
Introduction
Intracerebral hemorrhage (ICH), which accounts for 10-15% of stroke, is the most lethal form of stroke with more than 40% 30-day mortality compared to ischemic stroke [1, 2]. Hematoma expansion (HE), which occurs in approximately 33% ICH patients, is identified as one important independent predictor of early neurological deterioration and poor long-term clinical outcomes [3, 4]. However, several therapeutic treatments targeting HE, including early aggressive blood pressure lowering treatment and receiving recombinant activated factor VII (Ravia), have different conclusions of clinical outcome improvement in large randomized controlled trails [5, 6]. For example, in the Second Intensive Blood Pressure Reduction in Acute Cerebral Hemorrhage Trail (INTERACT2), lowering intensive blood pressure showed a borderline significant effect on better primary outcome. Their outcomes may result from the indiscriminate enrollment of patients with low rates of HE. In addition, some patients destined to experience HE were excluded from emergency surgery. Based on that supposition, more attention has been paid on better stratification of patients, especially patients destined to undergo HE. Therefore, it is important to find those HE predictors to stratify patients and tailor intensive therapies timely and effectively for high-risk patient. Several retrospective studies have identified different individual potential predictors using various methodologies, but results are conflicting (Table 1) [7-9]. In this review, we summarize and classify those potential predictors by clinical, laboratory, radiographic, and integrated score models. We discuss their underlying pathophysiology and future therapeutic strategies on HE prediction or prevention.
Table 1: Descriptive Summary of Predictors for HE of ICH patients.
Predictors |
References |
Recruitment Period |
Country |
Num |
Enrollment Window (CT scan) |
HE definition |
Sensitivity |
Specificity |
PPV |
NPV |
OR(95%CI) |
AUC |
||
Maximum SBP |
Ohwaki,2014[72] |
1998-2002 |
Japan |
76 |
second <48h |
>40% or >12.5 mL |
/ |
/ |
/ |
/ |
1.04(1.01-1.07) |
/ |
||
CRP>10mg/L |
Di Napo,2014l[100] |
2009-2011 |
International |
399 |
first <6h |
>33% or >12.5 mL |
/ |
/ |
/ |
/ |
4.71(2.75-8.06) |
/ |
||
C-Fn>6µg/mL |
Silva,2005[42] |
NA |
International |
183 |
symptom onset <12h |
>33% for <20mL; >10% for ≥20mL; |
/ |
/ |
/ |
/ |
92(22-381) |
/ |
||
IL-6>24 pg/mL |
/ |
/ |
/ |
/ |
16(2.3-119) |
/ |
||||||||
Density in CT |
Barras,2009[114] |
NA |
International |
90 |
first <3h |
>33% or >12.5 mL |
65.6% |
46.6% |
40.4% |
71.1% |
/ |
/ |
||
Shape in CT |
78.1% |
20.1% |
35.2% |
63.2% |
/ |
/ |
||||||||
mNIHSS |
Chan,2015[63] |
2008-2010 |
USA |
257 |
first <24h, second <48h |
>33% or >12.5 mL |
/ |
/ |
/ |
/ |
1.06 |
0·6712 |
||
Warfarin use |
/ |
/ |
/ |
/ |
1.9 |
0·6712 |
||||||||
Warfarin use |
Yaghi,2014[12] |
2009-2012 |
USA |
200 |
first <12h, second <24h |
>33% |
/ |
/ |
/ |
/ |
3.6(1.3-10.3) |
/ |
||
IVH |
/ |
/ |
/ |
/ |
5.7(1.5-20.9) |
/ |
||||||||
Spot sign |
Orito,2016[134] |
2012-2013 |
Japan |
80 |
NA |
>10% |
77.8% |
73.8% |
83.3% |
46.8% |
/ |
/ |
||
Spot sign |
Andrew,2012[17] |
2006-2010 |
6 countries |
268 |
first <6h |
>33% or >6 mL |
51% |
85% |
61% |
78% |
/ |
/ |
||
High HU of spot |
Kim,2014[143] |
2009-2011 |
Korea |
316 |
NA |
>33% or >6 mL |
/ |
/ |
/ |
/ |
1.048(1.01-1.09) |
/ |
||
Spot sign number |
≥1 |
Huynh 2013, [142] |
2006-2010 |
6 countries |
268 |
first <6h |
>33% or >6 mL |
51% |
85% |
61% |
78% |
/ |
/ |
|
≥2 |
32% |
92% |
64% |
74% |
||||||||||
≥3 |
12% |
97% |
64% |
70% |
||||||||||
≥4 |
3% |
99% |
50% |
68% |
||||||||||
Short initial time |
Kim,2014[143] |
2009-2011 |
Korea |
316 |
NA |
>33% or >6 mL |
/ |
/ |
/ |
/ |
0.197(0.06-0.61) |
/ |
||
Time from onset to CT |
0-2h |
Dowlatshahi,2016[109] |
1946-2016 |
International |
1039 |
NA |
>33% or >6 mL |
60% |
76% |
61% |
76% |
/ |
0.68 |
|
2-4h |
55% |
84% |
57% |
82% |
0.69 |
|||||||||
4-6h |
44% |
91% |
56% |
87% |
0.68 |
|||||||||
6-8h |
56% |
92% |
64% |
90% |
0.74 |
|||||||||
>8h |
30% |
90% |
33% |
89% |
0.60 |
|||||||||
CTP spot sign |
Koculym,2013[136] |
Six months |
Canada |
28 |
first <6h |
>30% or >6 mL |
78% |
100% |
100% |
71% |
/ |
/ |
||
Leakage sign |
Orito,2016[134] |
2012-2013 |
Japan |
80 |
NA |
>10% |
93.3% |
88.9% |
94.3% |
66.7% |
/ |
/ |
||
Spot & Leakage sign |
93.8% |
91.4% |
97.1% |
68.9% |
/ |
/ |
||||||||
Black hole sign |
Qi,2016[140] |
2011-2015 |
China |
206 |
first <6h, second <30h |
>33% or >12.5 mL |
31.9% |
94.1% |
73.3% |
73.2% |
/ |
/ |
||
Score Models |
9-point |
Huynh,2015[83] |
2006-2012 |
6 countries |
301 |
first <6h |
>33% or >6 mL |
/ |
/ |
/ |
/ |
/ |
0.761 |
|
24-point |
0.673 |
|||||||||||||
PREDICT A |
0.823 |
|||||||||||||
PREDICT B |
0.804 |
|||||||||||||
HE indicates hematoma expansion; PPV, positive predictive value; NPV, negative predictive value; SBP, systolic blood pressure; CT, computed tomography; CRP, C-Reactive Protein; C-Fn, cellular fibronectin;IL-6, interleukin-6;NA,not available; mNIHSS, modified National Institutes of Health Stroke Scale; IVH, intraventricular hemorrhage; HU, Hounsfield Unit; CTP, CT perfusion.
The Definition and Patho-physiology of Hematoma Expansion
The definition of HE varied in different literatures with a 40% relative volume increase or 12.6mL absolute volume increase in hematoma size from baseline CT to follow-up CT in one [10], a 33% relative increase in others [11, 12], or even a 50% relative increase and 2mL absolute increase in another [13]. One study found absolute HE definitions have higher positive predictive value for poor outcome than relative definitions. This could be explained because absolute HE is directly proportional to the truly damaged brain tissue [14]. Furthermore, they found that baseline hematoma size doesn’t influence the prediction of absolute and relative HE definition. This overturns the expectation that the absolute volume increase would be greater in large baseline size hemorrhages and absolute definition of HE would predict better in small baseline sizes. So using a method that assigns equal weight between absolute and relative definition could maximize sensitivity and specificity [15]. The INTERACT1 study categorized absolute and relative HE from minimal change (<5mL or <33%) to massive change (>12.5mL or >50%) [2]. Some studies on CT angiography contrast extravasation defined HE as a proportional increase of 33% or an absolute increase of more than 6mL [16-18]. One study mathematically derived “optimal” HE cutoff was ≥ 3mL for absolute growth (sensitivity 49%, specificity 81%) and ≥ 26% for relative growth (sensitivity 42%, specificity 80%) [14]. Interestingly, these “optimal” cut points may not be the most clinically suitable for use because a clinical relevant definition of HE should have a high positive predictive value, not equal weight of sensitivity and specificity. Thus, they assume the best clinical definition of HE was its dichotomized definition: relative (>33%) or absolute change (>12.5 mL) comparing with hematoma volume from initial to following CT slide, and applied in the large clinical trial like INTERACT2 and rFVIIa ICH trail [19, 20]. The relative cutoff was chosen prospectively for three reasons: First, a 10% change in diameter corresponds to a 33% growth in the sphere volume, which makes a clear difference to the naked eye of a physician viewing serial CT scans. Second, different positions and angles of the CT slice images could cause up to one-third “decrease” volume effect between the baseline and 1-hour CTs, especially for small hemorrhages. So the relative CT definition would represent the true HE, not volume variability of small hematoma in CT imaging [21, 22]. Third, this cutoff has been applied in the large clinical ATACH-I and II trials consistently [11]. The absolute cutoff was ≥12.5mL. Though absolute ≥ 12.5mL change was only seen in less 20% of patients, it’s a necessary trade-off having fewer patients for better positive predictive value, odds ratio, and detection. Despite the relative (>33%) or absolute change (>12.5 mL) combination definition was most used in recent studies, the preferred cutoff for clinically significant HE has not reached a consensus. Thus we believe mathematically or clinically optimal cutoffs could be validated, not only to lower the heterogeneity among studies, but also to better stratify high-risk patients.
