
INTRODUCTION
Normal embryonic development, as well as repair and dynamic maintenance of complex structures throughout the lifespan, both depends upon a set of signals that keeps individual cell activities orchestrated toward the large-scale anatomical goals of the host. Morphogenesis and remodeling can be challenged by cancer, which can be viewed as a process in which cells escape or become isolated from the normally tight morphogenetic control of the organism [1-6]. To achieve robust development, signaling pathways need to affect their target cells with sufficient spatiotemporal resolution to integrate organ sculpting, anatomical polarity, tissue identity, and growth rates appropriate to the large-scale order maintenance within the body. In the context of cancer, it has been well documented that healthy neighboring cells help to stabilize aberrant cell behavior [7-9] to control tumorigenesis by tissue-level organization that adheres to the proper patterning needs of the host [10-14]. Indeed, fascinating classical and recent data show that actively patterning environments, such as embryos and regenerating amphibian limbs, can normalize and reprogram tumors [15-31]. Thus, in addition to any cell-autonomous properties that may have gone awry in cancer cells, it is crucial to understand the non-cell-autonomous patterning signals that may be exploited to prevent and treat cancer.
Endogenous bioelectric signaling among all (not just excitable) cell types is one component of the microenvironment, and is now known to mediate instructive information for large-scale pattern formation [32, 33]. Bioelectrical processes, such as extracellular electric fields and transmembrane resting potentials produced by ion channel and pump proteins, regulate cell activity [34-36]. In particular, proliferation, differentiation, migration, and apoptosis are all important cell behaviors relevant to cancer that are guided partly by bioelectric signals, such as resting voltage potential (Vmem) in non-excitable cells [37-39]. It was recognized long ago that the electrical properties of mature tumors differ from that of healthy tissue [40-42], and we recently showed that monitoring of a distinctly depolarized resting potential allows early detection of tumors in vivo. Moreover, forced hyperpolarization via a number of different channels prevents those cells from forming tumors [43], revealing resting potential as an instructive cue and not merely a marker [39]. Indeed, a number of ion channels are now known to be bona fide oncogenes [44-54], and ion channel blockers are an important area for cancer drug development [55-58].
However, it is important to note that ion channels are not simply molecular markers or targets within the cancer cells themselves. Gradients of resting potential are a long-range, global system for exerting patterning control, and thus a tractable candidate for manipulation of the crosstalk by which microenvironment suppresses aberrant cell behavior [59]. For example, selective depolarization of glycine-gated chloride channel-expressing cells in the Xenopus model results in a metastatic conversion of melanocytes, which over-proliferate, acquire an arborized shape, and invade blood vessels and soft tissues in an MMP-dependent manner [46, 60, 61]. This effect occurs at considerable distance, via a serotonergic pathway, and demonstrates how disruption of long-range endogenous physiological signaling can activate a cancer phenotype in the absence of carcinogen exposure or DNA damage. The data suggest that bioelectric gradients are a fascinating new aspect of cancer:host interaction, and that the notion of microenvironment may need to be expanded to account for long-range interactions mediated by Vmem changes throughout tissues.
The existing literature on roles of ionic signaling in cancer leaves open a number of fundamental questions. What role might remote Vmem modulation play in oncogene-induced carcinogenesis? While distant changes of resting potential can trigger a metastatic conversion [60, 61], it is not known whether or how such signaling plays a role when canonical oncogenes initiate tumorigenesis. A better understanding of the microenvironment and long-range aspects of aberrant growth control by endogenous developmental patterning mechanisms would have significant implications for design of novel therapeutic approaches to prevent and reprogram cancer.
Here, we use mRNA encoding human tumor inducers (Gli1, XRel3, KRAS) in Xenopus laevis embryos to initiate growth of tumor like structures (ITLS) that highly resemble classic tumors. Remarkably, forced hyperpolarization (by misexpression of specific ion channels) is able to suppress ITLS formation, despite high levels of oncogene protein, even when the hyperpolarized cells are at a considerable distance from the oncogene-expressing tumor site. The suppression effect can also be exerted by native chloride intracellular channel 1 (CLIC1)-mediated hyperpolarization, revealing an endogenous target for bioelectric control of abnormal growth that does not require transgene expression. We show that the suppression effects of distant hyperpolarization are mediated by voltage control of a butyrate and histone deacetylase mechanism. Together, these data reveal the first mechanistic details of Vmem as a powerful, tractable regulator of long-range signaling between cancer cells and the host and suggest several new entry points for biomedical strategies.
RESULTS
Induced tumor-like structures (ITLS) exhibit striking key similarities to human tumors
To study the role of bioelectric events to oncogene-mediated tumorigenesis, we took advantage of Xenopus laevis embryos – a model system that is ideal for molecular biophysics approaches and also has been increasingly used for cancer research[43, 61-66]. We injected mRNAs encoding Xrel3 and human KRASG12D into Xenopus embryos; these oncogenes are known to cause morphologically apparent ITLS in up to 50% of injected embryos without any other developmental defects. ITLS have previously been characterized as having some of the hallmarks of human tumors – increased proliferation, vasculature attraction, lack of differentiation, invasiveness and transplantability to healthy recipients [43, 67-69]. We began by further investigating the pathology of the affected tissue to confirm and expand the relevance of Xenopus as a medically-relevant cancer model.
Clinically-relevant tumors exhibit a surplus of proliferative capacity [70]. To characterize the cell proliferation dynamics in ITLS loci, we monitored dynamic patterns of cell cycle progression in ITLS and unaffected regions using FUCCI (fluorescent ubiquitination-based cell cycle indicator) pair: mKO2-Cdt1 and mAG-Geminin [71, 72]. The accumulation of these two cell cycle regulators – in nuclei of transfected cells – in a mutually exclusive manner between G1(Cdt1) and G2/S/M (Geminin) phases, allowed us to monitor spatial dynamics of cell cycle progression: while the number of ITLS cells in G1 and control cells in G1 and G2/S/M phases are statistically the same, twice as many cells in G2/S/M phases were present in ITLS by comparison (N=8 per category, ANOVA, P<0.001) (Fig. 1A).
Next, we examined the extent of hypoxia within ITLS loci, since hypoxia is known to be present in tumors [73-75]. Treatment of ITLS bearing embryos (N=7) with the hypoxia marker pimonidazole revealed a 12-fold increase (t-test; p<0.0001) for immunoperoxidase staining for hypoxia (Fig. 1B). This is consistent with data obtained from pimonidazole-based qualitative and quantitative assessment of tumor hypoxia, where an increase in the frequency of detecting pimonidazole adducts is reported [76-78].
We next examined histological features of the ITLS using hematoxylin and eosin (H&E) [79]. Cells within the ITLS exhibited disorganized growth patterns, including misplaced mesodermal and endodermal cells, and mesodermal cells that were larger than those present in unperturbed regions, revealing features consistent with neoplasia (Fig. 1C).
We next examined a change in nuclear size, which is another well-known aspect of tumor tissue [80, 81]. Computer-aided image analysis of the area of 2371 nuclei each from 16 ITLS and 16 control regions of oncogene-injected tadpoles showed on average a 10% increase in nuclear size of ITLS, mirroring similar phenomenon observed between different stages of human breast cancer cells [82] (Fig. 1D). Another marker of tumorigenesis is the abnormal acidity of tumor tissue [83, 84]. Using BCECF, a dual excitation pH indicator, fluorescence intensity ratios at two different wavelengths of ITLS and control regions revealed that ITLS have a more acidic (pH 6.6) intracellular environment compared to that of control regions, which have a pH average of 7.41 (N=8 for both treatments, t-test, P<0.05) (Fig. 1E).
Finally, to investigate host immune response to tumor formation, immunohistochemistry with anti-XL2 (anti-Xenopus leukocytes) was performed on cross sections of tissue bearing ITLS. We observed recruitment of leukocytes to the site of transformed cells (Fig 1F), consistent with the known role of leukocyte migration as an anti-tumor immune response [85].
Taken together, these characteristics of ITLS are consistent with previous findings of increased proliferation within ITLS, ability to form in internal tissues (not just the epidermis), and propensity to attract vasculature[43], and demonstrate the relevance of human oncogene-induced tumors in Xenopus for understanding the basic mechanisms of carcinogenesis in vivo.
