
Introduction
The cell cycle is defined by a series of events that lead to a production of two identical daughter cells [12, 13]. It is divided into distinct temporal phases, Gap 1 (G1), Gap 0 (G0), Synthesis (S), Gap 2 (G2) and Mitosis (M) [1, 13]. Cyclins include cyclin A, B, D and E. D-type cyclins are important in regulating cell cycle progression and a variety of tumorigenic processes [1]. Cell cycle progression is mainly regulated by cyclins. They specifically interact with and activate serine/threonine kinases named cyclin-dependent kinases (Cdks). Therefore, the cyclin-Cdk complexes govern a linear progression of events that lead cells from a resting state, G0, through to G1, S, G2 and M phases [14].
Cyclin D1 promotes the G1/S-phase transition by binding and activating Cdk4 and Cdk6 [15]. Strong evidence implicates cyclin D1 overexpression as a collaborative oncogene in many types of cancers. Targeted expression of cyclin D1 to the mammary gland is sufficient for the induction of mammary tumorigenesis [16, 17]. Curiously, forced expression of a cyclin D1 mutant, defective in cdk binding, is also sufficient for the induction of mammary tumors with similar latency [17, 18] suggesting additional cdk-independent functions, such as the induction of chromosomal instability [19] may contribute to the pro-oncogenic phenotype [20, 21].
Cyclin-cdk complexes phosphorylate many substrates (pRB, NRF1, GCN5) [15, 22, 23] and cyclin D1 conveys cdk-independent functions which together regulate A) gene transcription factor interactions, such as Androgen Receptor (AR) [24], Estrogen Receptor alpha (ERα) [25], Peroxisome Proliferator-Activated Receptor gamma (PPARɣ), MyoD, B-Myb, DMP1 [26-29] and retinoblastoma (Rb) B) cellular metabolism through Nuclear Respiratory Factor 1 (NRF1) [22]; c) fat cell differentiation, through C/EBPβ and PPARɣ [30, 31] and d) cellular migration [32, 33]. Cyclin D1 regulates nuclear hormone receptors including the AR [24] and ERα [34, 35] both in tissue culture , and in vivo [25, 36]. Cyclin D1 enhances transcription of estrogen responsive genes in a kinase independent activity. Cyclin D1 selectively inhibits ligand-dependent AR function in several cell types, including breast cancer, bladder cancer, and androgen-independent prostate adenocarcinoma cell lines. Cyclin D1 forms a specific complex with the AR, requiring the C terminus of cyclin D1 [15].
Over the last three decades several different studies have assessed the role of cyclin D1 in DNA damage repair. Genomic DNA is continually subjected to insults by damaging ionizing radiation, chemical carcinogens, and reactive oxygen species (ROS) generated by cellular metabolism. Defects in the DDR can lead to genomic instability and cancer [10].
Given the key role for DNA Damage Response (DDR) and subsequent repair process in maintaining genomic integrity understanding the role of cyclin D1 in DNA damage and repair is important and is reviewed herein. Subsequent studies have focused on the role of cyclin D1 in the DNA damage repair associated with hormonal signaling. In this regard both estrogen and androgen signaling have been linked to DNA damage, and defective DNA repair has been linked to malignancies of both breast and prostate cancer. Herein, we summarize the latest findings on the role of cyclin D1 in mediating hormone-induced DDR.
The DNA Damage response in cancer
Thousands of DNA lesions occur per hour in each cell of the human body. In general, the DNA damage can be considered as either endogenous or exogenous in origin. The endogenous damages can be generated by reactive oxygen species (ROS) produced during normal cell metabolism or accumulation of replication errors during the DNA replication process. On the other hand, external agents, such as ultraviolet radiation, x-rays, gamma (γ) rays, mutagenic chemicals, plant toxins and viruses are responsible for exogenous damage. DNA damage can have deleterious effects, as it interferes with DNA replication and transcription, causing mutations and chromosomal aberrations. Genome integrity is preserved by the DNA damage repair machinery, which counteracts the adverse consequence of DNA lesions and prevents their transmission to daughter cells [37].
The DNA damage response (DDR) is a collective term for all intra- and inter-cellular signaling events and enzyme activities that result from the induction and detection of DNA damage. These include events that lead to cell-cycle arrest, regulation of DNA replication, and the repair or bypass of DNA damage [38]. DNA damage can induce a DNA break in a single strand (SSBs) or a double strand (DSBs). SSBs form upon oxidative attack of deoxyribose by cell metabolism products or through exposure to agents such as H2O2, ionizing radiation, and radiomimetic drugs. They also arise as intermediates during excision repair of damaged bases, and upon inhibition of topoisomerase I. In most eukaryotes, SSBs are initially detected by PARP-1, whose binding to DNA breaks triggers poly-ADP-ribosylation of nuclear proteins [39]. DSBs are generated in response to ionizing radiation or radiomimetic drugs by free radical attack of deoxyribose, and also arise in cells treated with topoisomerase II inhibitors. DSBs can be generated by other factors, including mechanical stress of chromosomes, DNA polymerase encountering a SSB and other DNA lesions [40]. This form of DNA lesion is most hazardous because it can lead to genomic rearrangement.
There are two major forms of repair when dealing with DNA double-strand breaks (DSBs): Homologous recombination repair (HRR) and non-homologous end-joining (NHEJ) pathways [41]. HRR is a relatively accurate and efficient repair pathway but depends upon the presence of an undamaged sister chromatid [42], while NHEJ pathways (Classical-NHEJ and alternative-NHEJ, C-NHEJ and alt-NHEJ respectively) are not dependent on the presence of a sister allele and, while still effective, are less accurate, potentially introducing DNA rearrangements [43, 44]. BRCA1 participates is homologous recombination, where the repair proteins utilize a template of the identical homologous intact sequence from a sister chromatid, from a homologous chromosome, or from the same chromosome, depending on cell cycle phase. BRCA1 also participates in termed mismatch repair. The BRCA1 protein interacts with RAD51 during repair of DNA double-strand breaks and co-localizes with γ-H2AX (histone H2AX phosphorylated on Serine-139) in DNA double-strand break repair foci. The BRCA2 protein, which has a function similar to that of BRCA1, also interacts with the RAD51 protein. RAD51 is a 339-amino acid protein that plays a major role in homologous recombination of DNA during double strand break repair. In this process, an ATP-dependent DNA strand exchange takes place in which a template strand invades base-paired strands of homologous DNA molecules.