HE presents the extended distribution of initial hemorrhage, including ventricle invasion, transition or rebleeding into compartments adjacent to the initial zone and intraparenchymal volume growth. HE occurs in about 73% of patients within 3 hours from symptom onset and clinically prominent expansion occurs in 35% of patients [13, 22, 23]. However, the pathophysiology of HE remains unclear and needs further investigation. Previous studies have proposed several mechanisms of HE, including ongoing bleeding, coagulopathy state within hematoma, as well as rupture of perihematomal vessels [24-27]. Leakage or rebleeding from one or more ruptured arteries may be the primary force of HE. In 1971, a study using microscopic serial sections on pontine and putamen hemorrhages have proposed an “avalanche” model for HE [28]. This model describes the HE as secondary mechanical shearing of periphery vessels caused by expansion of initial bleeding. Clinical observation, genetic data, radiologic evidence, and computational simulation supports this model [29-32]. Within the first hour, hematomas induce brain injury by neuronal and glial mechanical disruption, oligemia or ischemia, which induce neurotransmitters to release. In addition, mitochondrial failure, through glutamate release and calcium influx, resulted in sodium accumulation, cytotoxic edema, and necrosis within the first 4 hours [1, 33, 34]. Products of hemoglobin breakdown and coagulation, such as but not limited to thrombin, ferrous, iron, and heme initiate a secondary injury cascade [1, 33, 35]. In particular, thrombin activated microglia to release oxygen free radicals (OFR), tumor necrosis factor α (TNFα), interleukin-1β (IL-1β), matrix metalloproteinase (MMP), and complement factors to trigger blood-brain barrier (BBB) connective tissue breakdown, astrocytes aquaporin-4 (AQ-4) expression, and neuronal and glia apoptosis [36-39]. Those physiological processes induce vasogenic edema, polymorphonuclear neutrophil (PMN) and macrophage recruitment [1, 36, 40, 41]. Those ICH-initiated injury cascades: secondary inflammation elicited by degradation products and alterations in BBB basal membrane by MMP induction, are suggested to cause hematoma enlargement [36, 42, 43]. Furthermore, coagulopathies, oral anticoagulants, autoregulation failure, and uncontrolled perfusion pressure might account for HE by repeated or continuous bleeding. Since these mechanisms are all pathologically plausible, further investigation focusing on the precise mechanism of HE is required [44, 45] (Figure 1).
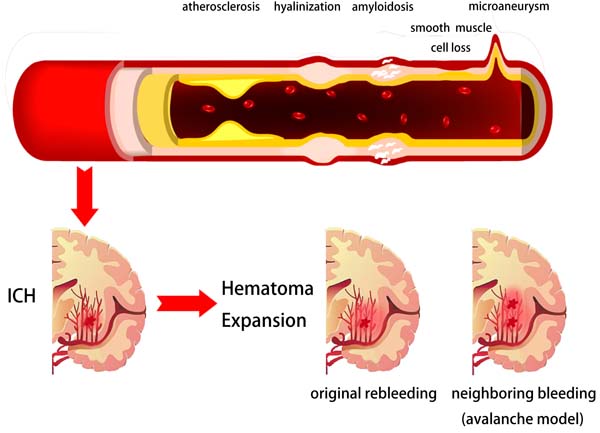
Figure 1: Potential pathophysiological mechanisms of intracerebral hemorrhage and hematoma expansion. Arterial pathophysiological changes like atherosclerosis, hyalinization, amyloidosis, smooth muscle cell loss and micro-aneurysm could be the underlying reasons of intracerebral hemorrhage. The original site ongoing bleeding or perihematomal vessels bleeding may be the primary force of hematoma expansion.
Clinical Features
Previous studies have demonstrated several clinical features related with HE, such as consciousness level, blood glucose, and blood pressure.
Consciousness level
Glasgow Coma Scale (GCS) and National Institutes of Health Stroke Scale scores (NIHSS) are identified as predictors of mortality and severe disability [13, 46, 47]. GCS and NIHSS, the routinely estimated clinical data, are also identified as HE predictors in some studies [13, 23, 46] although this relationship is infrequently reported in large trails [16, 48, 49]. We notice that consciousness disturbance is more likely normal course after ICH symptom onset and exacerbates along with HE. In addition, lower CGS and greater NIHSS could represent a number of factors, especially the larger initial ICH volume, a recognized predictor of HE [16, 49]. Furthermore, clinically emergent craniotomy and hematoma evacuation were performed for patients with poor neurological condition, like GCS score < 8 and not performed for patients with poorest consciousness level, like GCS score of three with bilateral dilated pupils. Despite remaining controversy in the relationship of HE and consciousness level, low consciousness level could already help surgeons select patients for more intensive strategies, regardless of whether or not HE occurs.
Blood glucose
More than 50% of patients with stroke have hyperglycemia [50]. Several studies have indicated relationship between high blood glucose at admission with high mortality, regardless of whether or not they are diabetic [51-53]; However, the relationship between elevated glucose and HE in ICH remains controversial. Some analysis found a univariate association between lowering serum glucose concentration and decreased HE, as well as perihematomal edema and outcome [54, 55]. Hyperglycemia increases the magnitude of HE in experimental ICH model [56, 57]. For the pathology of hyperglycemia, transient hyperglycemia following stroke is hypothesized as a transient stress reaction to ICH processes, inflammation, or some other mechanisms [58, 59]. Some studies show the deleterious effects of hyperglycemia are attributed to its secondary promotive effects of acidosis, free radical formation, and inflammatory cytokines release. Those secondary effects accelerated the BBB breakdown, impaired the integrity of adjacent vessels surrounding the initial bleeding site, and promoted emerging or continuous bleeding [53, 54, 56, 57, 60-62]. Those physiological processes are interrelated with HE as well and will be discussed below respectively. While other studies show different results [12, 63]. One study shows patients with or without HE have similar blood glucose in mg/dL (179±68 vs 153±71) and diabetes (14% vs 25%) [12]. Thus, the intrinsic mechanism of high serum glucose and HE needs to be explored in future studies. Whether hyperglycemia is the response or the cause of hematoma growth needs to be elucidated. Several studies have shown that not only hyperglycemia at admission, but hypoglycemia also occurred in intensive glucose control and could increase mortality among critically ill patients [64-67]. Thus, both clinical hypoglycemia and hyperglycemia should be avoided. The optimal management of hyperglycemia in ICH and the target glucose level call for extensive basic studies and large scale clinical trials [68]. The major problem to be revolved in future is the internal pathogenies between blood glucose and HE.
Blood pressure
Elevated blood pressure at admission of ICH patients is positively associated with poor outcomes, due to its automatic pressure to the arteries and elevation effect on intracranial pressure [69, 70]. Some studies found that hypertension is more likely to be associated with large hematoma volume and HE, which make early blood pressure a potential target for better outcomes by mitigating HE [6, 7, 71-74]. One genetic research study demonstrated that increasing numbers of high blood pressure-related alleles are associated with mean baseline hematoma volume and poor clinical outcome in ICH [75]. But unfortunately, several studies and large clinical trials INTERACT 1/2 failed to reach conventional statistical significance between lowering blood pressure earlier and improving primary outcome [5, 76-78]. Further analysis of INTERACT1 indicated that early treatment to lower blood pressure was able to attenuate HE at a 1.7 and a 3.4 mL absolute volume in HE within 6 and 4 hours respectively, which could be expected to result in at least 8% and 15% relative increased chances of better outcome. The main-phase study, INTERACT2 showed that the difference in HE between intensive-treatment group (with a target SBP <140 mmHg within 1 hour) and stand-treatment group (with a target SBP <180 mmHg) was not significant after adjusting the prognostic factors. INTERACT2 failed its primary endpoint of fatality and disability rate reduction, but in the key secondary endpoint of the distribution of Modified Rankin Scale (MRS), there was a significant favorable shift in intensive pressure control group [5]. The 5-year, multi-center, 1000 patients enrolled ATACH II clinical trial is expected to provide essential information regarding the efficacy of early intensive antihypertensive treatment using intravenous nicardipine. And the underlying mechanism for its expected beneficial effect is presumably mediated through reduction in the rate and magnitude of HE in acute ICH patients [11]. Recently, the ATACH II have reported that the primary outcome of death or disability was 38.7% in the intensive pressure reduction group (target of 110 to 139 mmHg) and 37.7% in the standard pressure reduction group (target of 140 to 179mmHg). In addition, the percentages of patients with HE didn’t differ significantly between two groups (18.9% in intensive group, 24.4% in standard group, adjusted risk ratio, 0.78;95%CI, 0.58-1.03) [79]. As for the clinical practice, early aggressive acute BP lowering is proven to be safe, with no more chance of exacerbation of cerebral ischemia, infarction, or perihematoma hypoperfusion [80-82]. In summary, the beneficial effect of early blood pressure control for outcome through HE prevention needs further elucidation.
Laboratory parameters
Laboratory parameters, which focus mainly on the aspects of coagulation status, inflammation, and microvascular integrity, draw more and more attention to reveal their relationships with HE based on the consideration of their mechanism rationality, detection applicability and simplicity.
Coagulation status
Altered coagulation status may increase the risk of surrounding vessel bleeding after injury and this influence will persist beyond 24 hours, which could result in volume growth at present and future observation [79]. The low level of fibrinogen, the high level of D-dimer and international normalized ratio (INR) >1.5 seem to be predictors of HE [13, 83]. Fibrinogen plays an important role in both primary (aggregate Glycoprotein IIa/IIIb coupled-platelet) and secondary hemostasis (cross-link each other to form fibrin polymers). D-dimer, as a marker of fibrin turnover, reflects disturbance in the coagulation and fibrinolysis pathways. Higher INR means longer prothrombin time, which means worse coagulation function and more time for plasma to clot after tissue factor addition.