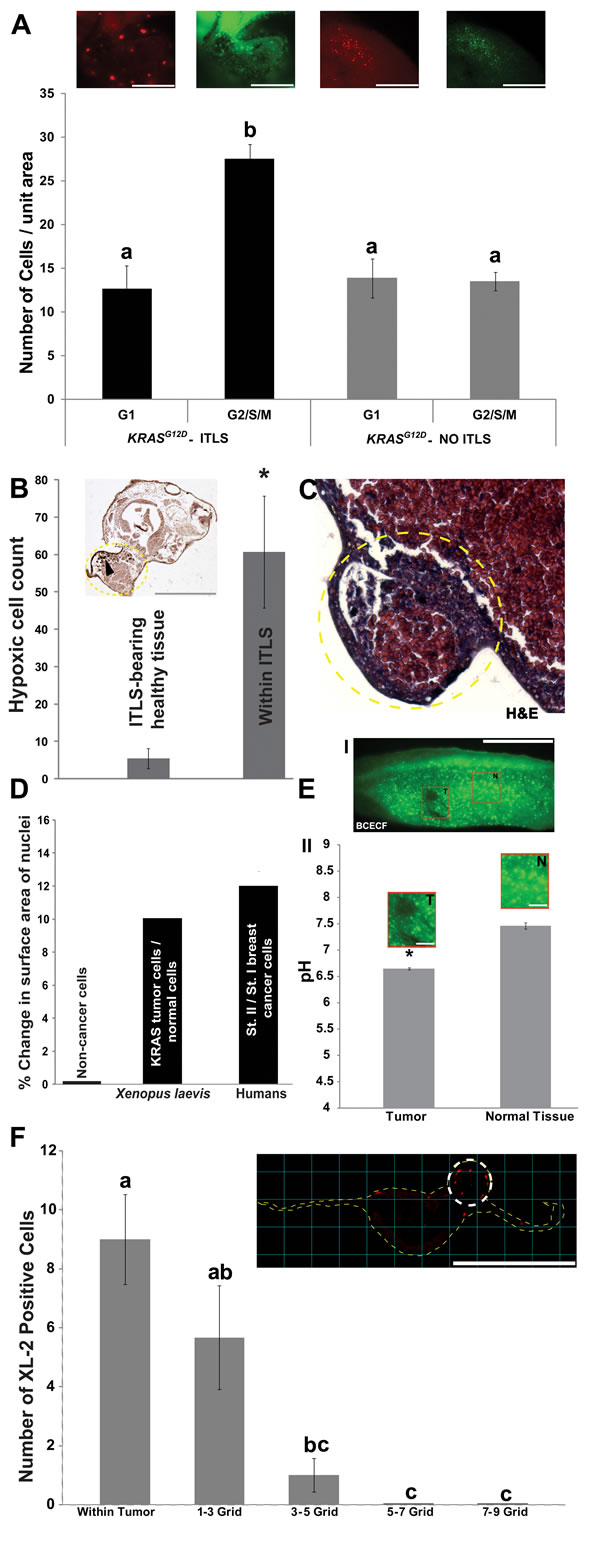
Figure 1: Induced tumor-like structures (ITLS) exhibit characteristics reminiscent of human tumors. (A) Rates of proliferation were analyzed in vivo in fluorescent cell cycle indicators (FUCCI pair: mKO2-zCdt1 and mAG-zGeminin) injected, ITLS-bearing embryos at stage 34. ITLS regions (black bars) have >65% more cells that are in G2/S/M phase (green insert, mAG-zGeminin) than there are in G1 phase (red insert, mKO2-zCdt1). Unperturbed regions (grey bars) showed no difference between the number of cells in G1 (red insert) and G2/S/M (green insert) phases. N=8 for all four categories. P<0.001, one-way ANOVA, tukey’s post hoc analysis; different letters indicate statistically significant difference; scale bar = 250 µm.(B) Immunoperoxidase analysis of hypoxia using detection of pimonidazole protein adducts (black arrowhead) in cells reveals a 12 fold increase for immunoperoxidase staining for hypoxia per unit area in ITLS (yellow traces) than in surrounding healthy tissue. N=7; *P<0.0001 Student’s t-test; scale bar = 500 µm.(C) H&E staining of KRASG12D ITLS sections (yellow circular trace, 40X) for evidence of features of neoplasia reveals disorganized growth patterns, including misplaced mesodermal and endodermal cells, and mesodermal cells that are larger than those present in unperturbed regions. (D) Automated analysis of the size of Hoechst Blue stained nuclei reveals a 10% increase in the size of tumor nuclei compared to nuclei from unaffected cells. A change in morpholology of the nuclei that includes a progressive increase in nucleus size has been documented in human breast cancer cells [82]. N=16 (2371 nuclei) for both ITLS and unaffected regions.(E) Intracellular pH measurements in ITLS and Control regions were made using the fluorescent pH reporter dye BCECF. (I) A decreased fluorescence is observed in ITLS cells (T) compared to a normal region (N). (II) Upon quantification, levels of fluorescence correspond to pH values of 6.6 in ITLS and 7.41 in control regions. N=8 for both treatments; *P<0.05 Student’s t-test; scale bar = 1mm in tail fragment; 150 µm in magnified inserts.(F) The response of innate immunity to ITLS formation was investigated using anti-XL2 immunohistochemistry to mark the presence of leukocytes: leukocytes are primarily present around ITLS (white circular trace) and 1-3 grid (300 µM) away from ITLSs. N=12; P<0.01, one-way ANOVA, Tukey’s post hoc analysis; different letters indicate statistically significant difference; scale bar = 500 µm. Error bars indicate ± 1 s.e.m in A, B, E, F.
Long-range hyperpolarization suppresses the formation of ITLS
To test the hypothesis that hyperpolarization affects oncogenic transformation of cells located at a distance, we performed functional experiments using sets of oncogenes and hyperpolarizing reagents. The use of multiple oncogenes and hyperpolarizing agents allowed us to confirm that there is a generalized suppression effect due to changes in Vmem, one that is not tied to a particular channel protein or ion. We used Xrel3 tagged with tdTomato to track the presence of oncogenic protein (Fig. 2A, I-III). To modulate Vmem, we used Kv1.5 (Potassium voltage-gated channel, shaker-related subfamily, member 5) [86], a well-characterized hyperpolarizing channel, whose overexpression in our model system has been used to alter developmental patterning [87].
To probe the effects of long-range hyperpolarization on tumor formation, we injected mRNA’s encoding Xrel3-tdTomato and Kv1.5-GFP3 in two separate blastomeres of 16-cell stage embryos, and scored for the presence of ITLS and hyperpolarizing channel/oncogene protein by stage 34. Compared to embryos receiving only oncogene injections, embryos with a remote hyperpolarized region (Fig. 2B, IV&V) often displayed lack of morphologically apparent ITLS (Fig. 2B, I) despite strong presence Xrel3-2A-tdTomato protein (Fig. 2B, II&III). Kv1.5-induced long-distance hyperpolarization was also able to significantly suppress Xrel3 and Gli1 ITLS formation by 34.9% (N=225; t-test, P<0.05) and 29.4% (N=292; t-test, P<0.05), respectively (Fig. 2C), suggesting the effect is not specific for just one type of oncogene. Treatment with high chloride (whose uptake by native Cl- channels hyperpolarizes cells) resulted in a 37.5% decrease (N=349; t-test P<0.05) in KRASG12D ITLS, demonstrating that the suppressive effect is not specific for Kv1.5 nor for potassium, as mediators of the crucial Vmem change (Fig. 2C).
Collectively, these data reveal a long-range signal that is initiated by hyperpolarization and has influence over tumorigenesis at remote sites in the tadpole. Moreover, these results demonstrate that ITLS suppression through Vmem modulation is possible to achieve by taking advantage of endogenous channels that are not necessarily located within oncogene expressing cells.
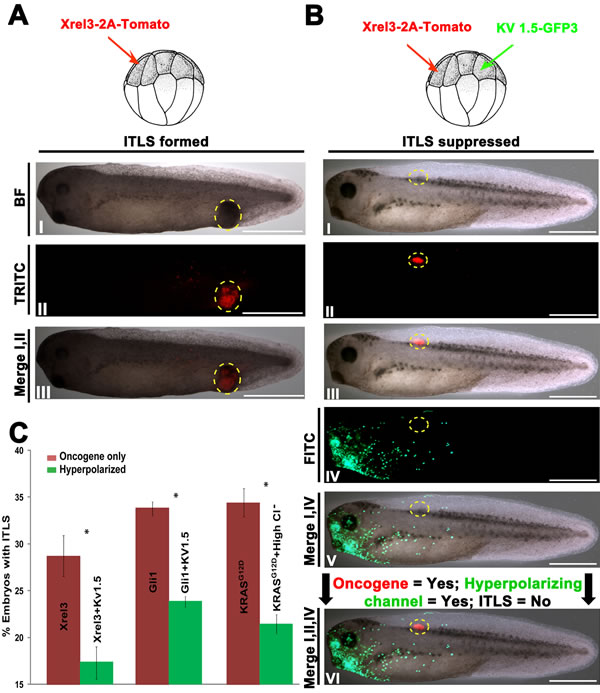
Figure 2: Long-range hyperpolarization suppresses the formation ITLSs. (A) To visualize and track ITLSs, mRNA encoding a fusion construct of Xrel3 and tdTomato was injected into a single blastomere of 16-cell stage embryos. (I, II) ITLSs resulting from Xrel3-2A-tdTomato injections were highly fluorescent when visualized under a TRITC filter set. (III) Overlay of bright field (BF) and TRITC images shows the co-localization of ITLS and red fluorescent signal, confirming that foci of oncogene expression are the cells that make up ITLS. Scale bar = 1 mm.(B) To test whether long-range hyperpolarization suppresses ITLS formation, a potassium based hyperpolarizing channel (Kv1.5-GFP3) and Xrel3-2A-tdTomato were injected in distantly separated blastomeres of a 16-cell stage embryo. (I) While morphologically-apparent ITLSs were often missing from these embryos, the oncogenic protein was present as evidenced by the tdTomato fluorescent signal (II, II). Robust expression of the hyperpolarizing channel protein (Kv1.5-GFP3) was observed in the head and gut regions (IV, V), and away from Xrel3-2A-tdTomato expressing cells (VI). Scale bar = 1 mm.(C) Fold change in ITLS formation for oncogene injected embryos with hyperpolarized treatments compared to oncogene-only injected embryos. Oncogene-only injected embryos with ITLS have a ratio of 1, and ratio values below and above one represent fewer and more embryos with ITLS, respectively. To show the effect of ITLS suppression due to change in Vmem, three different oncogenes and hyperpolarizing reagents (based on Cl- and K+) were used. When Kv1.5 (K+ hyperpolarizing channel) was introduced non-locally in Xrel3 and Gli1 injected embryos, 39.4% and 29.4% decreases, respectively, in ITLS formation were observed. A 37.5% decrease in ITLS formation was also achieved using KRASG12D as an oncogene and 70mM Cl- as a hyperpolarizing reagent. N=225-349 embryos for each treatment; *P<0.01 Student’s t-test. Error bars indicate ± 1 s.e.m.
