
INTRODUCTION
There is increasing evidence that annexin A5 (ANXA5) has roles in cytotoxicity [1, 2], apoptosis [3, 4], and anti-inflammatory effects [5]. However, the detailed molecular mechanisms of ANXA5 in cancer cells are not yet fully understood. Previously, we found that auranofin induces expression of ANXA5 in human prostate cancer cells and triggers apoptosis [3]. Auranofin is a well-known lipophilic gold compound that has anti-inflammatory effects and immunosuppressive properties. Consequently, auranofin has been widely used for the treatment of rheumatoid arthritis (RA). A number of studies showed that auranofin inhibits production of proinflammatory cytokines, such as tumor necrosis factor alpha (TNF-α), interleukin-1β (IL-1β), interleukin-6 (IL-6), and cyclooxygenase-2 (COX-2) expression [6-8]. In particular, the repressive effect of COX-2 has been previously emphasized [9, 10].
Several studies suggest a correlation between inflammation and prostate cancer. Nelson et al [11] and Dennis et al [12] showed that inflammation is a risk factor for development of prostate cancer. Moreover, Wang et al [13] has shown that COX-2 expression affects carcinogenic process and chemopreventive effects of anti-inflammatory drugs in prostate cancer cells. Therefore, it is important to demonstrate the detailed mechanism of COX-2 inhibitory effect by ANXA5 in prostate cancer cells for the improvement of prostate cancer chemotherapy.
Phosphorylation and activation of the p65 subunit of NF-κB have an essential role in the regulation of inflammatory cytokines [14-16]. Several researchers have shown that NF-κB activation induces the increase of COX-2 expression and inflammatory response both in vitro and in vivo by intra and extracellular stimuli [17-19]. The Ser536 residue of p65 is important for p65 translocation into the nucleus [20], being involved in IκB kinase (IKK)-mediated IκBα phosphorylation and subsequent ubiquitin-dependent degradation [21]. IKK activity in primary fibroblast-like synoviocytes was significantly increased within 10 min of stimulation by TNF-α and IL-1, accompanied by phosphorylation and degradation of IκBα [22]. Phosphorylation of p65 is not only related to activation of p65 nuclear translocation, but to induction of p65 transactivation activity. Phosphorylation of Ser311 has been shown to be important for interaction of p65 with the cAMP response element-binding protein (CBP)/p300 [23]. The interaction between p65 and CBP induces p65 transactivation and then promotes expression of inflammatory cytokines [24].
Previous studies have shown that PKC-ζ acts as a signaling component upstream of transforming growth factor β-activated kinase 1 (TAK1) and downstream of RhoA and induces activation of NF-κB in macrophages by lipopolysaccharide (LPS) [25]. PKC-ζ is one of the well-known kinase, which phosphorylates Ser536 and Ser311 residues of p65 to promote nuclear translocation and transactivation [24, 26].
ANXA5 has been shown to interact with some PKCs. PKC-α is directly coupled with ANXA5 to cause apoptotic events and PKC-δ translocation is upregulated through a direct interaction between ANXA5 and PKC-δ which participate in cellular signaling events [27-29], but interaction between ANXA5 and PKC-ζ has not yet been confirmed. In this study, we determined that inhibition of ANXA5 induces COX-2 expression in human prostate cancer cells and explored the correlation between PKC-ζ and ANXA5 on the regulation of COX-2 expression. Our results suggest that ANXA5 may act as a negative regulator for COX-2 expression by downregulating the PKC-ζ-NF-κB pathway in prostate cancer cells.
RESULTS
Auranofin inhibits COX-2 expression in PC-3 cells
Previously, we showed that auranofin induces ANXA5 in human prostate cancer PC-3 cells [3]. Auranofin is already known to inhibit COX-2 expression in synovial cells [10]. PC-3 cells were treated with auranofin in order to determine whether it is also able to inhibit COX-2 expression in PC-3 cells. After treatment with auranofin (0.25, 0.5, or 1 μM) for 24 h, COX-2 levels were measured by quantitative PCR (qPCR) and western blot (Figure 1A and 1B). Auranofin significantly inhibited COX-2 mRNA and protein expression, and this occurred in a time-dependent manner (Figure 1C and 1D). These data showed that auranofin suppresses COX-2 expression in PC-3 cells in concentration- and time-dependent manners.
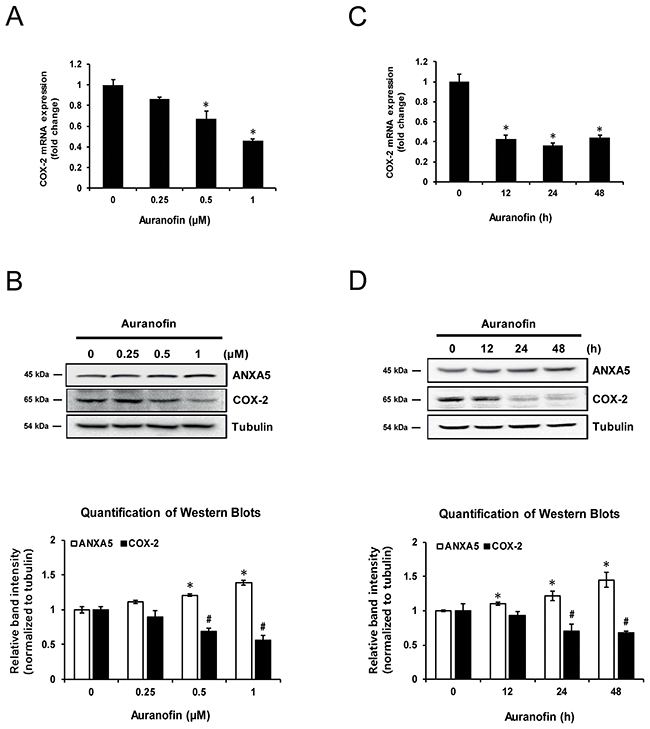
Figure 1: Inhibition of cyclooxygenase 2 (COX-2) expression by auranofin in PC-3 cells. (A-B) Cells were treated with the indicated concentration of auranofin for 24 h. (A) Expression of COX-2 mRNA was determined using qPCR. (B) COX-2 proteins were measured using western blot analysis. Quantification of western blots (n = 3). (C-D) Cells were treated with 1 μM auranofin for 12, 24, or 48 h. (C) Expression of COX-2 mRNA was determined using real-time qPCR. (D) COX-2 proteins were measured using western blot analysis. Quantification of western blots (n = 3). *, #P < 0.05.