Several retrospective studies demonstrated a negative relationship of prior antiplatelet use with HE and clinical outcomes [8, 49, 84, 85]. One study reported that aspirin use was associated with HE and mortality, but not with functional outcome at 3 months [85]. One retrospective analysis of 251 patients demonstrated that antiplatelet therapy was an independent predictor for the occurrence of hematoma enlargement, emergent death, and evacuation surgery [86]. While in the Cerebral Hemorrhage and NXY-059 Treatment (CHANT) trial, antiplatelet medication is not associated with increased hemorrhage volume, HE, or clinical outcome at 3 months [87]. Studies demonstrate that vitamin K antagonists are related to higher risk for HE and poorer clinical outcome [8, 49, 84, 85]. The measurements of anti-warfarin effect on HE, such as Prothrombin Complex Concentrates (PCC), Fresh Frozen Plasma (FFP) and targeted therapies (such as rFVIIa) are brought to the forefront. Some studies show PCC use in ICH patients could have faster INR reversal and less HE occurrence than FFP or vitamin K [88, 89] because FFP and vitamin K take nearly 24 hours to normalize INR [90-92]. Conversely, PCC could reverse INR within minutes, and rFVIIa may be faster than PCC [91-94]. The failure to reverse the INR within 2h is an independent predictor of death or severe disability, and rFVIIa could reverse INR within minutes [95]. Based on those considerations, rFVIIa was recommended as a potential ultra-early hemostatic intervention before coagulopathy and auto-regulation failure occurs. The rFVIIa ICH trail demonstrated early administration of rFVIIa (<4h of symptom onset) has a substantial reduction of HE at 24h (3.3mL, 4.5mL, and 5.8mL in the group 40μg, 80μg and 160μg rFVIIa per kilogram, respectively comparison with placebo group), better outcome at 3 months, and decreased mortality rate by 38% [6]. Based on those facts, we believe impaired coagulopathy increase the risk of HE and earlier recovery of it by faster INR normalization may represent less HE and better outcome. Theose safe and effective clinical interventions based on coagulopathy targets are the future directions.
Inflammation and microvascular integrity
Increasing evidence has shown that inflammatory responses and microvascular integrity damage participate in the pathophysiological processes of brain injury following ICH [96]. Multifocal bleeding around the circumstances of clot has induced hematoma growth [24]. Those clots, caused by the rupture of arterioles and venules, induce the thrombin generation. In experimental ICH model, thrombin activates the inflammatory cascade and the expression of matrix metalloproteinase (MMPs) [97, 98]. MMPs could cause the basal membrane components degradation, BBB damage, and brain edema. Inflammatory markers on admission, such as elevated white blood cell count, interleukin-6 (IL-6), C-reactive protein (CRP) are found to be associated with worse outcomes and HE [42, 99, 100]. One study has shown that patients with HE have significantly higher plasma concentrations of IL-6, TNF-α, MMP-9, and cellular fibronectin (c-Fn) than patients without HE [101]. c-Fn, as a glycoprotein, is one of basal membrane components, which is important for platelets’ adhesion to fibrin and blocking further bleeding. Notably, the IL-6 level (> 24 pg/mL) and c-Fn level (>6µg/mL) in the peripheral blood increased 16-fold and 92-fold the risk of HE, respectively [42]. Even more, IL-6 is the major inducer of CRP. Another study found CRP (>10mg/L) causes higher risk of HE (adjusted risk ratio, 4.7;95%CI, 2.8-8.1) than the spot sign (adjusted OR, 2.3;95%CI, 1.6-3.1) [100]. From the physiological standpoint, the acute reactant CRP can disrupt BBB to promote brain edema and HE formation. On one hand, the disappearance of basal membrane components and loss of microvascular integrity could aggravate inflammatory reaction [102-104]. On the other hand, it’s likely that even a mildly inflammatory status could damage coagulation function and vessel wall pathophysiology as well [99, 105]. Despite the underlying mechanisms aren’t fully understood so far, we believe inflammation and microvascular pathophysiology have mutual promotion effects and both can contribute to vessels’ persistent leakage, the formation of brain edema and HE subsequently. Based on the fact that those molecular signatures of vascular injury and inflammatory response are predictive of subsequent HE and easily measured in the first hours after symptom onset, we believe those molecular predictors may open new potential hemostatic and anti-inflammatory therapeutic strategies for ICH despite many underlying mechanisms still require future investigation.
Neuroradiological Criteria
The urgent, rapid-developing abnormalities and unconsciousness could suggest the diagnosis of ICH. But the imagings of the brain, particularly CT scan, are vital to diagnose ICH and distinguish ICH from cerebral infarction. The initial CT scan at admission could inform emergency surgeons the location and size of the hematoma, the presence of intraventricular hemorrhage and the occurrence of hydrocephalus. Conventional angiography is recommended to look for secondary causes of ICH, such as aneurysms and arteriovenous malformation. The follow-up CT, normally acquired within 6-72 h after the first CT, is usually used for ICH prognostication and HE diagnosis. In short, imaging characteristics on CT scan remain the hot research spot in the last decade.
The interval time from ICH onset
Computed tomographic angiography (CTA) and gadolinium-enhanced magnetic resonance imaging (enhanced MRI) studies confirm 46% and 36% of patients had active contrast extravasation within the hematoma, respectively. Those patients had a significantly less time interval between symptom onset and higher mortality (64% versus 16%) [106-108]. Some studies noticed that active bleeding seems to be stabilized within 6h and the incidence of HE decreases as the interval time from onset increases. It means that the interval time from ICH onset to admission or first CT scan is a strong predictor of HE [16, 109]. One prospective cohort study of 1000 patients proved that shorter time to initial CT (≤6 hours) means greater possibility of HE than later presentation (>6 hours) (adjusted odds ratio, 2.55;95%CI, 1.53-4.27) [16]. One explanation is that ICH is a dynamic illness. Those patients with HE may be still in the acute period of ICH and the bleeding is still ongoing. The contrast-enhanced imaging could indicate the active bleeding process in ICH. Initial CT scan could be applied before the hematoma formation and expansion stabilized, in order for those patients to be treated timely in their acute course of the fatal disease. In short, the earlier the first scan did, the greater the probability of later HE became. We think future research could focus on whether different interval time from onset to first scan and first scan to second scan influence HE definition and other factor’s predictive abilities.
The baseline ICH volume
ICH volume has significantly positive correlation with short term and long term mortality [47, 110-112]. Although the criteria for hematoma volume for favorable prognosis is still unclear, many studies demonstrated large volume (>30mL) is associated with poor outcomes at discharge and at 1 month follow-up [1, 47], while small volume (<30mL) means favorable outcomes [47, 113, 114]. Researchers propose that the internal connection may be the elevated intracranial pressure and cerebral edema, which are positively related to greater initial ICH volume. Unfortunately, despite perihematomal edema volume and baseline hematoma volume being intimately related, perihematomal edema volume has conflicting results on its independent predictive ability of clinical outcomes and HE [115-118]. One alternative assumption is that early HE, not perihematomal edema volume, may be the association between greater baseline ICH volume and higher morbidity and mortality. Large ICH volume (>30mL) is more likely to associate with HE, and HE is less likely in small hematoma (<10mL) [14]. From the multivariable analysis of the prediction score for HE, baseline ICH volume 30-60mL and >60mL have more adjusted odds ratio than ICH volume <30mL (1.64;95% CI, 1.04-2.59 and 2.10;95% CI, 1.25-3.55) [16]. A larger baseline ICH volume may indicate multiple bleeding artery points high systemic arterial pressure and further induction of perilesional hemorrhage through additional vessel shearing and the avalanche effect of HE [119]. Genetic studies have proven apolipoprotein E (APOE) ε2 allele one allele of the principal cholesterol carrier in the brain can promote vasculopathy in cerebral amyloid angiopathy and HE in ICH [120, 121]. A reasonable speculation is that this allele may increase the vascular walls’ fragility to the secondary neighboring vessel mechanical shearing effect, just like the strong correlation between this allele and an elevated disruption of vessel walls promoted by enhanced by cerebral amyloid angiopathy [122-125]. The increasing baseline ICH volume on initial CT scan is associated with higher likelihood of increased absolute ICH volume growth on the secondary scan [16]. We think those open problems could be solved in future studies: better ICH volume measurements, effective cutoff of large-small volume and internal mechanism of large volume.
The shape and density of hematoma on CT
Irregular shape and density of hematoma on CT scan seem to predict HE [13, 114]. Regular lesion edge indicates a solitary focus hematoma and irregular shape bespeaks the hematoma arises from multiple foci. In addition, heterogeneous density on CT also indicate active hemorrhage or multiple bleeding vessels, which is the underlying mechanism of HE. In one study, using 2 novel 5-point categorical scales reflecting the spectrum of appearance of ICH shape and Hounsfield Unit (HU) density variation, they found median growth(10-25mL) was higher in the heterogeneous group than the homogeneous group. And the irregular group had higher median growth than regular group. Furthermore, adjusting for the two independent predictors (baseline ICH volume and time to scan), density heterogeneity, not shape irregularity could predict HE on the continuous growth scale [114]. Considering the qualitative description of the shape and density on CT will vary greatly under different scan times, ICH initial volume, and observer diagnosis bias, not only time course of ICH and HE definition need to be elucidated, but the standardize morphology scale should be established for its feasibility and convenience for clinical practice [13, 22].
Intraventricular hemorrhage (IVH)
The risk of IVH becomes particular high when ICH has been noticed in close proximity to ventricular system, like thalamic hemorrhages [126]. Previous studies have found that the presence of IVH over the first few days after the symptom onset is associated with elevated mortality and worse clinical outcome [127-129].
One study demonstrates that patients with HE are more likely to have IVH (79% vs 45%) by univariate analysis. IVH is significantly predictive of HE (OR=5.7,95%CI:1.5-20.9) [12]. One hypothesis conveys that IVH tends to trigger pro-inflammatory cytokine activation combined with systematic and local homeostatic/fibrinolytic pathways alternation [25, 130, 131]. ICH patients with IVH have elevated levels of white blood cell count, thrombin-antithrombin complex, plasmin-antiplasmin complex and D-dimer, which collectively indicate the infection and stress states of the coagulation/fibrinolytic system [130]. The potential ultra-early hemostatic intervention, rFVIIa, can reduce the risk of HE in IVH (17% and 10% of placebo and rFVIIa treated patients) [126]. So far, the mechanism of systemic activation of hemostatic and inflammation systems in IVH patients and clinical measures to prevent HE based on this hypothesis need further studies to elucidate.
