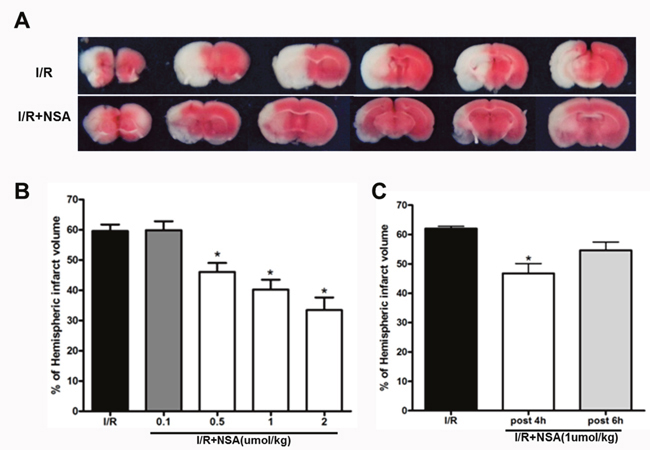
Figure 3: NSA treatment reduced infarct volume after cerebral I/R injury. (A) Representative TTC-stained coronal sections in I/R group and I/R+NSA (1 μmol/kg) group. (B) Infarct volume with different doses of NSA pre-treatment was analyzed. (C) NSA post-treatment (1 μmol/kg) on infarct volume was analyzed. *P<0.05, versus I/R group. Bars represent mean ± SEM of 5-8 brains.
NSA improved neurological functions
Given that MLKL expression increases after I/R injury, we speculated that MLKL inhibition will provide neuroprotection after ischemic injury. Therefore, we treated mice with NSA (2 μmol/kg, i.c.v.) 30 min before ischemia. After 24 h of reperfusion and 75 min of ischemia, neurological deficits were evaluated. As expected, cerebral ischemia injury caused significant neurological deficit scores in the I/R group. Importantly, we found that NSA treatment significantly improved neurological functions (P<0.05, Figure 4). Thus, MLKL seems to play a critical role in neuroprotection during brain injury.
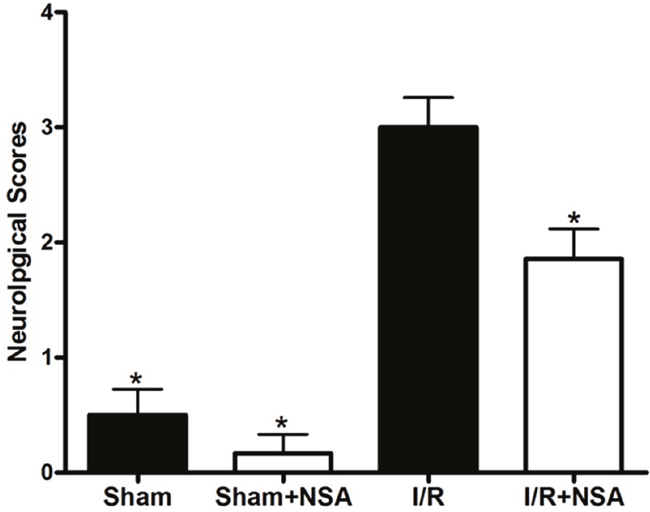
Figure 4: NSA treatment decreased neurological deficits after cerebral I/R injury. Mice were treated with NSA (1 μmol/kg) or vehicle 30 min before ischemia. After 24 h of reperfusion and 75 min of ischemia, neurological deficit scores were evaluated in four groups. *P<0.05, versus I/R group. Bars represent mean ± SEM for samples from 5 to 8 mice in each group.