Chloride-dependent hyperpolarization through endogenous chloride intracellular channel 1 (CLIC1)
Having shown a functional role of long-range hyperpolarization in the context of tumorigenesis, we proceeded to characterize the native channel responsible for chloride-based inhibition of ITLS formation. We performed an inverse drug screen [88] with chloride channel blockers to see which ones would abrogate the chloride-dependent ITLS suppression: Anthracene-9-Carboxylic Acid (ACA), inadyloxyacetic acid 94 (IAA), and 5-Nitro-2-(3-phenylpropylamino) benzoic acid (NPPB) [89-92] (Fig. 3A). After the injection of KRASG12D and treatment with high chloride (N=164), ITLS incidence dropped significantly (χ2, P<0.05) compared to their counterparts in a low chloride concentration (N=162). This anti-tumor effect of high chloride was completely abolished with IAA treatment (N=186; χ2, P<0.05) (Fig 3B), while the other two inhibitors did not significantly affect the rate of ITLS incidence. We then proceeded to validate this result using molecular reagents targeting IAA-sensitive channels.
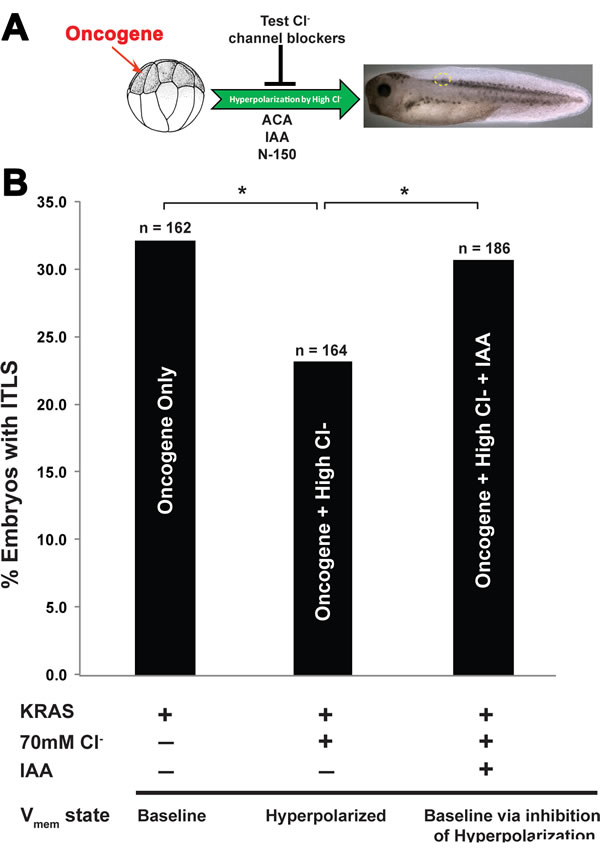
Figure 3: A suppression screen for chloride channels is employed to identify a channel responsible for long-range ITLS suppression. (A) Oncogene injected embryos were placed in a high chloride concentration (70mM) solution, followed immediately by separate treatments with very well-known chloride transport blockers: Anthracene-9-Carboxylic Acid (ACA), indanyloxyacetic acid (IAA) and 5-Nitro-2-(3-phenylpropylamino) benzoic acid (NPPB). (B) High chloride treatment of oncogene-injected embryos results in ~31%% decrease in KRASG12D ITLS incidence. While blocking chloride channels using ACA and NPPB did not affect the rate of this suppression, IAA treatments in the presence of high Cl- restored ITLS incidence back to oncogene-only levels. N=162, 164, 186 for oncogene only, oncogene + high Cl-, and oncogene + high Cl- + IAA, respectively; *P<0.05, χ2 compared to tumor incidence in KRASG12D only injected embryos.
A known target of IAA is the intracellular chloride channel 1 (CLIC1) [93]. Given its pharmacological profile and ubiquitous presence during every stage of the Xenopus laevis development [94], we focused on CLIC1 in this study. To test the hypothesis that CLIC1-mediated hyperpolarization affects oncogenic transformation of cells, we performed functional experiments involving CLIC1 (wildtype and mutant) and KRASG12D injections. Wildtype CLIC1 was tagged with GFP, while a dominant negative form of CLIC1 was generated by mutating cysteine 24 to serine, in order to form an inactive multimer channel [95], and tagged it with tdTomato to track localization of the protein product. mRNA’s encoding CLIC1-2A-GFP3/KRASG12D-tdTomato (Fig. 4B, I&II) and CLIC1C24S-2A-tdTomato/KRASG12D-GFP3 (Fig. 4B, III&IV) were injected in two different blastomeres of 16-cell stage embryos, treated with high Cl-, and scored for the presence of ITLS and CLIC1 channel/oncogene proteins. As a control to which CLIC injected embryos can be compared, KRASG12D-only injected embryos were also treated with high Cl- (N=372), resulting in 31.5% fold decrease in ITLS incidence when compared to KRASG12D-only injected embryos (N=341). The rate of ITLS suppression was further amplified (44.3% suppression) when the wildtype CLIC1 was overexpressed non-locally in oncogene-injected embryos that were then raised in 70mM Cl- (N=364). In contrast, overexpression of the mutant CLIC1 (N=336) effectively blocked ITLS suppression despite the presence of high Cl- (Fig 4C). Together, these data suggest a role for native CLIC1 in mediating hyperpolarization-induced long-range ITLS suppression.
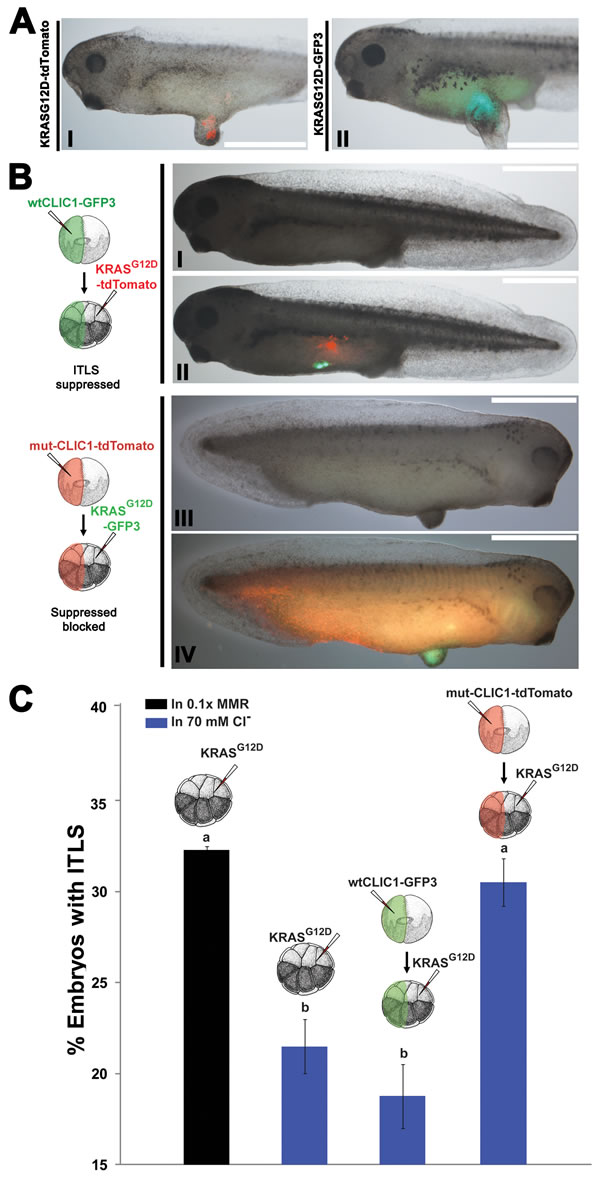
Figure 4: Hyperpolarization by the influx of Cl- through CLIC1 mediates long-range ITLS suppression. (A) To visualize and track ITLSs, KRASG12D mRNA was co-injected with GFP3 (I) and tdTomato (II) into a single blastomere of 16-cell stage embryos. In both cases, ITLS co-localize with the respective fluorescent signals, confirming that oncogene-expressing cells make up ITLS. Scale bar = 1 mm.(B) When mRNA encoding Xenopus wildtype CLIC1-GFP3 was injected at a distance from KRASG12D -tdTomato, morphologically-apparent ITLS formation was often suppressed despite the presence of oncogenic protein (III). Whereas overexpression of a CLIC1 dominant negative (CLIC1C24S-tdTomato) in non-oncogene expressing cells prevents ITLS suppression despite the presence of high Cl- in the media (IV). Scale bar = 1 mm. (C) Changes in ITLS formation incidence for oncogene and CLIC1 (mutant or wildtype) injected embryos in 70mM Cl- media. ITLS incidence from oncogene-only injection was normalized to 1 so that it can be used to measure against the effects of other treatments. Embryos injected with KRASG12D followed by a high Cl- treatment showed a 31.5% decrease in ITLS incidence. Overexpression of the wildtype CLIC1 and high Cl- bath also lowered the number of embryos with ITLS by 44.3% while resulting in smaller ITLS in escapees. Overexpression of the mutant CLIC1, which encodes for defective multimer channels, appeared to block ITLS suppression by a high Cl- treatment, implicating native CLIC1 channels in Vmem mediated, long-range suppression of ITLS. N= 336-364 embryos for each treatment; P=0.01, one-way ANOVA, Tukey’s post hoc analysis; different letters indicate statistically significant difference. Error bars indicate ± 1 s.e.m.
