
INTRODUCTION
Vascular remodeling is a common pathological occurrence in diabetes mellitus (DM) that causes complications such as atherosclerosis, leading to enhanced patient morbidity and mortality [1]. As a prominent component of the vascular wall, smooth muscle cells play a crucial role in the initiation and propagation of diabetic vascular complications including abnormal cell growth in the atherosclerotic arterial intimae. Due to the complexity of the pathogenic mechanisms underlines the process of vascular remodeling in diabetic conditions [2, 3], to further identify new molecules involved in hyperglycemia/high-glucose (HG)-induced vascular smooth muscle cell (VSMC) proliferation, related signaling pathways warrant significant attention.
Proviral integration site for Moloney murine leukemia virus 1 (Pim-1) is a serine/threonine kinase first reported in hematological malignancies [4]. Pim-1 belongs to the Pim family, which also includes Pim-2 and Pim-3, and all members are well conserved among vertebrates [5]. Both the human and orthologous mouse genes have been reported to encode 33-kDa and 44-kDa isoform resulting from the use of alternative in-frame translation initiation codons[6]. In human cells, the subcellular localization of human 44-kDa Pim-1 is primarily on the plasma membrane, while the 33-kDa isoform is present in both the cytosol and nucleus. Pim-1 may promote proliferation in the nuclear compartment while its cytoplasmic localization may promote survival, implying different Pim-1 roles depending on cellular location [7]. Universally, Pim-1 is broadly expressed in various cancer cells [5, 8-10], in neurons [11], and in liver and spleen tissues [10, 12], as well as in some stem/progenitor cells [13, 14]. Regarding the cardiovascular system, expression of Pim-1 is reported in cardiomyocytes [7, 15], endothelial cells [16] and VSMCs [17-20]. With focus on VSMCs, Pim-1 expression was discovered in the VSMCs of balloon-injured rat carotid arteries and human coronary arteries [17], whereas the Pim-1-specific inhibitor, quercetagetin, or adenovirally introduced Pim-1 shRNA, markedly suppressed VSMC proliferation [17, 18]. Thus, aberrant Pim-1 expression plays a crucial role in diseases of vascular remodeling involving abnormal proliferation of VSMCs.
Previous studies on Pim-1-mediated proliferation signaling pathways in different cell types highlight the complexity of pathogenic mechanisms. For example, Akt activation induces Pim-1 expression, which protects the infracted myocardium in mice [7]. Furthermore, Pim-1 overexpression promotes cell survival by mediating the activation of Bcl-2 and Bcl-XL, as well as phosphorylation of Bad [7, 21]. In hematologic malignancies, signal transducer and activator of transcription 3/5 (STAT3/5), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) or HOXA9 can transcriptionally activate Pim-1, supporting cellular proliferation through modification of cell cycle regulators, such as cooperating with c-Myc and c-Myb, enhancing Cdc25A and Cdc25C, and inactivating p21 and p27 [22]. STAT family members include seven transcription factors (STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6) that have multiple roles in cell proliferation and survival [23]. STAT3 was originally identified as an acute-phase response factor activated by many cytokines, with an important role in the development of cardiovascular diseases, including pulmonary arterial hypertension (PAH) [19, 24]. We hypothesized that aberrant STAT3/Pim-1 activation may contribute to vascular remodeling diseases such as DM-induced atherosclerosis.
Our previous study revealed abnormal proliferation of VSMCs in streptozotocin (STZ)-induced thoracic arteries of type I diabetic rats [25]. In the present study, we further sought to explore the role of aberrant Pim-1 expression in the proliferation of VSMCs in HG conditions. We found that significant upregulation of 33-kDa Pim-1 was detected in VSMCs from STZ-induced type I diabetic rat arteries in vivo and HG-treated VSMCs in vitro. Furthermore, inhibition of Pim-1 activity by quercetagetin significantly attenuated HG-induced VSMC growth, and more importantly HG-activated STAT3/Pim-1 signaling was found to contribute to VSMC proliferation. Our results provide novel insight into DM-induced vascular complications.
Results
Pim-1 and PCNA upregulation and alteration of phenotype-related gene expression in arterial samples of STZ-induced type I diabetic rats
Rats with a blood glucose level >/= 16.6 mmol/L were considered HG after intraperitoneal injection with STZ, while rats intraperitoneally injected with sodium citrate with normal blood glucose concentrations were served as normorglycemia (NG). Three weeks later, all rats were sacrificed and aortic arch, thoracic aorta and abdominal aorta were subjected to analyze of Pim-1 and PCNA expression. Immunohistochemistry showed weak expression of Pim-1 and PCNA protein in the VSMCs of NG rats; however, Pim-1 and PCNA protein expression levels were significantly enhanced in arterial-derived VSMCs from HG rats, and the positive signs of Pim-1 were in both nucleus and cytoplasm (Figure 1A). Immunofluorescence staining conformed that Pim-1 was co-located with α-SMA in the tunica media of thoracic aorta, which indicated that it was the VSMCs express Pim-1 protein (Supplementary Figure S1). Furthermore, HG clearly induced Pim-1 and PCNA mRNA expression compared with NG (Figure 1C). Additionally, mRNA levels of contractile markers of VSMCs such as SM22α (smooth muscle protein 22 alpha) and FHL2 (four and a half LIM domains 2) and myocardin (a nuclear factor that mediates part VSMC contractile markers) were reduced, while synthetic markers of VSMCs such as osteopontin, matrix metalloproteinase -2, -9 (MMP-2, MMP-9) were enhanced in HG rats (Supplementary Figure S2). These results indicated that 3-weeks of HG significantly induced the VSMC contractile phenotype to change into a synthetic phenotype in which Pim-1 upregulation and cell proliferation occurred.
