
INTRODUCTION
As an aggressive and usually fatal malignancy, malignant melanoma (MM) is one of the fastest rising cancer and has a poor prognosis [1]. At present, the main treatment of melanoma is surgical resection, but only patients in early stage can be cured [2]. Once it has progressed to the metastatic stage, it remains an incurable disease, with a 5-year survival rate of 16% [3]. Until now, most chemotherapeutics and immunotherapeutics as well as radiotherapeutics have failed to increase survival rates of patients with malignant melanoma [4, 5]. Therefore, investigating some novel target therapy with high efficiency for malignant melanoma are problems to be solved.
Compared with normal cells, there are significant differences of metabolism within tumor cells [6]. Pyrimidine bases are elementary precursors used in nucleic acids biosynthesis, and are considered to be important for cellular metabolism, especially in rapid growth cells, such as cancer cells. Therefore, pyrimidine bases are suggested to be ideal targets for cancer treatment. As a kind of iron flavin dependent enzyme, dihydroorotate dehydrogenase (DHODH, EC 1.3.3.1), which is localized at the inner mitochondrial membrane, is the fourth and a key enzyme in the de novo biosynthesis of pyrimidines [7]. DHODH catalyzes oxidation of dihydroorotate to orotate, which is precursors of uridines and cytidine nucleosides [8]. Recently, DHODH was reported to play essential roles during tumorigenesis and cancer development [9–11]. These evidences indicated that DHODH might be a potential target for drug intervention in cancer treatment.
Early in 1959, the anti-proliferative effect of DHODH inhibitors was applied in tumor cells [12]. During last decades, researchers had discovered multiple DHODH inhibitors, such as leflunomide, brequinar, teriflunomide (A77 1726), benzimidazole and so on [13, 14]. As classic DHODH inhibitors, leflunomide and its active metabolite A77 1726 have been demonstrated to suppress cell proliferation or to induce cell death in various tumors [15–17]. Importantly, DHODH inhibition by leflunomide induced a significant decrease in melanoma growth both in vitro and in vivo studies [18]. Several other studies also showed that teriflunomide could suppress growth of melanoma cells [16, 19]. However, the mechanisms underlying remained to be further explored.
In this paper, we confirmed the function of leflunomide in human melanoma cells. Our studies put forward that DHODH inhibition by leflunomide or shRNA knockdown suppressed tumor growth and induced apoptosis and autophagy in melanoma cells. Besides, we also explored the molecular mechanisms underlying. Our findings provided evidences for the potential of therapeutic leflunomide using as a novel agent for melanoma treatment.
RESULTS
DHODH inhibitor leflunomide inhibits cell proliferation and induces cell cycle arrest at S phase in melanoma cells
To explore the effect of DHODH inhibition by leflunomide, we detected cell growth and proliferation by cell counting method, MTT assay and Brdu assay in human melanoma A375 and MV3 cells after treatment of leflunomide. Under the microscope, cells dealt with different concentrations of leflunomide for 72 h, resulting in a significant reduction in the viable cell number in a dose-dependent manner (Supplementary Figure 1A and 1B). Then we implied MTT assay, and the results showed that cell proliferation was markedly decreased in 50 μM, 100 μM and 200 μM leflunomide-treated groups compared with DMSO-treated groups (Figure 1A). Brdu staining assay also showed that cells dealt with 100 μM leflunomide for 72 h resulted in a remarkable decrease in the percentage of Brdu positive cells, compared with control groups (Figure 1B). These results certified that leflunomide inhibited cell growth and proliferation in human melanoma cells.
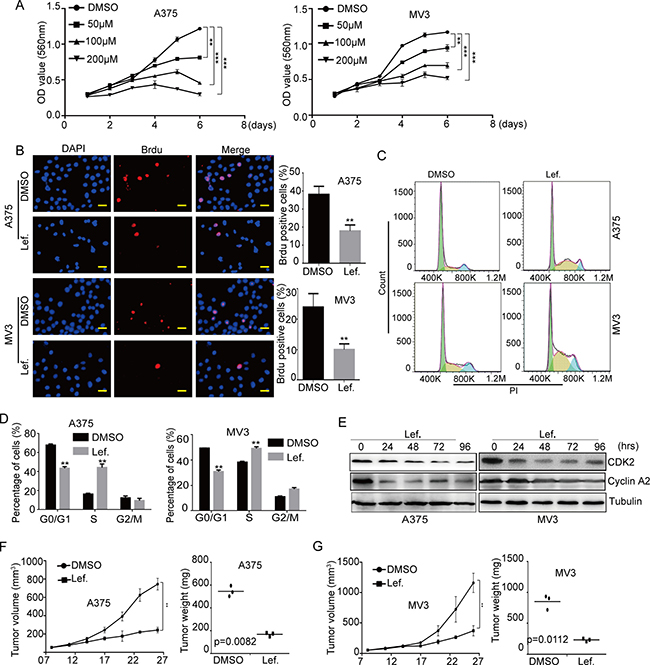
Figure 1: DHODH inhibitor leflunomide inhibits cell proliferation and induces cell cycle arrest at S phase in melanoma cells. (A) Cell growth was tested by the MTT assay in A375 and MV3 cells after treated DMSO or 50, 100, 200 μM leflunomide (Lef.) for 1–6 days. (B) Image and quantification of A375 and MV3 cells positive for Brdu staining after treating with DMSO or 100 μM leflunomide for 72 h, Scale bar, 20 μm. (C and D) The cell cycle of A375 and MV3 cells was analyzed by flow cytometry after treatment with DMSO or leflunomide. (E) Western blot assay was performed to assess the cell cycle-related protein levels in A375 and MV3 cells after treatment with leflunomide for 0, 24, 48, 72 and 96 hours. Tubulin was used as a loading control. (F and G) The six weeks old female nude mice (BALA/c) xenograft tumor after treatment with DMSO or 7.5 mg/kg leflunomide. Xenograft tumor volume and weight were analyzed. All data are shown as the mean ± SD, Student’s t-test was carried out. **p < 0.01, ***p < 0.001.
Since cell proliferation is usually regulated by cell cycle, we analyzed cell cycle of A375 and MV3 by flow cytometry to investigate whether leflunomide inhibited cell proliferation by inducing the cell cycle arrest. Cell cycle assessment showed that leflunomide-treated cells resulted into a distinct S phase arrest in A375 and MV3 cells, compared with the DMSO-treated group (Figure 1C and 1D). To confirm this result, we measured the expression of CDK2 and CyclinA2, which could promote cell cycle to go through the S phase. We found that the expression level of CDK2 and CyclinA2 were decreased in A375 and MV3 cells after leflunomide treatment in a time-course manner (Figure 1E). As DHODH inactivation blocked the catalyzation of dihydroorotate to orotate, which was precursor of uridine [8], we used 1 mM exogenous uridine to rescue nutritive deficiency of leflunomide-treated cells. The result showed that exogenous uridine could retrieve leflunomide induced cell cycle arrest (Supplementary Figure 1C and 1D). These results suggested that leflunomide induced cell cycle arrest by downregulating of CDK2-cyclin A2 complex in human melanoma.
Then soft agar assay was implemented to assess the effect of leflunomide in the self-renewal of melanoma cells. The result showed that A375 and MV3 dealt with leflunomide came into lesser and smaller colonies compared to DMSO group (Supplementary Figure 1E). To explore the effect of leflunomide in xenograft tumor, the melanoma A375 and MV3 cells were transplanted subcutaneously into female BALA/c nude mice, after seven days injected with 7.5 mg/kg leflunomide every three days for 12 days. At last the mice were sacrificed, and the formed tumors were removed. The results showed that leflunomide treatment significantly blocked tumor growth in both weight and size in nude mice (Figure 1F and 1G). These results indicated that leflunomide might be a potent target drug for melanoma treatment.
Leflunomide induces apoptosis and autophagy in melanoma cells
To detect whether viable cell number reduction after dealt with leflunomide was caused by apoptosis, cells were treated with 100 μM leflunomide or isometric DMSO for 72 h, then stained with AnnexinV-FITC/PI and analysed by flow cytometry. The results showed that leflunomide treatment brought out distinct apoptosis in A375 cells (Figure 2A). To further confirm it, we conducted Western blot assay and found that cleaved caspase-9 and cleaved caspase-3, which represented the caspase-dependent pathway of apoptosis, were increased compared with the control groups in A375 cells (Figure 2B and Supplementary Figure 2A). Besides, exogenous uridine also recovered leflunomide-induced apoptosis (Supplementary Figure 2B). These results showed that leflunomide induced apoptosis in A375 cells.