Changes in Vmem are transduced via HDAC1-dependent mechanisms
Bioelectric signals are transduced into transcriptional and epigenetics responses via a number of second-messenger pathways. In order to determine the transduction mechanism by which hyperpolarization inhibits tumorigenesis, we conducted a pharmacological suppression screen [63] and molecular-genetic loss-of-function of several well-characterized candidate pathways that have been shown to mediate the actions of Vmem change in other patterning contexts [96]. Our strategy was to block, one at a time, the candidate transduction mechanisms and see which ones abrogated the ability of hyperpolarization to reduce tumor incidence. Targeted pathways include: movement of serotonin through the Vmem-dependent serotonin transporter SERT [97] (inhibited via 10 µM fluoxetine); Vmem-guided transport of signaling molecules through gap junctions [98, 99] (disrupted by 0.5 ng injection of a dominant negative connexin H7); voltage gated calcium channels [100, 101] (blocked via 0.1 mM cadmium chloride), and Vmem-modulated transport of small molecule inhibitors of histone deacetylase 1 [102-104] (targeted by 0.5ng injection of mRNA encoding a dominant negative HDAC1) (Fig 5A). HDAC1 is an especially attractive target because it is known to control levels of histone acetylation, which determines cell cycle progression, rates of proliferation, apoptosis and differentiation of cancer cells [105-107]. Administering oncogene, hyperpolarizing agent, and Vmem-sensing pathway disruptors (Fig 5B) showed that blocking HDAC1 activity had the most significant effect (χ2, P<0.05) on CLIC1-mediated suppression of tumors: the rate of ITLS formation in DN-HDAC1 injected embryos in high Cl- was similar to that of oncogene-only injected embryos - introducing DN-HDAC1 non-locally to the site of KRASG12D increased ITLS formation incidence by 35.6% (χ2, P<0.05) (Fig. 5C), suggesting a role for histone deacetylase 1 activity controlling cell behavior at a distance.
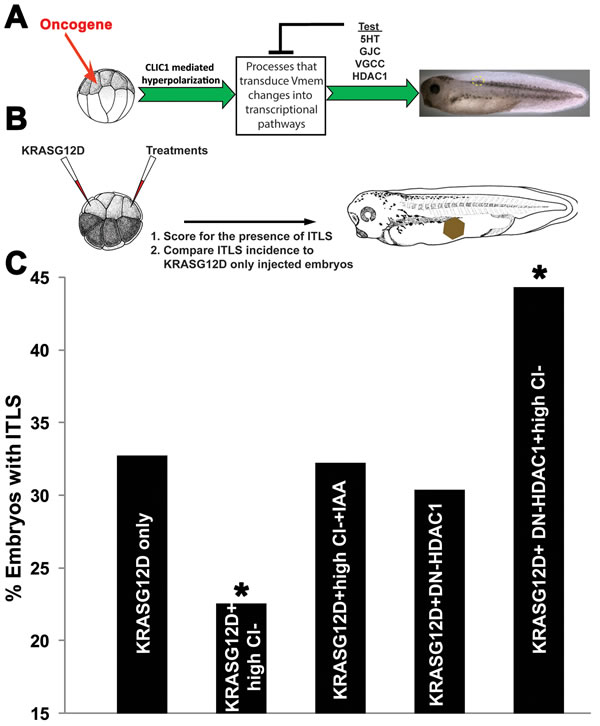
Figure 5: Long-range suppression of ITLS by hyperpolarization is HDAC1-dependent. (A)Hyperpolarization can be transduced into transcriptional pathways by processes that include Vmem-dependent transport of signaling molecules (e.g. serotonin transport through SERT), electrophoresis of morphogenes through gap junctions (GJC), calcium signaling via voltage-gated Ca++ channels (VGCC), and voltage dependent butyrate transport that results in HDAC inhibition. To identify the responsible transduction mechanism, each process was independently blocked: 10 µM fluoxetine shuts down SERT; H7, a dominant connexin disrupts the transport function of endogenous gap junctions; 0.1 mM cadmium chloride effectively blocks VGCCs; and a dominant negative 0.5 ng HDAC1 injection inactivates endogenous HDAC1. (B) In each case, a treatment used to probe a given transduction mechanism was targeted non-local to KRASG12D injected cells. KRASG12D injections and treatments were administered at the 16-cell stage, and embryos were then raised to stage 34 before scoring the presence of ITLS.(C) When analyzing ITLS incidences compared to oncogene only injected embryos – DN-HDAC1blocks the effects of CLIC1 mediated hyperpolarization, thus brining ITLS formation to the level of oncogene-only injection. Introducing DN-HDAC1 at a distance from KRASG12D without hyperpolarization increases ITLS incidence by 35.6%, implicating a role for HDAC1 in long-range ITLS suppression. N=164-251 embryos; *P<0.05, χ2 compared to tumor incidence in KrasG12D only injected embryos.
Having previously identified butyrate as an endogenous HDAC inhibitor involved in local ITLS suppression [43], we sought to identify the source of butyrate in this system. Reasoning that it would be produced by native bacteria, we conducted a screen for antibiotics that target butyrate producing bacteria. We first tested novobiocin, which has been shown to be effective against gram-positive bacteria [108]. At 78 μM, novobiocin sodium salt treatment significantly increased ITLS incidence by 32.1% (χ2, P<0.01) (Fig. 6). It has been determined in other studies that the mechanism of action for novobiocin involves the inhibition of the bacterial DNA gyrase [109, 110]. Given also nalidixic acid’s well-known inhibition of DNA gyrase [111], we tested its effect (at 43 μM) on ITLS incidence. While an 8.2% increase in ITLS was observed, it was not statistically significantly different from ITLS incidence in oncogene-only injected embryos (Fig. 6). Finally, a cocktail of antibiotics (gentamicin, metrodinazole, vancomycin hydrochloride, and clindamycin hydrochloride, all at 20 μM concentration) targeting gram-positive bacteria resulted in a significant increase in ITLS incidence by 26.2% (χ2, P<0.05) (Fig. 6). Bulk analysis of embryos exposed to this cocktail of antibiotics does indeed show a significant drop in total butyrate concentration (data not shown).
Together, these data suggest that the control of tumorigenesis by Vmem is mediated by HDAC1, via butyrate derived from gram-positive bacteria.
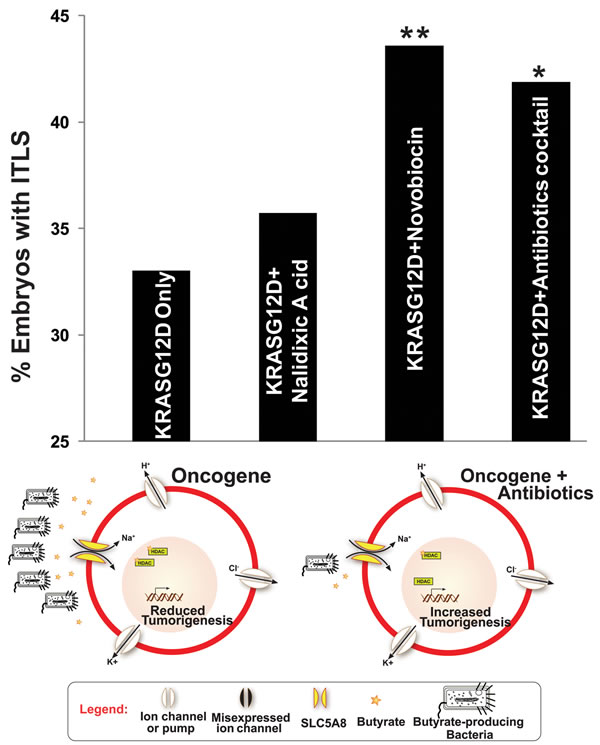
Figure 6: Targeted killing of butyrate producing Bacteria increases ITLS incidence To determine the source of butyrate, which is responsible for ITLS suppression through HDAC inhibition, variety of antibiotics were used to target butyrate-producing Bacteria. Compared to KRASG12D-only injected embryos, novobiocin and antibiotic cocktail (gentamicin, metrodinazole, vancomycin, and clindamycin) treatments significantly increased ITLS formation by 32.1% and 26.8%, respectively. Adding nalidixic acid, which works by inhibiting DNA gyrase as novobiocin does, did not significantly alter ITLS formation, showing only an 8.2% increase in ITLS incidence. N=537, 318, 431, 117 for KRASG12D, KRASG12D + Nalidiixic Acid, KRASG12D + Novobiocin, KRASG12D + antibiotics cocktail; *P<0.05, **P<0.01, χ2 compared to ITLS incidence in KRASG12D-only injected embryos.
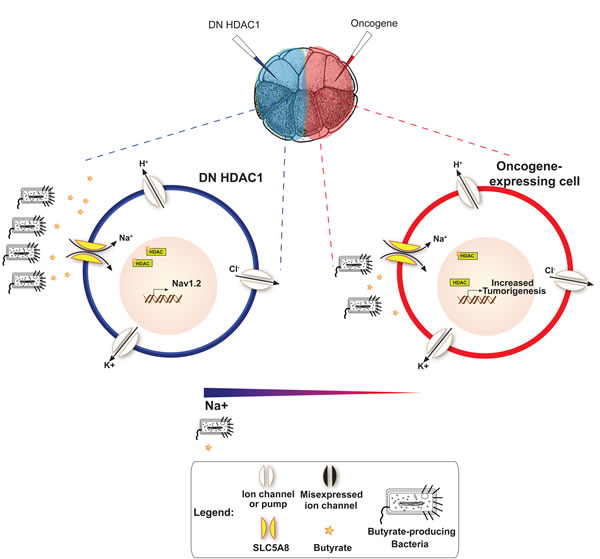
Figure 7: A model for Vmem – mediated, HDAC1-dependent, long-range control of cell dynamics during neoplastic transformation It is known that HDAC1 inhibition leads to the altered transcriptional of regulation of several ion channel genes including NaV 1.2 [197] – a voltage gated sodium channel encoding gene. One model consistent with our data is that the resulting Na+ gradient (more positive charge) would attract butyrate producing bacteria away from oncogene expressing cells (illustrated using Escherichia coli in Supp. Fig 1). Gram-positive bacteria are necessary for the production of butyrate, which gets imported into cells via SLC5A8 to reduce proliferation rate by inducing cell cycle arrest in tumor cells. Driving these bacteria away from ITLS cells would reduce the number of butyrate molecules available near oncogene cells, thus blocking ITLS suppression.