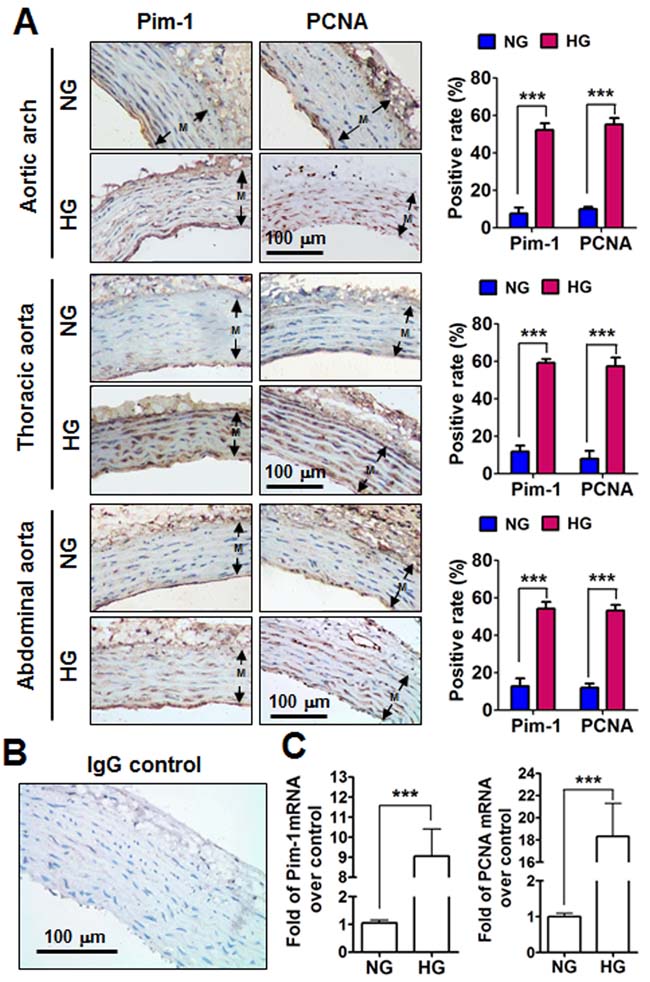
Figure 1: Pim-1 and PCNA expression in the tunica media (M) of the aortic arches, thoracic and abdominal arteries of STZ-induced hyperglycemia (HG) and normoglycemia (NG) rats. A. Representative photographs of Pim-1 and PCNA protein expression in rat aortic arches, thoracic and abdominal arteries. Immunohistochemical analysis was used to detect Pim-1 and PCNA protein expression in the tunica media of arteries. Antigens were localized by DAB and tissues were counterstained with hematoxylin. Positive rates for Pim-1 and PCNA expression were calculated using at least three representative visions (200×) for each arterial sample. Both Pim-1 and PCNA were localized in the cytoplasm and nucleus. B. Non-immune IgG was used as a negative control. Original magnification: 200×, Bar = 100 μm. C. Pim-1 and PCNA mRNA expression in the tunica media of thoracic arteries were examined by qRT-PCR. NG, Normoglycemia (n = 7); HG, Hyperglycemia (n = 10).
Upregulation of Pim-1 in HG-treated primary VSMCs in vitro
The Pim-1 gene encodes 33-kDa and 44-kDa isoforms. The role of HG in Pim-1 expression by VSMCs was explored in vitro. Primary VSMCs were treated in normal glucose (NG), mannitol (Mtol, osmotic control) and HG media, and 48 hours later, cells were analyzed for Pim-1 expression. Quantitative real time polymerase chain reaction (qRT-PCR) and western blotting showed that Mtol affected Pim-1 expression slightly when compared with NG (P > 0.05), while HG treatment led to moRE significant upregulation of the 33-kDa than the 44-kDa Pim-1 protein (Figures 2A & 2B). These results indicated that upregulation of Pim-1 was HG-dependent, and HG stimulation preferentially induced 33-kDa Pim-1 isoform expression. Furthermore, we found that HG induced Pim-1 expression in VSMCs in a manner of transient decline following sustained increase at both transcriptional and translational levels (Figure 2C-2D) and glucose concentration-dependent manner, with the optimal glucose concentration being 25 mmol/L (Figure 2E & 2F). Consistent with the in vivo trend, 48-hour of in vitro HG-treatment also led to upregulation of synthetic markers of VSMC (Supplementary Figure S3). Of notable, the bands for Pim-1 isoforms could be separated well using 12% SDS-PAGE; under 10% SDS-PAGE, the bands could not be separated well. Thus, Pim-1 bands within 10% SDS-PAGE represent the total proteins including two isoforms.
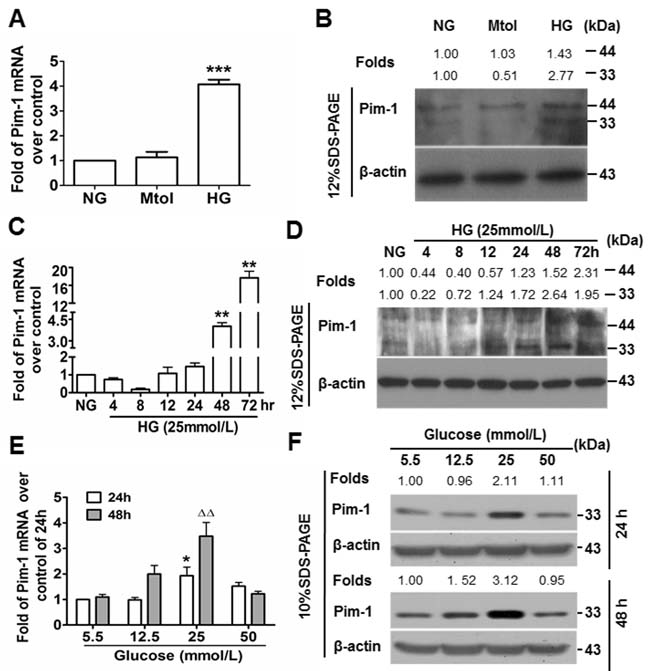
Figure 2: High glucose (HG) induces Pim-1 expression in cultured VSMCs. A. qRT-PCR and B. western blotting were used to detect Pim-1 expression in VSMCs maintained in media supplemented with normal glucose (NG, 5.5 mmol/L D-glucose), Mtol (25 mmol/L D-mannitol) and high glucose (HG, 25 mmol/L D-glucose) for 48 hours. ***P < 0.001 vs. NG or Mtol. C. qRT-PCR and D. western blotting were used to detect Pim-1 expression in VSMCs maintained in NG (5.5 mmol/L D-glucose, 24 hours) and HG (25 mmol/L D-glucose, 4, 8, 12, 24, 48,72 hours), **P < 0.01 vs. NG. E. qRT-PCR and F. western blotting were used to detect Pim-1 expression in VSMCs maintained in media supplemented with various concentration of D-glucose (5.5, 12.5, 25, and 50 mmol/L) for 24 or 48 hours, *P < 0.05 vs. 5.5 mmol/L D-glucose (NG). ∆∆P < 0.01 vs. 5.5 mmol/L D-glucose (NG). For the western blot, changes of Pim-1 isoform levels were depicted as folds over controls.