It has been reported that the major transducers involved in DDR are ataxia-telangiectasia mutated (ATM) and ATM- and Rad3-Related (ATR). ATM is activated by DSBs and triggers a cascade of events on the chromatin flanking DSBs. Upon DNA damage, ATM is activated by phosphorylation of Serine 1981, which causes dissociation of ATM homodimer [45]. Activated ATM is able to phosphorylate substrates such as p53 and H2AX. The histone variant H2AX is phosphorylated at Serine 139 (γH2AX). Mdc1 directly binds to γH2AX and is required for complete γH2AX foci formation [46-48]. Foci formation facilitates the recruitment of additional DNA repair components. In contrast to ATM, ATR responds to a broad spectrum of DNA damages, including DSBs and a variety of DNA lesions that interfere with replication. ATR is essential for the survival of proliferating cells and its deletion leads to early embryonic lethality in mouse and lethality of human cells [49-52].
E2F1 is induced in response to various DNA-damaging agents, including ionizing radiation, UV radiation, and a number of chemotherapeutic drugs [53]. Moreover, as shown by ultraviolet (UV) laser micro-irradiation and immunofluorescence staining, E2F1 accumulates at sites of DNA damage [54]. The mechanism by which E2F1 translocates to DSB lesions has been relatively well characterized showing that DNA topoisomerase II-beta protein (TopBP1) interacts with the E2F1 N-terminal domain and thereby enables E2F1 recruitment to DNA lesions [55].
It has also been reported that the action of DNA repair factors is tightly coordinated with cell cycle progression. For example, cells in G1 do not have an available sister chromatid and will therefore be dependent upon NHEJ pathways for the repair of DSBs. In addition, there are important differences in the primary role of checkpoints during stages of the cell cycle and in the participating DDR factors. For example, the G1/S checkpoint allows repair of DNA damage prior to the start of DNA replication, in order to remove obstacles to DNA synthesis, and key DDR factors regulating this checkpoint include ATM, CHK2, and p53. The intra-S phase checkpoint proteins ATR, CHK1, DNAPK, and WEE1 can delay replication origin firing to provide time to deal with any unrepaired DNA damage, thus preventing under-replicated DNA regions being conveyed beyond S-phase. The activities of the G2/M checkpoint proteins including CHK1, MYT1, and WEE1 lead to an increase in phosphorylated Cdk1, thereby maintaining an inactive state and delaying mitotic entry. The G2/M checkpoint represents the last major opportunity for preventing the passage of DNA lesions into mitosis, where unrepaired DSBs and under-replicated DNA may result in mitotic catastrophe and cell death [56]. This is of major importance since the DNA lesions promote genomic instability [57]. The cells possess a specific response mechanisms to avoid both accumulation of DNA damage and maintenance of genomic stability [58].
In contrast to normal cells, a universal hallmark of cancer cells is genomic instability due to defects in the mechanisms responsible for repair of DNA damage [57]. The pro-survival activities of DDR are realigned and misappropriated during cancer progression by activation of oncogenes such as Ras, Akt and Myc, which enhance replicative stress [57]. Replicative stress is defined as the harmful effect of DNA that is only partially replicated because of slow progression of the replication forks, which can be caused by oncogene-induced hyper-replication that activates multiple origins of replication per S phase, by nucleotide pool imbalance or by DNA damage [59]. This leads to increased DNA damage, which eventually increases genomic instability to a level that is incompatible with cell survival. However, induction of replicative stress by hyperactive growth factor signaling and oncogene signaling in established cancer can lead to compensatory upregulation of DNA repair pathways providing a new opportunity in cancer therapy [57, 59].
Historically, treatment of cancer by radiotherapy and DNA-damaging chemotherapy are based on the strategy of inducing DNA damage, promoting chromosomal catastrophe of transformed cells and ultimately the induction of cancer cell death. However, these approaches lead to significant collateral damage to normal tissue and unwanted side effects. Targeted therapy based on inhibiting DDR in cancers offers the potential for a greater therapeutic window by tailoring treatment to patients with tumors lacking specific DDR mechanisms [38].
DNA damage response and cyclin D1
In early studies cyclin D1 was shown to restrain the DNA repair process [60]. During repair DNA synthesis, subsequent to UV-induced DNA damage, G1 cells readily lost their cyclin D1 while proliferating cell nuclear antigen (PCNA) tightly associated with nuclear structures. Microinjection of cyclin D1 antisense accelerated DNA repair, whereas overexpression of cyclin D1 prevented DNA repair and the relocation of PCNA after DNA damage The abundance of cyclin D1 has been shown to play a role in cell-type dependent radiation-induced sensitivity. Cyclin D1−/− MEFs have enhanced apoptosis evoked by γ-irradiation [61] and cyclin D1 expression also inhibited UV induced apoptosis [62]. In contrast breast cancer cell lines overexpressing cyclin D1 showed enhanced apoptosis in response to γ irradiation [63, 64] suggesting cell type specific differences governing cyclin D1 mediated apoptosis. However, DNA damage induced rapid degradation of cyclin D1 [61]. These studies were consistent with a model in which cyclin D1 was considered to be a collaborative oncogene, and that restraining DNA repair would be consistent with such a function.