DISCUSSION
It has been well-documented that bioelectric properties control individual cell behaviors that are highly relevant to the processes of cancer [35, 36, 96, 112, 113]. For example, electric fields generated by voltage-gated sodium channels (VGSC) provide motility cues to highly metastatic rat prostate cells [114], and many cancer cells are galvanotactic [115-119]. Distinct from extracellular electric fields, resting membrane potentials established by ion channels and pumps control cell proliferation, transformation, and growth in number of tumor forming cells [120-125]. Moreover, the actions of these translocators have allowed for the development of predictive physiological markers [43, 61, 125] and control of proliferative and metastatic behaviors [39, 46, 60, 126]. Here, we show for the first time that resting potentials in remote cells have a profound influence on whether or not a cell expressing human oncogenes will go on to form a tumor in vivo.
The induced tumor like structures (ITLS) were shown to exhibit striking similarities with classic tumors, including increased proliferation as monitored by visualizing the cell cycle dynamics, disorganization of the normal developmental architecture revealed by H&E stains, increased hypoxia, increased nuclear size, acidic intracellular microenvironment, and ability to illicit innate immune response (Fig. 1A-F). Given the increased recognition of bioelectric signals as endogenous patterning cues that keep cellular activities orchestrated towards anatomical goals [32, 33, 127], our data confirm Xenopus embryos as a tractable model system in which to investigate the role of bioelectric signals in host:cancer interaction, and in which to ask fundamental questions about cancer as a disorder of developmental cues [5, 6, 10, 12, 128, 129].
Long-range control of tumorigenic process by Vmem
The spatial distance over which carcinogenesis can be predicted and controlled have been addressed in a few studies. One example is illustrated by the ability to assess cancer risk of readily-inaccessible organs using accessible surrogate sites – functionally or anatomically similar, but not necessarily contiguous with the target organ – that make up the cancer field [130-132]. Another is the activation of melanoma-like transformation in the melanocyte population [60] by Vmem modulation of even a few instructor cells at considerable distance from the melanocytes themselves [61]. However, to our knowledge this is the first time that modulation of distant cells’ resting potentials has been shown to impact the formation of discrete tumor foci. It is tempting to speculate that the long-range connections are bi-directional: not only does remote electrical state of tissue matter for tumor growth, but perhaps tumors also emit bioelectrical information that could be detectable at a distance. This is compatible with classical data [131, 133-135] and will be tested using molecular reagents in the future.
In our previous work [43], we showed that hyperpolarized cells were themselves resistant to transformation [136-138]. To analyze long-range effects of hyperpolarization, we established an assay in which Xenopus 16-cell embryos were injected with oncogene in one blastomere and subjected to a hyperpolarizing treatment, non-locally, via ectopic-expression of hyperpolarization channel or manipulation of ionic contents of the media. Remarkably, the data show that hyperpolarization significantly suppresses the formation of ITLS, regardless of which oncogene was used (Fig. 2B, C), even when the oncogene-bearing cells and the cells that have been hyperpolarized are on opposite ends of the body. Complete suppression of ITLS was not achieved, because our reagents had to be tittered down to avoid perturbing the normal developmental processes of the organism. Future work is needed to refine the strategy for optimal management of the bioelectric crosstalk that goes awry in cancer; it is likely that a more nuanced strategy, which manages spatial relationships at higher resolution, will be required in clinical practice. Another important area for future investigation is slow time-dependent variability of Vmem, as we only explored continuously-acting reagents and it is possible that important patterning information is encoded in the time profile of slowly-changing bioelectrical states [37, 139-142]. Optogenetics – the use of light to regulate ion channels with high spatio-temporal specificity - [143, 144] is a promising technology for both of these directions.
Resting potential changes can result from genomic, transcriptional, or post-translation control of ion channels and pumps. Importantly, we obtained tumor suppression using either Cl- or K+-based hyperpolarization, just as we showed previously for cell-autonomous tumorigenesis and metastatic activation by depolarization [43, 46, 60, 61]. This rules out signaling pathways triggered by specific ion channels or even limited to specific ion types. Thus, in complement to the gene-focused idea of specific ion channels being intrinsic oncogenes or tumor suppressors [44, 48, 145], we suggest that a systems-level physiological property – resting potential – can be a powerful causal factor in regulating these processes.
CLIC1-dependent hyperpolarization: a native target underlying suppression
A large body of literature reveals differential ion channel and transporter expression between tumor cells and their untransformed counterparts [146-150]. These native ion translocators are important not only as markers [52, 145, 151-153] but also as targets for interventions designed to manage the bioelectric state of cancer and surrounding tissue without needing gene therapy with heterologous channels. Exploiting native channels may allow manipulation of bioelectric signals merely by changing ionic composition of the cellular environment; indeed, we observed that hyperpolarization by high Cl- reduces ITLS incidences (Fig. 3B) without additional channel misexpression. The alteration of tumorigenic transformation by ionic conditions in the medium is consistent with previous data showing an association between the (depolarizing) level of sodium and cancer [154-156], the control of cell differentiation by sodium levels [157-159] and pH [160], and more recent work on stem cell reprogramming by acid baths and streptolysin-mediated membrane permeability change [161]. Reprogramming of cell state by physiological cues is certainly of relevance to cancer [162-165]. However, two important novel aspects of our data are the focus on Vmem per se, not specific ions as obligate signals, and the non-local nature of the suppression mechanism.
A pharmacological loss-of-function experiment using chloride channel blockers (Fig. 3A) and a gene-specific dominant negative (Fig. 4) implicated chloride intracellular chloride channel 1 (CLIC1) as a likely candidate for the channel required for hyperpolarization in the long-range ITLS suppression. Roles for CLIC1 in carcinogenesis – albeit cell autonomous – have been shown previously in several studies. A search for ion channels in the cancer profiling database, Oncomine, turns up CLIC1 as one of the most upregulated genes [166], and chloride current associated with increased CLIC1 expression is present in progenitor cells isolated from human glioblastomas, and is responsible for promoting proliferation, clonogenicity, and tumorigenic capacity [167]. Channels like this are of high interest as a therapeutic modality [56], but it should be noted that it is not enough to look for blockers (a standard loss-of-function genetic strategy) – the key is to modulate Vmem, which may mean opening or closing specific channels depending on the cells’ surrounding milieu and its ion gradients. An understanding of Vmem as a regulatory agent may also explain the anti-cancer activity of a number of agents such as ivermectin [57], bafilomycin [168, 169], and salinomycin [170].
In Xenopus, CLIC1 is expressed throughout development, with progressive increase in expression from mid-blastula transition to the tadpole stages; spatially, CLIC1 transcripts are present primarily in the ectoderm and ectodermally-originated organs [94]. Interestingly, CLIC1 expression is missing from the gut region where we frequently observe ITLS on the ectoderm. Thus, CLIC1 proteins are located at a distance from oncogene-induced foci, showing that they can act like the overexpressed potassium channels – in long-range hyperpolarization. By overexpressing wildtype Xenopus CLIC1 and raising extracellular chloride levels, we are able to achieve higher suppression of oncogenic transformation of cells located at a distance. Moreover, this suppression can be blocked – despite the presence of high extracellular chloride levels – by introducing a dominant negative chloride channel mutant away from oncogene-expressing cells (Fig. 4). Interestingly, another study from our lab has documented the endogenous presence of glycine-gated channels: though not present in melanocytes their depolarization confers highly proliferative and metastatic phenotype in melanocytes in a long-range, serotonergic signaling pathway [60, 61].
Transducing hyperpolarization into cell responses: HDAC1 inhibition
How do resting potential changes such as hyperpolarization impact on the transcriptional and epigenetic pathways of cancer? Recent studies have identified several mechanisms that transduce bioelectric signals at the cell membrane into biochemical responses (reviewed in [63, 104, 171]. Using a pharmacological suppression and molecular loss of function (Fig. 5A, B), we identified possible roles for gap junction communication (to be reported in a forthcoming study) and HDAC1 inhibition as signaling elements required for the long-range ITLS suppression. The introduction of a dominant negative HDAC1 mRNA [102] in a long-range manner was able to significantly increase neoplastic conversions of oncogene-expressing distant cells. The results are surprising relative to the well-known function of HDAC inhibitors as having antitumor activities, including reduced proliferation [172], increased differentiation/apoptosis [173], and that a number of them are on the clinical development pipeline for anti-cancer therapeutics [174-177]. In fact, variants of well-known HDAC inhibitors, such as vorinostat, (Merck) and romidepsin (Celgene) have already been developed and approved by the Food and Drug Administration for treating cutaneous T-cell lymphoma [178, 179]. However, consistent with our data, HDAC1 knock-down promotes early tumorigenesis in oncogene expressing cells [180], and genomic instability in a given cell can have a system-wide effect mediated by the immune system and other factors [181, 182]. Overall, this is consistent with our proposal that what is disrupted in cancer is a specific and highly modulated pattern of resting potentials - it is likely that the desired signaling cannot be achieved by universally increasing “anti-cancer” gene products. Depending on other factors that vary widely in different systems (such as ion concentrations, butyrate availability, and spatial arrangement of tumor and tissue), disbalance of voltage-dependent signaling in either direction could promote or suppress tumorigenesis.
We sought to understand how Vmem changes and acetylation state of chromatins could control tumor growth in a non-cell autonomous manner. Given the link between Vmem-guided butyrate transport and HDAC activities the control cell behavior during regeneration [62] and local tumorigenesis [43], we tested the effect of targeting butyrate-producing bacteria on ITLS formation. Both novobiocin, an antibiotic against gram-positive bacteria, and a cocktail of antibiotics (gentamicin, metrodinazole, vancomycin, and clindamycin, data not shown) that have been verified to reduce levels of butyrate, significantly increased the rate of ITLS formation (Fig. 6). Our data are consistent with, and provide a novel mechanism explaining, several studies that investigated the effects of antibiotics on the gut microbiota, including butyrate-producing bacteria species, and their subsequent implications in increased cancer risks and incidences [183-186].