The spot sign and modified spot sign
The phenomenon of ICH, spot sign, has been studied extensively in the last decade and might be the strongest individual predictor for subsequent growth of hematoma. Although spot sign has diverse definitions, it’s mostly denominated as at least 1 tiny focus of contrast pooling within the ICH and high Hounsfield Unit value (>120), discontinuous from normal or abnormal vasculature adjacent to the ICH and any size and morphology on CTA scan in most studies [132-134]. It has been validated that spot sign is a crucial imaging biomarker for HE and poor clinical outcome. Although the definition of spot sign and HE, even the timing of image acquisition, are not used constantly across literatures, CTA “spot sign” still had 51%-98% sensitivity and 50%-89% specificity in previous studies [9, 18, 135]. Further studies describe the spot sign as a dynamic entity. One plausible explanation is that spot sign might represent active bleeding or rebleeding through ruptured vessels, which reflects contrast extravasation on CT or MRI scan. Based on this mechanism, many modified spot signs were introduced, such as delayed CTA, venous phase CTA, dynamic CTA, post contrast CT (PCT), and CT perfusion (CTP) [135, 136]. Higher sensitivity and specificity was obtained by the combination of initial and modified CTA, regardless of the heterogeneity of secondary CT time and definition of spot sign. For example, CTP spot sign was proven as an independent predictor of HE and poor outcome (OR,13.7;95%CI,1.12-166) and higher sensitivity (78%) compared with CTA spot sign (44%) or PCT spot sign (50%). CTP’s improved sensitivity was mainly attributed to its moderate delay time (30-70 seconds for CTP, 20-26 seconds for CTA, and 300-360 seconds for PCT), which is enough for contrast to extravasate and not too long to diffuse or get subthreshold [136, 137]. Considering MRI can accurately detect acute and chronic ICH, one study investigated that spot sign detected on post-contrast T1-weighted and dynamic T1-weighted scan is also a potent predictor of the clinical outcome. But no significant HE differences were observed between spot sign and non-spot sign group (52.2% in spot sign group, 40.7% in non-spot sign group) [138]. Further research about MRI spot sign is needed due to this study’s small sample size, lack of long-term follow-up, and observational study design.
Based on the relatively lower sensitivity of spot sign and better results in modified spot signs (2 to 5 minutes delayed-phase CT) [31, 139], one study presents the leakage sign, which is defined as a 10% increase in HU of a 1-cm diameter restrictive area between CTA initial phase and delayed phase (specifically 5minutes after the initial phase). They found leakage sign has higher sensitivity (93.3%) and specificity (88.9%) for HE than the spot sign (77.8% sensitivity and 71.8% specificity) [134]. A study enrolling total 206 patients found that the black hole sign, 28 HU difference between the relatively hypo-attenuated area and adjacent hyperintense area on CT scan, is found more commonly in HE patients (31.9%) than those without HE (5.8%) [140]. The sensitivity and specificity value of black hole sign’ HE prediction are 31.9% and 94.1%, respectively. The black hole sign, a simple and easy-to-use CT predictor, has improved reliability and subjectivity compared with previous spot sign description and this improvement can be explained by the plausible pathophysiological process (hypo-attenuated fresh liquid blood turning to hyperintense “old blood” with clot retraction and serum dissociation) [27]. The optimal HU thresholds for leakage sign and black sign need further identification through more studies. More importantly, the spot sign score (SSS) is devised from three most prognostic radiological characteristics for HE: spot number (1-2 for 1 point, ≥3 for 2 points), maximum spot size (axial dimension 1-4mm for 0 point, ≥5mm for 1 point), and density heterogeneity. SSS reports an accurate risk stratification of hematoma expansion as well as a reliable independent prediction of mortality and poor clinical outcome [31, 141, 142]. Further multicenter external validation of the SSS from analysis of a multicenter prospective observational cohort study, the PREDICT study, demonstrates that spot sign number is the most predictive spot characteristic among the three radiological characteristics (p<0.001 for number, p=0.570 for maximum density and p=0.982 for maximum axial dimension by comparison HE group with non-HE group) [142]. Increased spot number shows significant positive association with absolute HE and relative HE (both p<0.001), whereas SSS category demonstrates no significant trends for absolute HE and relative HE (p=0.182 and 0.470, respectively). In conclusion, those useful radiological signs, such as spot sign, modified spot sign, leakage sign on CTA, black hole sign on CT, spot sign number are all the crucial predictors for HE and prognosis. And further exploration of other radiographic characteristics may establish a more reliable, objective, and convenient predictor.
Prediction Score Model
Some prediction scores have recently been developed and validated for HE prediction. A 9-point clinical prediction score is established on four recognized predictors: time to presentation, anticoagulation use, ICH volume, and CT angiography spot sign, all of which could be explained by the theoretical HE model described in 1971 [28]. 80% of patients with highest score of 9 have HE, while the incidence of HE reaches only 5.7% for patients with lowest number 0. The high score (4-9 vs 0-3 in the 9-point score) has strongly positive relation with greater risk of HE (OR, 4.59; 95%CI, 2.06-10.22) [16]. In-hospital and 3-month mortality are increased steadily with the higher prediction scores. This prediction score extends the enrollment time window for trials (>6 hours) and allows more patients to receive therapeutic benefit, without restricting intervention to the first hours after symptom onset. Similarly, a 24-point clinical prediction algorithm (BRAIN) is derived and validated from sub-studies of INTERACT1 and 2 [49]. This BRAIN score predicts the probability of HE (>6mL) within 24 hours after symptom onset, using multivariable logistic regression to identify 5 routinely assessed clinical factors: baseline ICH volume, recurrent ICH, anticoagulation with warfarin at symptom onset, IVH and time to have first-CT scan (only included patients within 6 hours), to predict the possibility of HE (3.4% for 0 point and 85.8% for 24 points). Of note, despite the most robust predictor of HE to date, spot sign on baseline CTA is not included, the 24-point prediction score still shows good discrimination and calibration (C-statistic, 0.73), comparable with 9-point score (0.72 for the development cohort and 0.77 for the validation cohort). Because GCS and NIHSS were often collected clinically and were also identified as HE predictors [13, 46], they were incorporated into two new HE prediction scores, PREDICT A and B, respectively [83]. Both prediction scores still included GCS or NIHSS, warfarin use or INR>1.5, spot sign number, and time from symptom onset to baseline CT. Compared with the 9-and 24-point scores, both PREDICT scores showed improved AUC for HE> 6mL or 33% discrimination compared with the 9- and 24-point scores (PREDICT A 0.78; 95%CI,0.73-0.84; PREDICT B 0.77; 95%CI,0.72-0.83; 9-point score 0.71; 95%CI,0.65-0.77; 24-point score0.76; 95%CI 0.70-0.82;). The delayed contrast enhanced image, like dynamic CTP and post-contrast CT, are still excluded in those prediction scores despite their improved sensitivity and specificity for HE prediction. To stratify patients accurately for current therapeutic intervention, further independent validation and modification of the aforementioned prediction scoring systems is required.
Conclusions
One third of ICH patients would may occur HE, which is strongly predictive of worse prognosis and potentially preventable. Potential HE predictors could help clinicians to better stratify patients, who are destined to undergo HE and tailor intensive therapies timely and effectively. In this review, we discuss the definition and pathophysiology of HE and summarize individual or combined predictors of HE, which include clinical (GCS, NIHSS, blood pressure, blood glucose), laboratory (anticoagulation, inflammation and microvascular integrity), and radiographic (interval time from onset, the baseline ICH volume, the shape and density on CT, IVH, spot sign parameters, modified spot signs) variables, as well as prediction scores (9-point, BRAIN, PREDICT A/B) (Figure 2). This review also discusses those predictors’ practical utility, internal pathogenesis and future research direction. Further elucidation of the pathogenesis and emergence of better predictors are still required to promote novel and aggressive medical therapeutic interventions via predicting and ameliorating HE after symptom onset.
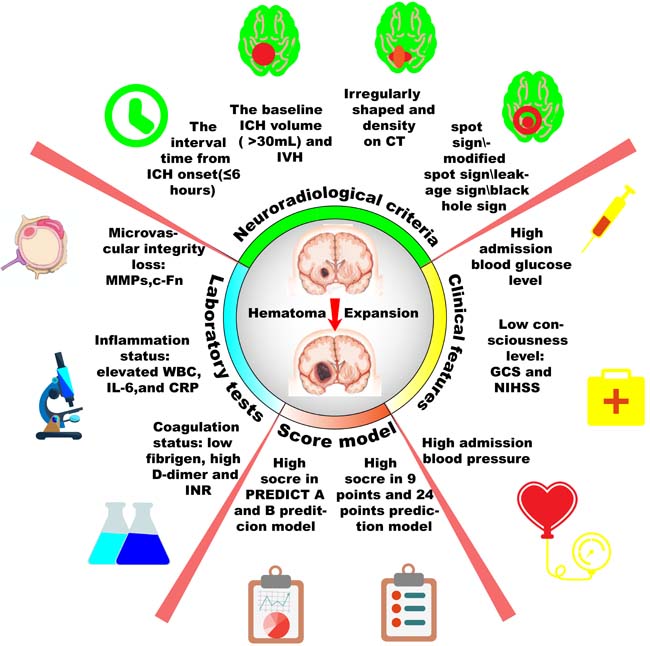
Figure 2: Scheme diagram of predictors for hematoma expansion after intracerebral hemorrhage. Hematoma expansion may be predicted by those four aspects: laboratory tests, clinical features, neuroradiological criteria and prediction score model. More accurate prediction may lead better stratification and intensive therapies to patients destined to poor prognosis.
ConflictS of Interest
There is no conflict of interest.
Funding
This study was supported by the National Natural Science Foundation of China (81500992), Natural Science Foundation of Zhejiang(LQ16H090002), Medical and health key project of Zhejiang Province (2016RCA015).