Quercetagetin and stattic repress HG-induced VSMC proliferation in vitro
Accumulating evidence, including our own, has previously confirmed that exposure to HG condition contributes to VSMC proliferation [25-28]. To explore the relationship between Pim-1 expression and HG-induced VSMC proliferation, we investigated the contribution of Pim-1 inhibition on the proliferation of VSMCs exposed to HG. After 48 hours exposure to HG, cell viability was assessed using the cell counting kit-8 (CCK-8) assay. Cell numbers were counted and qRT-PCR and western blotting were used to examine expression of PCNA. As shown in Figure 3, exposure of VSMCs to HG led to significantly enhanced OD values, cell numbers and PCNA expression levels compared with NG and Mtol groups. This indicated that treatment of VSMCs with HG enhanced cell proliferation in an HG-dependent manner. Addition of the Pim-1 blocker quercetagetin (5.5 μmol/L) to the HG medium resulted in decreased cell viability, cell number and PCNA expression (Figure 3). Thus, HG-induced VSMC proliferation may be partly due to HG-induced Pim-1 upregulation and activity maintenance. Interestingly, we also observed that stattic (10 μmol/L) also blocked HG-induced VSMC proliferation, implying a crucial role for the activation of STAT3 signaling in HG-induced VSMC proliferation (Figure 3).
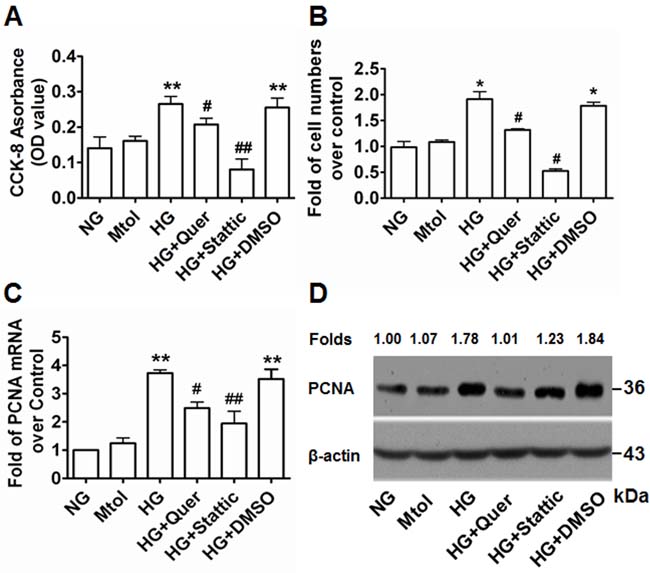
Figure 3: Quercetagetin and stattic attenuate HG-induced VSMC proliferation in vitro. A. Cells (2 × 103) were plated into 96-well plates and 6 hours later medium was changed to either NG (5.5 mmol/L D-glucose), Mtol (25 mmol/L D-mannitol), HG (25 mmol/L D-Glucose), HG+Quer (25 mmol/L D-Glucose + 5.5 μmmol/L quercetagetin), HG+Stattic (25 mmol/L D-Glucose + 10 μmol/L stattic), or HG+DMSO (25 mmol/L D-Glucose + 0.3% (v/v) DMSO) for 48 hours and the CCK-8 assay was used to detect cell viability. **P < 0.01 vs. NG or Mtol, #P < 0.05 vs. HG or HG+DMSO, ##P < 0.01 vs. HG or HG+DMSO. B. Cells (5×105) were plated into 6-well plates and 6 hours later the medium was changed as described above for an additional 48 hours and cell numbers were counted. *P < 0.05 vs. NG or Mtol; #P < 0.05 vs. HG or HG+DMSO. C. qRT-PCR and D. western blotting were used to examine the expression of PCNA. **P < 0.01 vs. NG or Mtol; #P < 0.05 vs. HG or HG+DMSO, ##P < 0.01 vs. HG or HG+DMSO. Quer, quercetagetin.
Quercetagetin and stattic mainly arrest VSMC cell cycle at G1 phase
VSMCs were serum-starved for 12 hours, then exposed to various treatments. Flow cytometric analysis showed that HG stimulated G1 to S phase progression to a greater extent compared with NG and Mtol groups. Furthermore, administration of quercatagetin (5.5 μmol/L) significantly repressed HG-induced G1 to S phase cell cycle progression, indicating that quercatagetin inhibited cell proliferation by suppressing DNA synthesis in the cells. Additionally, administration of stattic (10 μmol/L) elicited inhibitory effects on HG-induced VSMC accumulation in S phase (Figure 4).
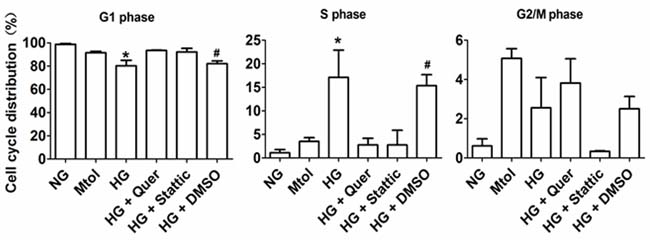
Figure 4: Cell cycle distribution of HG-treated VSMCs in vitro. Data represent means SD from three independent experiments. HG, 25 mmol/L D-glucose. *P < 0.05 vs. NG, Mtol, HG+Quer or HG+Stattic. #P < 0.05 vs. NG, Mtol, HG+Quer or HG+Stattic.
Quercetagetin inhibits Pim-1 protein activity but not gene expression in HG-treated VSMCs
After treating HG-exposed VSMCs with quercetagetin (5.5 μmol/L) for 48 hours, significant decreases of Pim-1 mRNA and protein expression were not observed (Figure 5A-5C), implying quercetagetin has no inhibitory effect on Pim-1 expression in VSMCs. We therefore detected the changes of Pim-1 protein activity. Because Pim-1 kinase promotes inactivation of Bad protein by its phosphorylation on Ser112 [18, 21], modulation of the level of p-Bad at Ser112 implies a change of Pim-1 kinase activity. As shown in Figures 5B and 5D, exposure of VSMCs to quercetagetin (5.5 μmol/L) increased Bad expression but decreased the level of p-Bad(T112). However, the ratio of p-Bad(T112)/Bad was decreased compared with the controls. Bad promotes apoptosis while p-Bad(T112) has an anti-apoptosis effect; consequently the decrease in ratio of p-Bad (T112)/Bad in quercetagetin-treated cells implies an inhibitory effect of quercetagetin on VSMC proliferation. These outcomes indicate that administration of quercetagetin decreased Pim-1 activity but not its expression in HG-treated VSMCs.