Our analysis was limited in two main ways: the lack of a technology for monitoring location of butyrate in vivo, and the inability to isolate and culture butyrate producing bacteria (which are highly anaerobic and do not grow under standard culture conditions [187]). Future work using GFP-expressing strains of appropriately engineered bacterial model species will further refine our understanding of the role of native microbiota on cancer progression and bioelectric signaling.
Collectively, the data presented here suggest a possible model (Fig. 7). Pharmacologically/molecular-genetically hyperpolarized cells have membrane that is negative on the inside, which creates a positively-charged cell surface environment. While bacteria – whether gram-positive or gram-negative – readily attach to a positively-charged surface, their viability is greatly diminished due to strong electrostatic interaction [188, 189]. As a result, distribution of butyrate producing-bacteria is skewed away from hyperpolarized cells in favor of oncogene-expressing cells, which are known to be depolarized pre-neoplastic transformation [43]. Availability of butyrate to these prospective tumor sites leads to HDAC inhibition and increased hyperacetylation of histones, which promote cell cycle arrest, leading to reduced rates of proliferation, apoptosis, and differentiation of cancer cells lines [105-107].
While the spatial dynamics of butyrate and bacteria remain to be investigated, our data reveal a novel long-range pathway regulating oncogene-mediated tumorigenesis at a distance. We do not yet know the maximum extent, in a large organism, of such long-range signaling by hyperpolarization. Future work must also examine the roles of remote endogenous bioelectric states in mammalian cancer models; if conserved, these data suggest a number of detection and treatment modalities focused on Vmem modulation. Because such strategies could be designed using ion channel drugs [190, 191] already approved for human use (as anti-epileptic agents), exciting opportunities for biomedicine may be presented by investigations of bioelectric signaling in cancer at a level beyond that of single cells.
MATERIALS AND METHODS
Animal Husbandry
Xenopus laevis embryos were collected and fertilized in vitro according to standard protocols [192], in 0.1X Modified Marc’s Ringers (MMR; pH 7.8) with 0.1% Gentamicin. Xenopus embryos were housed at 14-18°C and staged according to Nieuwkoop and Faber [193]. All experimental procedures involving the use of animals for experimental purposes were approved by the Institutional Animal Care and Use Committees (IACUC) and Tufts University Department of Lab Animal Medicine (DLAM) under the protocol number M2011-70.
Microinjection
Fertilized Xenopus embryos were transferred into mesh-bottomed dishes with 3% Ficoll and injected with capped, synthetic mRNAs (made using the Ambion Message Machine kit) dissolved in water at the stages indicated. 2 hours post injection, embryos were transferred into 0.75x MMR for 45 minutes before they were washed and cultured in 0.1X MMR until desired stage was reached. Constructs used included: FUCCI (fluorescent ubiquitination-based cell cycle indicator) pair: mKO2-Cdt1 and mAG-Geminin [71, 72]; Gli1 [68], Xrel3 [194], KRASG12D [69], and Kv1.5 [86]; CLIC1-2A-GFP3 and CLIC1C24S-tdTomato; DN-HDAC1 [102].
Drug treatments
Embryos were exposed in 0.1× MMR for the stages indicated to: Anthracene-9-Carboxylic Acid (ACA) 67 μM; indanyloxyacetic acid (IAA-94), 55 μM; 5-Nitro-2-(3-phenylpropylamino) benzoic acid (NPPB, Tocaris Bioscience, Bristol, UK), 8.5 nM; nalidixic acid 43 μM; novobiocin sodium salt 78 μM; gentamicin, 20 μM; metrodinazole, 20 μM; vancomycin hydrochloride (Santacruz Biotechnology, Texas, USA) 20 μM; Choline chloride, 70mM and clindamycin hydrochloride, 20 μM. All compounds were obtained from Sigma-Aldrich, St. Louis, MO unless otherwise noted.
Immunohistochemistry
Spatial analysis of leukocyte presence was performed by immunohistochemistry in paraffin sections, using an anti-XL2 antibody [195]. Briefly, embryos were fixed overnight in MEMFA [192], embedded in paraffin and sectioned at 5 μm using a Leica microtome. After deparaffinizing and rehydrating, tissue sections were permeabilized in phosphate buffered saline (PBS) + 0.1X Triton X-100 for 30 minutes, blocked with 10% goat serum in PBS + 0.1% tween-20 for 1 hour, and incubated at 40C overnight with anti-XL2 primary antibody. Sections were then washed six times with PBST (30 minutes each at room temperature) and incubated with Alexa-Fluor-555-conjugated secondary antibody at 1:1000 in PBST + 10% goat serum overnight at 40C. After five 30-minute washes in PBST, sections were mounted on a slide and photographed using the TRITC filter set on an Olympus BX61 spinning-disk confocal microscope with Hamamatsu ORCA digital CCD camera.
Hypoxia detection
Immunochemical detection of tissue hypoxia was performed by immunoperoxidase reaction in paraffin sections, using the HypoxyTM-1 Plus Kit (Hypoxyprobe, Inc, MA, USA). St.34 embryos with KRASG12D ITLS were incubated in 300 µM of pimonidazol HCl (HypoxyprobeTM-1). Paraffin sections were prepared for immunostaining as described above. Tissue sections were then incubated using 1:50 dilution of FITC-MAB1, which binds to protein adducts of pimonidazole in hypoxic regions, and counterstained with 1:50 dilution of horseradish peroxidase conjugated anti-FITC secondary.
pH measurements
pH measurements were calculated from BCECF fluorescent signals as described previously in [196]. Briefly, embryos were incubated in a 5 µM BCECF, AM (2’,7’-Bis-(2-Carboxyethyl)-5-(and-6)-Carboxyfluorescein, Acetoxymethyl Ester (Life Technologies, NY, USA ) solution. Excess dye was washed out, and embryos were anesthetized with MS222. To image BCECF, a dual excitation dye, filters were EX 450/20, D 460, EM 535/30 (the isobestic point) and EX 500/20, D 515, EM 535/30. To calibrate BCECF in our system, a prototonophore, CCCP, and NaOH were used to drive pH values to 4 (limiting minimum) and 9 (limiting maximum), respectively, by observing gradual decrease and increase of fluorescence. A plot profile of pixel intensities was then taken from ITLS and control regions, and conversions to pH values were made as described in [196]
Nuclear morphometry
Sections taken through ITLS and control regions were stained with Hoechst Blue (Life Technologies, NY, USA), imaged using the DAPI filter set on an Olympus BX61 spinning-disk confocal microscope with Hamamatsu ORCA digital CCD camera, and analyzed by using ImageJ. The threshold and analyze particle tools in ImageJ were used to interactively outline each nucleus and determine its size and shape.
Statistical analysis
All statistical analyses were performed using GraphPad InStat v. 3.10 (GraphPad Software, La Jolla, CA, USA). Data were expressed as the mean unless otherwise noted. Error bars represent standard error. The differences between treatment groups were analyzed using Student’s t-test, Chi-squared test or One-way ANOVA, (tukey’s post hoc comparisons), and the null hypothesis was rejected at the 0.05 level.
ACKNOWLEDGEMENTS
This paper is dedicated to Bjorn Nordenstrom, a pioneer of long-range bioelectrical signaling in cancer. We thank Leonard Zon for the KRAS mutant construct, Florian Lang for the Kv1.5 construct, Ariel Ruiz Altaba for the Gli1 construct, John Wallingford for the Xrel3 construct, Atsushi Miyawaki for the FUCCI plasmid pair (mKO2-zCdt1 and mAG-zGeminin), and Saburo Nagata for the XL-2 antibody. We thank Jeff Wolf for H&E analysis of ITLS sections. We thank Amber Currier, Erin Switzer, and Amanda Allen for general lab assistance and frog husbandry, and Joan Lemire and Jean-Francois Pare for molecular biology assistance. We are grateful to Douglas Brash, Robert Gillies, and the members of the Levin lab for many useful discussions on these topics. M.L. is grateful for support of the G. Harold and Leila Y. Mathers Charitable Foundation, NIH R01-AR055993, and of DARPA (subaward W911NF-09-1-0125).
CONFLICT OF INTEREST
The authors declare that there are no competing financial interests of the work described.
References
1. Soto AM and Sonnenschein C. The somatic mutation theory of cancer: growing problems with the paradigm? Bioessays. 2004; 26(10): p. 1097-107.
2. Rowlatt C. Some consequences of defining the neoplasm as focal self-perpetuating tissue disorganization. in New Frontiers in Cancer Causation. O.H. Iversen, Editor. 1994; Taylor & Francis: Washington, DC. p. 45–58.
3. Waddington CH. Cancer and the theory of organisers. Nature. 1935; 135: p. 606-608.
4. Clark WH, Jr. The nature of cancer: morphogenesis and progressive (self)-disorganization in neoplastic development and progression. Acta Oncol. 1995; 34(1): p. 3-21.
5. Rubin H. Cancer as a dynamic developmental disorder. Cancer Res. 1985; 45(7): p. 2935-42.
6. Levin M. Morphogenetic fields in embryogenesis, regeneration, and cancer: non-local control of complex patterning. Bio Systems. 2012; 109(3): p. 243-61.
7. Capp JP. Stochastic gene expression, disruption of tissue averaging effects and cancer as a disease of development. Bioessays. 2005; 27(12): p. 1277-85.
8. Potter JD. Morphostats: a missing concept in cancer biology. Cancer Epidemiol Biomarkers Prev. 2001; 10(3): p. 161-70.