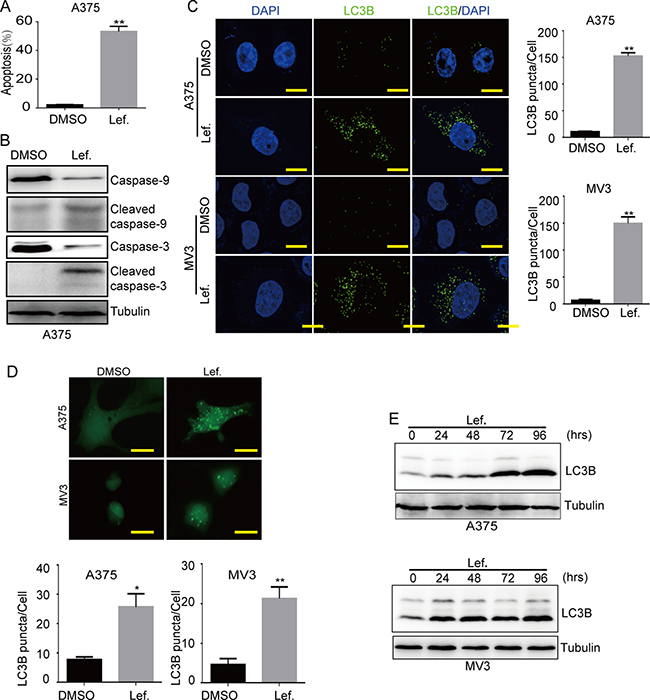
Figure 2: Leflunomide induces apoptosis and autophagy in melanoma cells. (A) The rate of apoptosis of A375 cells was analyzed using flow cytometry after treated with DMSO or 100 μM leflunomide for 72 h. (B) Western blot assay was performed to assess the cell apoptosis-related protein levels in A375 cells after treatment with DMSO or leflunomide for 72 h, respectively. Tubulin was used as a loading control. (C) Immunofluorescence staining with a LC3B antibody was performed to confirm the induction of autophagy after treated with DMSO or 100 μM leflunomide for 72 h. Representative LC3B-positive cells are shown. Scale bars, 10 μm. (D) Cells transfected with the GFP-LC3B plasmid after treated with DMSO or 100 μmol/L leflunomide for 72 h were examined using fluorescence microscopy. Scale bars, 10 μm. The quantification of LC3B-positive puncta is presented as a histogram. (E) The level of autophagy was evaluated by LC3B expression, as determined using Western blot in A375 and MV3 cells after treated with 100 μmol/L leflunomide for 0, 24, 48, 72 and 96 hours. Tubulin was used as the loading control. All data are shown as the mean ± SD, Student’s t-test was carried out. *p < 0.05,**p < 0.01.
At the same time, we observed that there were more vesicles in the cells treated with leflunomide compared to the control groups under microscope. It was reported that hallmarks of autophagosome formation include the conversion of LC3A (LC3-I, 16 kd) to phosphatidylethanolamine (PE)-conjugated LC3B (LC3-II, 14 kd) during autophagosome closure [20]. In our further research, immunofluorescence was performed to detect the formation of LC3B, the results presented that leflunomide-treated cells showed more aggregations of LC3B puncta in both MV3 and A375 cells compared to the control groups (Figure 2C). Then, we transiently transfected melanoma cells with GFP-LC3B plasmids in both A375 and MV3 cells and observed the cells under a fluorescence microscope, and the results presented that leflunomide-treated cells showed more aggregations of GFP-LC3B puncta compared to DMSO-treated groups (Figure 2D). According to these results, we tested the expression of LC3B by Western blot assay in both A375 and MV3 cells, and the results showed a persistently increased conversion of LC3B in leflunomide-treated cells in a time-dependent manner (Figure 2E). In addition, exogenous uridine retrieved aggregations of LC3B puncta induced by leflunomide in both MV3 and A375 cells (Supplementary Figure 2C and 2D). To explore whether autophagy induced by leflunomide treatment promoted cell survival or cell death, we used two autophagy inhibitors, 3-MA (10 mM) and chloroquine (CQ, 25 μM) to block autophagy in A375 and MV3 cells. The results showed that cell viabilities were partly rescued after 3-MA or CQ treatment in leflunomide-treated groups (Supplementary Figure 2E). All together, these observations suggested that DHODH inactivation induced autophagy and contributed to cell death in human melanoma cells.
DHODH knockdown suppresses cell proliferation and induces cell cycle arrest at S phase in melanoma cells
To address the importance of DHODH in cell proliferation and cell cycle, we used lentivirus carrying small hairpin RNA (shRNA) constructing against DHODH (shDHODH) or a scramble control to infect the human melanoma cells A375 and MV3, then cells were selected by puromycin. Western blot analysis showed that there was a significantly downregulation of DHODH in these two melanoma cells compared with scramble groups (Figure 3A). Then cell growth curvature was graphed according to the results of MTT assay after the knockdown of DHODH. The results showed that cell proliferation was distinctly reduced after DHODH deficiency compared with the scramble groups (Figure 3B). Brdu staining assay also showed that knockdown of DHODH resulted in a remarkable decrease in the percentage of Brdu positive cells compared with control groups (Figure 3C). These results indicated that DHODH was important for cell proliferation in human melanoma cells.
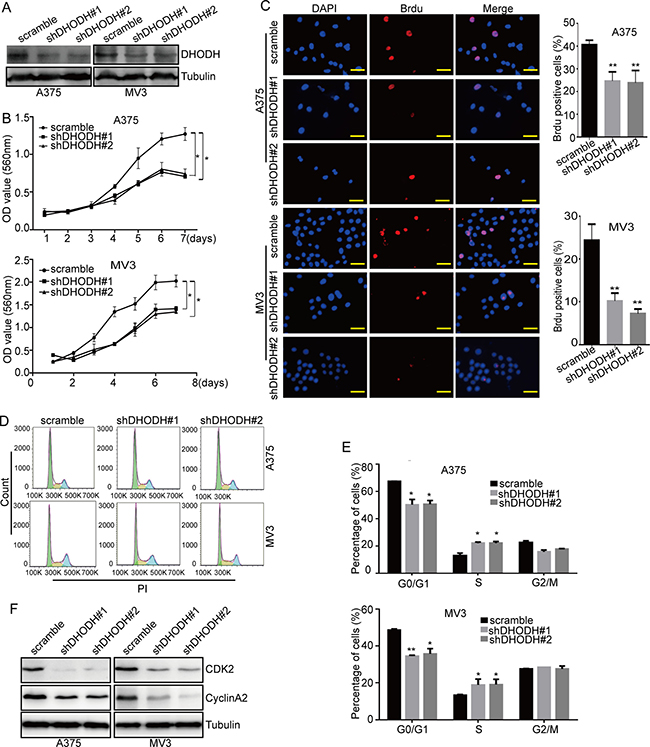
Figure 3: DHODH knockdown suppresses cell proliferation and induces cell cycle arrest at S phase in melanoma cells. (A) Western blot assay was used to characterize the expression of DHODH in DHODH-knockdown A375 and MV3 cells, scramble was used as the control. The proteins of stable DHODH knockdown cell lines were collected after 48 hours normal culture. Tubulin was used as a loading control. (B) Cell growth was tested by the MTT assay in A375 and MV3 cells after DHODH knockdown. (C) Image and quantification of A375 and MV3 cells positive for Brdu staining after DHODH knockdown, Scale bar, 40 μm. (D and E) The cell cycle of A375 and MV3 cells was analyzed by flow cytometry after DHODH knockdown. (F) Western blot assay was performed to assess the cell cycle-related protein levels in A375 and MV3 cells after DHODH knockdown. The proteins of stable DHODH knockdown cell lines were collected after 48 hours normal culture. Tubulin was used as a loading control. All data are shown as the mean ± SD, Student’s t-test was carried out. *p < 0.05,**p < 0.01.
Then, we analyzed the cell cycle by flow cytometry to investigate whether cell proliferation inhibition induced by DHODH downregulation was related to cell cycle progression. As expected, DHODH-silenced cells resulted into a remarkable S phase arrest in human melanoma cells, compared with the control groups (Figure 3D and 3E). To affirm the results, we measured the expression of CDK2 and CyclinA2 by Western blot assay and found that the expression levels of CDK2 and CyclinA2 were decreased in DHODH-silenced cells (Figure 3F). In addition, exogenous uridine also retrieved leflunomide-induced cell cycle arrest (Supplementary Figure 3A and 3B). These results suggested that knockdown of DHODH induced cell cycle arrest in human melanoma cells by downregulating of the Cyclin A2-CDK2 complex. Then, the role of DHODH in colony formation was evaluated by soft agar assay. The results showed that DHODH-silenced cells gave rise to lesser and smaller colonies compared with scramble group (Supplementary Figure 3C). These results demonstrated that DHODH was essential for melanoma progression in human melanoma cells.