The degradation of MLKL was mediated by the ubiquitination proteasome pathway
A previous study showed the degradation of MLKL occurs via the ubiquitination proteasome pathway [14]. Therefore, we investigated whether NSA promotes MLKL degradation through the same process. We used tandem-ubiquitin binding entities (TUBEs) to isolate ubiquitinated proteins. As expected, our results show that I/R injury increased ubiquitinated MLKL proteins (Figure 5A and 5B). NSA treatment further increased ubiquitinated MLKL (Figure 5, P<0.05). Most importantly, MLKL mRNA did not change after NSA treatment (P>0.05, Figure 6A and 6B). This clearly demonstrates that MLKL proteins were reduced with no reduction in MLKL transcription. Thus, our data indicated that NSA promoted the degradation of MLKL protein through the ubiquitination proteasome pathway.
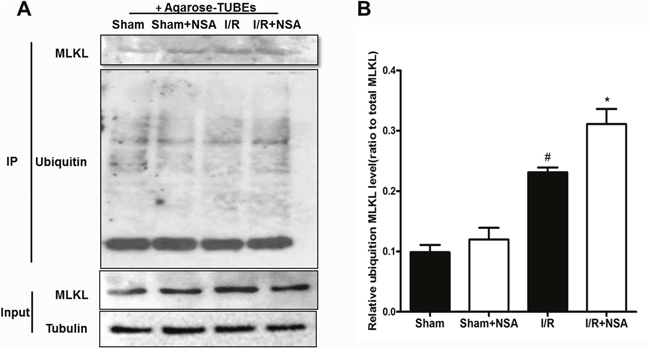
Figure 5: NSA treatment promoted degradation of MLKL via the ubiquitination proteasome pathway. Agrose-TUBE1 was used to isolate ubiquitinated proteins from brain tissues after I/R injury. Anti-MLKL antibody detected ubiquitinated MLKL and MLKL protein input. (A) The representative photographs showed ubiquitinated MLKL levels in ischemic brain tissues in four groups. (B) Quantitative analysis of ubiquitinated MLKL was performed. Bars represent mean ± SEM of 4-5 brains. #, P<0.05 versus sham group. *, P<0.05 versus I/R group.
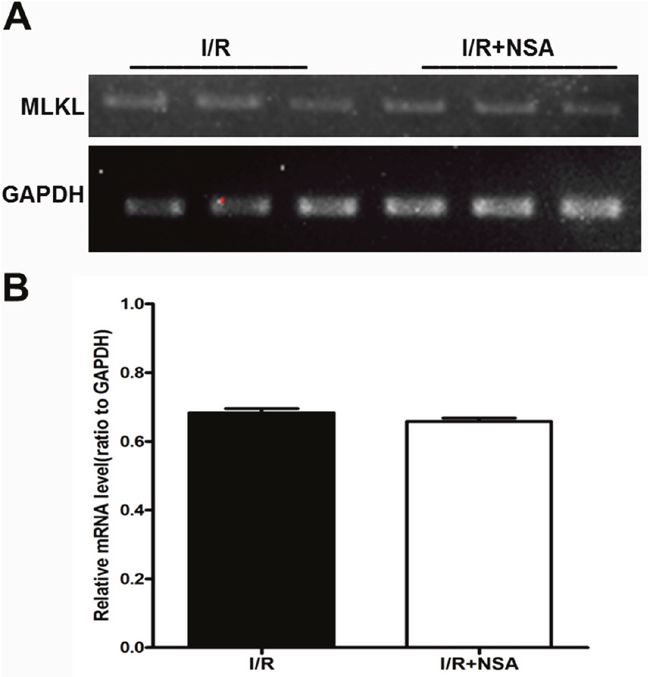
Figure 6: NSA did not affect the mRNA expression of MLKL after cerebral I/R injury. (A) Representative bands of mRNA level of MLKL I/R group and I/R+NSA (1 μmol/kg) group were showed. (B) Quantitative analysis of mRNA level of MLKL in two groups was performed. Bars represent mean ± SEM of 4-5 brains.
Reduced MLKL associated with increased cleaved PARP-1 level after I/R injury
MLKL is considered a critical player in the necrotic cell death pathway [6, 7, 10], but it is unclear whether MLKL affects the apoptotic cell death pathway. Because Poly(ADP-ribose) polymerase-1 (PARP-1) is a substrate of caspase-3, cleaved PARP-1 is considered a marker of caspase-3-dependent apoptosis [11]. Therefore, we examined the effect of MLKL reduction on cleaved PARP-1 after I/R injury.