Over the subsequent decade however it has become apparent that distinct pools of cyclin D1 exist within the cell, with distinct binding partners, that govern distinct functions [20, 65, 66]. Thus, the total abundance of cellular protein may not reflect an important role for cyclin D1 in a local site, such as chromatin with a transcription factor, or at focal contacts [67]. In 2005, cyclin D1 was shown to bind to BRCA1, both in subcellular foci and in the context of transcriptional regulatory complexes in chromatin (Figure 1) [6]. As noted above, BRCA1 functions as a scaffold protein organizing DNA damage sensors, thereby coordinating the repair of damaged DNA [68-70] The BRCA1 gene product, a nuclear polypeptide of 220 kDa, co-localizes in nuclear foci with RAD51, BRCA2, and RAD50. Protein complexes that regulate repair of DNA double-strand breaks, including hRAD50-hMre11-NBS1, MSH2, MSH6, and proliferating cell nuclear antigen, functionally interact with BRCA1 [71].
In 2010, cyclin D1 was shown to enhance the DNA damage response, and the mechanisms involved were characterized [7]. RAD51 is a key protein governing homologous DNA repair. RAD51 is involved in the search for homology and strand pairing stages of the process. This protein can interact with the ssDNA-binding proteins RPA, BRCA2, PALB2 and RAD52 a recombinase that drives the homologous recombination process. The studies by Li et al showed that the abundance of cyclin D1 determined the DNA damage response, as characterized by induction of γH2AX phosphorylation, the assembly of RAD51 DNA repair foci, and specific recruitment of DNA repair factors to chromatin. Secondly, Li showed, that cyclin D1 bound to RAD51. Thirdly it was shown that cyclin D1 recruited RAD51 and DNA repair factors in response to DNA damage, thereby enhancing γH2AX phosphorylation. Fourthly, these studies showed that cyclin D1, like DNA repair factors, elicited the DDR when stably associated with chromatin [7]. Cyclin D1 recruited DNA repair factors in a cyclin D1 isoform specific, occurring with the cyclin D1a but not the cyclin D1b isoform. As these two isoforms vary primarily in the carboxyl terminus these studies suggested a role for the cyclin D1a isoform C terminus in these events. Fifth, the enhancement of DNA damage induced γH2AX phosphorylation by cyclin D1 involved specific kinases (DNA-PK, JNK and Casein Kinase 2). These studies demonstrated the importance of cyclin D1a in the DDR using distinct activators of the DDR, including γ irradiation, double stranded-DNA damage inducing agents (doxorubicin, 5-FU) and the targeting of single DNA repair factors to chromatin.
In 2011, a proteomic screen for cyclin D1 protein binding partners by Jirawatnotai et al, identified RAD51 and many other binding proteins. These studies extended our understanding of cyclin D1 in the DDR by showing the recruitment of cyclin D1 to sites of DNA damage was BRCA2-dependent [8]. Furthermore, these studies showed that cyclin D1 regulated not only the DNA damage response and the creation of DNA damage repair foci, but showed that cyclin D1 enhanced DNA repair [8]. Given that cyclin D1 is thought to be a collaborative oncogene, and the DNA repair defects often are associated with predisposition to cancer, it was considered “surprising, that this protein also mediates the repair of damaged DNA, a mechanism that commonly prevents cancer” [72].
These functional studies [7, 8] were however consistent with several gene expression studies in which cyclin D1 expression was shown to induce DDR gene expression. Genome wide microarray analysis demonstrated cyclin D1 induced expression of genes involved in DNA replication and DNA damage checkpoints suggesting a role for cyclin D1 in the DDR. Cyclin D1a induced the mRNA expression of Minichromosome maintenance (MCM)3, MCM4, replication factor C (activator 1) 4 (Rfc4), cell division cycle 6 homology (Cdc6), cell division cell associated 7(Cdc7) and H2Afx (H2A Histone family member x) in MEFs [73]. Mammary gland targeted cyclin D1 inducible antisense transgenic demonstrated endogenous cyclin D1 maintained expression of MCM2, Rfc2, Cdc20, Rad51 and histone 1 in the mammary epithelium [74]. In subsequent studies forced expression of cyclin D1 induced expression of genes regulating the DDR, including MCM3 and Cdc7 [75]. Expression were increased in mammary tumors derived from mammary epithelial cell targeted cyclin D1a transgenic mice. Furthermore, forced expression of a degradation defective cyclin D1a mutant induced the DDR in murine lymphoid tissue [76]. Thus, prior indirect evidence implicated cyclin D1 in enhancing gene expression governing the DDR in a number of different cell types
Hormonally-mediated DNA damage response
Steroid hormones are systemically circulating small molecules that elicit autocrine, paracrine, and endocrine functions in physiology and pathology, most often through the binding to, and regulation of cognate nuclear receptors (NR) [77]. Steroid hormones influence distinct and important cancer-associated phenotypes, including, but not limited to cellular proliferation, apoptosis, migration, and invasion. Steroid-induced biological outcomes occur in a context-dependent manner in multiple malignancies, including breast (BrCa) and prostate (PCa) cancers. As such, therapy for selected hormone-dependent cancers focuses on diminishing availability of ligand or direct antagonism of NRs.
While the biological implications remain uncertain, steroid hormones (e.g., estrogens, androgens, glucocorticoids) have also been associated with the induction of genotoxic stress via multiple mechanisms, such as formation of DNA adducts, or generation of reactive oxygen species (ROS). These effects can be exacerbated by both genetic aberrations, such as mutation of important DNA repair genes, as well as chemical perturbations, or through the use of NR antagonists. Conflicting data exists as to whether hormones are carcinogenic or cancer-protective, and the discrepancies are context-, model-, and hormone-specific. However, steroid hormones have been shown to promote transformation, as well as generate complex genomic rearrangements through induction of DSBs that are associated with transformation. Conversely, hormone signaling has also been shown to stabilize DNA and be chemopreventive. Additionally, some NR antagonists (e.g., tamoxifen) have also been demonstrated to induce DNA damage, adding further complexity [77].