DHODH downregulation sensitizes cells to drug-induced apoptosis and induces autophagy in melanoma cells
To detect whether DHODH knockdown induced apoptosis in melanoma cells, we conducted flow cytometry to analyze apoptosis in DHODH knockdown cells. As showed in Supplementary Figure 4A, there was no significant apoptosis in A375 and MV3 cells after DHODH knockdown. So we supposed that DHODH might sensitize drug-induced apoptosis. Actinomycin D and doxorubicin were commonly used as apoptosis inducers, while temozolomide was used as a chemotherapy drug in melanoma treatment. To further explored whether DHODH downregulation affected apoptosis induced by these drugs, DHODH-silenced human melanoma cells were dealt with 1 nM actinomycin D for 24 h, 0.5 μM doxorubicin for 12 h, or 400 μM temozolomide for 72 h severally (DMSO as the control), then analyzed by flow cytometry. In comparison, both actinomycin D and doxorubicin were able to induce more pronounced cell apoptosis in A375 cells (Figure 4A). Accordingly, we tested the expression levels of cleaved caspase-9 and cleaved caspase-3 by Western blot assay and found that cleaved caspase-3 was increased in A375 cells after DHODH knockdown compared with the control group (Figure 4B). When exogenous uridine was added in medium, apoptosis induced by these drugs treatment was abrogated in DHODH-knockdown cells (Supplementary Figure 4B). These results showed that DHODH downregulation sensitized drug-induced apoptosis in A375 melanoma cells.
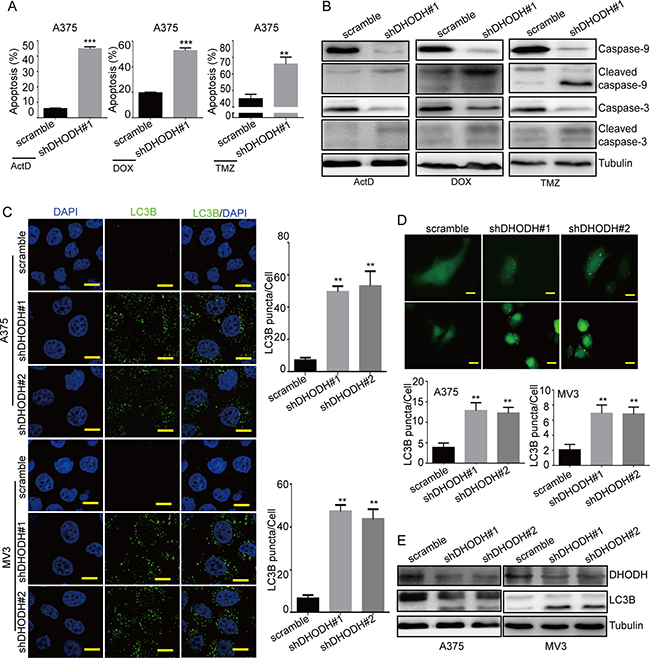
Figure 4: DHODH downregulation sensitizes drug-induced apoptosis and induces autophagy in melanoma cells. (A) The rate of apoptosis was analyzed using flow cytometry in DHODH-knockdown A375 cells in the presence of 1 nM actinomycin D (ActD) for 24 h, 0.5 μM doxorubicin (DOX) for 12 h, or 400 μM temozolomide (TMZ) for 72 h in culture medium. (B) Western blot assay was performed to assess the cell apoptosis-related protein levels in DHODH-knockdown A375 cells in the presence of 1 nM actinomycin D (ActD) for 24 h, 0.5 μM doxorubicin (DOX) for 12 h, or 400 μM temozolomide (TMZ) for 72 h in culture medium. Tubulin was used as a loading control. (C) Immunofluorescence staining with a LC3B antibody was performed to confirm the induction of autophagy in A375 and MV3 cells after DHODH knockdown. Representative LC3B-positive cells are shown. Scale bars, 10 μm. (D) A375 and MV3 cells transfected with the GFP-LC3B plasmid after DHODH knockdown were examined using fluorescence microscopy. Scale bars, 10 μm. The quantification of LC3B-positive puncta is presented as a histogram. (E) The level of autophagy was evaluated by LC3B expression, as determined using Western blot in A375 and MV3 cells after DHODH knockdown. Tubulin was used as a loading control. All data are shown as the mean ± SD, Student’s t-test was carried out. **p < 0.01, ***p < 0.001.
Furthermore, we evaluated the autophagy level in DHODH-silenced human melanoma cells. By immunofluorescent assay, we found that DHODH-silenced cells showed more puncta aggregations of LC3B, which was different from the diffuse LC3B observed in the control groups (Figure 4C and 4D). Then we detected the expression of LC3B and the result showed that LC3B expression was also increased in DHODH-knockdown cells than that in control groups (Figure 4E). Besides, increased LC3B puncta aggregations induced by DHODH deficiency could be rescued by exogenous uridine (Supplementary Figure 4C and 4D). In conclusions, these clues indicated that DHODH deficiency induced autophagy in A375 and MV3 melanoma cells.
BCL-2 is a switch of apoptosis induced by DHODH inhibition
During our experiments, we found that there was no apoptosis in MV3, neither in leflunomide-treated group nor apoptosis inducers-treated group (Figure 5A and 5B). Accidently, we found that there was a higher expression of BCL-2 in MV3cells than that in A375 cells (Figure 5C). As a big family in mitochondria, BCL-2 protein family, which contained at least 16 members, played an important role in the modulation of intrinsic mitochondrial apoptosis pathway [21]. They were categorized into three functional groups: the pro-survival BCL-2-like family members, including BCL-2, MCL-2, BCL-XL, BFL1 and BCL-w; the multi-BH-domain pro-apoptotic members, such as BAX and BAK; and the pro-apoptotic BH3-only proteins, such as BIM, BID, BIK, PUMA, NOXA, BAD, HRK, BLK and BMF [22]. We conducted a qRT-PCR to detect the different expression of these members in MV3 and A375 cells, and the results showed that there was less bcl-2 expression in A375 cells (Supplementary Figure 5A). These results indicated that BCL-2 might be a critical factor of apoptosis induced by DHODH inhibition in human melanoma cells.
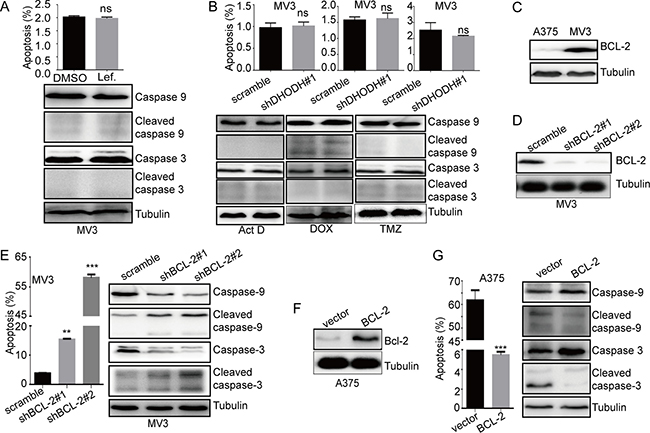
Figure 5: BCL-2 is a switch of apoptosis induced by DHODH inhibition. (A) The rate of apoptosis of MV3 cells was analyzed using flow cytometry after treated with 100 μM leflunomide, and Western blot assay was performed to assess the cell apoptosis-related protein levels, respectively. (B) The rate of apoptosis of MV3 cells was analyzed by using flow cytometry after DHODH knockdown in the presence of 1nM actinomycin D (ActD) for 24 h, 0.5 μM doxorubicin (DOX) for 12 h, or 400 μM temozolomide (TMZ) for 72 h in medium. Western blot assay was performed to assess the cell apoptosis-related protein levels, respectively. (C) The expressiom of BCL-2 in human melanoma cells A375 and MV3 was measured by Western blot assay. (D) Western blot assay was performed to assess the protein levels of BCL-2 in BCL-2-knockdown MV3 cells. (E) The rate of apoptosis analyzed by flow cytomentry in BCL-2-knockdown MV3 cells after dealt with 100 μM leflunomide, and Western blot assay was performed to assess the cell apoptosis-related protein levels, respectively. (F) Western blot assay was performed to assess the protein levels of BCL-2 in BCL-2-overexpressed A375 cells. (G) The rate of apoptosis in BCL-2-overexpressed A375 cells after dealt with 100 μM leflunomide, and Western blot assay was performed to assess the cell apoptosis-related protein levels, respectively. All data are shown as the mean ± SD, Student’s t-test was carried out. **p < 0.01, ***p < 0.001, ns, no sense.