We found a robust increase in cleaved PARP-1 at 24 h after I/R injury in the I/R group (P<0.05; Figure 7A). Interestingly, cleaved PARP-1 levels were even higher after NSA treatment (Figure 7A; P<0.05, Figure 7B). These data demonstrated the reduction of MLKL by NSA treatment blocks necrosis, but promotes apoptotic cell death.
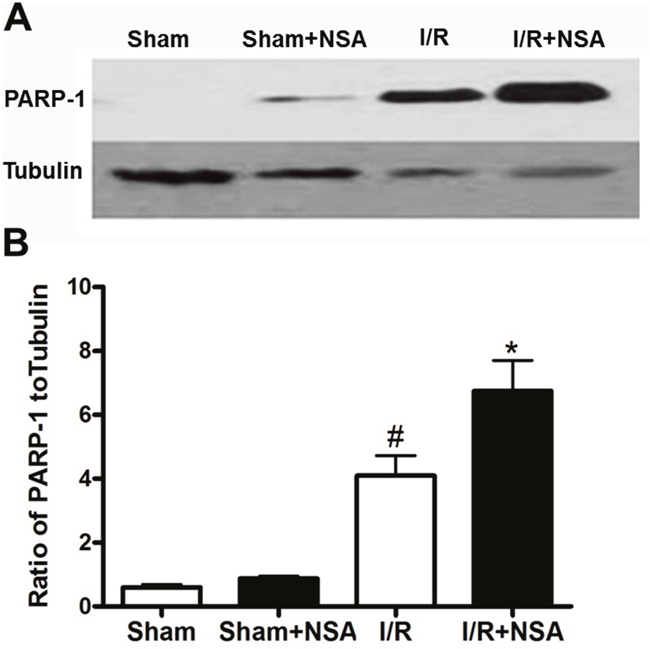
Figure 7: NSA treatment increased cleaved PARP-1 level after I/R injury. Mice were treated with NSA (1 μmol/kg) or vehicle and the ischemic brain tissues were collected to determine cleaved PARP-1 level. (A) Representative bands of cleaved PARP-1 in four groups were showed. (B) Quantitative analysis of cleaved PARP-1 in four groups was performed. Bars represent mean ± SEM of 4-5 brains. #, P<0.05 versus sham group. *, P<0.05 versus I/R group.
DISCUSSION
We demonstrate here for the first time that MLKL expression increased in ischemic brains. We also show that NSA significantly inhibits MLKL expression. In addition, reduced MLKL is associated with reduced infarct volume and improved neurological deficits after ischemic injury. Our findings suggest NSA reduced MLKL protein levels through the ubiquitination proteasome pathway and did not inhibit MLKL mRNA transcription. Interestingly, NSA also increased cleaved PARP-1, an apoptosis marker. Together, our findings suggest that MLKL protein promotes ischemia-induced cell death through a necroptotic pathway, but not an apoptotic pathway. Thus, MLKL may be an interesting therapeutic target for brain injury.
MLKL protein is a downstream target of RIP3 and is considered to be involved in ROS generation [12]. It forms a complex with RIP1 and RIP3 to induce necroptosis [7]. Kim et al suggest palmitate-induced, RIP1/RIP3/MLKL-dependent cell death might not only occur via mitochondrial ROS generation but also could be induced through other potential mechanisms such as pore forming in the plasma membrane [13]. It’s clear that RIPK3 and MLKL play critical roles in necroptosis [14, 15]. Chen et al reported that knockdown of RIPK3 or MLKL increased cell survival [16]. From these data we inferred that MLKL plays a key role in ischemic brain injury. Our experimental results support our hypothesis, showing that MLKL expression increases significantly after I/R injury. Both pre-treatment and post-treatment with NSA significantly reduced infarct volume and improved neurological deficits after ischemia. NSA interacts with human MLKL through the N-terminal region, but the region is absent in mouse MLKL [7]. Therefore, NSA should have no protection on MLKL-induced cell death in mice. However, our findings suggest that promoting degradation of MLKL is the key to the neuroprotection. We demonstrated that NSA decreased MLKL levels via the ubiquitination proteasome pathway which did not inhibit RNA transcription. The details on how NSA promotes degradation warrants further investigation.