Inhibition of ANXA5 induces COX-2 expression in PC-3 cells
To determine whether ANXA5 exhibits an inhibitory effect on COX-2 expression, PC-3 cells were transfected with ANXA5-specific siRNA. Knockdown of ANXA5 increased COX-2 mRNA and protein levels by 3-fold and 1.5-fold, respectively (Figure 2A and 2B). However, in other cancer cell lines, including human hepatoma cells Hep3B and HuH-7, human cervical cancer cells HeLa, and human breast cancer cells MCF-7 and MDA-MB-231, ANXA5 knockdown showed no significant effects on COX-2 expression (Supplementary Figure 1). Prostaglandin E2 (PGE2) is a well-known metabolite produced by COX-2. A number of studies confirmed the alteration of PGE2 content in association with COX-2 expression [30-33]. Thus, to demonstrate whether the change in COX-2 expression induced by ANXA5 siRNA influenced prostaglandin metabolism, alteration in PGE2 production in cell culture media was measured. As shown in Figure 2C, PGE2 production was increased 1.4-fold by ANXA5 siRNA. Additionally, to determine whether ANXA5 controlled COX-2 promoter activity, a dual-luciferase reporter assay was performed (Figure 2D). Similar to previous data, COX-2 promoter activity was increased by more than 80% compared to the control when cells were treated with ANXA5 siRNA (37.5 nM) for 48 h.
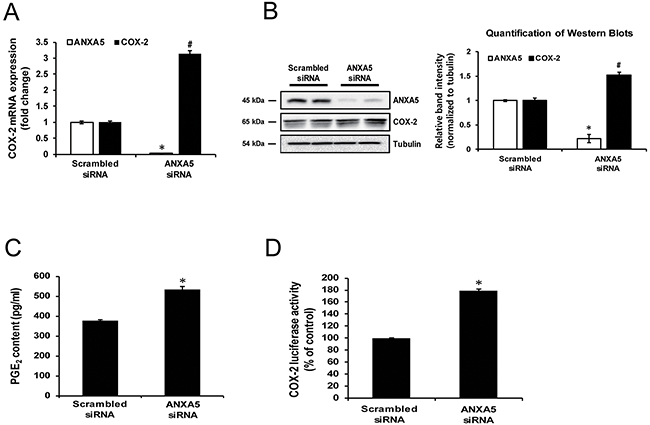
Figure 2: Effect of annexin A5 knockdown by siRNA on cyclooxygenase 2 (COX-2) expression. (A-D) PC-3 cells were transfected with scrambled or annexin A5 siRNA for 48 h. (A) Expression of COX-2 mRNA was determined using qPCR. (B) COX-2 proteins were measured using western blot analysis. Quantification of western blots (n = 3). (C) PGE2concentration in cell supernatants was measured by ELISA. (D) COX-2 promoter activity was determined using dual luciferase assay. *, #P < 0.05.
Induction of ANXA5 by auranofin inhibits COX-2 expression in prostate cancer cells
We next determined whether the inhibition of COX-2 expression by auranofin was rescued by ANXA5 knockdown in prostate cancer cells. PC-3, LNCaP-LN3, and DU145 cells were transfected with ANXA5 siRNA (37.5 nM) for 48 h in the presence of auranofin (1 μM) (Figure 3A). Western blot analysis revealed that COX-2 expression was rescued in ANXA5 knockdown PC-3 and LNCaP-LN3 cells compared to auranofin-only-treated cells. The data from confocal microscopy analysis also showed that COX-2 levels decreased by auranofin were recovered by ANXA5 knockdown (Figure 3B). In contrast with the results in PC-3 and LNCaP-LN3 cells, ANXA5 siRNA did not show any significant effect in DU145 cells. Taken together, our data demonstrate that auranofin-mediated induction of ANXA5 may play a crucial role in the inhibition of COX-2 expression in PC-3 and LNCaP-LN3 cells.
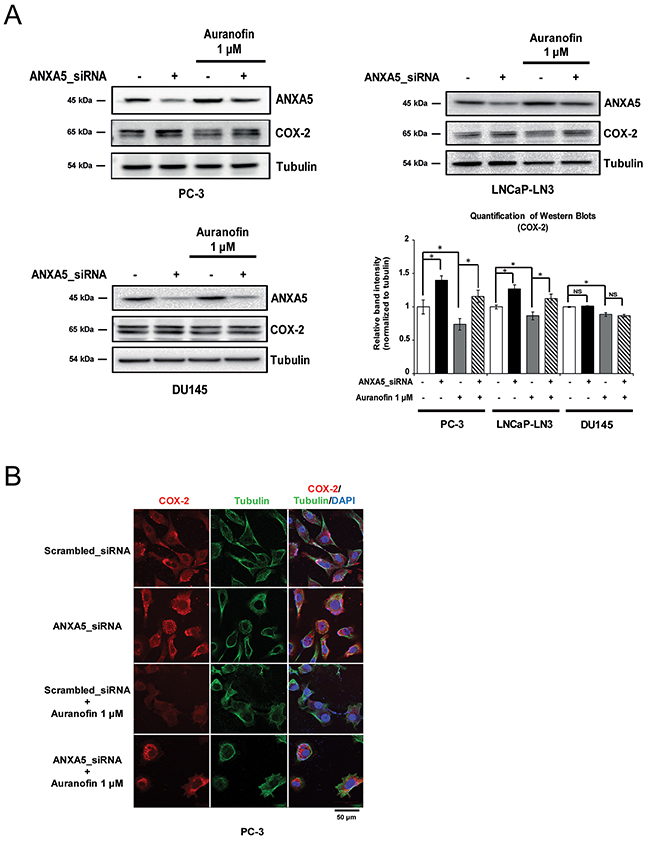
Figure 3: Rescue of cyclooxygenase 2 (COX-2) expression in auranofin-treated prostate cancer cells by inhibition of annexin A5. (A-B) Cells were transfected with annexin A5 siRNA and then treated with auranofin (1 μM) for 24 h. (A) Western blot analysis was performed using whole cell lysates of PC-3, LNCaP-LN3, and DU145 cells. Quantification of western blots (n = 3). (B) Confocal analysis was performed to detect COX-2 expression. Microscopy scale bar = 50 μm. NS = not significant, *P < 0.05.