Estrogen is necessary for the physiological growth and development of breast tissue. Increased levels of estrogens are one of the major risk factors for breast cancer. It has been shown that estrogens can contribute to breast cancer by inducing DNA damage [4]. High levels of estrogens in serum are associated with a 2–to 2.5- fold greater risk of breast cancer [4]. Early breast cancer lesions exhibit chromosomal instability and aneuploidy, and the induction of DNA damage by estrogen leads to double stranded DNA breaks and genomic instability. Estrogen serves as a ligand for ERα, inducing nuclear receptor activity and also participates in an acute cytoplasmic membrane-associated activity (Reviewed in [78]). The ERα regulates nuclear gene expression via binding to both canonical DNA and non-canonical DNA sequences in the promoter of target genes. Extra-nuclear pools of ERα have been identified in the plasma membranes [79]. Estrogen can induce DNA damage via the production of oxidative metabolites that cause DNA adducts, or other oxidative DNA damage, evidenced by in vitro and animal model studies [4]. Secondly, hyper-activated estrogen signaling provokes excessive proliferation when pathways become dysregulated, and this theory has strong support from in vitro modeling and gene signatures in breast cancer [80]. Excessive proliferation promotes DNA damage accumulation due to insufficient timely repair, leading to replication fork stalling and possibly even double stranded DNA breaks [81]. Furthermore, E2 is known to delay the assembly and prolongs the resolution of γH2AX and RAD51 foci through inhibition of ATR kinase signaling [3].
Knowing that cyclin D1 interacted with the estrogen receptor, Li et al. examined the role for cyclin D1 in E2-mediated DNA damage signaling (Figure 1). These studies showed that cyclin D1 governs an essential role in the E2-dependent DDR in human breast cancer cells [9]. The ability to distinguish nuclear from extra-nuclear ERα signaling has been enabled through the generation of 17β-estradiol dendrimer conjugates (EDC), which are localized to the extra-nuclear compartment [82]. Using estrogen dendrimers that are excluded from the nucleus, Li et al showed that E2-mediated cyclin D1 dependent DDR occurred via a novel extra-nuclear function. Their findings revealed that ERα-cyclin D1 binding at the cytoplasmic membrane augmented AKT phosphorylation (Ser473) and γH2AX foci formation. In the nucleus, cyclin D1 enhanced homology-directed DNA repair. Cyclin D1 was recruited to γH2AX foci by E2 and induced RAD51 expression [9]. Given the pro-survival and proliferative function of Akt, the augmentation of Akt1 phosphorylation by cyclin D1 may contribute to aberrant growth signaling and is consistent with in vivo studies illustrating the importance of cyclin D1 in E2-mediated growth signaling in vivo [25].
Recent studies have shown androgens restrain DNA damage and enhance DNA repair [5, 83]. Sawyers et al showed that prostate cancer cells treated with IR plus androgen demonstrate enhanced DNA repair and decreased DNA damage and furthermore that antiandrogen treatment causes increased DNA damage and decreased clonogenic survival of prostate cancer cells [5]. Antiandrogen treatment resulted in decreased classical non-homologous end joining. These studies began with an unbiased query of gene expression in a clinically validated xenograft model of castration-resistant prostate cancer. The authors defined an “AR-associated DNA repair gene” signature of 144 DNA repair genes that were significantly associated with canonical AR output [5]. In a related study, DNA damage induced repair, was delayed in cultured cells deprived of androgens. Furthermore, the AR recruitment to the XRCC2 gene after γ irradiation was said to be delayed. A 20% induction of DNA-PKcs was observed after 24 hours and phosphorylation of DNA-PKcs (Ser2056) was reduced by enzalutamide, leading to a proposed model in which liganded AR governs DNA repair via DNA-PKcs phosphorylation [83]. Curiously although it was proposed γ irradiation induced AR signaling, KLK3/PSA was not induced, suggesting further complexities remain to be understood [83]. Nonetheless together these studies were consistent with a model in which androgens enhance the repair of damaged DNA.
Like androgens, cyclin D1 promotes prostate cellular proliferation [36]. Cyclin D1 binds to the androgen receptor [24] and augments prostate stem cell expansion [36]. Furthermore, the cyclin D1-mediated gene expression signature predicts poor outcome in human prostate cancer [36]. The role of cyclin D1 in androgen-dependent signaling was therefore assessed [10]. Casimiro et al. found that endogenous cyclin D1 diminished further the dihydrotestosterone (DHT)-dependent reduction of γH2AX foci. They also showed that cyclin D1 was required for the androgen-mediated DNA damage response both in vitro and in vivo. These findings suggest that the hormone-mediated recruitment of cyclin D1 to sites of DDR may facilitate the resistance of prostate cancer cells to radiotherapy (Figure 1) [10]. In support of these studies, Marampon F. et al. found that silencing cyclin D1 leads to an impairment of DNA DSBs repair. Furthermore, they showed a physical interaction between cyclin D1 and activated ATM, DNA-PKcs and RAD51. Taken together these data suggest a key role for cyclin D1 in regulating the androgen-dependent DDR response [11].
The last decade has demonstrated an important role for cyclin D1-dependent kinase inhibitors in the treatment of patients with ERα+ breast cancers who are concurrently taking other medications, such as Fulvestrant [1, 2]. At the same time, new evidence has accrued that cyclin D1 plays an important role in the DNA damage response to a variety of therapeutic agents. Furthermore, the hormone-mediated DNA damage effects of estradiol and androgen directly involves cyclin D1. Together these studies suggest careful timing of multiple therapies may be important in optimizing therapeutic outcomes. The finding that distinct domains of cyclin D1 participate in the pRB phosphorylation function, vs the transcription factor interaction domains raises the possibility that targeting of multiple distinct domains of cyclin D1 may potentially also improve therapeutic specificity.