To confirm this result, MV3 cells was infected with lentivirus which carrying small hairpin RNA (shRNA) constructing against BCL-2 (shBCL-2) or a scramble control, subsequently selected by puromycin. Western blot analysis showed that BCL-2 was significantly downregulated after knockdown by BCL-2 shRNAs in MV3 cells (Figure 5D). Then cell apoptosis was confirmed by flow cytometry assay in BCL-2-silenced MV3 cells, the result showed that BCL-2 knockdown didn’t induce apoptosis (Supplementary Figure 5B). However, leflunomide treatment induced marked apoptosis in BCL-2-silenced MV3 cells contrasted with scramble groups (Figure 5E). Besides, we measured the expression of cleaved caspase-9 and cleaved caspase-3 and the result showed that the spliceosomes of caspase-9 and caspase-3 were increased in BCL2-silenced MV3 cell which dealt with leflunomide (Figure 5E). In addition, similar results also showed in MV3 cells treated with ABT, a specific BCL-2 inhibitor (Supplementary Figure 5C and 5D). At the same time, A375 cells were infected with lentivirus encoding BCL-2, subsequently selected by puromycin. Western blot analysis showed that BCL-2 was significantly upregulated in A375 cells (Figure 5F). Similarly, we conducted cell apoptosis by flow cytometry, and measured the expression of caspase-3 by Western blot assay. The results showed that overexpression of BCL-2 didn’t affect apoptosis in normal culture condition (Supplementary Figure 5E), but rescued apoptosis induced by leflunomide in A375 cells (Figure 5G). In conclusions, these results indicated that BCL-2 was a switch of apoptosis induced by DHODH inhibition.
DHODH inactivation/deficiency-induced cell cycle arrest and autophagy are correlated with BCL-2 expression
Recently, BCL-2 was shown to play essential roles during cell cycle and autophagy regulation [23–25]. So we supposed that BCL-2 might also play some roles in the regulation of cell cycle arrest and autophagy induced by DHODH inactivation/deficiency. Western blot showed that CDK2 was increased in BCL-2 knockdown MV3 cells, while decreased in BCL-2 overexpressed A375 cells, in the presence of 100 μM leflunomide (Figure 6A and 6B). Flow cytometry results also showed that cell cycle arrest at S phase was also aggravated in BCL-2 overexpressed A375 cells, while remitted in BCL-2 knockdown MV3 cells, in the presence of 100 μM leflunomide (Figure 6C and 6D). These results indicated that BCL-2 inhibited cell cycle progression in melanoma cells. In addition, LC3B expression increased in BCL-2 knockdown MV3 cells, while decreased in BCL-2 overexpressed A375 cells, in the presence of 100 μM leflunomide (Figure 6A and 6B). Besides, LC3B puncta observed under laser confocal microscope showed similar results (Figure 6E and 6F). These results indicated that BCL-2 was an autophagy inhibitor in leflunomide-treated melanoma cells. In summary, BCL-2 was a key regulator of autophagy and cell cycle arrest induced by DHODH inhibition in melanoma cells.
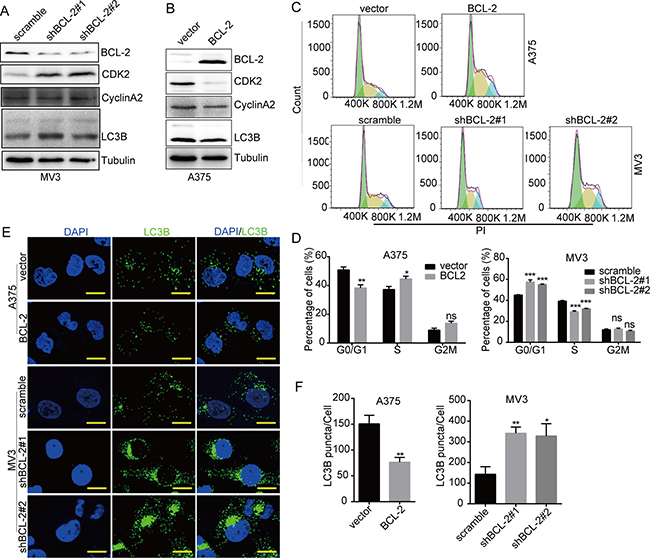
Figure 6: DHODH inhibition-induced cell cycle arrest and autophagy are regulated by BCL-2. (A) Western blot assay was performed to assess the protein levels of cell cycle-related proteins and LC3B in BCL-2 knockdown MV3 cells in the presence of 100 μM leflunomide in medium for 72 h. (B) Western blot assay was performed to assess the protein levels of cell cycle-related proteins and LC3B in BCL-2 overexpressed A375 cells in the presence of 100 μM leflunomide in medium for 72 h. (C and D) The cell cycle of BCL-2 overexpressed A375 and BCL-2 knockdown of MV3 cells was analyzed by flow cytometry in the presence of 100 μM leflunomide in medium for 72 h. (E and F) Immunofluorescence staining with a LC3B antibody was performed to confirm the induction of autophagy in BCL-2 overexpressed A375 and BCL-2 knockdown of MV3 cells in the presence of 100 μM leflunomide in medium for 72 h. Representative LC3B-positive cells are shown. Scale bars, 10 μm. All data are shown as the mean ± SD, Student’s t-test was carried out. *p < 0.05,**p < 0.01, ***p < 0.001, ns, no sense.
DHODH inactivation/deficiency induces autophagy via AMPK-Ulk axis and BCL-2 phosphorylation
It was reported that starvation could induce activation of AMPK, and the subsequent phosphorylation (Ser 555) and activation of Ulk1, and in turn the induction of autophagy [26–31]. So we want to detect whether AMPK-Ulk axis was essential during autophagy caused by DHODH inhibition-induced nucleotides starvation. As expected, AMPK was activated by phosphorylation of Thr 172 in MV3 and A375 cells after treated with 100 μM leflunomide for 72 hours (Figure 7A) or DHODH knockdown (Figure 7B). Besides, Ulk was inactivated by phosphorylation of Ser 555 in MV3 and A375 cells after treated with 100 μM leflunomide for 72 hours or DHODH knockdown (Figure 7A and 7B). These evidences indicated that AMPK-Ulk axis participated into DHODH inhibition-induced autophagy. It was reported that BCL-2 interacted with Beclin1 and inhibited its downstream activation in autophagy [32]. While phospho-JNK could phosphorylate BCL-2 and inhibit it to interact with Beclin1 [32]. So we detected whether BCL-2 was phosphorylated during this process. As expected, BCL-2 was further phosphorylated at Thr 87 in MV3 and A375 cells after treated with 100 μM leflunomide for 72 hours or DHODH knockdown (Figure 7A and 7B). Besides, the interaction between BCL-2 and Beclin1 was weakened after treated with 100 μM leflunomide for 72 hours or DHODH knockdown (Figure 7C and 7D). These evidences indicated that BCL-2 was also involved into DHODH inhibition-induced autophagy. In summary, DHODH inactivation/deficiency might induce autophagy through AMPK-Ulk axis and JNK-BCL-2 axis (Figure 7E).
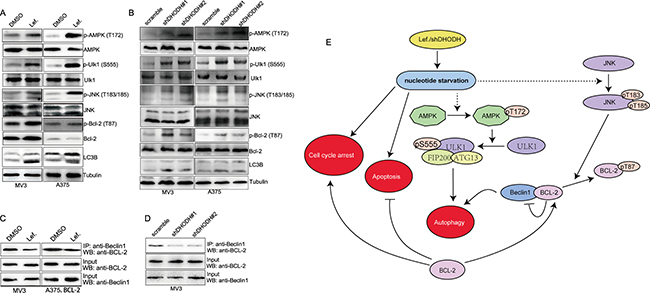
Figure 7: DHODH inhibition induces autophagy via AMPK-Ulk axis and BCL-2 phosphorylation. (A) Western blot assay was performed to assess the protein levels of AMPK-Ulk axis and BCL-2 phosphorylation in MV3 and A375 cells treated with 100 μM leflunomide or DMSO for 72 h. (B) Western blot assay was performed to assess the protein levels of AMPK-Ulk axis and BCL-2 phosphorylation in MV3 and A375 cells after DHODH knockdown. (C) co-Immunoprecipitation assay was performed to detect the interaction between BCL-2 and Beclin1 in MV3 and BCL-2 overexpressed A375 cells treated with 100 μM leflunomide or DMSO for 72 h. (D) Co-Immunoprecipitation assay was performed to detect the interaction between BCL-2 and Beclin1 in MV3 cells after DHODH knockdown. (E) Overview of the mechanism of DHODH inhibition induced cell cycle arrest, apoptosis and autophagy. DHODH knockdown or leflunomide treatment induced nucleotide starvation in melanoma cells, and induced cell cycle arrest, apoptosis, and autophagy, which could be influenced by BCL-2 levels in cells. In this process, AMPK-Ulk axis was activated to induce autophagy and JNK and BCL-2 was phosphorylated to decrease the interaction between BCL-2 and Beclin1, thus induced autophagy.