Overexpression of ANXA5 represses TNF-α-mediated COX-2 induction
Next, we assessed the contribution of ANXA5 to the regulation of COX-2 expression by overexpressing ANXA5. Although there was no significant change in COX-2 mRNA and protein levels (Figure 4A and 4B), ANXA5 overexpression inhibited COX-2 expression when cells were treated with TNF-α (20 ng/mL), which is known to induce COX-2 mRNA expression in various prostate cancer cells, including PC-3, LNCaP-LN3, and DU145 [34]. COX-2 protein levels were also decreased by ANXA5 overexpression in TNF-α-stimulated cells (Figure 4B). Confocal microscopy analysis confirmed that ANXA5 overexpression suppressed TNF-α-induced COX-2 expression (Figure 4C). These results indicate that ANXA5 may downregulate TNF-α-induced COX-2 expression in prostate cancer cells.
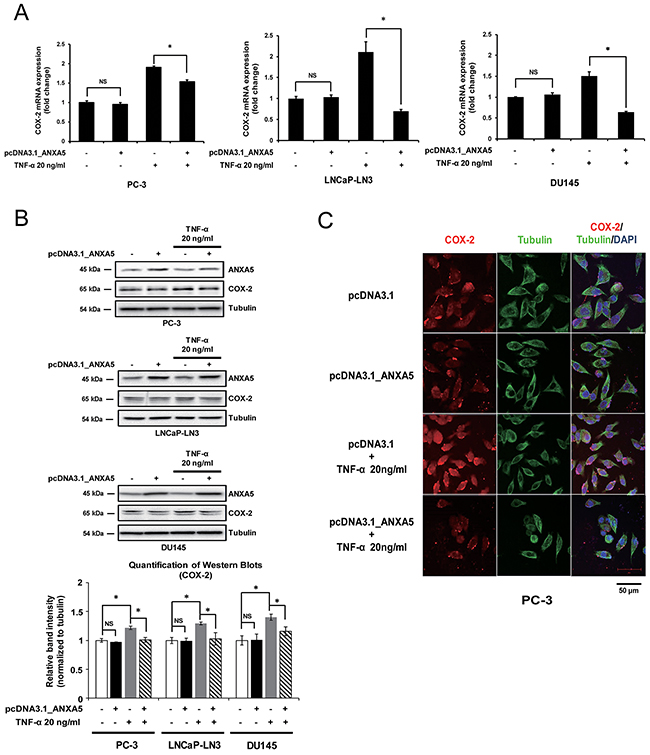
Figure 4: Suppression of tumor necrosis factor α (TNF-α)-induced cyclooxygenase 2 (COX-2) expression by overexpression of annexin A5. (A-B) PC-3, LNCaP-LN3, and DU145 cells were transfected with annexin A5 expression vector and then treated with TNF-α (20 ng/μL) for 4, 6, and 1 h, respectively. (A) qPCR was performed to detect the expression of COX-2 mRNA. (B) Western blot analysis was performed by using whole cell lysates. Quantification of western blots (n = 3). (C) Confocal analysis was performed to detect COX-2 expression. Microscopy scale bar = 50 μm. NS = not significant, *P < 0.05.
ANXA5 may block NF-κB p65 activation
NF-κB is one of the most well-known proinflammatory transcription factors. Therefore, to clarify the molecular mechanism of ANXA5 downregulation of COX-2 expression, we first identified the alteration of NF-κB p65 subunit level using whole cell lysates. Previous studies have shown that auranofin reduced NF-κB p65 expression in RAW 264.7 cells [35]. As shown in Figure 5A, our data also confirmed that auranofin (1 μM) inhibits p65 expression in PC-3 cells. We found that treatment with ANXA5 siRNA did not change the total p65 level. However, phosphorylation of p65 on Ser536 and Ser311 was increased by ANXA5 knockdown, and auranofin-mediated suppression of p65 phosphorylation was rescued by ANXA5 siRNA (Figure 5A). According to previous studies, these phosphorylations of p65 increase the translocation of p65 into the nucleus and induce the transcription of proinflammatory genes such as IL-6, TNF-α, and COX-2 [20, 24]. To identify whether p65 translocation was increased by inhibition of ANXA5, we compared the p65 levels in cytosolic and nuclear fractions (Figure 5B). ANXA5 siRNA increased the p65 level and phosphorylation of p65 on Ser536 in the nucleus, whereas the cytosolic p65 level was decreased. Confocal microscopic analysis also showed nuclear translocation of p65 in PC-3 cells (Figure 5C). To investigate whether p65 phosphorylation by ANXA5 knockdown leads to activation of p65 transcriptional activity, NF-κB p65 binding activity was measured after transfection with ANXA5 siRNA in auranofin-treated cells (Figure 5D). Auranofin significantly inhibited the binding activity of p65; however, ANXA5 knockdown recovered the p65 binding activity decreased by auranofin. These results suggested that inhibition of ANXA5 induces p65 phosphorylation and subsequently activates p65 transcriptional activity in PC-3 cells.
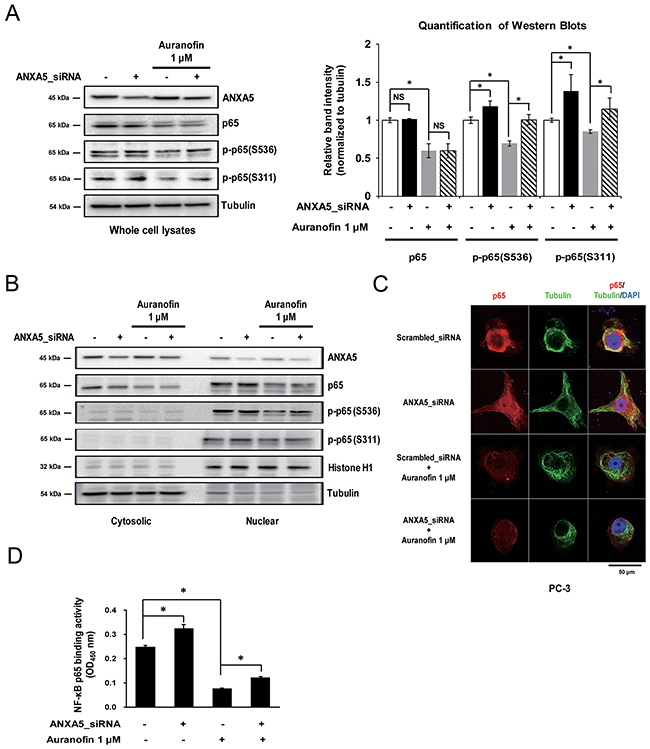
Figure 5: Phosphorylation of the nuclear factor-κB (NF-κB) p65 subunit by inhibition of annexin A5. (A-D) PC-3 cells were transfected with annexin A5 siRNA and then treated with auranofin (1 μM) for 24 h. (A) Whole cell lysates were subjected to western blot analysis for p65, p-p65 (Ser536), and p-p65 (Ser311). Quantification of western blots (n = 3). (B) Nuclear and cytosolic lysates were subjected to western blot analysis for p65, p-p65 (Ser536), and p-p65 (Ser311). (C) Binding activity assay of the NF-κB p65 subunit was performed by using the nuclear lysates. (D) Confocal analysis was performed to detect p65 expression and location. Microscopy scale bar = 50 μm. NS = not significant, *P < 0.05.