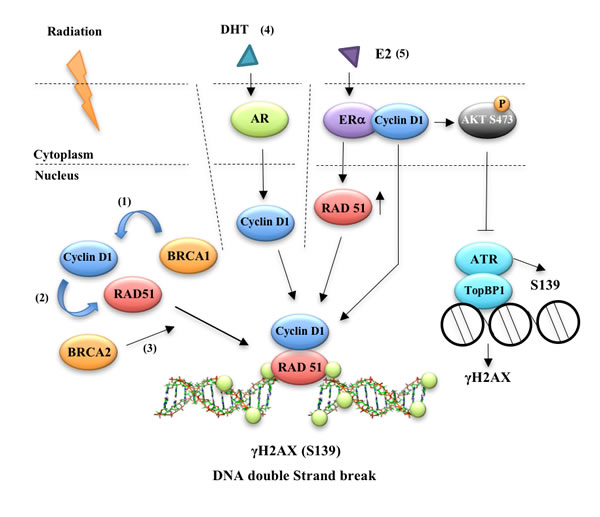
Figure 1: Cyclin D1 participates in estrogen and androgen mediated DNA damage repair responses. Cyclin D1 binds (1) BRCA1 [6], (2) RAD51 ([7, 8]) and (3) BRCA2 [8]. Cyclin D1 was required for the (4) androgen-mediated DNA damage response both in vitro and in vivo suggesting hormone-mediated recruitment of cyclin D1 to sites of DDR may facilitate the resistance of prostate cancer cells to radiotherapy [10]. Silencing cyclin D1 leads to an impairment of DNA DSBs repair and a physical interaction was identified between cyclin D1 and activated ATM, DNA-PKcs and RAD51. Taken together these data suggest a key role for cyclin D1 in regulating the androgen-dependent DDR response [11]. (5) Using estrogen dendrimers that are excluded from the nucleus, Li et al showed that E2-mediated cyclin D1 dependent DDR occurred via a novel extra-nuclear function. Their findings revealed that ERα-cyclin D1 binding at the cytoplasmic membrane augmented AKT phosphorylation (Ser473) and γH2AX foci formation. In the nucleus, cyclin D1 enhanced homology-directed DNA repair. Cyclin D1 was recruited to γH2AX foci by E2 and induced Rad51 expression [9].
Conflicts of interests
The authors disclose no potential conflicts of interest.
Funding
This work was supported in part by NIH grants R01CA132115 (R.G.P.); the Breast Cancer Research Foundation (R.G.P.), generous grants from the Dr. Ralph and Marian C. Falk Medical Research Trust (R.G.P.) and the Pennsylvania Department of Health (R.G.P.), and an American-Italian Cancer Foundation Post-doctoral Research Fellowship (G.D.). There are no conflicts of interest associated with this manuscript. The Department of Health specifically disclaims responsibility for analyses, interpretations or conclusions.
References
1. Casimiro MC, Velasco-Velazquez M, Aguirre-Alvarado C, Pestell RG. Overview of cyclins D1 function in cancer and the CDK inhibitor landscape: past and present. Expert Opinion on Investigational Drugs. 2014; 23: 295-304. doi: 10.1517/13543784.2014.867017.
2. Sherr CJ, Beach D, Shapiro GI. Targeting CDK4 and CDK6: From Discovery to Therapy. Cancer Discov. 2015. doi: 10.1158/2159-8290.CD-15-0894.
3. Pedram A, Razandi M, Evinger AJ, Lee E, Levin ER. Estrogen inhibits ATR signaling to cell cycle checkpoints and DNA repair. Mol Biol Cell. 2009; 20: 3374-89. doi: 10.1091/mbc.E09-01-0085.
4. Caldon CE. Estrogen signaling and the DNA damage response in hormone dependent breast cancers. Front Oncol. 2014; 4: 106. doi: 10.3389/fonc.2014.00106.
5. Polkinghorn WR, Parker JS, Lee MX, Kass EM, Spratt DE, Iaquinta PJ, Arora VK, Yen WF, Cai L, Zheng D, Carver BS, Chen Y, Watson PA, et al. Androgen Receptor Signaling Regulates DNA Repair in Prostate Cancers. Cancer Discov. 2013. doi: 10.1158/2159-8290.CD-13-0172.
6. Wang C, Fan S, Li Z, Fu M, Rao M, Ma Y, Lisanti MP, Albanese C, Katzenellenbogen BS, Kushner PJ, Weber B, Rosen EM, Pestell RG. Cyclin D1 antagonizes BRCA1 repression of estrogen receptor alpha activity. Cancer Res. 2005; 65: 6557-67.
7. Li Z, Jiao X, Wang C, Shirley LA, Elsaleh H, Dahl O, Wang M, Soutoglou E, Knudsen ES, Pestell RG. Alternative cyclin d1 splice forms differentially regulate the DNA damage response. Cancer Res. 2010; 70: 8802-11. doi: 10.1158/0008-5472.CAN-10-0312.
8. Jirawatnotai S, Hu Y, Michowski W, Elias JE, Becks L, Bienvenu F, Zagozdzon A, Goswami T, Wang YE, Clark AB, Kunkel TA, van Harn T, Xia B, et al. A function for cyclin D1 in DNA repair uncovered by protein interactome analyses in human cancers. Nature. 2011; 474: 230-4. doi: 10.1038/nature10155.
9. Li Z, Chen K, Jiao X, Wang C, Willmarth NE, Casimiro MC, Li W, Ju X, Kim SH, Lisanti MP, Katzenellenbogen JA, Pestell RG. Cyclin D1 integrates estrogen-mediated DNA damage repair signaling. Cancer Res. 2014; 74: 3959-70. doi: 10.1158/0008-5472.CAN-13-3137.
10. Casimiro MC, Di Sante G, Ju X, Li Z, Chen K, Crosariol M, Yaman I, Gormley M, Meng H, Lisanti MP, Pestell RG. Cyclin D1 Promotes Androgen-Dependent DNA Damage Repair in Prostate Cancer Cells. Cancer Res. 2016; 76: 329-38. doi: 10.1158/0008-5472.CAN-15-0999.
11. Marampon F, Gravina GL, Ju X, Vetuschi A, Sferra R, Casimiro MC, Pompili S, Festuccia C, Colapietro A, Gaudio E, Di Cesare E, Tombolini V, Pestell RG. Cyclin D1 silencing suppresses tumorigenicity, impairs DNA double strand break repair and thus radiosensitizes androgenindependent prostate cancer cells to DNA damage. Oncotarget. 2016; 7:5383-5400. doi: 10.18632/oncotarget.6579.