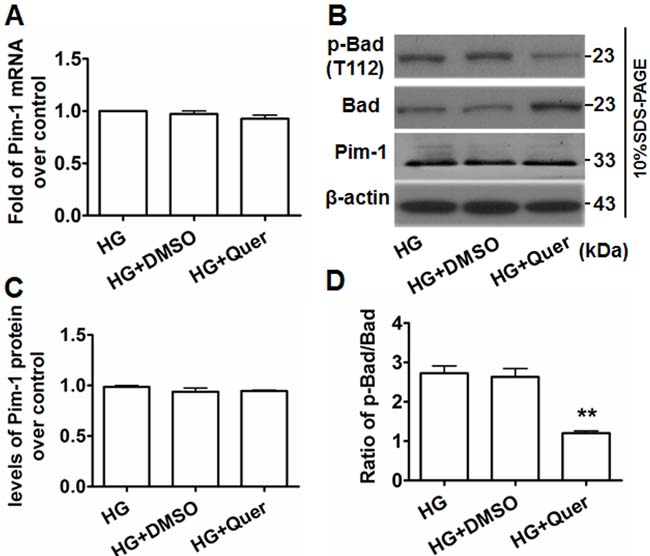
Figure 5: Quercetagetin inhibits Pim-1 activity but not its expression in HG-treated VSMCs. A. qRT-PCR was used to detect Pim-1 mRNA expression in HG-treated VSMCs. B. Western blotting was used to detect Pim-1, Bad and p-Bad(T112) protein expression in HG-treated VSMCs. For qRT-PCR and western blotting, VSMCs were maintained in media supplemented with HG (25 mmol/L D-glucose), HG+Quer (25 mmol/L D-Glucose + 5.5 μmol/L quercetagetin), or HG+DMSO (25 mmol/L D-Glucose + 0.3% (v/v) DMSO) for 48 hours, then cells were harvested and subjected to analysis. C. Levels of Pim-1 protein in HG-treated VSMCs. D. Ratio of p-Bad/Bad in HG-treated VSMCs. **P < 0.01 vs. HG or HG+DMSO.
HG induces VSMC proliferation via activation of STAT3/Pim-1 signaling
Western blotting showed that upregulation of p-STAT3(Y705) was observed in the HG-treated but not the NG- or Mtol-treated VSMCs, and that administration of stattic (10 μmol/L) decreased the levels of p-STAT3(Y705) (Figures 6A & 6B). Consequently, attenuated expression of translational Pim-1 and PCNA were observed (Figures 6C & 6D, Figure 3D). These results showed that HG induced VSMC proliferation partly through HG-activated STAT3/Pim-1 signaling.
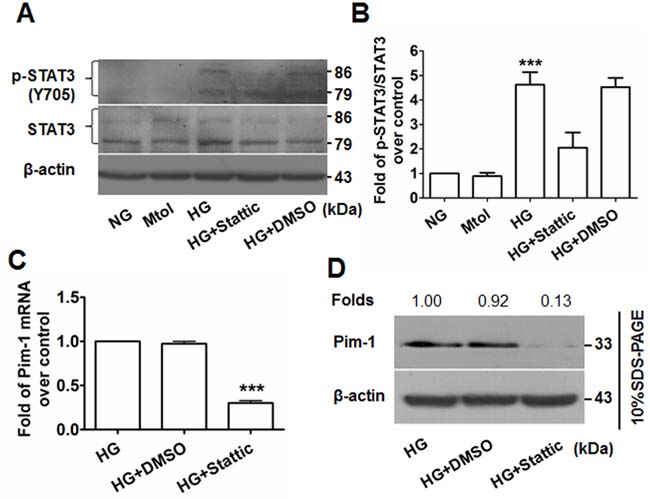
Figure 6: Stattic blocks STAT3 activation and results in attenuated Pim-1 expression in HG-treated VSMCs. A. p-STAT3(Y705) and STAT3 expression in VSMCs were detected by western blotting. B. Ratio of p-STAT3(Y705)/STAT3. ***P < 0.001 vs. NG, Mtol, or HG+Stattic. C. qRT-PCR and D. western blotting were used to examine Pim-1 mRNA and protein expression in HG-treated VSMCs, respectively. ***P < 0.001 vs. HG or HG+DMSO.
Discussion
Excessive proliferation of VSMCs derived from the tunica media is a major factor involved in DM-induced atherosclerosis. Moreover, the factors involved in VSMC proliferation are complex and unsolved. We have previously proven that HG induces VSMC proliferation and migration via SDF1/CXCR4/PI3K/Akt signaling [25]. Consistent with our previous study, we report here that STZ-induced HG led to significantly increased proliferation of rat aortic VSMCs in vivo. This was evidenced by the upregulation of PCNA expression, with in vitro HG stimulation resulting in a significant increase of VSMC growth, as indicated by the cell viability, cell number and PCNA expression. Furthermore, HG stimuli altered the expression of contractile and synthetic markers of VSMCs. Changes in expression of the contractile and synthetic markers of VSMCs support the phenotypic changes of VSMCs under HG conditions [29], which implying one of the reasons for DM-induced atherosclerosis. Interestingly, upregulation of Pim-1 expression was observed in both STZ-induced diabetes and HG-treated VSMCs. Besides, we revealed that HG enhanced both 33-kDa and 44-kDa Pim-1 isoform in VSMCs in a time-dependent manner, which indicated HG condition not only favors VSMCs proliferation but also inhibits apoptosis. Moreover, HG condition prominently favored the 33-kDa but not the 44-kDa isoform. In addition, 25 mmol/L D-glucose was optimal for Pim-1 expression. Interestingly, the Pim-1 inhibitor quercetagetin attenuated HG-induced VSMC proliferation. These findings are consistent with previous reports that Pim-1 is expressed in the VSMCs of balloon-injured rat carotid arteries and human coronary arteries [17], and in the VSMCs of PAH patients [19]. To the best of our knowledge, the present study provides details regarding aberrant Pim-1 expression in HG-insulted VSMCs.