Inhibition of ANXA5 affects COX-2 expression by increasing the PKC-ζ protein level
It has been reported that phosphorylation of p65 on Ser536 and Ser311 is affected by PKC-ζ [24, 26]. Therefore, to identify whether the phosphorylation of p65 after ANXA5 knockdown occurs via the induction of PKC-ζ expression, we first examined the alteration of PKC-ζ protein levels under the same conditions shown in Figure 5A. PKC-ζ protein levels were increased by ANXA5-knockdown in PC-3 and LNCaP-LN3 cells (Figure 6A). Confocal microscopic analysis showed that ANXA5 siRNA recovered the reduced PKC-ζ expression seen in auranofin-treated cells (Figure 6B). However, in DU145 cells, PKC-ζ expression was not affected by ANXA5 knockdown, although, when auranofin-treated DU145 cells were transfected with ANXA5 siRNA, the PKC-ζ protein level was slightly increased (Figure 6A). Because phosphorylation and degradation of IκBα may induce phosphorylation of p65 at Ser536 [22], it might be similarly induced by ANXA5 siRNA if PKC-ζ promotes phosphorylation of IκBα. As expected, increased phosphorylation of IκBα was shown by ANXA5 knockdown (Figure 6A). Interestingly, although PKC-ζ protein levels were increased by ANXA5 knockdown, PKC-ζ mRNA levels were not affected, indicating that ANXA5 may control PKC-ζ expression at the post-translational level (Figure 6C). To understand whether induction of COX-2 expression by ANXA5 siRNA is promoted via increased PKC-ζ expression, PC-3 cells were transfected with ANXA5 and PKC-ζ siRNA. PKC-ζ siRNA repressed the induction of COX-2 expression triggered by ANXA5 knockdown (Figure 6D). Therefore, these results suggest that inhibition of ANXA5 promotes COX-2 expression via induction of increased PKC-ζ protein levels.
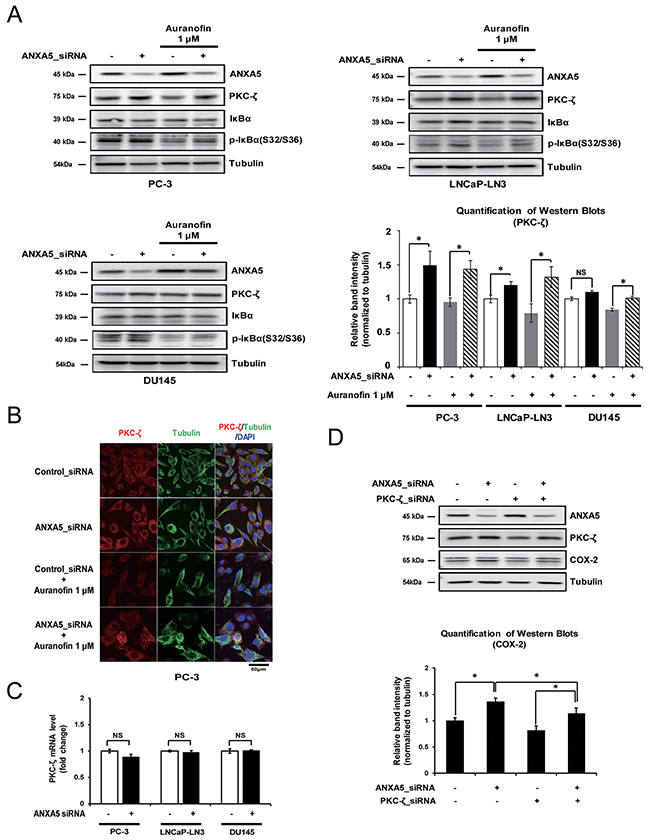
Figure 6: Effect of annexin A5 on protein kinase c (PKC)-ζ expression. (A-B) Cells were transfected with annexin A5 siRNA and then treated with auranofin (1 μM) for 24 h. (A) Western blot analysis was performed to detect PKC-ζ expression by using PC-3, LNCaP-LN3, and DU145 whole cell lysates. Quantification of western blots (n = 3). (B) Confocal analysis was performed to detect PKC-ζ expression. (C) PC-3, LNCaP-LN3, and DU145 cells were transfected with annexin A5 siRNA, and qPCR was performed to detect the expression of PKC-ζ mRNA. (D) PC-3 cells were cotransfected with annexin A5 and PKC-ζ siRNA, and western blot analysis was performed to detect COX-2 expression by using the whole cell lysate. Microscopy scale bar = 50 μm. NS = not significant, *P < 0.05.
DISCUSSION
ANXA5 is widely distributed and abundantly expressed in cancer cells. In previous studies, ANXA5 stimulated various cellular responses, such as apoptosis and anti-inflammation [2, 3, 5]. However, the detailed role of ANXA5 in anti-inflammation and the signaling pathways involved have not been fully elucidated. Here we demonstrate that ANXA5 knockdown induces COX-2 expression, mediated by phosphorylation of p65 and ANXA5 overexpression inhibits TNF-α-induced COX-2 expression. Previous study showed that TNF-α induces phosphorylation of IκBα on Ser32 and Ser36 and induces p65 phosphorylation on Ser536 [36]. Therefore, ANXA5 overexpression may suppress TNF-α-induced COX-2 expression through interfering the IκBα phosphorylation and degradation.