REFERENCES
1. Qureshi AI, Mendelow AD, Hanley DF. Intracerebral haemorrhage. The Lancet. 2009; 373:1632-1644.
2. Delcourt C, Huang Y, Arima H, Chalmers J, Davis SM, Heeley EL, Wang J, Parsons MW, Liu G, Anderson CS, Investigators I. Hematoma growth and outcomes in intracerebral hemorrhage: the INTERACT1 study. Neurology. 2012; 79:314-319.
3. Daverat P, Castel JP, Dartigues JF, Orgogozo JM. Death and functional outcome after spontaneous intracerebral hemorrhage a prospective study of 166 cases using multivariate analysis. Stroke. 1991; 22:1-6.
4. Cheung RTF, Zou LY. Use of the original, modified, or new intracerebral hemorrhage score to predict mortality and morbidity after intracerebral hemorrhage. Stroke. 2003; 34:1717-1722.
5. Anderson CS, Heeley E, Huang Y, Wang J, Stapf C, Delcourt C, Lindley R, Robinson T, Lavados P, Neal B, Hata J, Arima H, Parsons M, et al. Rapid blood-pressure lowering in patients with acute intracerebral hemorrhage. The New England journal of medicine. 2013; 368:2355-2365.
6. Mayer SA, Brun NC, Begtrup K, Broderick J, Davis S, Diringer MN, Skolnick BE, Steiner T. Recombinant activated factor VII for acute intracerebral hemorrhage. New England Journal of Medicine. 2005; 352:777-785.
7. Broderick JP, Diringer MN, Hill MD, Brun NC, Mayer SA, Steiner T, Skolnick BE, Davis SM. Determinants of intracerebral hemorrhage growth: An exploratory analysis. Stroke. 2007; 38:1072-1075.
8. Flibotte JJ, Hagan N, O’Donnell J, Greenberg SM, Rosand J. Warfarin, hematoma expansion, and outcome of intracerebral hemorrhage. Neurology. 2004; 63:1059-1064.
9. Goldstein JN, Fazen LE, Snider R, Schwab K, Greenberg SM, Smith EE, Lev MH, Rosand J. Contrast extravasation on CT angiography predicts hematoma expansion in intracerebral hemorrhage. Neurology. 2007; 68:889-894.
10. Kazui S, Naritomi H, Yamamoto H, Sawada T, Yamaguchi T. Enlargement of Spontaneous Intracerebral Hemorrhage: Incidence and Time Course. Stroke. 1996; 27:1783-1787.
11. Qureshi AI, Palesch YY. Antihypertensive treatment of acute cerebral hemorrhage (ATACH) II: Design, methods, and rationale. Neurocritical Care. 2011; 15:559-576.
12. Yaghi S, Dibu J, Achi E, Patel A, Samant R, Hinduja A. Hematoma expansion in spontaneous intracerebral hemorrhage: Predictors and outcome. International Journal of Neuroscience. 2014; 124:890-893.
13. Fujii Y, Takeuchi S, Sasaki O, Minakawa T, Tanaka R. Multivariate analysis of predictors of hematoma enlargement in spontaneous intracerebral hemorrhage. Stroke. 1998; 29:1160-1166.
14. Dowlatshahi D, Demchuk AM, Flaherty ML, Ali M, Lyden PL, Smith EE. Defining hematoma expansion in intracerebral hemorrhage: Relationship with patient outcomes. Neurology. 2011; 76:1238-1244.
15. Perkins NJ, Schisterman EF. The Inconsistency of “optimal” cutpoints obtained using two criteria based on the receiver operating characteristic curve. American Journal of Epidemiology. 2006; 163:670-675.
16. Brouwers HB, Chang Y, Falcone GJ, Cai X, Ayres AM, Battey TWK, Vashkevich A, McNamara KA, Valant V, Schwab K, Orzell SC, Bresette LM, Feske SK, et al. Predicting hematoma expansion after primary intracerebral hemorrhage. JAMA Neurology. 2014; 71:158-164.
17. Demchuk AM, Dowlatshahi D, Rodriguez-Luna D, Molina CA, Blas YS, Dzialowski I, Kobayashi A, Boulanger JM, Lum C, Gubitz G, Padma V, Roy J, Kase CS, et al. Prediction of haematoma growth and outcome in patients with intracerebral haemorrhage using the CT-angiography spot sign (PREDICT): A prospective observational study. The Lancet Neurology. 2012; 11:307-314.
18. Wada R, Aviv RI, Fox AJ, Sahlas DJ, Gladstone DJ, Tomlinson G, Symons SP. CT angiography “spot sign” predicts hematoma expansion in acute intracerebral hemorrhage. Stroke. 2007; 38:1257-1262.
19. Broderick JP, Diringer MN, Hill MD, Brun NC, Mayer SA, Steiner T, Skolnick BE, Davis SM. Determinants of intracerebral hemorrhage growth: an exploratory analysis. Stroke. 2007; 38:1072-1075.
20. Anderson CS, Qureshi AI. Implications of INTERACT2 and other clinical trials: blood pressure management in acute intracerebral hemorrhage. Stroke. 2015; 46:291-295.
21. Brott T, Broderick J, Kothari R, Barsan W, Tomsick T, Sauerbeck L, Spilker J, Duldner J, Khoury J. Early hemorrhage growth in patients with intracerebral hemorrhage. Stroke. 1997; 28:1-5.
22. Fujii Y, Tanaka R, Takeuchi S, Koike T, Minakawa T, Sasaki O. Hematoma enlargement in spontaneous intracerebral hemorrhage. Journal of Neurosurgery. 1994; 80:51-57.
23. Davis SM, Broderick J, Hennerici M, Brun NC, Diringer MN, Mayer SA, Begtrup K, Steiner T. Hematoma growth is a determinant of mortality and poor outcome after intracerebral hemorrhage. Neurology. 2006; 66:1175-1181.
24. Mayer SA, Lignelli A, Fink ME, Kessler DB, Thomas CE, Swarup R, Van Heertum RL. Perilesional blood flow and edema formation in acute intracerebral hemorrhage: a SPECT study. Stroke. 1998; 29:1791-1798.
25. Martí-Fàbregas J, Borrell M, Silva Y, Delgado-Mederos R, Martínez-Ramírez S, de Juan-Delago M, Tirado I, Alejaldre A, Marín R, Martí-Vilalta JL, Fontcuberta J. Hemostatic proteins and their association with hematoma growth in patients with acute intracerebral hemorrhage. Stroke. 2010; 41:2976-2978.
26. Takahashi H, Urano T, Nagai N, Takada Y, Takada A. Progressive expansion of hypertensive intracerebral hemorrhage by coagulopathy. American journal of hematology. 1998; 59:110-114.
27. Schlunk F, Greenberg SM. The pathophysiology of intracerebral hemorrhage formation and expansion. Translational Stroke Research. 2015; 6:257-263.
28. Fisher CM. Pathological observations in hypertensive cerebral hemorrhage. Journal of Neuropathology and Experimental Neurology. 1971; 30:536-550.
29. Greenberg SM, Nandigam RN, Delgado P, Betensky RA, Rosand J, Viswanathan A, Frosch MP, Smith EE. Microbleeds versus macrobleeds: evidence for distinct entities. Stroke. 2009; 40:2382-2386.
30. Brouwers HB, Biffi A, Ayres AM, Schwab K, Cortellini L, Romero JM, Rost NS, Viswanathan A, Greenberg SM, Rosand J. Apolipoprotein E genotype predicts hematoma expansion in lobar intracerebral hemorrhage. Stroke. 2012; 43:783–789.
31. Delgado Almandoz JE, Yoo AJ, Stone MJ, Schaefer PW, Goldstein JN, Rosand J, Oleinik A, Lev MH, Gonzalez RG, Romero JM. Systematic characterization of the computed tomography angiography spot sign in primary intracerebral hemorrhage identifies patients at highest risk for hematoma expansion: the spot sign score. Stroke. 2009; 40:2994-3000.
32. Greenberg CH, Frosch MP, Goldstein JN, Rosand J, Greenberg SM. Modeling intracerebral hemorrhage growth and response to anticoagulation. PLoS ONE. 2012; 7.
33. Qureshi AI, Tuhrim S, Broderick JP, Batjer HH, Hondo H, Hanley DF. Spontaneous intracerebral hemorrhage. New England Journal of Medicine. 2001; 344:1450-1460.
34. Li P, Meng X, Bian H, Burns N, Zhao KS, Song R. Activation of sirtuin 1/3 improves vascular hyporeactivity in severe hemorrhagic shock by alleviation of mitochondrial damage. Oncotarget. 2015; 6:36998-37011. doi: 10.18632/oncotarget.6076.
35. Dang G, Yang Y, Wu G, Hua Y, Keep RF, Xi G. Early Erythrolysis in the hematoma after experimental intracerebral hemorrhage. Translational Stroke Research. 2016:1-9.
36. Alvarez-Sabín J, Delgado P, Abilleira S, Molina CA, Arenillas J, Ribó M, Santamarina E, Quintana M, Monasterio J, Montaner J. Temporal profile of matrix metalloproteinases and their inhibitors after spontaneous intracerebral hemorrhage: Relationship to clinical and radiological outcome. Stroke. 2004; 35:1316-1322.
37. Gong C, Boulis N, Qian J, Turner DE, Hoff JT, Keep RF. Intracerebral hemorrhage-induced neuronal death. Neurosurgery. 2001; 48:882-883.
38. Wan S, Cheng Y, Jin H, Guo D, Hua Y, Keep RF, Xi G. Microglia activation and polarization after intracerebral hemorrhage in mice: the role of protease-activated receptor-1. Translational Stroke Research. 2016:1-10.
39. Chen M, Sun J, Lu C, Chen X, Ba H, Lin Q, Cai J, Dai J. The impact of neuronal Notch-1/JNK pathway on intracerebral hemorrhage-induced neuronal injury of rat model. Oncotarget. 2016; 7:73903-73911. doi: 10.18632/oncotarget.12094.