Pim-1 contributes mainly to cell cycle progression and anti-apoptotic signaling. Reports have documented that Pim-1 kinases are phosphorylated and down-regulated by p21/27Kip1 at transcriptional and posttranscriptional levels [30, 31]. Cdc25A/C, a direct transcriptional target for c-Myc, physically interacts with and is phosphorylated by Pim-1 [32]. Furthermore, Pim-1 promotes formation between NuMA, HP1beta, dynein, and dynactin, a complex that is necessary for mitosis [33]. The transcriptional co-activator p100, which interacts with the c-Myb transcription factor, binds to and is phosphorylated by Pim-1 [34]. Moreover, it has been shown that Pim-1 plays a pivotal role in the regulation of survival signaling through modulation of Bcl-2 family members [35]. As a highly selective Pim-1 inhibitor, quercetagetin interacts with the ATP binding site of the Pim-1 protein, leading to impaired Pim-1 activity but not impaired expression. The outcomes of the present investigation show that administration of HG-treated VSMCs with quercetagetin resulted in changes in the ratio of p-Bad(T112)/Bad but not Pim-1 expression. Consequently, attenuated HG-induced VSMC proliferation and cell cycle progression occurred. Our results are consistent with other reports in that treatment with Pim-1 inhibitors effectively induces cell cycle arrest [36-38], and administration of quercetagetin or Pim-1 siRNA inhibits PDGFbb-induced VSMC proliferation [18].
Pim-1 kinase is constitutively active when expressed and its regulation occurs primarily via transcription and protein stabilization [39]. Many mitogenic stimuli including growth factors such as interleukins, GM-CSF and G-CSF, and interferon regulate Pim-1 expression at transcriptional level [40]. The majority of these factors transduce their primary signals through the JAK/STAT pathway, indicating that this cascade is essential for regulating the expression of Pim genes [41], which is one of reason why we focused on STAT signaling presently. As a member of the STAT family, STAT3 was originally identified as an acute-phase response factor. It is reported that STAT3 binds to the Pim-1 promoter at the ISFR/GAS-sequence (IFN-γ activation sequence), implying direct regulation of Pim-1 expression [42]. Paulin et al. [19] have reported that STAT3/Pim-1 activation promotes pulmonary artery smooth muscle cell proliferation. In our present study, HG stimuli activated STAT3, evidenced by the upregulation of p-STAT3(Y705). Moreover, administration of stattic, an inhibitor of STAT3, significantly attenuated HG-induced STAT3 activation and VSMC proliferation. Interestingly, stattic-inhibited STAT3 activation led to a down-regulation of Pim-1 expression. Thus, the present study suggests that HG-induced VSMC proliferation might partly be due to the activation of STAT3/Pim-1 signaling, consistent with other reports [19, 43, 44].
Previous reports have indicated that Pim-1 is down-regulated in cardiomyocytes of SZT-induced hyperglycemic rats, especially in females [45]. On the contrary, forced expression of Pim-1 would be beneficial for curing diabetic cardiomyopathy [46]. Recently, a report also indicated HG suppressed Pim-1 expression in vascular endothelial cells (EPCs) [47]. However, our current study indicated that HG conditions induced Pim-1 expression in VSMCs at the manner of transient decline following sustained increase. Because HG condition favors VSMCs proliferation while attenuates EPCs viability [48, 49], it seems that VSMCs displays various responses post-HG insults when compared with EPCs and cardiomyocytes, the underlying mechanisms remain to be further elucidated. Report [45] also indicated Pim-1 was downregulated in ex vivo aortic rings of diabetic mice, which is inconsistent with our current results. The reasons are unknown. EPCs also participate in the pathological process of DM-induced vascular remodeling; we just focused on VSMCs in the current investigation. Because the complex role of HG in Pim-1 expression in VSMCs and EPCs, we need another model to interpret the interaction between VSMCs and EPCs in HG-induced vascular remodeling process in future.
Taken together, the present study provides the first evidence that HG treatment can induce VSMC proliferation, which is partly mediated by HG-activated STAT3/Pim-1 signaling. Therefore, Pim-1 overexpression plays a pivotal role in DM-induced vascular complications. Our study provides novel insight regarding DM-related vascular remolding diseases.
MATERIALS AND METHODS
Establishing a rat model of type I diabetes
Male Sprague Dawley (SD) rats weighing 250 ± 50 g were purchased from the Center of Experimental Animals of Guangdong Medical University, Zhanjiang, China. All procedures were performed in accordance with the Guidelines of the Guangdong Council of Animal Care and approved by the Animal Use Subcommittee at Guangdong Medical University. Diabetes was induced in rats by single intraperitoneal injection of STZ (55 mg⁄kg, Sigma-Aldrich, Shanghai, China) as previously described [25]. Blood glucose was monitored for up to 3 weeks, and only rats with blood glucose levels >/= 16.6 mmol/L were used in the study. Three weeks after STZ injection, rats were sacrificed and aortic arch, thoracic and abdominal arterial samples were obtained. The tunica media was quickly immersed in liquid nitrogen for later use; for paraffin-embedded sections, samples were fixed in neutral formalin.
Cell isolation and culture
Primary VSMCs were isolated from SD rat aortic arteries and identified as described previously [25]. VSMCs were maintained in Dulbecco’s Modified Eagle Medium (DMEM, HyCloneTM, GE Healthcare Life Sciences, Utah, USA) containing 5.5 mmol/L glucose and 10% fetal bovine serum (FBS, HyCloneTM). All cells were maintained at 37°C in a 5% CO2 atmosphere. DMEM containing 12.5, 25 and 50 mmol/L D-glucose was used as HG in all experiments, while DMEM containing 5.5 mmol/L glucose (normal glucose, NG) and DMEM containing 25 mmol/L D-mannitol (Mtol) were used as the NG control and osmotic control, respectively. Cells at passages 4-6 were used for experimentation. In all experiments, cells were serum-starved for 12 hours with DMEM containing 0.5% FBS and then subjected to the different treatments.
Determining cell viability and cell number
In vitro cell proliferation assays were performed as described previously [50, 51]. Briefly, VSMCs (2 × 103 /well) were seeded into 96-well plates (Corning, Lowell, MA, USA). Cells were cultured in NG, Mtol and HG media supplemented with quercetagetin (5.5 μmol/L, Pim-1 inhibitor, Merck, Germany), stattic (10 μmol/L, STAT3 inhibitor, Merck) and dimethylsulphoxide (DMSO, ≤0.3%, v/v) for 48 hours. After washing twice with 1×PBS, a total of 100 μL DMEM plus 10 μL CCK-8 reagents (Beyotime Institute of Biotechnology, Jiangsu, China) were added to each well and the optical density (OD) was measured 4 hours later using a microplate reader (Multiskan MKS, Thermo Scientific, MA, USA) at dual wavelength (450/630 nm). Each group was duplicated in six wells and each assay was performed in triplicate.
Cell number was determined as described previously [52]. Briefly, VSMCs were seeded into 6-well plates (5 × 105 per well). After overnight recovery in an incubator at 37°C/5% CO2, the cells were serum-deprived for 12 hours and exposed to the aforementioned conditions for 48 hours and then counted using a hemocytometer ( n = 3 wells / group).