Arnold et al. [5] showed that recombinant human ANXA5 inhibits LPS binding to thetoll-like receptor 4 (TLR4)/MD-2 receptor complex, leading to reduced Akt and NF-κB signaling. However, the detailed mechanism remained unclear. In our study, we showed that ANXA5 knockdown induces phosphorylation of the p65 subunit. Although there are several phosphorylation sites in p65, we confirmed that phosphorylation of p65 at Ser536 and Ser311 may play an important role in COX-2 expression [24, 37]. In particular, the phosphorylation of p65 at Ser536 is associated with translocation into the nucleus [20]. Confocal microscopic analysis and western blot data revealed that the nuclear p65 level is increased by ANXA5 knockdown.
PKC-ζ is able to induce phosphorylation of p65 at both Ser536 and Ser311 [24, 26] and increases the induction of COX-2 expression in response to proinflammatory stimuli [38]. In the present study, we found that ANXA5 knockdown increases PKC-ζ protein levels, and suppression of PKC-ζ by siRNA decreases the induction of COX-2 expression caused by ANXA5 knockdown. Thus, we believe that ANXA5 may play protective roles in the NF-κB-PKC-ζ pathway, causing suppression of COX-2.
Unlike other prostate cancer cells, no noticeable alteration of PKC-ζ expression level by ANXA5 knockdown was observed in DU145 cells, which is similar to the pattern of COX-2 expression. Previous studies show that differences in the basal expression level of specific genes in cells obtained from the same organ indicate different responses to extracellular stimuli [39-41]. Shabbeer et al [42] showed that differences in the basal expression levels of proliferating cell nuclear antigen between PC-3 and DU145 cells affect relatively high resistance of PC-3 cells to valproic acid as compared to DU145 cells. We found that DU145 cells expressed higher levels of PKC-ζ than PC-3 and LNCaP-LN3 cells (Supplementary Figure 2). Accordingly, we hypothesized that the increased expression of PKC-ζ in DU145 explains their weak response to ANXA5 knockdown-derived COX-2 and PKC-ζ expression. In addition, PKC-ζ mRNA levels were not affected by ANXA5 knockdown in PC-3, LNCaP-LN3, or DU145 cells.
In a recent study, Xin et al. [43] showed that mammalian target of rapamycin complex (mTORC)-2 phosphorylates the PKC-ζ turn motif, which protects against proteasome-mediated degradation and induces kinase activity. Notably, these effects are associated with phosphorylation at Thr560, a site known to be regulated by autophosphorylation [44]. Chou et al. [45] showed that phosphoinositide-dependent kinase-1 (PDK-1) activates phosphorylation of PKC-ζ at the activation loop (Thr410) and subsequently induces PKC-ζ autophosphorylation at Thr560 and PKC-ζ activity. We also found increased phosphorylation of PKC-ζ at Thr410 after ANXA5 knockdown (data not shown). Thus, we suggest that ANXA5 may inhibit phosphorylation of the PKC-ζ activation loop, subsequently interfering with PKC-ζ autophosphorylation at Thr560 to cause PKC-ζ degradation and reduce NF-κB signaling. The possibility that ANXA5 directly interacts with PKC-ζ remains to be elucidated.
Unlike PDK-1-dependent activation, Standaert et al. [46] showed that phosphatidylinositol 3,4,5 trisphosphate (PIP3) activates PKC-ζ autophosphorylation at Thr560 independent of phosphorylation of the activation loop. In addition, a previous report showed that p38 kinase also regulates PKC-ζ autophosphorylation at Thr560 [47]. Activated p38 can interact with the PKC-ζ regulatory domain and inhibits autophosphorylation. Although the mechanism of regulation of PKC-ζ translation and/or stability by ANXA5 has not been completely demonstrated, our data suggest that ANXA5 may regulate several kinases, including p38, which interact with PKC-ζ phosphorylation sites. Thus, future studies will be needed to understand the control mechanisms of PKC-ζ protein levels by ANXA5 without transcriptional changes, and elucidate whether differences in reactivity in response to proinflammatory stimuli are affected by the basal level of PKC-ζ expression in other cancer cells.
In summary, we demonstrated that ANXA5 suppresses COX-2 expression via inhibition of NF-κB p65 phosphorylation, which is regulated by alteration of PKC-ζ protein levels in prostate cancer cells. The scheme in Figure 7 summarizes these signaling pathway. Our results indicate that ANXA5 may have a crucial protective role in the cellular inflammatory response to proinflammatory stimuli. Therefore, we anticipate that applying the anti-inflammatory effect of ANXA5 to prostate cancer therapy will have a better effect on inhibition of cancer development and improvement of drug resistance.
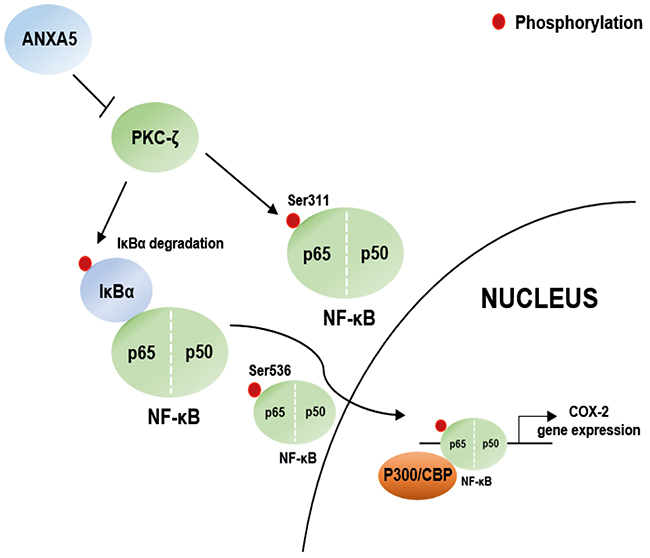
Figure 7: Scheme for the novel mechanisms of ANXA5. Scheme for ANXA5 suppressed COX-2 expression via PKC-ζ – NF-κB signaling in prostate cancer cells.