DISCUSSION
At present, melanoma is one of the most aggressive and drug-resistant human tumors. However, the genetic events that driving melanoma progression are less understood, and much efforts are required to identify driving factors and susceptible factors that could be pharmaceutical targets of melanoma. Since cancerous cells need more nutrient substance to produce new cells, metabolic signaling pathways are suggested to be essential during carcinogenesis [33]. As a rate-limiting enzyme of nucleotide metabolism pathway, DHODH seemed to contribute to malignant characteristics of various tumors, including multiple myeloma [16], chronic lymphocytic leukemia [34–37], neuroblastoma [38], colon cancer [9], prostate cancer [39], and so on.
Importantly, DHODH seemed to be a potential target in human malignant cutaneous tumors, especially melanoma. Expression level of DHODH was higher in precancerous cells and malignant cutaneous keratinocytes, compared to that in normal cutaneous cells [40]. Besides, DHODH inhibition by teriflunomide encouraged cytostatic and apoptotic effects in premalignant and malignant cutaneous keratinocytes [41]. Importantly, DHODH inhibition by leflunomide suppressed melanoma formation and growth in both zebrafish and mouse model [18]. However, the exact molecular mechanism of DHODH inhibition-induced cell proliferation suppression in melanoma was not fully elucidated.
Herein, we applied experiments to explore the function of DHODH in melanoma by using both a commonly used clinical DHODH inhibitor leflunomide and shRNAs of DHODH. As expected, DHODH inactivation/deficiency induced significant cell proliferation suppression in human A375 and MV3 melanoma cells (Figure 1A and 3B). Besides, Brdu positive cells were also reduced in these cells after DHODH inhibition (Figure 1B and 3C). Besides, tumor volume and weight was also reduced in A375 and MV3 melanoma xenograft mouse models (Figure 1F and 1G). These results consisted with previous reports [18], and showed that DHODH inhibition suppressed melanoma proliferation both in vitro and in vivo, indicating that DHODH might be a potential target in melanoma treatment.
As DHODH is one of the important rate-limiting enzyme during biological de novo synthesis of nucleotides, its inactivation/deficiency might reduce the abundance of nucleotides in the cells, thus affected cell cycle progression. Previous reports showed that A77 1726 induced G1 cell cycle arrest via modulation of cyclin D2 and pRb expression in myeloma cells [17]. Our results showed that DHODH inactivation/deficiency induced cell cycle arrest at S phase in A375 and MV3 cells (Figures 1C, 1D, 3D and 3E). In addition, CDK2 and CyclinA2, key regulators of S phase progression [42], were also dramatically reduced after DHODH inactivation/deficiency (Figures 1E and 3F). Consistently, Doscas ME et al. also found that A77 1726 accelerated cell cycle entry into the S phase through ERK1/2 activation in A375 cells and that pyrimidine nucleotide depletion halted cell cycle progression [16]. Our results further confirmed that DHODH inactivation/deficiency suppressed cell proliferation through S phase arrest.
Programmed cell death (PCD), including apoptosis, autophagy and programmed necrosis, is death of a cell mediated by intracellular programs. It decides the fate of cancer cells and its dysfunction provides an important clue to treat tumors [43]. Among them, apoptosis is the most common type of PCD, which characterized by nuclear fragmentation and pyknosis, chromatin condensation, as well as caspases activation, DNA breakdown and modifications of membrane surface that allow phagocytic cells to recognize and engulf the apoptotic cells [44]. Apoptosis is mainly triggered by two major mechanisms: one is extrinsic pathway, in which death ligands bind to death receptors; another is intrinsic mitochondrial pathway, in which cells are triggered to release cytochrome c from mitochondria to cytoplasm and caspase-9 is cleaved and auto-activated [45]. In both two apoptotic pathways, executioner caspase-3 is cleaved and activated to induce cell death [46].
In our results, DHODH inactivation by leflunomide also induced apoptosis in A375 melanoma cells, and both caspase-9 and caspase-3 were cleaved (Figure 2B), which indicating that DHODH inactivation induced intrinsic mitochondrial pathway of apoptosis in melanoma. However, DHODH knockdown couldn’t induce significant apoptosis (Supplementary Figure 4A). These might be a result of a not so efficient knockdown by shRNAs. Because DHODH was an enzyme, a little expression was enough for a cell to use under normal culture condition, but might be not in stressful environments. Our results showed that DHODH knockdown sensitized A375 cells to apoptosis induced by extracellular stress, such as actinomycin D, doxorubicin, as well as temozolomide (Figure 4A and 4B). These results indicated that DHODH inhibitors combined with other chemotherapeutic drugs might be a novel therapeutic method for melanoma treatment.
Autophagy is a cellular catabolic process with degrading and recycling intracellular components in response to cell stress [47]. During this process, cytoplasmic components, such as organelles and cytosolic molecules, are engulfed by autophagosomes. Meanwhile, LC3-I is conjugated to phosphatidylethanolamine to form LC3-II, which is recruited to the membrane of autophagosomes [48]. Defects in the autophagy machinery are associated to the pathogenesis of many diseases including autoimmune, neurodegenerative, heart and liver disorders as well as cancer [49]. It was reported that autophagy played a dual role in cancer: on the one hand it could inhibit the occurrence and development of tumor, on the other hand it could also make cancer cells to adapt to adverse metabolic environment leading to the survival of tumor cells [50]. Our results also showed that DHODH inactivation/deficiency induced a persistently increased LC3B expression in A375 and MV3 melanoma cells in a time-course manner (Figure 2E and 4E). Besides, laser confocal microscopic images showed that LC3B puncta aggregations in cells were increased after DHODH inactivation/deficiency (Figure 2C and 4C). After transfected exogenous GFP-linked LC3B into cells, cells also showed more puncta aggregations of LC3B, which was different from the diffuse LC3B observed in the control groups (Figure 2D and 4D). In addition, autophagy inhibitor 3-MA and chloroquine partly rescued leflunomide-induced cell viability (Supplementary Figure 2E). These results indicated that autophagy might contribute to cell death induced by DHODH inhibition in melanoma cells.
To explore the mechanism underlying, we detected several essential regulators in autophagy pathway, including Erk1/2, DAPK1, p62 and Beclin1, the results showed that no significant expression changes of these proteins (Data not shown). However, we found that AMPK was phosphorylated at Thr 172, a prerequisite for AMPK activity, after DHODH inactivation/deficiency (Figure 7A and 7B). There were several mechanisms by which AMPK could promote autophagy. Historically, AMPK was an established negative regulator of the mTOR signaling cascade [51]. This could be accomplished by AMPK-mediated phosphorylation of the TSC complex which was a negative regulator of mTORC1 activation at the lysosome [52]. Alternatively, AMPK could directly phosphorylate the Raptor subunit of the mTORC1 complex, which induced 14-3-3 binding and inhibited mTORC1 target phosphorylation [53]. However, in 2010, Behrends et al. first discovered the interaction between AMPK and Ulk1/2 in a global proteomic analysis of the human autophagy network [54]. Subsequently, different groups confirmed the direct interaction between these two kinases [26–31]. Some of them additionally reported the AMPK-mediated phosphorylation of Ulk1. Phosphorylated Ulk activated itself and subsequently phosphorylated Beclin-1 on Ser 14, thereby enhancing the activity of the ATG14L-containing VPS34 complexes, thus induced autopghagy [55]. So we showed that Ser 555 phosphorylation of Ulk was increased after DHODH inhibition (Figure 7A and 7B). Our results indicated that AMPK-Ulk axis was essential in DHODH inhibition-induced autophagy (Figure 7E).
In addition, we found that DHODH inactivation/deficiency-induced cell cycle arrest, apoptosis and autophagy could be rescued by adding exogenous uridine in the medium (Supplementary Figures 1C, 1D, 2B, 2C, 2D, 3A, 3B, 4B,4C and 4D). These results indicated that DHODH inhibition-induced uridine deficiency contributed to cell cycle arrest, apoptosis and autophagy.
Interestingly, we found that DHODH inactivation/deficiency-induced apoptosis didn’t occur in MV3 cells (Figure 5A and 5B). And we found that the expression of BCL-2 was much higher in MV3 cells than that in A375 cells (Figure 5C). Further qRT-PCR analysis also revealed that there was little bcl-2 expression in A375 cells (Supplementary Figure 5C). As one of the core member of BCL-2 families, BCL-2 is localized to the inner mitochondrial membrane [56] or the plasma membrane [57] and is an essential anti-apoptotic factor in the intrinsic mitochondrial pathway of apoptosis [21]. BCL-2 could keep pro-apoptotic BAX and BAK in check, preventing them causing mitochondrial outer membrane permeabilization (MOMP) of apoptogenic factors such as cytochrome c into the cytoplasm [58]. Besides, BCL-2 was considered as an oncogene in multiple tumors, including melanoma, and contributed to cancer cell survival [59–61]. In addition, bfl1 expression in A375 cells was extremely higher than MV3 cells (Supplementary Figure 5A). However, it was considered as an anti-apoptotic factor. So we supposed that BCL-2 was one of the most essential members for the survival of melanoma cells, and we knocked down BCL-2 in MV3 cells. The result showed that leflunomide induced remarkable apoptosis and activated caspase-9 and caspase-3 activities after BCL-2 knockdown (Figure 5D and 5E). In addition, BCL-2 overexpression retrieved leflunomide-induced apoptosis in A375 cells (Figure 5F and 5G). These results showed that BCL-2 was a switch of DHODH inactivation/deficiency-induced apoptosis.