12. Wang JD, Levin PA. Metabolism, cell growth and the bacterial cell cycle. Nat Rev Microbiol. 2009; 7: 822-7. doi: 10.1038/nrmicro2202.
13. Schafer KA. The cell cycle: a review. Vet Pathol. 1998; 35: 461-78.
14. Casimiro MC, Crosariol M, Loro E, Li Z, Pestell RG. Cyclins and cell cycle control in cancer and disease. Genes Cancer. 2012; 3: 649-57. doi: 10.1177/1947601913479022.
15. Fu M, Wang C, Li Z, Sakamaki T, Pestell RG. Minireview: Cyclin D1: Normal and Abnormal Functions. Endocrinology. 2004; 145: 5439-47.
16. Wang TC, Cardiff RD, Zukerberg L, Lees E, Arnold A, Schmidt EV. Mammary hyperplasia and carcinoma in MMTV-cyclin D1 transgenic mice. Nature. 1994; 369: 669-71. doi: 10.1038/369669a0.
17. Casimiro MC, Di Sante G, Crosariol M, Loro E, Dampier W, Ertel A, Yu Z, Saria EA, Papanikolaou A, Li Z, Wang C, Addya S, Lisanti MP, et al. Kinase-independent role of cyclin D1 in chromosomal instability and mammary tumorigenesis. Oncotarget. 2015; 6: 8525-38. doi: 10.18632/oncotarget.3267.
18. Casimiro MC, Arnold A, Pestell RG. Kinase independent oncogenic cyclin D1. Aging (Albany NY). 2015; 7: 455-6. doi: 10.18632/aging.100773.
19. Casimiro MC, Crosariol M, Loro E, Ertel A, Yu Z, Dampier W, Saria EA, Papanikolaou A, Stanek TJ, Li Z, Wang C, Fortina P, Addya S, et al. ChIP sequencing of cyclin D1 reveals a transcriptional role in chromosomal instability in mice. The Journal of Clinical Investigation. 2012; 122: 833-43. doi: 10.1172/JCI60256.
20. Pestell RG. New roles of cyclin D1. Am J Pathol. 2013; 183: 3-9. doi: 10.1016/j.ajpath.2013.03.001.
21. Casimiro MC, Pestell RG. Cyclin d1 induces chromosomal instability. Oncotarget. 2012; 3: 224-5. doi: 10.18632/oncotarget.476.
22. Wang C, Li Z, Lu Y, Du R, Katiyar S, Yang J, Fu M, Leader JE, Quong A, Novikoff PM, Pestell RG. Cyclin D1 repression of nuclear respiratory factor 1 integrates nuclear DNA synthesis and mitochondrial function. Proc Natl Acad Sci U S A. 2006; 103: 11567-72. doi: 10.1073/pnas.0603363103.
23. Lee Y, Dominy JE, Choi YJ, Jurczak M, Tolliday N, Camporez JP, Chim H, Lim JH, Ruan HB, Yang X, Vazquez F, Sicinski P, Shulman GI, et al. Cyclin D1-Cdk4 controls glucose metabolism independently of cell cycle progression. Nature. 2014; 510: 547-51. doi: 10.1038/nature13267.
24. Reutens AT, Fu M, Wang C, Albanese C, McPhaul MJ, Sun Z, Balk SP, Janne OA, Palvimo JJ, Pestell RG. Cyclin D1 binds the androgen receptor and regulates hormone-dependent signaling in a p300/CBP-associated factor (P/CAF)-dependent manner. Mol Endocrinol. 2001; 15: 797-811.
25. Casimiro MC, Wang C, Li Z, Di Sante G, Willmart NE, Addya S, Chen L, Liu Y, Lisanti MP, Pestell RG. Cyclin D1 determines estrogen signaling in the mammary gland in vivo. Mol Endocrinol. 2013; 27: 1415-28. doi: 10.1210/me.2013-1065.
26. Wang C, Li Z, Fu M, Bouras T, Pestell RG. Signal transduction mediated by cyclin D1: from mitogens to cell proliferation: A molecular target with therapeutic potential. Cancer Treat Res. 2004; 119: 217-37.
27. Zhang JM, Wei Q, Zhao X, Paterson BM. Coupling of the cell cycle and myogenesis through the cyclin D1-dependent interaction of MyoD with cdk4. EMBO J. 1999; 18: 926-33. doi: 10.1093/emboj/18.4.926.
28. Horstmann S, Ferrari S, Klempnauer KH. Regulation of B-Myb activity by cyclin D1. Oncogene. 2000; 19: 298-306. doi: 10.1038/sj.onc.1203302.
29. Inoue K, Sherr CJ. Gene expression and cell cycle arrest mediated by transcription factor DMP1 is antagonized by D-type cyclins through a cyclin-dependent-kinase-independent mechanism. Mol Cell Biol. 1998; 18: 1590-600.
30. Wang C, Pattabiraman N, Zhou JN, Fu M, Sakamaki T, Albanese C, Li Z, Wu K, Hulit J, Neumeister P, Novikoff PM, Brownlee M, Scherer PE, et al. Cyclin D1 repression of peroxisome proliferator-activated receptor gamma expression and transactivation. Molecular and Cell Biology. 2003; 23: 6159-73.
31. Fu M, Rao M, Bouras T, Wang C, Wu K, Zhang X, Li Z, Yao TP, Pestell RG. Cyclin D1 inhibits peroxisome proliferator-activated receptor gamma-mediated adipogenesis through histone deacetylase recruitment. J Biol Chem. 2005; 280: 16934-41. doi: 10.1074/jbc.M500403200.
32. Neumeister P, Pixley FJ, Xiong Y, Xie H, Wu K, Ashton A, Cammer M, Chan A, Symons M, Stanley ER, Pestell RG. Cyclin D1 governs adhesion and motility of macrophages. Molecular Biology of the Cell. 2003; 14: 2005-15.