MATERIALS AND METHODS
Cell culture
Human prostate cancer cell lines PC3, DU145, and LNCaP-LN3, and the human cervical cancer cell line HeLa were obtained from the Korean Cell Line Bank (Seoul, Korea). Human hepatocellular carcinoma cell lines Hep3B and HuH-7, and human breast cancer cell lines MDA-MB-231 and MCF-7 were obtained from the American Type Culture Collection (Manassas, VA). All cells were cultured in RPMI 1640 supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin. Cells were maintained at 37 °C in a humidified atmosphere of 5% CO2.
Reagents
Auranofin was purchased from Sigma-Aldrich (St. Louis, MO). A solution of auranofin was prepared in dimethyl sulfoxide, stored as small aliquots at -20 °C, and then diluted as needed in cell culture medium. FBS and RPMI 1640 medium were purchased from HyClone (Logan, UT). The Neon transfection system was from Life Technologies (Carlsbad, CA). The bicinchoninic acid (BCA) protein assay kit and ECL kit were from Thermo Scientific (Waltham, MA). PGE2 ELISA assay kit was purchased from Enzo Life Science (Farmingdale, NY). Antibodies against tubulin (sc-8035) and goat anti-rabbit IgG-Texas Red (sc-2780), and Ultra CruzTM mounting medium (sc-24941) were from Santa Cruz Biotechnology (Santa Cruz, CA). COX-2 antibody (A303-600A-M) was from Bethyl (Montgomery, TX). NF-κB (p65) (CSB-PA003436), p-p65 (S536) (CSB-PA000586), p-p65 (S311) (CSB-PA000719), and PKC-ζ (CSB-PA003805) antibodies were from Flarebio (College Park, MD). All other chemicals were of the highest purity or molecular biology grade and were obtained from commercial sources.
Transient transfection
ANXA5 SMART pool siRNA (Dharmacon, Lafayette, CO) and the overexpression vector pcDNA3.1/Zeo+ containing the ANXA5-encoding sequence were used for transfection. Cells were transfected with 37.5 nM siRNA or 5 μg plasmid with the Neon Transfection System (Life Technologies) and cultured in 60-mm dishes in antibiotic-free RPMI with 10% FBS for 48 h.
Quantitative PCR
Total RNA was extracted using a Ribospin™ kit (GeneALL, Seoul, Korea). Total RNA (0.5 μg) was reverse-transcribed at 37 °C for 1 h in 20 μL total volume containing 5× RT buffer, 10 mM dNTPs, 40 U RNase inhibitor, 200 U Moloney murine leukemia virus reverse transcriptase, and 100 pmol oligo-dT primer. qPCR was performed using the Rotor-Gene SYBR1 PCR Kit, as recommended by the manufacturer, and analyzed by using QIAGEN Rotor-Gene Q Series software. Each reaction contained 10 μL of 2×SYBR1 Green PCR Master Mix, 1 μM oligonucleotide primers, and 2 μL of cDNA in a final volume of 20 μL. Amplification was conducted as follows: one cycle at 95 °C for 5 min, followed by 40 cycles of denaturation at 95 °C for 5 s and annealing/extension at 56 °C for 10 s. The following primer sets were used for qPCR: COX-2, 5′-CTTGGGCACAGAGAGCA-3′ and 5′-AACTGCTCATCACCCCATTC-3′; 18S rRNA, 5′-GTAACCCGTTGAACCCCATT-3′ and 5′-CCATCCAAT CGGTAGTAGCG-3′; PKC-ζ, 5′-AGAGCCTCCAGTAGA CGACAA-3′ and 5′-CGGGATGAGGAAATGTAAG CAA-3′.
Western blot analysis
Cells were solubilized with ice-cold lysis buffer containing 50 mM Tris-HCl (pH 7.4), 1% NP-40, 150 mM NaCl, 0.5% Na-deoxycholate, 2 mM EDTA, 0.1% SDS, and 50 mM NaF. The extracted proteins (20 μg) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on 10–12% polyacrylamide gels, and electrophoretically transferred onto 0.45 μm PVDF membranes. Membranes were blocked with 5% (w/v) bovine serum albumin in Tris-buffered saline for 2 h at 4 °C and then incubated overnight with primary antibodies at a 1:1000 dilution in 5% (w/v) Tris-buffered saline containing 0.1% Tween-20. After incubation with secondary antibody for 2 h, proteins were visualized by ECL, and the band intensity was analyzed by using a ChemiDoc XRS densitometer and quantified by Quantity One software (Bio-Rad, Richmond, CA). Protein concentrations were estimated using the BCA method according to the supplier’s recommendations, by using bovine serum albumin as a standard.
Immunofluorescence
Cells grown on poly d-lysine-coated coverslips were treated with auranofin and/or ANXA5 siRNA in growth medium. The cells were fixed with 4% (w/v) paraformaldehyde in PBS, pH 7.4, for 30 min at 24 °C. After washing with PBS, the cells were blocked for 30 min in PBS containing 5% goat serum and 0.2% Triton X-100, then incubated with primary antibody (1:250) at 4 °C overnight, washed extensively, and stained for 6 h with goat anti-rabbit IgG-Texas Red (1:250). After further washes, the coverslips were mounted on glass slides using Ultra CruzTM mounting medium. Fluorescence signals were analyzed using an LSM 800 confocal laser scanning microscope (Carl Zeiss, Germany).
Dual luciferase reporter assay
Cells (1 × 106 cells/well) were cotransfected with 2.5 μg of human COX-2 promoter luciferase constructs and pRL-renilla vectors (Promega, Madison, WI), according to the manufacturer’s protocol, by using the NeonTM transfection system. Luciferase activities were measured consecutively by using the Dual Luciferase Assay System (Promega) with a FilterMax F3 microplate reader (Molecular Devices, CA).
NF-κB p65 transcription factor assay
Nuclear proteins were extracted from cells by using NE-PERTM Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific). NF-κB binding activity was measured by using the NF-κB p65 Transcription Factor Assay Kit (Abcam, UK). The assay was carried out as recommended in the assay kit manual.
PGE2 ELISA assay
Cells were transfected with ANXA5 siRNA and incubated for 48 h in 60-mm plates. The culture supernatants were used for measuring the PGE2 concentration. PGE2 concentration in the culture supernatants were determined by PGE2 high sensitivity ELISA kit (Enzo Life Science).