40. Aronowski J, Zhao X. Molecular pathophysiology of cerebral hemorrhage: secondary brain injury. Stroke. 2011; 42:1781-1786.
41. Gong C, Hoff JT, Keep RF. Acute inflammatory reaction following experimental intracerebral hemorrhage in rat. Brain Research. 2000; 871:57-65.
42. Silva Y, Leira R, Tejada J, Lainez JM, Castillo J, Dávalos A. Molecular signatures of vascular injury are associated with early growth of intracerebral hemorrhage. Stroke. 2005; 36:86-91.
43. Wang J, Tsirka SE. Neuroprotection by inhibition of matrix metalloproteinases in a mouse model of intracerebral haemorrhage. Brain. 2005; 128:1622-1633.
44. Jiang B, Li L, Chen Q, Tao Y, Yang L, Zhang B, Zhang JH, Feng H, Chen Z, Tang J, Zhu G. Role of glibenclamide in brain injury after intracerebral hemorrhage. Translational Stroke Research. 2016:1-11.
45. Shi Y, Leak RK, Keep RF, Chen J. Translational stroke research on blood-brain barrier damage: challenges, perspectives, and goals. Translational Stroke Research. 2016; 7:89-92.
46. Sorimachi T, Fujii Y, Morita K, Tanaka R. Predictors of hematoma enlargement in patients with intracerebral hemorrhage treated with rapid administration of antifibrinolytic agents and strict blood pressure control. Journal of Neurosurgery. 2007; 106:250-254.
47. Kim KH. Predictors of 30-day mortality and 90-day functional recovery after primary intracerebral hemorrhage: Hospital based multivariate analysis in 585 patients. Journal of Korean Neurosurgical Society. 2009; 45:341-349.
48. Steiner T, Bösel J. Options to restrict hematoma expansion after spontaneous intracerebral hemorrhage. Stroke. 2010; 41:402-409.
49. Wang X, Arima H, Al-Shahi Salman R, Woodward M, Heeley E, Stapf C, Lavados PM, Robinson T, Huang Y, Wang J, Delcourt C, Anderson CS. Clinical prediction algorithm (BRAIN) to determine risk of hematoma growth in acute intracerebral hemorrhage. Stroke. 2015; 46:376-381.
50. Scott JF, Robinson GM, French JM, O’Connell JE, Alberti KG, Gray CS. Prevalence of admission hyperglycaemia across clinical subtypes of acute stroke. Lancet (London, England). 1999; 353:376-377.
51. Fogelholm R, Murros K, Rissanen A, Avikainen S. Admission blood glucose and short term survival in primary intracerebral haemorrhage: A population based study. Journal of Neurology, Neurosurgery and Psychiatry. 2005; 76:349-353.
52. Kimura K, Iguchi Y, Inoue T, Shibazaki K, Matsumoto N, Kobayashi K, Yamashita S. Hyperglycemia independently increases the risk of early death in acute spontaneous intracerebral hemorrhage. Journal of the Neurological Sciences. 2007; 255:90-94.
53. Koga M, Yamagami H, Okuda S, Okada Y, Kimura K, Shiokawa Y, Nakagawara J, Furui E, Hasegawa Y, Kario K, Arihiro S, Sato S, Homma K, et al. Blood glucose levels during the initial 72 h and 3-month functional outcomes in acute intracerebral hemorrhage: the SAMURAI-ICH study. J Neurol Sci. 2015; 350:75-78.
54. Qureshi AI, Palesch YY, Martin R, Novitzke J, Cruz-Flores S, Ehtisham A, Ezzeddine MA, Goldstein JN, Kirmani JF, Hussein HM, Suri MF, Tariq N, Liu Y. Association of serum glucose concentrations during acute hospitalization with hematoma expansion, perihematomal edema, and three month outcome among patients with intracerebral hemorrhage. Neurocrit Care. 2011; 15:428-435.
55. Kazui S, Minematsu K, Yamamoto H, Sawada T, Yamaguchi T. Predisposing factors to enlargement of spontaneous intracerebral hematoma. Stroke. 1997; 28:2370-2375.
56. Liu J, Gao BB, Clermont AC, Blair P, Chilcote TJ, Sinha S, Flaumenhaft R, Feener EP. Hyperglycemia-induced cerebral hematoma expansion is mediated by plasma kallikrein. Nature medicine. 2011; 17:206-210.
57. Zheng Y, Hu Q, Manaenko A, Zhang Y, Peng Y, Xu L, Tang J, Tang J, Zhang JH. 17beta-Estradiol attenuates hematoma expansion through estrogen receptor alpha/silent information regulator 1/nuclear factor-kappa b pathway in hyperglycemic intracerebral hemorrhage mice. Stroke. 2015; 46:485-491.
58. Weir CJ, Murray GD, Dyker AG, Lees KR. Is hyperglycaemia an independent predictor of poor outcome after acute stroke? Results of a long term follow up study. British Medical Journal. 1997; 314:1303-1306.
59. Slowik A, Turaj W, Pankiewicz J, Dziedzic T, Szermer P, Szczudlik A. Hypercortisolemia in acute stroke is related to the inflammatory response. Journal of the Neurological Sciences. 2002; 196:27-32.
60. Esposito K, Nappo F, Marfella R, Giugliano G, Giugliano F, Ciotola M, Quagliaro L, Ceriello A, Giugliano D. Inflammatory cytokine concentrations are acutely increased by hyperglycemia in humans: role of oxidative stress. Circulation. 2002; 106:2067-2072.
61. Allen CL, Bayraktutan U. Antioxidants attenuate hyperglycaemia-mediated brain endothelial cell dysfunction and blood-brain barrier hyperpermeability. Diabetes, obesity & metabolism. 2009; 11:480-490.
62. Feng W, Tauhid S, Goel S, Sidorov EV, Selim M. Hyperglycemia and outcome in intracerebral hemorrhage: from bedside to bench-more study is needed. Transl Stroke Res. 2012; 3:113-118.
63. Chan S, Conell C, Veerina KT, Rao VA, Flint AC. Prediction of intracerebral haemorrhage expansion with clinical, laboratory, pharmacologic, and noncontrast radiographic variables. International Journal of Stroke. 2015; 10:1057-1061.
64. Finfer S, Bellomi R, Blair D, Su SYS, Foster D, Dhingra V, Cook D, Dodek P, Henderson WR, Hébert PC, Heyland DK, McArthur C, McDonald E, et al. Intensive versus conventional glucose control in critically Ill patients. New England Journal of Medicine. 2009; 360:1283-1297.
65. Godoy DA, Di Napoli M, Rabinstein AA. Treating hyperglycemia in neurocritical patients: benefits and perils. Neurocrit Care. 2010; 13:425-438.
66. Oddo M, Schmidt JM, Carrera E, Badjatia N, Connolly ES, Presciutti M, Ostapkovich ND, Levine JM, Roux PL, Mayer SA. Impact of tight glycemic control on cerebral glucose metabolism after severe brain injury: A microdialysis study. Critical Care Medicine. 2008; 36:3233-3238.
67. Vespa P, Boonyaputthikul R, McArthur DL, Miller C, Etchepare M, Bergsneider M, Glenn T, Martin N, Hovda D. Intensive insulin therapy reduces microdialysis glucose values without altering glucose utilization or improving the lactate/pyruvate ratio after traumatic brain injury. Critical Care Medicine. 2006; 34:850-856.
68. Hemphill JC, Greenberg SM, Anderson CS, Becker K, Bendok BR, Cushman M, Fung GL, Goldstein JN, Macdonald RL, Mitchell PH, Scott PA, Selim MH, Woo D. Guidelines for the management of spontaneous intracerebral hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2015; 46:2032-2060.
69. Sakamoto Y, Koga M, Yamagami H, Okuda S, Okada Y, Kimura K, Shiokawa Y, Nakagawara J, Furui E, Hasegawa Y, Kario K, Arihiro S, Sato S, et al. Systolic blood pressure after intravenous antihypertensive treatment and clinical outcomes in hyperacute intracerebral hemorrhage: The stroke acute management with urgent risk-factor assessment and improvement-intracerebral hemorrhage study. Stroke. 2013; 44:1846-1851.
70. Patarroyo SXF, Anderson C. Blood pressure lowering in acute phase of stroke: Latest evidence and clinical implications. Therapeutic Advances in Chronic Disease. 2012; 3:163-171.
71. Qureshi AI, Palesch YY, Martin R, Novitzke J, Cruz-Flores S, Ehtisham A, Ezzeddine MA, Goldstein JN, Hussein HM, Suri FK, Tariq N. Effect of systolic blood pressure reduction on hematoma expansion, perihematomal edema, and 3-month outcome among patients with intracerebral hemorrhage: Results from the antihypertensive treatment of acute cerebral hemorrhage study. Archives of neurology. 2010; 67:570-576.
72. Ohwaki K, Yano E, Nagashima H, Hirata M, Nakagomi T, Tamura A. Blood pressure management in acute intracerebral hemorrhage: Relationship between elevated blood pressure and hematoma enlargement. Stroke. 2004; 35:1364-1367.
73. Worster A, Keim SM, Carpenter CR, Adeoye O. Does early intensive lowering of blood pressure reduce hematoma volume and improve clinical outcome after acute cerebral hemorrhage? Journal of Emergency Medicine. 2009; 37:433-438.
74. Maruishi M, Shima T, Okada Y, Nishida M, Yamane K. Involvement of fluctuating high blood pressure in the enlargement of spontaneous intracerebral hematoma. Neurologia Medico-Chirurgica. 2001; 41:300-304.
75. Falcone GJ, Biffi A, Devan WJ, Brouwers HB, Anderson CD, Valant V, Ayres AM, Schwab K, Rost NS, Goldstein JN, Viswanathan A, Greenberg SM, Selim M, et al. Burden of blood pressure–related alleles is associated with larger hematoma volume and worse outcome in intracerebral hemorrhage. Stroke. 2013; 44:321-326.