It was reported that Bcl-2 proteins bound to Beclin1 through a BH3 domain and disrupt its autophagy function [24]. Our results showed that LC3B expression increased in BCL-2 knockdown MV3 cells, while decreased in BCL-2 overexpressed A375 cells (Figure 6A and 6B). Besides, LC3B puncta aggregations observed under laser confocal microscope showed similar results (Figure 6E and 6F). These results indicated that BCL-2 inhibited autophagy in melanoma cells after leflunomide treatment. Besides, the interaction between BCL-2 and Beclin1 was decreased after DHODH inhibition (Figure 7C and 7D). It was reported that nutrient starvation could activate JNK by its Thr 87 phosphorylation directly or through AMPK activiation, and phospho-JNK could phosphorylate BCL-2 and inhibit it to interact with Beclin1 [32]. Our results showed that Thr 87 phosphorylation of BCL-2 was increased in MV3 and A375 cells after DHODH inactivation/deficiency (Figure 7A and 7B). Our results provided an alternative underlying mechanism of DHODH inactivation/deficiency -induced autophagy.
It was reported that BCL-2 could inhibit cell cycle entry by facilitating G0 [23]. However, BCL2 didn’t significantly affect growth rates under optimal conditions, but prolonged G1 in suboptimal conditions [62]. Our results showed that CDK2 was increased in BCL-2 knockdown MV3 cells, while decreased in BCL-2 overexpressed A375 cells (Figure 6A and 6B). The results of flow cytometry also showed that BCL-2 promoted cell cycle arrest in melanoma cells (Figure 6C and 6D). These results indicated that BCL-2 inhibited cell cycle in leflunomide-treated melanoma cells. Our results indicated that combination inhibition of BCL-2 and DHODH might be a new therapeutic regimen for malignant melanoma treatment. However, much more need to be considered in this therapeutic regimen because inhibiting BCL-2 might promote cell cycle progression.
In conclusions, our data showed that DHODH inhibition by leflunomide or shRNAs suppressed cell proliferation, induced cell cycle arrest at S phase, as well as promoted programmed cell death including intrinsic apoptosis and autophagy in human melanoma cells. And we found the molecular mechanisms dictating cell cycle arrest and programed cell death induced by DHODH inactivation/deficiency. Our results provided clues for leflunomide and DHODH target used in malignant melanoma treatments.
MATERIALS AND METHODS
Cell culture
The human melanoma cells A375 were purchased from American Type Culture Collection (ATCC, Rockville, MD, USA). MV3 were purchased from the Third Military Medical University. Cell line A375 was cultured in Dulbecco’s modified Eagle’s medium (DMEM, Life Technologies, Grand Island, NY, USA) and MV3 was grown in Roswell Park Memorial Institute-1640 (RPMI-1640, Life Technologies, Grand Island, NY, USA), simultaneously mixed with 10% fetal bovine serum (FBS, Life Technologies, Grand Island, NY, USA) as a supplement. All cells were incubated at 37°C in humidified incubator with 5% CO2.
Reagents
DHODH inhibitor leflunomide (Sigma), apoptotic inducer actinomycin D (Sigma) and doxorubicin (Sigma), antitumor drug temozolomide (TMZ, Sigma), 3-MA (Sigma), chloroquine (CQ, Sigma), BCL-2 inhibitor ABT-199 (Sigma) were dissolved in dimethyl sulfoxide (DMSO, Sigma). Uridine (Sigma) was dissolved in ddH2O. In our experiments, DMSO was used as control (untreated).
Transfection and infection assay
The cDNA sequences of DHODH (NM 001361.4) and BCL-2 (NM 000633.2) were gained from NCBI. The RNAi candidate target sequences as follows: shDHODH#1 (AAGTGAGAGTTCTGGGCCATAAATTCC), shDHO DH#2 (AATTGCTGCAGGATTTGACAAGCATGG), shBCL-2#1 (AACCGGGAGATA GTGATGAAGTAC ATC), shBCL-2#2 (AAGTACATCCATTATAAGCTG TCGCAG). These sequences were inserted into the pLKO.1 vector. Human full length BCL-2 cDNA fragment was then cloned into PCDH-CMV-MCS-EF1-puro vector. The GFP-LC3B plasmid was a gift by Prof. Ning Gao at the Third Military Medical University, China. The shRNA plasmid with the packaging plasmids pLP1, pLP2, and VSVG (Invitrogen) mixed with Lipofectamine 2000, then this compound was transfected to 293FT cells. Afterwards virus-containing supernatants which used for cell infections were collected. After infection, the aim cells were selected in the presence of 4 μg/mL puromycin for 3 days, and drug-resistant cells were collected for the following experiment.
MTT assay
The cell growth was of human melanoma cell lines A375and MV3 was evaluated by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT, Sigma) assay. That is, about 2000 cells/well were evenly distributed in 96-well plate. After dealt with leflunomide at 50 μM, 100 μM and 200 μM concentration respectively, and DMSO was used as a control for the indicated time, 20 μl MTT was added to each well, then incubated at 37°C for 2 h. Finally, the absorbance value was measured at a wavelength of 560 nm after shaking for 10 min.
Brdu staining assay
Cells were seeded on 24-well plate and treated with 100 μM leflunomide (and equal volume of DMSO as control) for 72 h. After that, incubated with 10 μg/ml thymidine analog 5-bromo-2 deoxyuridine (Brdu; Sigma) for 30 min at indoor temperature, then discarded the supernatant and washed thrice with PBS, anchored by 4% paraformaldehyde for 15 min, permeabilized with 0.5% Triton X-100 for 10 min after pre-treated with 2 M HCl for 10 min, blocked with 10% goat serum for 1 h, followed by incubated a monoclonal rat primary antibody against Brdu (1:300, Sigma) overnight, followed by Alexa FluorR® 594 goat anti-rat IgG secondary antibody (H+L; Invitrogen). 500 μl Hoechest33342 was used for nucleus staining. In the end, the percentage of Brdu was calculated under more than 8 microscopic fields (Nikon 80i, Nikon Corporation, Tokyo, Japan).
Cell cycle assay
Cells were plated in 60 mm plates and treated with 100 μM leflunomide (and equal volume of DMSO as control) for 72 h. Afterwards cells were washed by pre-cooling PBS, anchored by 70 % ethanol for 24 h. After that, cells was stained with propidium iodide (PI) at 37°C for 1 h in dark. Finally collected cells by the FACS C6 (BD Biosciences, San Jose, CA, USA) and data was analyzed with FlowJo6.0 software.
Cell apoptosis assay
Cells were plated in 60 mm plates and treated with 100 μM leflunomide (and equal volume of DMSO as control) for 72 h. Afterwards cells were washed by pre-cooling PBS and collected. Cells were resuspended in 100 μl binding buffer, then stained with 5 μl propidium iodide (PI) and 5 μl Annexin V-FITC at room temperature for 20 min in dark. Finally collected cells by the FACS C6 (BD Biosciences, San Jose, CA, USA) and data was analyzed with FlowJo6.0 software.
Western blot assay
Cells were treated with leflunomide for 72 h, then cells were collected, after that proteins were extracted by RIPA Lysis Buffer which contained 1% Phenyl methane sulfonyl fluoride (PMSF), stockpiled at −80°C. The proteins were separated by SDS-PAGE, subsequently, transfered onto PVDF membranes (Millipore, USA). The PVDF membranes were blocked with 5% BSA for 2 h, and incubated with a primary antibody against human Tubulin (1:1000, Beyotime), LC3B (1:1000, Cell Signaling), CDK2 (1:1000, Cell Signaling), CyclinA2 (1:1000, Cell Signaling), DHODH (1:1000, Cell Signaling), BCL-2 (1:1000, Cell Signaling), Caspase-9 (1:1000, Cell Signaling), Caspase-3 (1:1000, Cell Signaling), pT172-AMPK (1:800, Cell Signaling), AMPK (1:800, Cell Signaling), pSer555-Ulk (1:1000, Cell Signaling), Ulk (1:1000, Cell Signaling), pThr 183/186-JNK (1:2000, Abcam), JNK (1:1000, Abcam), BCL-2 (1:1000, Cell Signaling) and Beclin1 (1:1000, Cell Signaling) at 4°C overnight. Then incubated with homologous secondary antibodies HRP-labeled goat anti-rabbit IgG (H+L) (1:2000, Beyotime) or goat anti-mouse IgG (H+L) (1:2000, Beyotime) for 2 h. Finally, the signal was captured by the ECL reagent (Beyotime) and visualized by Western blotting detection instruments (Clinx Science).