Nec-1 blocks translocation of apoptotic inducing factor (AIF) from the mitochondria to the nucleus and suppresses the activation of PARP-1 [5, 17]. In addition, Nec-1 reverses traumatic brain injury-induced Caspase-3 activation and Bcl-2 down-regulation [18]. Nec-1 may influence different proteins involved in necrosis and apoptosis. For NSA, we speculated it would have an indirect effect on apoptosis. Due to the existence of multiple cell death pathways during ischemia, we examined the effect of NSA on cleaved PARP-1, an apoptotic marker. Surprisingly, NSA increased cleaved PARP-1, suggesting a shift from necrosis to apoptosis. Our data indicate that NSA triggered protection of necrotic cells and promoted apoptosis after I/R injury. NSA inhibits MLKL-induced necrosis and in turn drives cells to use apoptosis as an alternative cell death mechanism. The phenomenon is similar with caspase inhibition blocking apoptotic pathways that induces necrosis via TNF-α stimulus [1, 19]. This observation underscores the well-known phenomena that cells alternate between cell death systems under various environments [20].
In summary, our data demonstrate that MLKL expression was significantly increased after I/R injury. Inhibition of MLKL expression markedly reduced infarct volume and improved neurological deficits. Thus, MLKL may be a potential therapeutic target for stroke. Promoting the degradation of MLKL may represent a novel avenue for reducing necrotic cell death after ischemic brain injury.
MATERIALS AND METHODS
Animals
Male ICR mice weighing about 25g were purchased from SLAC Company (Shanghai, China). All mouse procedures were approved by the University Committee on Animal Care of Soochow University and carried out in strict accordance with the guidelines of Animal Use and Care of the National Institutes of Health. All surgery was performed under chloral hydrate anesthesia, and all efforts were made to minimize suffering.
Middle cerebral artery occlusion (MCAO) model
MCAO was performed according to the method described previously [21]. Briefly, mice were anesthetized intraperitoneally (i.p.) with 4% chloral hydrate, (0.1 ml/10g body weight). We dissected the right common carotid artery (CCA), the right external carotid artery (ECA) and the internal carotid artery (ICA) through a midline neck incision. Blood flow through the CCA was then blocked via a twisted silk suture. A 6-0 nylon monofilament coated with silicon resin was introduced into the right ECA and advanced 9-11 mm. Reperfusion was achieved by withdrawing the suture after 30 min or 75 min of MCAO. Body temperature was maintained at 36.5-37.5°C by using a heating blanket throughout the procedure from the start of the surgery until mice recovered from anesthesia.
Intracerebroventricular administration of NSA
Necrosulfonamide (NSA, Toronto, Ontario, Canada) was dissolved in dimethylsulfoxide (DMSO) for stock concentration of 10 mM. NSA was diluted to different concentrations with saline. NSA was injected via a hole (0.5 mm posterior and 1.0 mm lateral to the Bregma) by a needle with a brain stereotaxic instrument.
2, 3, 5-Triphenyltetrazolium chloride (TTC) staining
After neurological deficit evaluation, brains were removed quickly from the skull, cut into 6 slices and incubated with 0.2% TTC (Sigma-Aldrich, USA) at 37°C for 30 min, and fixed in 4% paraformaldehyde overnight. AlphaEase Image Analysis Software V3.1.2 (Alpha Innotech Corp., San Leandro, CA, USA) was used to analyze the infarct area in each slice. The percentage of hemispheric infarction volume was calculated as described in our previous study [22, 23].