76. Zahuranec DB, Wing JJ, Edwards DF, Menon RS, Fernandez SJ, Burgess RE, Sobotka IA, German L, Trouth AJ, Shara NM, Gibbons MC, Boden-Albala B, Kidwell CS. Poor long-term blood pressure control after intracerebral hemorrhage. Stroke. 2012; 43:2580-2585.
77. Koch S, Romano JG, Forteza AM, Otero CM, Rabinstein AA. Rapid blood pressure reduction in acute intracerebral hemorrhage: Feasibility and safety. Neurocritical Care. 2008; 8:316-321.
78. Anderson CS, Huang Y, Wang JG, Arima H, Neal B, Peng B, Heeley E, Skulina C, Parsons MW, Kim JS, Tao QL, Li YC, Jiang JD, et al. Intensive blood pressure reduction in acute cerebral haemorrhage trial (INTERACT): a randomised pilot trial. The Lancet Neurology. 2008; 7:391-399.
79. Qureshi AI, Palesch YY, Barsan WG, Hanley DF, Hsu CY, Martin RL, Moy CS, Silbergleit R, Steiner T, Suarez JI, Toyoda K, Wang Y, Yamamoto H, et al. Intensive blood-pressure lowering in patients with acute cerebral hemorrhage. New England Journal of Medicine. 2016; 375:1033-1043.
80. Gould B, McCourt R, Gioia LC, Kate M, Hill MD, Asdaghi N, Dowlatshahi D, Jeerakathil T, Coutts SB, Demchuk AM, Emery D, Shuaib A, Butcher K. Acute blood pressure reduction in patients with intracerebral hemorrhage does not result in borderzone region hypoperfusion. Stroke. 2014; 45:2894-2899.
81. Butcher KS, Jeerakathil T, Hill M, Demchuk AM, Dowlatshahi D, Coutts SB, Gould B, McCourt R, Asdaghi N, Findlay JM, Emery D, Shuaib A. The Intracerebral Hemorrhage Acutely Decreasing Arterial Pressure Trial. Stroke. 2013; 44:620-626.
82. Gould B, McCourt R, Asdaghi N, Dowlatshahi D, Jeerakathil T, Kate M, Coutts SB, Hill MD, Demchuk AM, Shuaib A, Emery D, Butcher K. Autoregulation of cerebral blood flow is preserved in primary intracerebral hemorrhage. Stroke. 2013; 44:1726-1728.
83. Huynh TJ, Aviv RI, Dowlatshahi D, Gladstone DJ, Laupacis A, Kiss A, Hill MD, Molina CA, Rodriguez-Luna D, Dzialowski I, Silva Y, Kobayashi A, Lum C, et al. Validation of the 9-point and 24-point hematoma expansion prediction scores and derivation of the PREDICT A/B Scores. Stroke. 2015; 46:3105-3110.
84. Cucchiara B, Messe S, Sansing L, Kasner S, Lyden P. Hematoma growth in oral anticoagulant related intracerebral hemorrhage. Stroke. 2008; 39:2993-2996.
85. Saloheimo P, Ahonen M, Juvela S, Pyhtinen J, Savolainen ER, Hillbom M. Regular aspirin-use preceding the onset of primary intracerebral hemorrhage is an independent predictor for death. Stroke. 2006; 37:129-133.
86. Toyoda K, Okada Y, Minematsu K, Kamouchi M, Fujimoto S, Ibayashi S, Inoue T. Antiplatelet therapy contributes to acute deterioration of intracerebral hemorrhage. Neurology. 2005; 65:1000-1004.
87. Sansing LH, Messe SR, Cucchiara BL, Cohen SN, Lyden PD, Kasner SE. Prior antiplatelet use does not affect hemorrhage growth or outcome after ICH. Neurology. 2009; 72:1397-1402.
88. Huttner HB, Schellinger PD, Hartmann M, Köhrmann M, Juettler E, Wikner J, Mueller S, Meyding-Lamade U, Strobl R, Mansmann U, Schwab S, Steiner T. Hematoma growth and outcome in treated neurocritical care patients with intracerebral hemorrhage related to oral anticoagulant therapy: Comparison of acute treatment strategies using vitamin K, fresh frozen plasma, and prothrombin complex concentrates. Stroke. 2006; 37:1465-1470.
89. Fredriksson K, Norrving B, Stromblad LG. Emergency reversal of anticoagulation after intracerebral hemorrhage. Stroke. 1992; 23:972-977.
90. Steiner T, Rosand J, Diringer M. Intracerebral hemorrhage associated with oral anticoagulant therapy: Current practices and unresolved questions. Stroke. 2006; 37:256-262.
91. Frontera JA, Lewin JJ 3rd, Rabinstein AA, Aisiku IP, Alexandrov AW, Cook AM, del Zoppo GJ, Kumar MA, Peerschke EIB, Stiefel MF, Teitelbaum JS, Wartenberg KE, Zerfoss CL. Guideline for reversal of antithrombotics in intracranial hemorrhage: a statement for healthcare professionals from the Neurocritical Care Society and Society of Critical Care Medicine. Neurocritical Care. 2016; 24:6-46.
92. Steiner T, Poli S, Griebe M, Hüsing J, Hajda J, Freiberger A, Bendszus M, Bösel J, Christensen H, Dohmen C, Hennerici M, Kollmer J, Stetefeld H, et al. Fresh frozen plasma versus prothrombin complex concentrate in patients with intracranial haemorrhage related to vitamin K antagonists (INCH): A randomised trial. The Lancet Neurology. 2016; 15:566-573.
93. Sarode R, Matevosyan K, Bhagat R, Rutherford C, Madden C, Beshay JE. Rapid warfarin reversal: A 3-factor prothrombin complex concentrate and recombinant factor VIIa cocktail for intracerebral hemorrhage: Clinical article. Journal of Neurosurgery. 2012; 116:491-497.
94. Pinner NA, Hurdle AC, Oliphant C, Reaves A, Lobo B, Sills A. Treatment of warfarin-related intracranial hemorrhage: A comparison of prothrombin complex concentrate and recombinant activated factor VII. World Neurosurgery. 2010; 74:631-635.
95. Ilyas C, Beyer GM, Dutton RP, Scalea TM, Hess JR. Recombinant factor VIIa for warfarin-associated intracranial bleeding. Journal of clinical anesthesia. 2008; 20:276-279.
96. Chen S, Yang Q, Chen G, Zhang JH. An update on inflammation in the acute phase of intracerebral hemorrhage. Translational Stroke Research. 2015; 6:4-8.
97. Lee KR, Colon GP, Betz AL, Keep RF, Kim S, Hoff JT. Edema from intracerebral hemorrhage: The role of thrombin. Journal of Neurosurgery. 1996; 84:91-96.
98. Xi G, Wagner KR, Keep RF, Hua Y, De Courten-Myers GM, Broderick JP, Brott TG, Hoff JT. Role of blood clot formation on early edema development after experimental intracerebral hemorrhage. Stroke. 1998; 29:2580-2586.
99. Kuhlmann CRW, Librizzi L, Closhen D, Pflanzner T, Lessmann V, Pietrzik CU, De Curtis M, Luhmann HJ. Mechanisms of C-reactive protein-induced blood-brain barrier disruption. Stroke. 2009; 40:1458-1466.
100. Di Napoli M, Parry-Jones AR, Smith CJ, Hopkins SJ, Slevin M, Masotti L, Campi V, Singh P, Papa F, Popa-Wagner A, Tudorica V, Godoy DA. C-reactive protein predicts hematoma growth in intracerebral hemorrhage. Stroke. 2014; 45:59-65.
101. Behrouz R. Re-exploring tumor necrosis factor alpha as a target for therapy in intracerebral hemorrhage. Translational Stroke Research. 2016; 7:93-96.
102. Lee KR, Kawai N, Kim S, Sagher O, Hoff JT. Mechanisms of edema formation after intracerebral hemorrhage: Effects of thrombin on cerebral blood flow, blood-brain barrier permeability, and cell survival in a rat model. Journal of Neurosurgery. 1997; 86:272-278.
103. Horstmann S, Kalb P, Koziol J, Gardner H, Wagner S. Profiles of matrix metalloproteinases, their inhibitors, and laminin in stroke patients: Influence of different therapies. Stroke. 2003; 34:2165-2170.
104. Aoki T, Sumii T, Mori T, Wang X, Lo EH. Blood-brain barrier disruption and matrix metalloproteinase-9 expression during reperfusion injury mechanical versus embolic focal ischemia in spontaneously hypertensive rats. Stroke. 2002; 33:2711-2717.
105. Roberts CJ, Birkenmeier TM, McQuillan JJ, Akiyama SK, Yamada SS, Chen WT, Yamada KM, McDonald JA. Transforming growth factor β stimulates the expression of fibronectin and of both subunits of the human fibronectin receptor by cultured human lung fibroblasts. Journal of Biological Chemistry. 1988; 263:4586-4592.
106. Becker KJ, Baxter AB, Bybee HM, Tirschwell DL, Abouelsaad T, Cohen WA. Extravasation of radiographic contrast is an independent predictor of death in primary intracerebral hemorrhage. Stroke. 1999; 30:2025-2032.
107. Morgenstern LB, Demchuk AM, Kim DH, Frankowski RF, Grotta JC. Rebleeding leads to poor outcome in ultra-early craniotomy for intracerebral hemorrhage. Neurology. 2001; 56:1294-1299.
108. Kaneko M, Tanaka K, Shimada T, Sato K, Uemura K. Long-term evaluation of ultra-early operation for hypertensive intracerebral hemorrhage in 100 cases. Journal of Neurosurgery. 1983; 58:838-842.
109. Dowlatshahi D, Brouwers HB, Demchuk AM, Hill MD, Aviv RI, Ufholz LA, Reaume M, Wintermark M, Hemphill JC, Murai Y, Wang Y, Zhao X, Wang Y, et al. Predicting intracerebral hemorrhage growth with the spot sign: The effect of onset-to-scan time. Stroke. 2016; 47:695-700.