Flow cytometric determination of cell cycles
VSMCs were seeded into 6-well plates and treated as described above (n = 3 wells/group). Forty-eight hours later, harvested cells were stained with propidium iodide (PI) and were subjected to flow cytometric analysis (BD FACS Canto II).
RNA extraction, reverse transcription (RT) and quantitative PCR
RNA extraction, RT and quantitative PCR were performed as described previously [53]. Primer pairs (5´-3´) were synthesized by Sangon Biotech Co., Ltd. (Shanghai, China) as follows: Pim-1 (NM_017034.1) forward, ctgctcaaggacacagtctaca; Pim-1 reverse, agggacaggcaccatctaataa; PCNA (NM_022381.1) forward, gggtgaagttttctgcgagt; PCNA reverse, cagtggagtggcttttgtga; β-actin (NM_031144) forward, cccatctatgagggttacgc, β-actin reverse, tttaatgtcacgcacgatttc. PCR was conducted using a LightCycler480 II instrument (Roche Ltd., Guangzhou, China). Relative abundances of target mRNAs were determined from the CT values and plotted as the fold-change compared with the control groups.
Western blotting
Western blot analyses were performed using protein extracts from VSMCs as previously described [54, 55]. A total of 30 - 75 μg of protein was transferred onto PVDF membranes by electrophoretic transfer following electrophoretic separation by 10% - 12% SDS-polyacrylamide gel electrophoresis (SDS-PAGE). The antibodies used were Pim-1, PCNA, Bad, p-Bad(T112), STAT3, p-STAT3(Y705), Myocardin, FHL2, Osteopontin, α-SMA and β-actin. Details of these antibodies are included in Supplementary Table S1.
Immunohistochemistry
Immunohistochemical staining was performed on rat arterial sections. The 5 μM-thick paraffin sections were deparaffinized and re-hydrated according to standard protocols. Heat-induced antigen retrieval was then performed in sodium citrate buffer (10 mmol/L, pH 6.0) and endogenous peroxidases were blocked by incubation in 0.3% H2O2. Sections were then incubated with primary antibodies against Pim-1 (1:50) and PCNA (1:100) at 4°C overnight and non-immune IgG was used as a negative control. Antigenic sites were localized using the SP9000 or SP9003 kit and 3,3´-diaminobenzidine (DAB) kit (ZSGB-BIO, Beijing, China). Brown coloration in the VSMCs was considered as positive expression.
Statistical analyses
Statistical analyses were carried out using PRISM Software (GraphPad Software, CA, USA). Data were expressed as means ± standard deviation (SD) from three independent experiments. For analysis of differences between two groups, the Student’s t-test was performed. For multiple groups, analysis of variance (ANOVA) was carried out followed by the Student-Newman-Keuls test. The level of statistical significance was set at P < 0.05.
Author contributions
KW, XD, YJ, RD, XL, SW, YH and YW performed the experiments. KW, ZS and YK collected and analyzed the data. RL sectioned the samples. JG and WJ conceived and coordinated the study, WJ wrote the paper. All authors read and approved the final manuscript.
ACKNOWLEDGMENTS
This study was supported by grants from the National Natural Science Foundation of China (81170121, 81460042, 81670254), the YangFan Plan of Guangdong Province (4YF16007G), the Youth Innovative Talents Project of Hainan Association for Science and Technology (2015-16), the Scientific Research Foundation for the 2015 Returned Overseas of Ministry of Human Society, and Hainan Provincial Science & Technology Key Project (ZDYF2016168 and ZDYF2016224), the Doctoral Research Program of Guangdong Medical University (B2010013), the Innovation Training Projects for College Students in Guangdong Province (1057112015), and the Innovative Research Projects for Undergraduates of Guangdong Medical University (2011ZZZF001).
CONFLICTS OF INTERESTS
The authors declare that they have no competing interests.
REFERENCES
1. Nathan DM, Lachin J, Cleary P, Orchard T, Brillon DJ, Backlund JY, O’Leary DH, Genuth S. Intensive diabetes therapy and carotid intima-media thickness in type 1 diabetes mellitus. N Engl J Med. 2003; 348:2294-2303.
2. Popov D, Constantinescu E. Arterial smooth muscle cells dysfunction in hyperglycaemia and hyperglycaemia associated with hyperlipidaemia: from causes to effects. Arch Physiol Biochem. 2008; 114:150-160.
3. Rizzoni D, Rosei EA. Small artery remodeling in diabetes mellitus. Nutr Metab Cardiovasc Dis. 2009; 19:587-592.
4. Cuypers HT, Selten G, Quint W, Zijlstra M, Maandag ER, Boelens W, van Wezenbeek P, Melief C, Berns A. Murine leukemia virus-induced T-cell lymphomagenesis: integration of proviruses in a distinct chromosomal region. Cell. 1984; 37:141-150.
5. Narlik-Grassow M, Blanco-Aparicio C, Carnero A. The PIM family of serine/threonine kinases in cancer. Med Res Rev. 2014; 34:136-159.
6. Saris CJ, Domen J, Berns A. The pim-1 oncogene encodes two related protein-serine/threonine kinases by alternative initiation at AUG and CUG. EMBO J. 1991; 10:655-664.
7. Muraski JA, Rota M, Misao Y, Fransioli J, Cottage C, Gude N, Esposito G, Delucchi F, Arcarese M, Alvarez R, Siddiqi S, Emmanuel GN, Wu W, et al. Pim-1 regulates cardiomyocyte survival downstream of Akt. Nat Med. 2007; 13:1467-1475.
8. Santio NM, Salmela M, Arola H, Eerola SK, Heino J, Rainio EM, Koskinen PJ. The PIM1 kinase promotes prostate cancer cell migration and adhesion via multiple signalling pathways. Exp Cell Res. 2016; 342:113-124.
9. Merkel AL, Meggers E, Ocker M. PIM1 kinase as a target for cancer therapy. Expert Opin Investig Drugs. 2012; 21:425-436.
10. Leung CO, Wong CC, Fan DN, Kai AK, Tung EK, Xu IM, Ng IO, Lo RC. PIM1 regulates glycolysis and promotes tumor progression in hepatocellular carcinoma. Oncotarget. 2015; 6:10880-10892. doi: 10.18632/oncotarget.3534.
11. Zhang Y, Parsanejad M, Huang E, Qu D, Aleyasin H, Rousseaux MW, Gonzalez YR, Cregan SP, Slack RS, Park DS. Pim-1 kinase as activator of the cell cycle pathway in neuronal death induced by DNA damage. J Neurochem. 2010; 112:497-510.