Immunofluorescence staining assay
Cells were seeded on cleaned and autoclaved glass slide, exposured to 100 μM Leflunomide for 72 h (DMSO as control). After that, anchored by 4% paraformaldehyde for 15 min, permeabilized with 0.5% Triton X-100 for 10 min after pre-treated with 2 M HCl for 10 min, blocked with 10% goat serum for 1 h. Then a monoclonal rat primary antibody against LC3B (1:200, Cell Signaling) vernight at 4°C. Followed by incubation with Alexa Fluor 488 goat anti-rabbit IgG (H+L) (1:2000, Beyotime), which as a secondary antibody. Then Hoechst 33342 (Sigma-Aldrich, USA) staining nuclei for 20 min. Fluorescent images were captured with a Zeiss LSM laser scanning confocal microscope.
Co-Immunoprecipitation assay
Cell lysates was pretreated by adding 20 μl Protein A+G Agarose (Beyotime, Jiangsu, China) for 3 hours at 4°C. Precleared lysates were incubated with anti-Beclin1 antibodies (Abcam) or normal Ig G (Beyotime) overnight at 4°C. And then associated proteins with each antibody were precipitated with protein agarose A+G for 6 hours at 4°C. Immunoprecipitates were resolved by polyacrylate gel electrophoresis and analyzed by western blotting.
Tumor xenografts assay
For tumor xenografts, 4 weeks old female nude mice (BALA/c) were purchased from Beijing laboratory animal research center. They were allowed to acclimate for 1 week in the SPF room. Cells were grown to 70–80% confluence collected and resuspended. 1 × 106 cells in 100 ul DMEM or 1640 were used in subcutaneous injection. A week later, the mice were divided into two groups randomly. One group delt with leflunomide at 7.5 mg/kg, in the meantime, the other group was injected with equal volume of DMSO as a control for 12 days. Tumor growth was measured by vernier caliper every three day, then tumor volume was calculated by (small diameter)2 × (large diameter)/2. Finally, tumors were stripped from the mice and weighed. All animal experiments were pre-approved by the Institutional Animal Care and Use Committees of the Southwest University.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This work is supported by the National Key Research and Development Program of China (2016YFC1302204), the National Natural Science Foundation of China (81672502, 81602479), Chongqing University Innovation Team Building Program funded projects (CXTDX201601010) and China Postdoctoral Science Foundation (2017T100669).
REFERENCES
1. DeSantis CE, Lin CC, Mariotto AB, Siegel RL, Stein KD, Kramer JL, Alteri R, Robbins AS, Jemal A. Cancer treatment and survivorship statistics, 2014. CA Cancer J Clin. 2014; 64:252–71. https://doi.org/10.3322/caac.21235.
2. Schadendorf D, Hauschild A. Melanoma in 2013: Melanoma–the run of success continues. Nat Rev Clin Oncol. 2014; 11:75–6. https://doi.org/10.1038/nrclinonc.2013.246.
3. Balch CM, Gershenwald JE, Soong SJ, Thompson JF, Atkins MB, Byrd DR, Buzaid AC, Cochran AJ, Coit DG, Ding S, Eggermont AM, Flaherty KT, Gimotty PA, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009; 27:6199–206. https://doi.org/10.1200/jco.2009.23.4799.
4. Roesch A. Tumor heterogeneity and plasticity as elusive drivers for resistance to MAPK pathway inhibition in melanoma. Oncogene. 2015; 34:2951–7. https://doi.org/10.1038/onc.2014.249.
5. Shtivelman E, Davies MQ, Hwu P, Yang J, Lotem M, Oren M, Flaherty KT, Fisher DE. Pathways and therapeutic targets in melanoma. Oncotarget. 2014; 5:1701–52. https://doi.org/10.18632/oncotarget.1892.
6. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–74. https://doi.org/10.1016/j.cell.2011.02.013.
7. Loffler M, Fairbanks LD, Zameitat E, Marinaki AM, Simmonds HA. Pyrimidine pathways in health and disease. Trends Mol Med. 2005; 11:430–7. https://doi.org/10.1016/j.molmed.2005.07.003.
8. Liu S, Neidhardt EA, Grossman TH, Ocain T, Clardy J. Structures of human dihydroorotate dehydrogenase in complex with antiproliferative agents. Structure. 2000; 8:25–33. https://doi.org/10.1016/S0969-2126(00)00077-0.
9. He T, Haapa-Paananen S, Kaminskyy VO, Kohonen P, Fey V, Zhivotovsky B, Kallioniemi O, Perala M. Inhibition of the mitochondrial pyrimidine biosynthesis enzyme dihydroorotate dehydrogenase by doxorubicin and brequinar sensitizes cancer cells to TRAIL-induced apoptosis. Oncogene. 2014; 33:3538–49. https://doi.org/10.1038/onc.2013.313.
10. Vyas VK, Ghate M. Recent developments in the medicinal chemistry and therapeutic potential of dihydroorotate dehydrogenase (DHODH) inhibitors. Mini Rev Med Chem. 2011; 11:1039–55. https://doi.org/10.2174/138955711797247707.
11. Hail N Jr, Chen P, Kepa JJ, Bushman LR. Evidence supporting a role for dihydroorotate dehydrogenase, bioenergetics, and p53 in selective teriflunomide-induced apoptosis in transformed versus normal human keratinocytes. Apoptosis. 2012; 17:258–68. https://doi.org/10.1007/s10495-011-0667-0.
12. Smith LH Jr, Baker FA. Pyrimidine metabolism in man. I. The biosynthesis of orotic acid. J Clin Invest. 1959; 38:798–809. https://doi.org/10.1172/jci103862.
13. Munier-Lehmann H, Vidalain PO, Tangy F, Janin YL. On dihydroorotate dehydrogenases and their inhibitors and uses. J Med Chem. 2013; 56:3148–67. https://doi.org/10.1021/jm301848w.
14. Evers DL, Wang X, Huong SM, Andreoni KA, Huang ES. Inhibition of human cytomegalovirus signaling and replication by the immunosuppressant FK778. Antiviral Res. 2005; 65:1–12. https://doi.org/10.1016/j.antiviral.2004.03.007.
15. Fairbanks LD, Bofill M, Ruckemann K, Simmonds HA. Importance of ribonucleotide availability to proliferating T-lymphocytes from healthy humans. Disproportionate expansion of pyrimidine pools and contrasting effects of de novo synthesis inhibitors. J Biol Chem. 1995; 270:29682–9. https://doi.org/10.1074/jbc.270.50.29682.
16. Doscas ME, Williamson AJ, Usha L, Bogachkov Y, Rao GS, Xiao F, Wang Y, Ruby C, Kaufman H, Zhou J, Williams JW, Li Y, Xu X. Inhibition of p70 S6 kinase (S6K1) activity by A77 1726 and its effect on cell proliferation and cell cycle progress. Neoplasia. 2014; 16:824–34. https://doi.org/10.1016/j.neo.2014.08.006.
17. Baumann P, Mandl-Weber S, Volkl A, Adam C, Bumeder I, Oduncu F, Schmidmaier R. Dihydroorotate dehydrogenase inhibitor A771726 (leflunomide) induces apoptosis and diminishes proliferation of multiple myeloma cells. Mol Cancer Ther. 2009; 8:366–75. https://doi.org/10.1158/1535-7163.mct-08-0664.
18. White RM, Cech J, Ratanasirintrawoot S, Lin CY, Rahl PB, Burke CJ, Langdon E, Tomlinson ML, Mosher J, Kaufman C, Chen F, Long HK, Kramer M, et al. DHODH modulates transcriptional elongation in the neural crest and melanoma. Nature. 2011; 471:518–22. https://doi.org/10.1038/nature09882.
19. O'Donnell EF, Kopparapu PR, Koch DC, Jang HS, Phillips JL, Tanguay RL, Kerkvliet NI, Kolluri SK. The aryl hydrocarbon receptor mediates leflunomide-induced growth inhibition of melanoma cells. PLoS One. 2012; 7:e40926. https://doi.org/10.1371/journal.pone.0040926.
20. Tasdemir E, Galluzzi L, Maiuri MC, Criollo A, Vitale I, Hangen E, Modjtahedi N, Kroemer G. Methods for assessing autophagy and autophagic cell death. Methods Mol Biol. 2008; 445:29–76. https://doi.org/10.1007/978-1-59745-157-4_3.
21. Delbridge AR, Grabow S, Strasser A, Vaux DL. Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies. Nat Rev Cancer. 2016; 16:99–109. https://doi.org/10.1038/nrc.2015.17.
22. Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002; 2:647–56. https://doi.org/10.1038/nrc883.
23. Zinkel S, Gross A, Yang E. BCL2 family in DNA damage and cell cycle control. Cell Death Differ. 2006; 13:1351–9. https://doi.org/10.1038/sj.cdd.4401987.
24. Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005; 122:927–39. https://doi.org/10.1016/j.cell.2005.07.002.
25. Vairo G, Innes KM, Adams JM. Bcl-2 has a cell cycle inhibitory function separable from its enhancement of cell survival. Oncogene. 1996; 13:1511–9.
26. Shang L, Chen S, Du F, Li S, Zhao L, Wang X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc Natl Acad Sci USA. 2011; 108:4788–93. https://doi.org/10.1073/pnas.1100844108.
27. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011; 13:132–41. https://doi.org/10.1038/ncb2152.
28. Chan EY. mTORC1 phosphorylates the ULK1-mAtg13-FIP200 autophagy regulatory complex. Sci Signal. 2009; 2:pe51. https://doi.org/10.1126/scisignal.284pe51.
29. Lee JW, Park S, Takahashi Y, Wang HG. The association of AMPK with ULK1 regulates autophagy. PLoS One. 2010; 5:e15394. https://doi.org/10.1371/journal.pone.0015394.
30. Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, Asara JM, Fitzpatrick J, Dillin A, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011; 331:456–61. https://doi.org/10.1126/science.1196371.
31. Loffler AS, Alers S, Dieterle AM, Keppeler H, Franz-Wachtel M, Kundu M, Campbell DG, Wesselborg S, Alessi DR, Stork B. Ulk1-mediated phosphorylation of AMPK constitutes a negative regulatory feedback loop. Autophagy. 2011; 7:696–706. https://doi.org/10.4161/auto.7.7.15451.
32. Cherra SJ 3rd, Kulich SM, Uechi G, Balasubramani M, Mountzouris J, Day BW, Chu CT. Regulation of the autophagy protein LC3 by phosphorylation. J Cell Biol. 2010; 190:533–9. https://doi.org/10.1083/jcb.201002108.
33. Svensson RU, Shaw RJ. Cancer metabolism: Tumour friend or foe. Nature. 2012; 485:590–1. https://doi.org/10.1038/485590a.
34. Lang R, Wagner H, Heeg K. Differential effects of the immunosuppressive agents cyclosporine and leflunomide in vivo. Leflunomide blocks clonal T cell expansion yet allows production of lymphokines and manifestation of T cell-mediated shock. Transplantation. 1995; 59:382–9. https://doi.org/10.1097/00007890-199502150-00013.
35. Ringshausen I, Oelsner M, Bogner C, Peschel C, Decker T. The immunomodulatory drug Leflunomide inhibits cell cycle progression of B-CLL cells. Leukemia. 2008; 22:635–8. https://doi.org/10.1038/sj.leu.2404922.
36. Anderson LW, Strong JM, Cysyk RL. Cellular pharmacology of DUP-785, a new anticancer agent. Cancer Commun. 1989; 1:381–7. https://doi.org/10.1007/978-1-4615-3492-1_16.
37. Dietrich S, Kramer OH, Hahn E, Schafer C, Giese T, Hess M, Tretter T, Rieger M, Hullein J, Zenz T, Ho AD, Dreger P, Luft T. Leflunomide induces apoptosis in fludarabine-resistant and clinically refractory CLL cells. Clin Cancer Res. 2012; 18:417–31. https://doi.org/10.1158/1078-0432.ccr-11-1049.
38. Zhu S, Yan X, Xiang Z, Ding HF, Cui H. Leflunomide reduces proliferation and induces apoptosis in neuroblastoma cells in vitro and in vivo. PLoS One. 2013; 8:e71555. https://doi.org/10.1371/journal.pone.0071555.
39. Hail N Jr, Chen P, Bushman LR. Teriflunomide (leflunomide) promotes cytostatic, antioxidant, and apoptotic effects in transformed prostate epithelial cells: evidence supporting a role for teriflunomide in prostate cancer chemoprevention. Neoplasia. 2010; 12:464–75. https://doi.org/10.1593/neo.10168.
40. Hail N Jr, Chen P, Kepa JJ, Bushman LR, Shearn C. Dihydroorotate dehydrogenase is required for N-(4-hydroxyphenyl)retinamide-induced reactive oxygen species production and apoptosis. Free Radic Biol Med. 2010; 49:109–16. https://doi.org/10.1016/j.freeradbiomed.2010.04.006.
41. Hail N Jr, Chen P, Rower J, Bushman LR. Teriflunomide encourages cytostatic and apoptotic effects in premalignant and malignant cutaneous keratinocytes. Apoptosis. 2010; 15:1234–46. https://doi.org/10.1007/s10495-010-0518-4.
42. Lundberg AS, Weinberg RA. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol Cell Biol. 1998; 18:753–61.
43. Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT, Liu B, Bao JK. Programmed cell death pathways in cancer: a review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012; 45:487–98. https://doi.org/10.1111/j.1365-2184.2012.00845.x.
44. Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS, Golstein P, Green DR, Hengartner M, Knight RA, Kumar S, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009; 16:3–11. https://doi.org/10.1038/cdd.2008.150.
45. Koff JL, Ramachandiran S, Bernal-Mizrachi L. A time to kill: targeting apoptosis in cancer. Int J Mol Sci. 2015; 16:2942–55. https://doi.org/10.3390/ijms16022942.
46. Tait SW, Green DR. Mitochondrial regulation of cell death. Cold Spring Harb Perspect Biol. 2013; 5. https://doi.org/10.1101/cshperspect.a008706.
47. Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res. 2014; 24:24–41. https://doi.org/10.1038/cr.2013.168.
48. Tanida I, Ueno T, Kominami E. (2008). LC3 and Autophagy. In: Deretic V, ed. Autophagosome and Phagosome. (Totowa, NJ: Humana Press), pp. 77-88.
49. Kroemer G. Autophagy: a druggable process that is deregulated in aging and human disease. J Clin Invest. 2015; 125:1–4. https://doi.org/10.1172/jci78652.
50. Wu WK, Coffelt SB, Cho CH, Wang XJ, Lee CW, Chan FK, Yu J, Sung JJ. The autophagic paradox in cancer therapy. Oncogene. 2012; 31:939–53. https://doi.org/10.1038/onc.2011.295.
51. Russell RC, Yuan HX, Guan KL. Autophagy regulation by nutrient signaling. Cell Res. 2014; 24:42–57. https://doi.org/10.1038/cr.2013.166.
52. Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003; 115:577–90. https://doi.org/10.1016/S0092-8674(03)00929-2.
53. Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008; 30:214–26. https://doi.org/10.1016/j.molcel.2008.03.003.
54. Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature. 2010; 466:68–76. https://doi.org/10.1038/nature09204.
55. Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013; 15:741–50. https://doi.org/10.1038/ncb2757.
56. Hockenbery D, Nunez G, Milliman C, Schreiber RD, Korsmeyer SJ. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature. 1990; 348:334–6. https://doi.org/10.1038/348334a0.
57. Haldar S, Beatty C, Tsujimoto Y, Croce CM. The bcl-2 gene encodes a novel G protein. Nature. 1989; 342:195–8. https://doi.org/10.1038/342195a0.
58. Rosse T, Olivier R, Monney L, Rager M, Conus S, Fellay I, Jansen B, Borner C. Bcl-2 prolongs cell survival after Bax-induced release of cytochrome c. Nature. 1998; 391:496–9. https://doi.org/10.1038/35160.
59. Juin P, Geneste O, Gautier F, Depil S, Campone M. Decoding and unlocking the BCL-2 dependency of cancer cells. Nat Rev Cancer. 2013; 13:455–65. https://doi.org/10.1038/nrc3538.
60. Loria R, Bon G, Perotti V, Gallo E, Bersani I, Baldassari P, Porru M, Leonetti C, Di Carlo S, Visca P, Brizzi MF, Anichini A, Mortarini R, et al. Sema6A and Mical1 control cell growth and survival of BRAFV600E human melanoma cells. Oncotarget. 2015; 6:2779–93. https://doi.org/10.18632/oncotarget.2995.
61. Mohana-Kumaran N, Hill DS, Allen JD, Haass NK. Targeting the intrinsic apoptosis pathway as a strategy for melanoma therapy. Pigment Cell Melanoma Res. 2014; 27:525–39. https://doi.org/10.1111/pcmr.12242.
62. Simpson NH, Singh RP, Emery AN, Al-Rubeai M. Bcl-2 over-expression reduces growth rate and prolongs G1 phase in continuous chemostat cultures of hybridoma cells. Biotechnol Bioeng. 1999; 64:174–86. https://doi.org/10.1002/(SICI)1097-0290(19990720)64:2<174::AID-BIT6>3.0.CO;2-D.