110. Broderick JP, Brott TG, Duldner JE, Tomsick T, Huster G. Volume of intracerebral hemorrhage: A powerful and easy-to-use predictor of 30-day mortality. Stroke. 1993; 24:987-993.
111. Zubkov AY, Mandrekar JN, Claassen DO, Manno EM, Wijdicks EFM, Rabinstein AA. Predictors of outcome in warfarin-related intracerebral hemorrhage. Archives of neurology. 2008; 65:1320-1325.
112. Jaffe J, Alkhawam L, Du H, Tobin K, O’Leary J, Pollock G, Batjer HH, Awad IA. Outcome predictors and spectrum of treatment eligibility with prospective protocolized management of intracerebral hemorrhage. Neurosurgery. 2009; 64:436-445.
113. Rathor MY, Rani MFA, Jamalludin AR, Amran M, Shahrin TCA, Shah A. Prediction of functional outcome in patients with primary intracerebral hemorrhage by clinicalcomputed tomographic correlations. Journal of Research in Medical Sciences. 2012; 17:1056-1062.
114. Barras CD, Tress BM, Christensen S, MacGregor L, Collins M, Desmond PM, Skolnick BE, Mayer SA, Broderick JP, Diringer MN, Steiner T, Davis SM. Density and shape as CT predictors of intracerebral hemorrhage growth. Stroke. 2009; 40:1325-1331.
115. Yang J, Arima H, Wu G, Heeley E, Delcourt C, Zhou J, Chen G, Wang X, Zhang S, Yu S, Chalmers J, Anderson CS. Prognostic significance of perihematomal edema in acute intracerebral hemorrhage: Pooled analysis from the intensive blood pressure reduction in acute cerebral hemorrhage trial studies. Stroke. 2015; 46:1009-1013.
116. Appelboom G, Bruce SS, Hickman ZL, Zacharia BE, Carpenter AM, Vaughan KA, Duren A, Hwang RY, Piazza M, Lee K, Claassen J, Mayer S, Badjatia N, et al. Volume-dependent effect of perihaematomal oedema on outcome for spontaneous intracerebral haemorrhages. Journal of Neurology, Neurosurgery and Psychiatry. 2013; 84:488-493.
117. Thiex R, Tsirka SE. Brain edema after intracerebral hemorrhage: mechanisms, treatment options, management strategies, and operative indications. Neurosurgical focus. 2007; 22.
118. Selim M, Sheth KN. Perihematoma edema: A potential translational target in intracerebral hemorrhage? Translational Stroke Research. 2015; 6:104-106.
119. Dowlatshahi D, Smith EE, Flaherty ML, Ali M, Lyden P, Demchuk AM. Small intracerebral haemorrhages are associated with less haematoma expansion and better outcomes. International Journal of Stroke. 2011; 6:201-206.
120. Greenberg SM, Vonsattel JP, Segal AZ, Chiu RI, Clatworthy AE, Liao A, Hyman BT, Rebeck GW. Association of apolipoprotein E epsilon2 and vasculopathy in cerebral amyloid angiopathy. Neurology. 1998; 50:961-965.
121. Pang J, Wu Y, Peng J, Yang P, Kuai L, Qin X, Cao F, Sun X, Chen L, Vitek MP, Jiang Y. Potential implications of Apolipoprotein E in early brain injury after experimental subarachnoid hemorrhage: Involvement in the modulation of blood-brain barrier integrity. Oncotarget. 2016; 7:56030-56044. doi: 10.18632/oncotarget.10821.
122. Brouwers HB, Biffi A, Ayres AM, Schwab K, Cortellini L, Romero JM, Rost NS, Viswanathan A, Greenberg SM, Rosand J. Apolipoprotein E genotype predicts hematoma expansion in lobar intracerebral hemorrhage. Stroke. 2012; 43:1490-1495.
123. Mccarron MO, Nicoll JA, Stewart J, Ironside JW, Mann DM, Love S, Graham DI, Dewar D. The apolipoprotein E epsilon2 allele and the pathological features in cerebral amyloid angiopathy-related hemorrhage. Journal of Neuropathology & Experimental Neurology. 1999; 58:711-718.
124. Tzourio C, Arima H, Harrap S, Anderson C, Godin O, Woodward M, Neal B, Bousser MG, Chalmers J, Cambien F, MacMahon S. APOE genotype, ethnicity, and the risk of cerebral hemorrhage. Neurology. 2008; 70:1322-1328.
125. Pang J, Chen Y, Kuai L, Yang P, Peng J, Wu Y, Chen Y, Vitek MP, Chen L, Sun X, Jiang Y. Inhibition of blood-brain barrier disruption by an apolipoprotein e-mimetic peptide ameliorates early brain injury in experimental subarachnoid hemorrhage. Translational Stroke Research. 2016:1-16.
126. Steiner T, Diringer MN, Schneider D, Mayer SA, Begtrup K, Broderick J, Skolnick BE, Davis SM. Dynamics of intraventricular hemorrhage in patients with spontaneous intracerebral hemorrhage: Risk factors, clinical impact, and effect of hemostatic therapy with recombinant activated factor VII. Neurosurgery. 2006; 59:767-773.
127. Li Q, Huang YJ, Zhang G, Lv FJ, Wei X, Dong MX, Chen JJ, Zhang LJ, Qin XY, Xie P. Intraventricular hemorrhage and early hematoma expansion in patients with intracerebral hemorrhage. Scientific Reports. 2015; 5.
128. Tuhrim S, Horowitz DR, Sacher M, Godbold JH. Volume of ventricular blood is an important determinant of outcome in supratentorial intracerebral hemorrhage. Critical Care Medicine. 1999; 27:617-621.
129. Maas MB, Nemeth AJ, Rosenberg NF, Kosteva AR, Prabhakaran S, Naidech AM. Delayed intraventricular hemorrhage is common and worsens outcomes in intracerebral hemorrhage. Neurology. 2013; 80:1295-1299.
130. Fujii Y, Takeuchi S, Harada A, Abe H, Sasaki O, Tanaka R. Hemostatic activation in spontaneous intracerebral hemorrhage. Stroke. 2001; 32:883-890.
131. Garton T, Keep RF, Wilkinson DA, Strahle JM, Hua Y, Garton HJL, Xi G. Intraventricular hemorrhage: The role of blood components in secondary injury and hydrocephalus. Translational Stroke Research. 2016; 7:447-451.
132. Li N, Wang Y, Wang W, Ma L, Xue J, Weissenborn K, Dengler R, Worthmann H, Wang DZ, Gao P, Liu L, Wang Y, Zhao X. Contrast extravasation on computed tomography angiography predicts clinical outcome in primary intracerebral hemorrhage: A prospective study of 139 cases. Stroke. 2011; 42:3441-3446.
133. Brouwers HB, Goldstein JN, Romero JM, Rosand J. Clinical applications of the computed tomography angiography spot sign in acute intracerebral hemorrhage: a review. Stroke. 2012; 43:3427-3432.
134. Orito K, Hirohata M, Nakamura Y, Takeshige N, Aoki T, Hattori G, Sakata K, Abe T, Uchiyama Y, Sakamoto T, Morioka M. Leakage sign for primary intracerebral hemorrhage: A novel predictor of hematoma growth. Stroke. 2016; 47:958-963.
135. Ciura VA, Brouwers HB, Pizzolato R, Ortiz CJ, Rosand J, Goldstein JN, Greenberg SM, Pomerantz SR, Gonzalez RG, Romero JM. Spot sign on 90-second delayed computed tomography angiography improves sensitivity for hematoma expansion and mortality: Prospective Study. Stroke. 2014; 45:3293-3297.
136. Koculym A, Huynh TJ, Jakubovic R, Zhang L, Aviv RI. CT perfusion spot sign improves sensitivity for prediction of outcome compared with CTA and postcontrast CT. American Journal of Neuroradiology. 2013; 34:965-970.
137. Mir DI, Gupta A, Dunning A, Puchi L, Robinson CL, Epstein HA, Sanelli PC. CT perfusion for detection of delayed cerebral ischemia in aneurysmal subarachnoid hemorrhage: a systematic review and meta-analysis. AJNR American journal of neuroradiology. 2014; 35:866-871.
138. Schindlbeck KA, Santaella A, Galinovic I, Krause T, Rocco A, Nolte CH, Villringer K, Fiebach JB. Spot sign in acute intracerebral hemorrhage in dynamic t1-weighted magnetic resonance imaging. Stroke. 2016; 47:417-423.
139. D’Esterre CD, Chia TL, Jairath A, Lee TY, Symons SP, Aviv RI. Early rate of contrast extravasation in patients with intracerebral hemorrhage. American Journal of Neuroradiology. 2011; 32:1879-1884.
140. Qi L, Gang Z, Xin X, Wang XC, Yang WS, Li KW, Xiao W, Peng X. Black hole sign: novel imaging marker that predicts hematoma growth in patients with intracerebral hemorrhage. Stroke. 2016; 47:1777-81.
141. Delgado Almandoz JE, Yoo AJ, Stone MJ, Schaefer PW, Oleinik A, Brouwers HB, Goldstein JN, Rosand J, Lev MH, Gonzalez RG, Romero JM. The spot sign score in primary intracerebral hemorrhage identifies patients at highest risk of in-hospital mortality and poor outcome among survivors. Stroke. 2010; 41:54-60.
142. Huynh TJ, Demchuk AM, Dowlatshahi D, Gladstone DJ, Krischek O, Kiss A, Hill MD, Molina CA, Rodriguez-Luna D, Dzialowski I, Silva Y, Czlonkowska A, Lum C, et al. Spot sign number is the most important spot sign characteristic for predicting hematoma expansion using first-pass computed tomography angiography: Analysis from the PREDICT study. Stroke. 2013; 44:972-977.
143. Kim SH, Jung HH, Whang K, Kim JY, Pyen JS, Woong Oh J. Which emphasizing factors are most predictive of hematoma expansion in spot sign positive intracerebral hemorrhage? Journal of Korean Neurosurgical Society. 2014; 56:86-90.