33. Li Z, Wang C, Jiao X, Katiyar S, Casimiro MC, Prendergast GC, Powell MJ, Pestell RG. Alternate cyclin D1 mRNA splicing modulates p27KIP1 binding and cell migration. J Biol Chem. 2008; 283: 7007-15. doi: 10.1074/jbc.M706992200.
34. Zwijsen RM, Wientjens E, Klompmaker R, van der Sman J, Bernards R, Michalides RJ. CDK-independent activation of estrogen receptor by cyclin D1. Cell. 1997; 88: 405-15.
35. Neuman E, Ladha MH, Lin N, Upton TM, Miller SJ, DiRenzo J, Pestell RG, Hinds PW, Dowdy SF, Brown M, Ewen ME. Cyclin D1 stimulation of estrogen receptor transcriptional activity independent of cdk4. Mol Cell Biol. 1997; 17: 5338-47.
36. Ju X, Casimiro MC, Gormley M, Meng H, Jiao X, Katiyar S, Crosariol M, Chen K, Wang M, Quong AA, Lisanti MP, Ertel A, Pestell RG. Identification of a cyclin D1 network in prostate cancer that antagonizes epithelial-mesenchymal restraint. Cancer Res. 2014; 74: 508-19. doi: 10.1158/0008-5472.CAN-13-1313.
37. Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001; 411: 366-74. doi: 10.1038/35077232.
38. O’Connor MJ. Targeting the DNA Damage Response in Cancer. Mol Cell. 2015; 60: 547-60. doi: 10.1016/j.molcel.2015.10.040.
39. Swindall AF, Stanley JA, Yang ES. PARP-1: Friend or Foe of DNA Damage and Repair in Tumorigenesis? Cancers (Basel). 2013; 5: 943-58. doi: 10.3390/cancers5030943.
40. Jackson SP. Sensing and repairing DNA double-strand breaks. Carcinogenesis. 2002; 23: 687-96.
41. Shibata A, Jeggo PA. DNA double-strand break repair in a cellular context. Clin Oncol (R Coll Radiol). 2014; 26: 243-9. doi: 10.1016/j.clon.2014.02.004.
42. Moynahan ME, Jasin M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol. 2010; 11: 196-207. doi: 10.1038/nrm2851.
43. Ceccaldi R, Liu JC, Amunugama R, Hajdu I, Primack B, Petalcorin MI, O’Connor KW, Konstantinopoulos PA, Elledge SJ, Boulton SJ, Yusufzai T, D’Andrea AD. Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature. 2015; 518: 258-62. doi: 10.1038/nature14184.
44. Lieber MR, Gu J, Lu H, Shimazaki N, Tsai AG. Nonhomologous DNA end joining (NHEJ) and chromosomal translocations in humans. Subcell Biochem. 2010; 50: 279-96. doi: 10.1007/978-90-481-3471-7_14.
45. Kitagawa R, Kastan MB. The ATM-dependent DNA damage signaling pathway. Cold Spring Harb Symp Quant Biol. 2005; 70: 99-109. doi: 10.1101/sqb.2005.70.002.
46. Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature. 2003; 421: 961-6. doi: 10.1038/nature01446.
47. Lee MS, Edwards RA, Thede GL, Glover JN. Structure of the BRCT repeat domain of MDC1 and its specificity for the free COOH-terminal end of the gamma-H2AX histone tail. J Biol Chem. 2005; 280: 32053-6. doi: 10.1074/jbc.C500273200.
48. Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 2005; 123: 1213-26. doi: 10.1016/j.cell.2005.09.038.
49. Brown EJ, Baltimore D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 2000; 14: 397-402.
50. Brown EJ, Baltimore D. Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Dev. 2003; 17: 615-28. doi: 10.1101/gad.1067403.
51. de Klein A, Muijtjens M, van Os R, Verhoeven Y, Smit B, Carr AM, Lehmann AR, Hoeijmakers JH. Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr Biol. 2000; 10: 479-82.
52. Cortez D, Guntuku S, Qin J, Elledge SJ. ATR and ATRIP: partners in checkpoint signaling. Science. 2001; 294: 1713-6. doi: 10.1126/science.1065521.
53. Blattner C, Sparks A, Lane D. Transcription factor E2F-1 is upregulated in response to DNA damage in a manner analogous to that of p53. Mol Cell Biol. 1999; 19: 3704-13.
54. Guo R, Chen J, Zhu F, Biswas AK, Berton TR, Mitchell DL, Johnson DG. E2F1 localizes to sites of UV-induced DNA damage to enhance nucleotide excision repair. J Biol Chem. 2010; 285: 19308-15. doi: 10.1074/jbc.M110.121939.
55. Liu K, Lin FT, Ruppert JM, Lin WC. Regulation of E2F1 by BRCT domain-containing protein TopBP1. Mol Cell Biol. 2003; 23: 3287-304.
56. Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: a molecular definition. Oncogene. 2004; 23: 2825-37. doi: 10.1038/sj.onc.1207528.
57. Karanika S, Karantanos T, Li L, Corn PG, Thompson TC. DNA damage response and prostate cancer: defects, regulation and therapeutic implications. Oncogene. 2015; 34: 2815-22. doi: 10.1038/onc.2014.238.
58. Nowsheen S, Yang ES. The intersection between DNA damage response and cell death pathways. Exp Oncol. 2012; 34: 243-54.
59. Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer. 2012; 12: 801-17. doi: 10.1038/nrc3399.
60. Pagano M, Theodoras AM, Tam SW, Draetta GF. Cyclin D1-mediated inhibition of repair and replicative DNA synthesis in human fibroblasts. Genes Dev. 1994; 8: 1627-39.
61. Agami R, Bernards R. Distinct initiation and maintenance mechanisms cooperate to induce G1 cell cycle arrest in response to DNA damage. Cell. 2000; 102: 55-66. doi: 10.1016/S0092-8674(00)00010-6.