12. Narimatsu M, Maeda H, Itoh S, Atsumi T, Ohtani T, Nishida K, Itoh M, Kamimura D, Park SJ, Mizuno K, Miyazaki J, Hibi M, Ishihara K, et al. Tissue-specific autoregulation of the stat3 gene and its role in interleukin-6-induced survival signals in T cells. Mol Cell Biol. 2001; 21:6615-6625.
13. An N, Lin YW, Mahajan S, Kellner JN, Wang Y, Li Z, Kraft AS, Kang Y. Pim1 serine/threonine kinase regulates the number and functions of murine hematopoietic stem cells. Stem Cells. 2013; 31:1202-1212.
14. Samse K, Emathinger J, Hariharan N, Quijada P, Ilves K, Volkers M, Ormachea L, De La Torre A, Orogo AM, Alvarez R, Din S, Mohsin S, Monsanto M, et al. Functional Effect of Pim1 Depends upon Intracellular Localization in Human Cardiac Progenitor Cells. J Biol Chem. 2015; 290:13935-13947.
15. Sussman MA. Mitochondrial integrity: preservation through Akt/Pim-1 kinase signaling in the cardiomyocyte. Expert Rev Cardiovasc Ther. 2009; 7:929-938.
16. Zippo A, De Robertis A, Bardelli M, Galvagni F, Oliviero S. Identification of Flk-1 target genes in vasculogenesis: Pim-1 is required for endothelial and mural cell differentiation in vitro. Blood. 2004; 103:4536-4544.
17. Katakami N, Kaneto H, Hao H, Umayahara Y, Fujitani Y, Sakamoto K, Gorogawa S, Yasuda T, Kawamori D, Kajimoto Y, Matsuhisa M, Yutani C, Hori M, et al. Role of pim-1 in smooth muscle cell proliferation. J Biol Chem. 2004; 279:54742-54749.
18. Willert M, Augstein A, Poitz DM, Schmeisser A, Strasser RH, Braun-Dullaeus RC. Transcriptional regulation of Pim-1 kinase in vascular smooth muscle cells and its role for proliferation. Basic Res Cardiol. 2010; 105:267-277.
19. Paulin R, Courboulin A, Meloche J, Mainguy V, Dumas de la Roque E, Saksouk N, Cote J, Provencher S, Sussman MA, Bonnet S. Signal transducers and activators of transcription-3/pim1 axis plays a critical role in the pathogenesis of human pulmonary arterial hypertension. Circulation. 2011; 123:1205-1215.
20. Qian Z, Zhang L, Chen J, Li Y, Kang K, Qu J, Wang Z, Zhai Y, Li L, Gou D. MiR-328 targeting PIM-1 inhibits proliferation and migration of pulmonary arterial smooth muscle cells in PDGFBB signaling pathway. Oncotarget. 2016; 7:54998-55011. doi: 10.18632/oncotarget.10714.
21. Aho TL, Sandholm J, Peltola KJ, Mankonen HP, Lilly M, Koskinen PJ. Pim-1 kinase promotes inactivation of the pro-apoptotic Bad protein by phosphorylating it on the Ser112 gatekeeper site. FEBS letters. 2004; 571:43-49.
22. Brault L, Gasser C, Bracher F, Huber K, Knapp S, Schwaller J. PIM serine/threonine kinases in the pathogenesis and therapy of hematologic malignancies and solid cancers. Haematologica. 2010; 95:1004-1015.
23. Paukku K, Silvennoinen O. STATs as critical mediators of signal transduction and transcription: lessons learned from STAT5. Cytokine Growth Factor Rev. 2004; 15:435-455.
24. Boengler K, Hilfiker-Kleiner D, Drexler H, Heusch G, Schulz R. The myocardial JAK/STAT pathway: from protection to failure. Pharmacol Ther. 2008; 120:172-185.
25. Jie W, Wang X, Zhang Y, Guo J, Kuang D, Zhu P, Wang G, Ao Q. SDF-1alpha/CXCR4 axis is involved in glucose-potentiated proliferation and chemotaxis in rat vascular smooth muscle cells. Int J Exp Pathol. 2010; 91:436-444.
26. Li H, Peng W, Zhuang J, Lu Y, Jian W, Wei Y, Li W, Xu Y. Vaspin attenuates high glucose-induced vascular smooth muscle cells proliferation and chemokinesis by inhibiting the MAPK, PI3K/Akt, and NF-kappaB signaling pathways. Atherosclerosis. 2013; 228:61-68.
27. Maile LA, Allen LB, Hanzaker CF, Gollahon KA, Dunbar P, Clemmons DR. Glucose regulation of thrombospondin and its role in the modulation of smooth muscle cell proliferation. Exp Diabetes Res. 2010; 2010.
28. Wang L, Zhu LH, Jiang H, Tang QZ, Yan L, Wang D, Liu C, Bian ZY, Li H. Grape seed proanthocyanidins attenuate vascular smooth muscle cell proliferation via blocking phosphatidylinositol 3-kinase-dependent signaling pathways. J Cell Physiol. 2010; 223:713-726.
29. Carrillo-Sepulveda MA, Matsumoto T. Phenotypic modulation of mesenteric vascular smooth muscle cells from type 2 diabetic rats is associated with decreased caveolin-1 expression. Cell Physiol Biochem. 2014; 34:1497-1506.
30. Zhang Y, Wang Z, Magnuson NS. Pim-1 kinase-dependent phosphorylation of p21Cip1/WAF1 regulates its stability and cellular localization in H1299 cells. Mol Cancer Res. 2007; 5:909-922.
31. Morishita D, Katayama R, Sekimizu K, Tsuruo T, Fujita N. Pim kinases promote cell cycle progression by phosphorylating and down-regulating p27Kip1 at the transcriptional and posttranscriptional levels. Cancer Res. 2008; 68:5076-5085.
32. Mochizuki T, Kitanaka C, Noguchi K, Muramatsu T, Asai A, Kuchino Y. Physical and functional interactions between Pim-1 kinase and Cdc25A phosphatase. Implications for the Pim-1-mediated activation of the c-Myc signaling pathway. J Biol Chem. 1999; 274:18659-18666.
33. Bhattacharya N, Wang Z, Davitt C, McKenzie IF, Xing PX, Magnuson NS. Pim-1 associates with protein complexes necessary for mitosis. Chromosoma. 2002; 111:80-95.