Statistical analysis
Statistical analysis was performed using one-way analysis of variance, followed by Dunnett’s pairwise multiple comparison t-test, with GraphPad Prism software (GraphPad Software Inc., CA) when appropriate. Differences were considered statistically significant at P < 0.05.
ACKNOWLEDGMENTS
This research was supported by the National Research Foundation of Korea (NRF) funded by the Korean government (MSIP) (NRF-2015R1A2A2A01003865 and NRF-2015R1A5A1008958).
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest with the contents of this article.
REFERENCES
1. Jeong JJ, Park N, Kwon YJ, Ye DJ, Moon A, Chun YJ. Role of annexin A5 in cisplatin-induced toxicity in renal cells: molecular mechanism of apoptosis. J Biol Chem. 2014; 289: 2469-2481.
2. Simsek B, Buyukcelik M, Soran M, Bayazit AK, Noyan A, Seydaoglu G, Anarat A. Urinary annexin V in children with nephrotic syndrome: a new prognostic marker? Pediatr Nephrol. 2008; 23: 79-82.
3. Park N, Chun YJ. Auranofin promotes mitochondrial apoptosis by inducing annexin A5 expression and translocation in human prostate cancer cells. J Toxicol Environ Health A. 2014; 77: 1467-1476.
4. Shin DW, Kwon YJ, Ye DJ, Baek HS, Lee JE, Chun YJ. Auranofin suppresses plasminogen activator inhibitor-2 expression through annexin A5 induction in human prostate cancer cells. Biomol Ther (Seoul). 2017; 25: 177-185
5. Arnold P, Lu X, Amirahmadi F, Brandl K, Arnold JM, Feng Q. Recombinant human annexin A5 inhibits proinflammatory response and improves cardiac function and survival in mice with endotoxemia. Crit Care Med. 2014; 42: e32-41.
6. Yamada R, Sano H, Hla T, Hashiramoto A, Fukui W, Miyazaki S, Kohno M, Tsubouchi Y, Kusaka Y, Kondo M. Auranofin inhibits interleukin-1beta-induced transcript of cyclooxygenase-2 on cultured human synoviocytes. Eur J Pharmacol. 1999; 385: 71-79.
7. Kim NH, Lee MY, Park SJ, Choi JS, Oh MK, Kim IS. Auranofin blocks interleukin-6 signalling by inhibiting phosphorylation of JAK1 and STAT3. Immunology. 2007; 122: 607-614.
8. Bondeson J, Sundler R. Auranofin inhibits the induction of interleukin 1 beta and tumor necrosis factor alpha mRNA in macrophages. Biochem Pharmacol. 1995; 50: 1753-1759.
9. Youn HS, Lee JY, Saitoh SI, Miyake K, Hwang DH. Auranofin, as an anti-rheumatic gold compound, suppresses LPS-induced homodimerization of TLR4. Biochem Biophys Res Commun. 2006; 350: 866-871.
10. Kobayashi H, Takeno M, Saito T, Takeda Y, Kirino Y, Noyori K, Hayashi T, Ueda A, Ishigatsubo Y. Regulatory role of heme oxygenase 1 in inflammation of rheumatoid arthritis. Arthritis Rheum. 2006; 54: 1132-1142.
11. Nelson WG, DeWeese TL, DeMarzo AM. The diet, prostate inflammation, and the development of prostate cancer. Cancer Metastasis Rev. 2002; 21: 3-16.
12. Dennis LK, Lynch CF, Torner JC. Epidemiologic association between prostatitis and prostate cancer. Urology. 2002; 60: 78-83.
13. Wang W, Bergh A, Damber JE. Chronic inflammation in benign prostate hyperplasia is associated with focal upregulation of cyclooxygenase-2, Bcl-2, and cell proliferation in the glandular epithelium. Prostate. 2004; 61: 60-72.
14. Lawrence T. The nuclear factor NF-κB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009; 1: a001651.
15. Tak PP, Firestein GS. NF-κB: a key role in inflammatory diseases. J Clin Invest. 2001; 107: 7-11.
16. Lee JY, Lee BH. Gambogic acid disrupts toll-like receptor4 activation by blocking lipopolysaccharides binding to myeloid differentiation factor 2. Toxicol Res. 2015; 31: 11-16.
17. Plummer SM, Holloway KA, Manson MM, Munks RJ, Kaptein A, Farrow S, Howells L. Inhibition of cyclo-oxygenase 2 expression in colon cells by the chemopreventive agent curcumin involves inhibition of NF-κB activation via the NIK/IKK signalling complex. Oncogene. 1999; 18: 6013-6020.
18. Wu D, Marko M, Claycombe K, Paulson KE, Meydani SN. Ceramide-induced and age-associated increase in macrophage COX-2 expression is mediated through up-regulation of NF-κB activity. J Biol Chem. 2003; 278: 10983-10992.
19. Ki SH, Choi MJ, Lee CH, Kim SG. Gα12 specifically regulates COX-2 induction by sphingosine 1-phosphate. Role for JNK-dependent ubiquitination and degradation of IκBα. J Biol Chem. 2007; 282: 1938-1947.
20. Mattioli I, Sebald A, Bucher C, Charles RP, Nakano H, Doi T, Kracht M, Schmitz ML. Transient and selective NF-κB p65 serine 536 phosphorylation induced by T cell costimulation is mediated by IκB kinase β and controls the kinetics of p65 nuclear import. J Immunol. 2004; 172: 6336-6344.
21. Aupperle KR, Bennett BL, Boyle DL, Tak PP, Manning AM, Firestein GS. NF-κB regulation by IκB kinase in primary fibroblast-like synoviocytes. J Immunol. 1999; 163: 427-433.
22. Chen ZJ, Parent L, Maniatis T. Site-specific phosphorylation of IκBα by a novel ubiquitination-dependent protein kinase activity. Cell. 1996; 84: 853-862.
23. Zhong H, May MJ, Jimi E, Ghosh S. The phosphorylation status of nuclear NF-κB determines its association with CBP/p300 or HDAC-1. Mol Cell. 2002; 9: 625-636.
24. Duran A, Diaz-Meco MT, Moscat J. Essential role of RelA Ser311 phosphorylation by ζPKC in NF-κB transcriptional activation. EMBO J. 2003; 22: 3910-3918.