Neurological deficit evaluation
Neurological deficits were evaluated after 75 min of ischemia and 24 h of reperfusion based on a 5-point scale system described in our previous study [24].
Western blot analysis
After 30 min of MCAO and 48 h of reperfusion, the whole right hemisphere tissue was collected and stored at −80°C. The 30 min time point of ischemia was chosen because minimal cell death was detected by TTC staining. Brain tissues were solubilized in lysis buffer and sonicated on ice. The tissue samples were then centrifuged and the supernatants used for Western blot analysis. The same amount of total proteins (approximately 30-40μg) were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and then transferred to nitrocellulose membranes. After blocking with 5% dry milk in phosphate buffered saline/0.1% Tween 20 (PBST) for 2 h, blots were incubated with primary antibodies in PBST overnight at 4°C. The blots were washed with PBST and incubated with horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature [25]. After washing three times, the blots were developed with ECL chemiluminescence and the immunoreactive bands captured on autoradiographic films. The densitometry of the bands was analyzed with Alpha Ease Image Analysis Software V3.1.2. Anti-MLKL goat antibody (Santa Cruz Biotechnology, USA), anti-PARP-1 rabbit antibody (ImmunoWay Biotechnology, USA), anti-ubiquitin rabbit antibody (Millipore Corporation, USA) and anti-β-tubulin mouse antibody (Sigma-Aldrich, USA) were used.
Fluorescent immunostaining
Frozen brain sections were mounted on glass slides and blocked with buffer containing 10% goat serum, 1% bovine serum albumin (BSA), and 0.3% Triton X-100 in PBS for 1 h. The sections were then incubated with MLKL antibody in the blocking buffer overnight at 4°C. After washing with PBS for 3 times, the sections were incubated with anti-goat fluorescent secondary antibodies (Jackson ImmunoResesrch Laboratories, PA, USA) in blocking buffer for 1 h [26]. Each group contained 3 mice; every mouse had 6 slides processed; brain slices with same position from four groups were selected. All slides were examined under a fluorescence microscope (AXIO SCOPE A1, ZEISS, Germany).
Tandem ubiquitin binding entity (TUBE) purification
Brain samples were homogenized in lysis buffer, centrifuged at 160,000g for 30 min at 4°C and then the supernatants were collected for TUBE purification. Samples were adjusted to 500 μl at a concentration of 1 μg/μl and pre-absorbed by agarose-TUBE1 (LifeSensors Inc, USA) for 12 h at 4 °C with gentle shaking. Samples were spun for 5 min at 1,000×g at 4°C. Beads were further washed with lysis buffer for 3 times. The bound proteins were eluted with sample buffer containing 100 mM Tis-HCl (pH 6.8), 200 mM dithiothreitol, 4% sodium dodecyl sulfate, 0.2% bromochlorophenol blue, and 20% glycerol. The samples were heated at 96°C for 10 min and were collected for Western blot analysis.
Total RNA extraction and RT-PCR
Brain tissues were collected and solubilized by sonication for 3 times at 5 sec each on ice in 1 ml TRIpure [27]; then 200μl of chloroform was added to the tubes. After 15 sec of the sonication and 15 min of the incubation on ice, the mixture was centrifuged at 14,000g for 15 min at 4°C. The upper solutions were collected in new tubes and mixed with equal amount isopropanol. The tubes were blended for 15 sec, then the mixture was centrifuged at 14,000g for 10 min at 4°C and isopropanol was decanted. Ice-cold 75% ethanol was added to the RNA pellet for two gentle washes. After centrifuging for 10 min, ethanol was removed and RNA pellets were dried at room temperature for 5 to 10 min. Finally, 25μl DEPC water was added to the tubes [28]. cDNA Reverse transcription was performed using the Transcriptor First Strand cDNA Synthesis Kit (Roche, Germany), and cDNA was used for PCR. GAPDH was used as an endogenous control. Primers were: MLKL (5′-GCTGCTGCTTCAGGTTTATC-3′, 5′-CGCAAGATGTTGGGATC-3′); GAPDH (5′-CATGGCCTTCCGTGTTCCTA-3′, 5′-CTTCACCACCTTCTTGATGTCATC-3′).