34. Leverson JD, Koskinen PJ, Orrico FC, Rainio EM, Jalkanen KJ, Dash AB, Eisenman RN, Ness SA. Pim-1 kinase and p100 cooperate to enhance c-Myb activity. Molecular cell. 1998; 2:417-425.
35. Lilly M, Sandholm J, Cooper JJ, Koskinen PJ, Kraft A. The PIM-1 serine kinase prolongs survival and inhibits apoptosis-related mitochondrial dysfunction in part through a bcl-2-dependent pathway. Oncogene. 1999; 18:4022-4031.
36. Lin YW, Beharry ZM, Hill EG, Song JH, Wang W, Xia Z, Zhang Z, Aplan PD, Aster JC, Smith CD, Kraft AS. A small molecule inhibitor of Pim protein kinases blocks the growth of precursor T-cell lymphoblastic leukemia/lymphoma. Blood. 2010; 115:824-833.
37. Beharry Z, Zemskova M, Mahajan S, Zhang F, Ma J, Xia Z, Lilly M, Smith CD, Kraft AS. Novel benzylidene-thiazolidine-2,4-diones inhibit Pim protein kinase activity and induce cell cycle arrest in leukemia and prostate cancer cells. Mol Cancer Ther. 2009; 8:1473-1483.
38. Keeton EK, McEachern K, Dillman KS, Palakurthi S, Cao Y, Grondine MR, Kaur S, Wang S, Chen Y, Wu A, Shen M, Gibbons FD, Lamb ML, et al. AZD1208, a potent and selective pan-Pim kinase inhibitor, demonstrates efficacy in preclinical models of acute myeloid leukemia. Blood. 2014; 123:905-913.
39. Amaravadi R, Thompson CB. The survival kinases Akt and Pim as potential pharmacological targets. J Clin Invest. 2005; 115:2618-2624.
40. Wang Z, Bhattacharya N, Weaver M, Petersen K, Meyer M, Gapter L, Magnuson NS. Pim-1: a serine/threonine kinase with a role in cell survival, proliferation, differentiation and tumorigenesis. J Vet Sci. 2001; 2:167-179.
41. Nawijn MC, Alendar A, Berns A. For better or for worse: the role of Pim oncogenes in tumorigenesis. Nat Rev Cancer. 2011; 11:23-34.
42. Blanco-Aparicio C, Carnero A. Pim kinases in cancer: diagnostic, prognostic and treatment opportunities. Biochem Pharmacol. 2013; 85:629-643.
43. Iwakura T, Mohri T, Hamatani T, Obana M, Yamashita T, Maeda M, Katakami N, Kaneto H, Oka T, Komuro I, Azuma J, Nakayama H, Fujio Y. STAT3/Pim-1 signaling pathway plays a crucial role in endothelial differentiation of cardiac resident Sca-1+ cells both in vitro and in vivo. J Mol Cell Cardiol. 2011; 51:207-214.
44. Hofmann AD, Takahashi T, Duess J, Gosemann JH, Puri P. Increased expression of activated pSTAT3 and PIM-1 in the pulmonary vasculature of experimental congenital diaphragmatic hernia. J Pediatr Surg. 2015; 50:908-911.
45. Moore A, Shindikar A, Fomison-Nurse I, Riu F, Munasinghe PE, Ram TP, Saxena P, Coffey S, Bunton RW, Galvin IF, Williams MJ, Emanueli C, Madeddu P, et al. Rapid onset of cardiomyopathy in STZ-induced female diabetic mice involves the downregulation of pro-survival Pim-1. Cardiovasc Diabetol. 2014; 13:68.
46. Katare R, Caporali A, Zentilin L, Avolio E, Sala-Newby G, Oikawa A, Cesselli D, Beltrami AP, Giacca M, Emanueli C, Madeddu P. Intravenous gene therapy with PIM-1 via a cardiotropic viral vector halts the progression of diabetic cardiomyopathy through promotion of prosurvival signaling. Circ Res. 2011; 108:1238-1251.
47. Chen M, Yi B, Zhu N, Wei X, Zhang GX, Huang S, Sun J. Pim1 kinase promotes angiogenesis through phosphorylation of endothelial nitric oxide synthase at Ser-633. Cardiovasc Res. 2016; 109:141-150.
48. Zhang W, Chen S, Zhang Z, Wang C, Liu C. FAM3B mediates high glucose-induced vascular smooth muscle cell proliferation and migration via inhibition of miR-322-5p. Sci Rep. 2017; 7:2298.
49. Hou Q, Lei M, Hu K, Wang M. The effects of high glucose levels on reactive oxygen species-induced apoptosis and involved signaling in human vascular endothelial cells. Cardiovasc Toxicol. 2015; 15:140-146.
50. Shen Z, Zeng Y, Guo J, Wu Y, Jiang X, Ding R, Wu C, Li R, Luo B, Zeng C, Jiang H, Jie W. Over-expression of the special AT rich sequence binding protein 1 (SATB1) promotes the progression of nasopharyngeal carcinoma: association with EBV LMP-1 expression. J Transl Med. 2013; 11:217.
51. Shen Z, Jiang X, Zeng C, Zheng S, Luo B, Zeng Y, Ding R, Jiang H, He Q, Guo J, Jie W. High expression of ubiquitin-conjugating enzyme 2C (UBE2C) correlates with nasopharyngeal carcinoma progression. BMC cancer. 2013; 13:192.
52. Jie W, Guo J, Shen Z, Wang X, Zheng S, Wang G, Ao Q. Contribution of myocardin in the hypoxia-induced phenotypic switching of rat pulmonary arterial smooth muscle cells. Exp Mol Pathol. 2010; 89:301-306.
53. Ding R, Jiang X, Ha Y, Wang Z, Guo J, Jiang H, Zheng S, Shen Z, Jie W. Activation of Notch1 signalling promotes multi-lineage differentiation of c-Kit(POS)/NKX2.5(POS) bone marrow stem cells: implication in stem cell translational medicine. Stem Cell Res Ther. 2015; 6:91.
54. Guo J, Jie W, Shen Z, Li M, Lan Y, Kong Y, Guo S, Li T, Zheng S. SCF increases cardiac stem cell migration through PI3K/AKT and MMP2/9 signaling. Int J Mol Med. 2014; 34:112-118.
55. Wu Y, Shen Z, Wang K, Ha Y, Lei H, Jia Y, Ding R, Wu D, Gan S, Li R, Luo B, Jiang H, Jie W. High FMNL3 expression promotes nasopharyngeal carcinoma cell metastasis: role in TGF-beta1-induced epithelia-to-mesenchymal transition. Sci Rep. 2017; 7:42507.