25. Huang X, Chen LY, Doerner AM, Pan WW, Smith L, Huang S, Papadimos TJ, Pan ZK. An atypical protein kinase C (PKCζ) plays a critical role in lipopolysaccharide-activated NF-κB in human peripheral blood monocytes and macrophages. J Immunol. 2009; 182: 5810-5815.
26. Diaz-Meco MT, Dominguez I, Sanz L, Dent P, Lozano J, Municio MM, Berra E, Hay RT, Sturgill TW, Moscat J. ζ PKC induces phosphorylation and inactivation of IκB-α in vitro. EMBO J. 1994; 13: 2842-2848.
27. Dubois T, Mira JP, Feliers D, Solito E, Russo-Marie F, Oudinet JP. Annexin V inhibits protein kinase C activity via a mechanism of phospholipid sequestration. Biochem J. 1998; 330: 1277-1282.
28. Kheifets V, Bright R, Inagaki K, Schechtman D, Mochly-Rosen D. Protein kinase C δ (δPKC)-annexin V interaction: a required step in δPKC translocation and function. J Biol Chem. 2006; 281: 23218-23226.
29. Hoque M, Rentero C, Cairns R, Tebar F, Enrich C, Grewal T. Annexins - scaffolds modulating PKC localization and signaling. Cell Signal. 2014; 26: 1213-1225.
30. Yang CM, Lee IT, Lin CC, Yang YL, Luo SF, Kou YR, Hsiao LD. Cigarette smoke extract induces COX-2 expression via a PKCα/c-Src/EGFR, PDGFR/PI3K/Akt/NF-κB pathway and p300 in tracheal smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2009; 297: L892-902.
31. Onodera Y, Teramura T, Takehara T, Shigi K, Fukuda K. Reactive oxygen species induce Cox-2 expression via TAK1 activation in synovial fibroblast cells. FEBS Open Bio. 2015; 5: 492-501.
32. Jung SK, Ha SJ, Kim YA, Lee J, Lim TG, Kim YT, Lee NH, Park JS, Yeom MH, Lee HJ, Lee KW. MLK3 is a novel target of dehydroglyasperin D for the reduction in UVB-induced COX-2 expression in vitro and in vivo. J Cell Mol Med. 2015; 19: 135-142.
33. Gutierrez-Venegas G, Gonzalez-Rosas Z. Apigenin reduce lipoteichoic acid-induced inflammatory response in rat cardiomyoblast cells. Arch Pharm Res. 2017; 40: 240-249.
34. Subbarayan V, Sabichi AL, Llansa N, Lippman SM, Menter DG. Differential expression of cyclooxygenase-2 and its regulation by tumor necrosis factor-α in normal and malignant prostate cells. Cancer Res. 2001; 61: 2720-2726.
35. Jeon KI, Jeong JY, Jue DM. Thiol-reactive metal compounds inhibit NF-κB activation by blocking I kappa B kinase. J Immunol. 2000; 164: 5981-5989.
36. Sakurai H, Suzuki S, Kawasaki N, Nakano H, Okazaki T, Chino A, Doi T, Saiki I. Tumor necrosis factor-α-induced IKK phosphorylation of NF-κB p65 on serine 536 is mediated through the TRAF2, TRAF5, and TAK1 signaling pathway. J Biol Chem. 2003; 278: 36916-36923.
37. Moles A, Sanchez AM, Banks PS, Murphy LB, Luli S, Borthwick L, Fisher A, O’Reilly S, van Laar JM, White SA, Perkins ND, Burt AD, Mann DA, et al. Inhibition of RelA-Ser536 phosphorylation by a competing peptide reduces mouse liver fibrosis without blocking the innate immune response. Hepatology. 2013; 57: 817-828.
38. He D, Natarajan V, Stern R, Gorshkova IA, Solway J, Spannhake EW, Zhao Y. Lysophosphatidic acid-induced transactivation of epidermal growth factor receptor regulates cyclo-oxygenase-2 expression and prostaglandin E(2) release via C/EBPβ in human bronchial epithelial cells. Biochem J. 2008; 412: 153-162.
39. Metzger R, Danenberg K, Leichman CG, Salonga D, Schwartz EL, Wadler S, Lenz HJ, Groshen S, Leichman L, Danenberg PV. High basal level gene expression of thymidine phosphorylase (platelet-derived endothelial cell growth factor) in colorectal tumors is associated with nonresponse to 5-fluorouracil. Clin Cancer Res. 1998; 4: 2371-2376.
40. Sierra A, Gottfried-Blackmore AC, McEwen BS, Bulloch K. Microglia derived from aging mice exhibit an altered inflammatory profile. Glia. 2007; 55: 412-424.
41. Zurney J, Howard KE, Sherry B. Basal expression levels of IFNAR and Jak-STAT components are determinants of cell-type-specific differences in cardiac antiviral responses. J Virol. 2007; 81: 13668-13680.
42. Shabbeer S, Kortenhorst MS, Kachhap S, Galloway N, Rodriguez R, Carducci MA. Multiple Molecular pathways explain the anti-proliferative effect of valproic acid on prostate cancer cells in vitro and in vivo. Prostate. 2007; 67: 1099-1110.
43. Li X, Gao T. mTORC2 phosphorylates protein kinase Cζ to regulate its stability and activity. EMBO Rep. 2014; 15: 191-198.
44. Le Good JA, Ziegler WH, Parekh DB, Alessi DR, Cohen P, Parker PJ. Protein kinase C isotypes controlled by phosphoinositide 3-kinase through the protein kinase PDK1. Science. 1998; 281: 2042-2045.
45. Chou MM, Hou W, Johnson J, Graham LK, Lee MH, Chen CS, Newton AC, Schaffhausen BS, Toker A. Regulation of protein kinase C ζ by PI 3-kinase and PDK-1. Curr Biol. 1998; 8: 1069-1077.
46. Standaert ML, Bandyopadhyay G, Kanoh Y, Sajan MP, Farese RV. Insulin and PIP3 activate PKC-ζ by mechanisms that are both dependent and independent of phosphorylation of activation loop (T410) and autophosphorylation (T560) sites. Biochemistry. 2001; 40: 249-255.
47. Kim JS, Park ZY, Yoo YJ, Yu SS, Chun JS. p38 kinase mediates nitric oxide-induced apoptosis of chondrocytes through the inhibition of protein kinase C ζ by blocking autophosphorylation. Cell Death Differ. 2005; 12: 201-212.