Statistical analysis
All data are expressed as mean ± SEM. One-way analysis of variance followed by Tukey multiple-comparison test were used to compare differences among groups. P<0.05 was considered statistically significant.
Author contributions
Zhong Zhao conceived and supervised the project. Zhigang Miao, Yanlong Zhou and Beiqun Zhou wrote the manuscript and generated the final figures. All experimental procedures and data analysis were carried out by Yanlong Zhou, Beiqun Zhou and Hui Tu. Yan Tang and Chen Xu contribute to MCAO models. Yanbo Chen contributed to data analysis.
ACKNOWLEDGMENTS
This work was supported by the grant from the National Natural Science Foundation of China (81601154), the grants from Jiangsu Province Health and Family Planning Commission (H201651) and Suzhou Science and Technology Bureau (Applied Basic Research Programs, SYS201650), and the Priority Academic Program Development of Jiangsu Higher Education Institutions of China.
CONFLICTS OF INTEREST
The authors disclose no potential conflicts of interest.
REFERENCES
1. Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005; 1:112-119.
2. Degterev A, Hitomi J, Germscheid M, Ch'en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008; 4:313-321.
3. Wang X, Jiang W, Yan Y, Gong T, Han J, Tian Z, Zhou R. RNA viruses promote activation of the NLRP3 inflammasome through a RIP1-RIP3-DRP1 signaling pathway. Nat Immunol. 2014; 15:1126-1133.
4. Chen WW, Yu H, Fan HB, Zhang CC, Zhang M, Zhang C, Cheng Y, Kong J, Liu CF, Geng D, Xu X. RIP1 mediates the protection of geldanamycin on neuronal injury induced by oxygen-glucose deprivation combined with zVAD in primary cortical neurons. J Neurochem. 2012; 120:70-77.
5. Xu X, Chua CC, Kong J, Kostrzewa RM, Kumaraguru U, Hamdy RC, Chua BH. Necrostatin-1 protects against glutamate-induced glutathione depletion and caspase-independent cell death in HT-22 cells. J Neurochem. 2007; 103:2004-2014.
6. Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J, Liu ZG. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Nat Acad Sci U S A. 2012; 109:5322-5327.
7. Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, Wang X. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012; 148:213-227.
8. Wu J, Huang Z, Ren J, Zhang Z, He P, Li Y, Ma J, Chen W, Zhang Y, Zhou X, Yang Z, Wu SQ, Chen L, Han J. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res. 2013; 23:994-1006.
9. He L, Peng K, Liu Y, Xiong J, Zhu FF. Low expression of mixed lineage kinase domain-like protein is associated with poor prognosis in ovarian cancer patients. Onco Targets Ther. 2013; 6:1539-1543.
10. Zhang J, Yang Y, He W, Sun L. Necrosome core machinery: MLKL. Cell Mol Life Sci. 2016; 73:2153-2163.
11. Zhang Y, Zhang X, Park TS, Gidday JM. Cerebral endothelial cell apoptosis after ischemia-reperfusion: role of PARP activation and AIF translocation. J Cereb Blood Flow Metab. 2005; 25:868-877.
12. Wang ZG, Jiang H, Chen S, Du FH, Wang XD. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell. 2012; 148:228-243.
13. Kim SK, Yun M, Seo G, Lee JY, Lee SB. Palmitate induces RIP1/RIP3-dependent necrosis via MLKL-mediated pore formation in the plasma membrane of RAW 264.7 cells. Biochem Biophys Res Commun. 2017; 482:359-365.
14. Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010; 11:700-714.
15. Moriwaki K, Bertin J, Gough PJ, Orlowski GM, Chan FK. Differential roles of RIPK1 and RIPK3 in TNF-induced necroptosis and chemotherapeutic agent-induced cell death. Cell Death Dis. 2015; 6:e1636.