62. Albanese C, D’Amico M, Reutens AT, Fu M, Watanabe G, Lee RJ, Kitsis RN, Henglein B, Avantaggiati M, Somasundaram K, Thimmapaya B, Pestell RG. Activation of the cyclin D1 gene by the E1A-associated protein p300 through AP-1 inhibits cellular apoptosis. J Biol Chem. 1999; 274: 34186-95.
63. Coco Martin JM, Balkenende A, Verschoor T, Lallemand F, Michalides R. Cyclin D1 overexpression enhances radiation-induced apoptosis and radiosensitivity in a breast tumor cell line. Cancer Res. 1999; 59: 1134-40.
64. Zhou Q, Fukushima P, DeGraff W, Mitchell JB, Stetler Stevenson M, Ashkenazi A, Steeg PS. Radiation and the Apo2L/TRAIL apoptotic pathway preferentially inhibit the colonization of premalignant human breast cells overexpressing cyclin D1. Cancer Res. 2000; 60: 2611-5.
65. Xu H, Yu S, Liu Q, Yuan X, Mani S, Pestell RG, Wu K. Recent advances of highly selective CDK4/6 inhibitors in breast cancer. J Hematol Oncol. 2017; 10: 97. doi: 10.1186/s13045-017-0467-2.
66. Hydbring P, Malumbres M, Sicinski P. Non-canonical functions of cell cycle cyclins and cyclin-dependent kinases. Nat Rev Mol Cell Biol. 2016; 17: 280-92. doi: 10.1038/nrm.2016.27.
67. Meng H, Tian L, Zhou J, Li Z, Jiao X, Li WW, Plomann M, Xu Z, Lisanti MP, Wang C, Pestell RG. PACSIN 2 represses cellular migration through direct association with cyclin D1 but not its alternate splice form cyclin D1b. Cell Cycle. 2011; 10: 73-81.
68. Brown JS, O’Carrigan B, Jackson SP, Yap TA. Targeting DNA Repair in Cancer: Beyond PARP Inhibitors. Cancer Discov. 2017; 7: 20-37. doi: 10.1158/2159-8290.CD-16-0860.
69. Densham RM, Morris JR. The BRCA1 Ubiquitin ligase function sets a new trend for remodelling in DNA repair. Nucleus. 2017; 8: 116-25. doi: 10.1080/19491034.2016.1267092.
70. Kolinjivadi AM, Sannino V, de Antoni A, Techer H, Baldi G, Costanzo V. Moonlighting at replication forks - a new life for homologous recombination proteins BRCA1, BRCA2 and RAD51. FEBS Lett. 2017; 591: 1083-100. doi: 10.1002/1873-3468.12556.
71. Wang Y, Cortez D, Yazdi P, Neff N, Elledge SJ, Qin J. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev. 2000; 14: 927-39.
72. Bartek J, Lukas J. DNA repair: Cyclin D1 multitasks. Nature. 2011; 474: 171-2. doi: 10.1038/474171a.
73. Fu M, Wang C, Rao M, Wu X, Bouras T, Zhang X, Li Z, Jiao X, Yang J, Li A, Perkins ND, Thimmapaya B, Kung AL, et al. Cyclin D1 represses p300 transactivation through a cyclin-dependent kinase-independent mechanism. J Biol Chem. 2005; 280: 29728-42. doi: 10.1074/jbc.M503188200.
74. Sakamaki T, Casimiro MC, Ju X, Quong AA, Katiyar S, Liu M, Jiao X, Li A, Zhang X, Lu Y, Wang C, Byers S, Nicholson R, et al. Cyclin D1 determines mitochondrial function in vivo. Mol Cell Biol. 2006; 26: 5449-69. doi: 10.1128/MCB.02074-05.
75. Lin DI, Lessie MD, Gladden AB, Bassing CH, Wagner KU, Diehl JA. Disruption of cyclin D1 nuclear export and proteolysis accelerates mammary carcinogenesis. Oncogene. 2008; 27: 1231-42. doi: 10.1038/sj.onc.1210738
76. Aggarwal P, Lessie MD, Lin DI, Pontano L, Gladden AB, Nuskey B, Goradia A, Wasik MA, Klein-Szanto AJ, Rustgi AK, Bassing CH, Diehl JA. Nuclear accumulation of cyclin D1 during S phase inhibits Cul4-dependent Cdt1 proteolysis and triggers p53-dependent DNA rereplication. Genes Dev. 2007; 21: 2908-22. doi: 10.1101/gad.1586007.
77. Schiewer MJ, Knudsen KE. Linking DNA Damage and Hormone Signaling Pathways in Cancer. Trends Endocrinol Metab. 2016; 27: 216-25. doi: 10.1016/j.tem.2016.02.004.
78. Levin ER. Minireview: Extranuclear Steroid Receptors: Roles in Modulation of Cell Functions. Mol Endocrinol. 2010. doi: 10.1210/me.2010-0284.
79. Levin ER, Pietras RJ. Estrogen receptors outside the nucleus in breast cancer. Breast Cancer Res Treat. 2008; 108: 351-61. doi: 10.1007/s10549-007-9618-4.
80. Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer. 2009; 9: 631-43. doi: 10.1038/nrc2713.
81. Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008; 319: 1352-5. doi: 10.1126/science.1140735.
82. Harrington WR, Kim SH, Funk CC, Madak-Erdogan Z, Schiff R, Katzenellenbogen JA, Katzenellenbogen BS. Estrogen dendrimer conjugates that preferentially activate extranuclear, nongenomic versus genomic pathways of estrogen action. Mol Endocrinol. 2006; 20: 491-502. doi: 10.1210/me.2005-0186.
83. Goodwin JF, Schiewer MJ, Dean JL, Schrecengost RS, de Leeuw R, Han S, Ma T, Den RB, Dicker AP, Feng FY, Knudsen KE. A Hormone-DNA Repair Circuit Governs the Response to Genotoxic Insult. Cancer Discov. 2013. doi: 10.1158/2159-8290.CD-13-0108.