9. Potter JD. Morphogens, morphostats, microarchitecture and malignancy. Nat Rev Cancer. 2007; 7(6): p. 464-74.
10. Soto AM and Sonnenschein C. The tissue organization field theory of cancer: a testable replacement for the somatic mutation theory. Bioessays. 2011; 33(5): p. 332-40.
11. Soto AM and Sonnenschein C. Emergentism as a default: cancer as a problem of tissue organization. J Biosci. 2005; 30(1): p. 103-18.
12. Tarin D. Clinical and Biological Implications of the Tumor Microenvironment. Cancer microenvironment : official journal of the International Cancer Microenvironment Society. 2012.
13. Ingber DE. Can cancer be reversed by engineering the tumor microenvironment? Semin Cancer Biol. 2008; 18(5): p. 356-64.
14. Bissell MJ and Labarge MA. Context, tissue plasticity, and cancer: are tumor stem cells also regulated by the microenvironment? Cancer Cell. 2005; 7(1): p. 17-23.
15. Mintz B and Illmensee K. Normal genetically mosaic mice produced from malignant teratocarcinoma cells. Proc Natl Acad Sci U S A. 1975; 72(9): p. 3585-9.
16. Illmensee K and Mintz B. Totipotency and normal differentiation of single teratocarcinoma cells cloned by injection into blastocysts. Proc Natl Acad Sci U S A. 1976; 73(2): p. 549-53.
17. Astigiano S, Damonte P, Fossati S, Boni L, and Barbieri O. Fate of embryonal carcinoma cells injected into postimplantation mouse embryos. Differentiation. 2005; 73(9-10): p. 484-90.
18. Webb CG, Gootwine E, and Sachs L. Developmental potential of myeloid leukemia cells injected into midgestation embryos. Dev Biol. 1984; 101(1): p. 221-4.
19. Li L, Connelly MC, Wetmore C, Curran T, and Morgan JI. Mouse embryos cloned from brain tumors. Cancer Res. 2003; 63(11): p. 2733-6.
20. Pierce GB and Wells RS. The blastocyst in control of embryonal carcinoma. Prog Clin Biol Res. 1982; 85 Pt B: p. 593-600.
21. Pierce GB, Arechaga J, and Wells RS. Embryonic control of cancer. Prog Clin Biol Res. 1986; 226: p. 67-77.
22. Podesta AH, Mullins J, Pierce GB, and Wells RS. The neurula stage mouse embryo in control of neuroblastoma. Proc Natl Acad Sci U S A. 1984; 81(23): p. 7608-11.
23. Pierce GB, Aguilar D, Hood G, and Wells RS. Trophectoderm in control of murine embryonal carcinoma. Cancer Res. 1984; 44(9): p. 3987-96.
24. Pierce GB. The cancer cell and its control by the embryo. Rous-Whipple Award lecture. Am J Pathol. 1983; 113(1): p. 117-24.
25. Pierce GB, Pantazis CG, Caldwell JE, and Wells RS. Specificity of the control of tumor formation by the blastocyst. Cancer Res. 1982; 42(3): p. 1082-7.
26. Haldi M, Ton C, Seng WL, and McGrath P. Human melanoma cells transplanted into zebrafish proliferate, migrate, produce melanin, form masses and stimulate angiogenesis in zebrafish. Angiogenesis. 2006; 9(3): p. 139-51.
27. Lee LM, Seftor EA, Bonde G, Cornell RA, and Hendrix MJ. The fate of human malignant melanoma cells transplanted into zebrafish embryos: assessment of migration and cell division in the absence of tumor formation. Dev Dyn. 2005; 233(4): p. 1560-70.
28. Kasemeier-Kulesa JC, Teddy JM, Postovit LM, Seftor EA, Seftor RE, Hendrix MJ, and Kulesa PM. Reprogramming multipotent tumor cells with the embryonic neural crest microenvironment. Dev Dyn. 2008; 237(10): p. 2657-66.
29. Hendrix MJ, Seftor EA, Seftor RE, Kasemeier-Kulesa J, Kulesa PM, and Postovit LM. Reprogramming metastatic tumour cells with embryonic microenvironments. Nat Rev Cancer. 2007; 7(4): p. 246-55.
30. Kulesa PM, Kasemeier-Kulesa JC, Teddy JM, Margaryan NV, Seftor EA, Seftor RE, and Hendrix MJ. Reprogramming metastatic melanoma cells to assume a neural crest cell-like phenotype in an embryonic microenvironment. Proc Natl Acad Sci U S A. 2006; 103(10): p. 3752-7.
31. Coleman WB, Wennerberg AE, Smith GJ, and Grisham JW. Regulation of the differentiation of diploid and some aneuploid rat liver epithelial (stemlike) cells by the hepatic microenvironment. Am J Pathol. 1993; 142(5): p. 1373-82.
32. Tseng A and Levin M. Cracking the bioelectric code: Probing endogenous ionic controls of pattern formation. Communicative & Integrative Biology. 2013; 6(1): p. 1-8.
33. Levin M. Reprogramming cells and tissue patterning via bioelectrical pathways: molecular mechanisms and biomedical opportunities. Wiley Interdisciplinary Reviews: Systems Biology and Medicine. 2013; 5(6): p. 657-676.
34. McCaig CD, Rajnicek AM, Song B, and Zhao M. Controlling cell behavior electrically: current views and future potential. Physiol Rev. 2005; 85(3): p. 943-78.
35. Levin M. Bioelectric mechanisms in regeneration: Unique aspects and future perspectives. Semin Cell Dev Biol. 2009; 20(5): p. 543-56.
36. McCaig CD, Song B, and Rajnicek AM. Electrical dimensions in cell science. J Cell Sci. 2009; 122(Pt 23): p. 4267-76.
37. Blackiston DJ, McLaughlin KA, and Levin M. Bioelectric controls of cell proliferation: ion channels, membrane voltage and the cell cycle. Cell Cycle. 2009; 8(21): p. 3519-28.
38. Sundelacruz S, Levin M, and Kaplan DL. Role of membrane potential in the regulation of cell proliferation and differentiation. Stem Cell Rev Rep. 2009; 5(3): p. 231-46.
39. Yang M and Brackenbury WJ. Membrane potential and cancer progression. Frontiers in physiology. 2013; 4: p. 185.
40. Fricke H and Morse S. The Electric Capacity of Tumors of the Breast. The Journal of Cancer Research. 1926; 10(3): p. 340-376.
41. Binggeli R and Cameron IL. Cellular potentials of normal and cancerous fibroblasts and hepatocytes. Cancer Research. 1980; 40(6): p. 1830-5.
42. Morimoto T, Kimura S, Konishi Y, Komaki K, Uyama T, Monden Y, Kinouchi Y, and Iritani T. A study of the electrical bio-impedance of tumors. J Invest Surg. 1993; 6(1): p. 25-32.
43. Chernet BT and Levin M. Transmembrane voltage potential is an essential cellular parameter for the detection and control of tumor development in a Xenopus model. Dis Model Mech. 2013; 6(3): p. 595-607.
44. Than BL, Goos JA, Sarver AL, O’Sullivan MG, Rod A, Starr TK, Fijneman RJ, Meijer GA, Zhao L, Zhang Y, Largaespada DA, Scott PM, and Cormier RT. The role of KCNQ1 in mouse and human gastrointestinal cancers. Oncogene. 2013.
45. Roepke TK, Purtell K, King EC, La Perle KM, Lerner DJ, and Abbott GW. Targeted deletion of Kcne2 causes gastritis cystica profunda and gastric neoplasia. PLoS One. 2010; 5(7): p. e11451.
46. Morokuma J, Blackiston D, Adams DS, Seebohm G, Trimmer B, and Levin M. Modulation of potassium channel function confers a hyperproliferative invasive phenotype on embryonic stem cells. Proc Natl Acad Sci U S A. 2008; 105(43): p. 16608-13.
47. Lallet-Daher H, Wiel C, Gitenay D, Navaratnam N, Augert A, Le Calve B, Verbeke S, Carling D, Aubert S, Vindrieux D, and Bernard D. Potassium channel KCNA1 modulates oncogene-induced senescence and transformation. Cancer research. 2013; 73(16): p. 5253-65.
48. House CD, Vaske CJ, Schwartz AM, Obias V, Frank B, Luu T, Sarvazyan N, Irby R, Strausberg RL, Hales TG, Stuart JM, and Lee NH. Voltage-gated Na+ channel SCN5A is a key regulator of a gene transcriptional network that controls colon cancer invasion. Cancer Res. 2010; 70(17): p. 6957-67.
49. Weksberg R, Shuman C, Caluseriu O, Smith AC, Fei YL, Nishikawa J, Stockley TL, Best L, Chitayat D, Olney A, Ives E, Schneider A, Bestor TH, Li M, Sadowski P, and Squire J. Discordant KCNQ1OT1 imprinting in sets of monozygotic twins discordant for Beckwith-Wiedemann syndrome. Human Molecular Genetics. 2002; 11(11): p. 1317-25.
50. Pei L, Wiser O, Slavin A, Mu D, Powers S, Jan LY, and Hoey T. Oncogenic potential of TASK3 (Kcnk9) depends on K+ channel function. Proc Natl Acad Sci U S A. 2003; 100(13): p. 7803-7.
51. Pardo LA, del Camino D, Sanchez A, Alves F, Bruggemann A, Beckh S, and Stuhmer W. Oncogenic potential of EAG K(+) channels. Embo J. 1999; 18(20): p. 5540-7.
52. Onkal R and Djamgoz MB. Molecular pharmacology of voltage-gated sodium channel expression in metastatic disease: Clinical potential of neonatal Nav1.5 in breast cancer. Eur J Pharmacol. 2009.