16. Chen S, Lv X, Hu B, Shao Z, Wang B, Ma K, Lin H, Cui M. RIPK1/RIPK3/MLKL-mediated necroptosis contributes to compression-induced rat nucleus pulposus cells death. Apoptosis. 2017; 22:626-638.
17. Xu X, Chua CC, Zhang M, Geng D, Liu CF, Hamdy RC, Chua BH. The role of PARP activation in glutamate-induced necroptosis in HT-22 cells. Brain Res. 2010; 1343:206-212.
18. Wang YQ, Wang L, Zhang MY, Wang T, Bao HJ, Liu WL, Dai DK, Zhang L, Chang P, Dong WW, Chen XP, Tao LY. Necrostatin-1 suppresses autophagy and apoptosis in mice traumatic brain injury model. Neurochem Res. 2012; 37:1849-1858.
19. Behrouz R. Re-exploring tumor necrosis factor alpha as a target for therapy in intracerebral hemorrhage. Transl Stroke Res. 2016; 7:93-96.
20. Duprez L, Bertrand MJ, Vanden Berghe T, Dondelinger Y, Festjens N, Vandenabeele P. Intermediate domain of receptor-interacting protein kinase 1 (RIPK1) determines switch between necroptosis and RIPK1 kinase-dependent apoptosis. J Biol Chem. 2012; 287:14863-14872.
21. Zhang G, Chen L, Yang L, Hua X, Zhou B, Miao Z, Li J, Hu H, Namaka M, Kong J, Xu X. Combined use of spatial restraint stress and middle cerebral artery occlusion is a novel model of post-stroke depression in mice. Sci Rep. 2015; 5:16751.
22. Yu H, Zhang ZL, Chen J, Pei A, Hua F, Qian X, He J, Liu CF, Xu X. Carvacrol, a food-additive, provides neuroprotection on focal cerebral ischemia/reperfusion injury in mice. PLoS One. 2012; 7:e33584.
23. Liu NW, Ke CC, Zhao Y, Chen YA, Chan KC, Tan DT, Lee JS, Chen YY, Hsu TW, Hsieh YJ, Chang CW, Yang BH, Huang WS, Liu RS. Evolutional characterization of photochemically induced stroke in rats: a multimodality imaging and molecular biological study. Transl Stroke Res. 2017; 8:244-256.
24. Xu X, Chua KW, Chua CC, Liu CF, Hamdy RC, Chua BH. Synergistic protective effects of humanin and necrostatin-1 on hypoxia and ischemia/reperfusion injury. Brain Res. 2010; 1355:189-194.
25. Jiang B, Li L, Chen Q, Tao Y, Yang L, Zhang B, Zhang JH, Feng H, Chen Z, Tang J, Zhu G. Role of glibenclamide in brain injury after intracerebral hemorrhage. Transl Stroke Res. 2017; 8:183-193.
26. Li AL, Zhang JD, Xie W, Strong JA, Zhang JM. Inflammatory changes in paravertebral sympathetic ganglia in two rat pain models. Neurosci Bull. 2017. https://doi.org/10.1007.
27. Yen TL, Chen RJ, Jayakumar T, Lu WJ, Hsieh CY, Hsu MJ, Yang CH, Chang CC, Lin YK, Lin KH, Sheu JR. Andrographolide stimulates p38 mitogen-activated protein kinase-nuclear factor erythroid-2-related factor 2-heme oxygenase 1 signaling in primary cerebral endothelial cells for definite protection against ischemic stroke in rats. Transl Res. 2016; 170:57-72.
28. Miao X, Huang Y, Liu TT, Guo R, Wang B, Wang XL, Chen LH, Zhou Y, Ji RR, Liu T. TNF-alpha/TNFR1 signaling is required for the full expression of acute and chronic itch in mice via peripheral and central mechanisms. Neurosci Bull. 2017. https://doi.org/10.1007.