53. Teh JL and Chen S. Glutamatergic signaling in cellular transformation. Pigment Cell & Melanoma Research. 2012; 25(3): p. 331-42.
54. Saito T, Schlegel R, Andresson T, Yuge L, Yamamoto M, and Yamasaki H. Induction of cell transformation by mutated 16K vacuolar H+-atpase (ductin) is accompanied by down-regulation of gap junctional intercellular communication and translocation of connexin 43 in NIH3T3 cells. Oncogene. 1998; 17(13): p. 1673-80.
55. Frede J, Fraser SP, Oskay-Ozcelik G, Hong Y, Ioana Braicu E, Sehouli J, Gabra H, and Djamgoz MB. Ovarian cancer: Ion channel and aquaporin expression as novel targets of clinical potential. European Journal of Cancer. 2013; 49(10): p. 2331-44.
56. Arcangeli A, Pillozzi S, and Becchetti A. Targeting ion channels in leukemias: a new challenge for treatment. Current Medicinal Chemistry. 2012; 19(5): p. 683-96.
57. Sharmeen S, Skrtic M, Sukhai MA, Hurren R, Gronda M, Wang X, Fonseca SB, Sun H, Wood TE, Ward R, Minden MD, Batey RA, Datti A, Wrana J, Kelley SO, and Schimmer AD. The antiparasitic agent ivermectin induces chloride-dependent membrane hyperpolarization and cell death in leukemia cells. Blood. 2010; 116(18): p. 3593-603.
58. Arcangeli A, Crociani O, Lastraioli E, Masi A, Pillozzi S, and Becchetti A. Targeting ion channels in cancer: a novel frontier in antineoplastic therapy. Current medicinal chemistry. 2009; 16(1): p. 66-93.
59. Chernet B and Levin M. Bioelectric signaling in cancer. Journal of Experimental and Clinical Oncology. 2014; in press.
60. Blackiston D, Adams DS, Lemire JM, Lobikin M, and Levin M. Transmembrane potential of GlyCl-expressing instructor cells induces a neoplastic-like conversion of melanocytes via a serotonergic pathway. Dis Model Mech. 2011; 4(1): p. 67-85.
61. Lobikin M, Chernet B, Lobo D, and Levin M. Resting potential, oncogene-induced tumorigenesis, and metastasis: the bioelectric basis of cancer in vivo. Phys Biol. 2012; 9(6): p. 065002.
62. Tseng AS and Levin M. Transducing bioelectric signals into epigenetic pathways during tadpole tail regeneration. Anatomical Record. 2012; 295(10): p. 1541-51.
63. Adams DS and Levin M. Endogenous voltage gradients as mediators of cell-cell communication: strategies for investigating bioelectrical signals during pattern formation. Cell Tissue Res. 2013; 352(1): p. 95-122.
64. Nutt LK. The Xenopus oocyte: a model for studying the metabolic regulation of cancer cell death. Semin Cell Dev Biol. 2012; 23(4): p. 412-8.
65. Nedelkovska H and Robert J. Hsp72 mediates stronger antigen-dependent non-classical MHC class Ib anti-tumor responses than hsc73 in Xenopus laevis. Cancer Immun. 2013; 13: p. 4.
66. Liu X, Ji XM, Du XN, Zong XC, Liang DF, Ma L, Wu HT, and Zhang SQ. Molecular cloning, expression, bioinformatics analysis, and bioactivity of TNFSF13 (APRIL) in the South African clawed frog (Xenopus laevis): a new model to study immunological diseases. OMICS. 2013; 17(7): p. 384-92.
67. Wallingford JB, Seufert DW, Virta VC, and Vize PD. p53 activity is essential for normal development in Xenopus. Curr Biol. 1997; 7(10): p. 747-57.
68. Dahmane N, Lee J, Robins P, Heller P, and Ruiz i Altaba A. Activation of the transcription factor Gli1 and the Sonic hedgehog signalling pathway in skin tumours. Nature. 1997; 389(6653): p. 876-81.
69. Le X, Langenau DM, Keefe MD, Kutok JL, Neuberg DS, and Zon LI. Heat shock-inducible Cre/Lox approaches to induce diverse types of tumors and hyperplasia in transgenic zebrafish. Proc Natl Acad Sci U S A. 2007; 104(22): p. 9410-5.
70. Bast RC and American Cancer Society. Cancer medicine 5. 2000, Hamilton, Ont. ; London: B.C. Decker. xxix, 2546 p., 48 p. of plates.
71. Sugiyama M, Sakaue-Sawano A, Iimura T, Fukami K, Kitaguchi T, Kawakami K, Okamoto H, Higashijima S, and Miyawaki A. Illuminating cell-cycle progression in the developing zebrafish embryo. Proc Natl Acad Sci U S A. 2009; 106(49): p. 20812-7.
72. Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, Imamura T, Ogawa M, Masai H, and Miyawaki A. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 2008; 132(3): p. 487-98.
73. Hockel M and Vaupel P. Biological consequences of tumor hypoxia. Semin Oncol. 2001; 28(2 Suppl 8): p. 36-41.
74. Vaupel P, Kallinowski F, and Okunieff P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res. 1989; 49(23): p. 6449-65.
75. Ryan HE, Poloni M, McNulty W, Elson D, Gassmann M, Arbeit JM, and Johnson RS. Hypoxia-inducible factor-1alpha is a positive factor in solid tumor growth. Cancer Res. 2000; 60(15): p. 4010-5.
76. Varia MA, Calkins-Adams DP, Rinker LH, Kennedy AS, Novotny DB, Fowler WC, Jr., and Raleigh JA. Pimonidazole: a novel hypoxia marker for complementary study of tumor hypoxia and cell proliferation in cervical carcinoma. Gynecol Oncol. 1998; 71(2): p. 270-7.
77. Kaanders JH, Wijffels KI, Marres HA, Ljungkvist AS, Pop LA, van den Hoogen FJ, de Wilde PC, Bussink J, Raleigh JA, and van der Kogel AJ. Pimonidazole binding and tumor vascularity predict for treatment outcome in head and neck cancer. Cancer Res. 2002; 62(23): p. 7066-74.
78. McPhail LD and Robinson SP. Intrinsic susceptibility MR imaging of chemically induced rat mammary tumors: relationship to histologic assessment of hypoxia and fibrosis. Radiology. 2010; 254(1): p. 110-8.
79. Ross MH and Pawlina W. Histology : a text and atlas : with correlated cell and molecular biology. 6th ed. 2010, Philadelphia, Pa. ; London: Lippincott Williams & Wilkins.
80. Pienta KJ and Coffey DS. Correlation of nuclear morphometry with progression of breast cancer. Cancer. 1991; 68(9): p. 2012-6.
81. Baak JP, Van Dop H, Kurver PH, and Hermans J. The value of morphometry to classic prognosticators in breast cancer. Cancer. 1985; 56(2): p. 374-82.
82. Abdalla F, Boder J, Markus R, Hashmi H, Buhmeida A, and Collan Y. Correlation of nuclear morphometry of breast cancer in histological sections with clinicopathological features and prognosis. Anticancer Res. 2009; 29(5): p. 1771-6.
83. De Milito A and Fais S. Tumor acidity, chemoresistance and proton pump inhibitors. Future Oncol. 2005; 1(6): p. 779-86.
84. Tannock IF and Rotin D. Acid pH in tumors and its potential for therapeutic exploitation. Cancer Res. 1989; 49(16): p. 4373-84.
85. Entschladen F, Lang K, Drell TL, Joseph J, and Zaenker KS. Neurotransmitters are regulators for the migration of tumor cells and leukocytes. Cancer Immunol Immunother. 2002; 51(9): p. 467-82.
86. Strutz-Seebohm N, Gutcher I, Decher N, Steinmeyer K, Lang F, and Seebohm G. Comparison of potent Kv1.5 potassium channel inhibitors reveals the molecular basis for blocking kinetics and binding mode. Cell Physiol Biochem. 2007; 20(6): p. 791-800.
87. Pai VP, Aw S, Shomrat T, Lemire JM, and Levin M. Transmembrane voltage potential controls embryonic eye patterning in Xenopus laevis. Development. 2011.
88. Adams DS and Levin M. Inverse drug screens: a rapid and inexpensive method for implicating molecular targets. Genesis. 2006; 44(11): p. 530-40.
89. Landry DW, Reitman M, Cragoe EJ, Jr., and Al-Awqati Q. Epithelial chloride channel. Development of inhibitory ligands. J Gen Physiol. 1987; 90(6): p. 779-98.
90. Welsh MJ. Anthracene-9-carboxylic acid inhibits an apical membrane chloride conductance in canine tracheal epithelium. J Membr Biol. 1984; 78(1): p. 61-71.
91. Pope AJ, Richardson SK, Ife RJ, and Keeling DJ. Characterisation of the effects of anthranilic and (indanyloxy) acetic acid derivatives on chloride transport in membrane vesicles. Biochim Biophys Acta. 1991; 1067(1): p. 51-63.
92. Keeling DJ, Taylor AG, and Smith PL. Effects of NPPB (5-nitro-2-(3-phenylpropylamino)benzoic acid) on chloride transport in intestinal tissues and the T84 cell line. Biochim Biophys Acta. 1991; 1115(1): p. 42-8.
93. Li X and Weinman SA. Chloride channels and hepatocellular function: prospects for molecular identification. Annu Rev Physiol. 2002; 64: p. 609-33.
94. Shorning BY, Wilson DB, Meehan RR, and Ashley RH. Molecular cloning and developmental expression of two Chloride Intracellular Channel (CLIC) genes in Xenopus laevis. Dev Genes Evol. 2003; 213(10): p. 514-8.
95. Singh H and Ashley RH. Redox regulation of CLIC1 by cysteine residues associated with the putative channel pore. Biophys J. 2006; 90(5): p. 1628-38.
96. Levin M and Stevenson CG. Regulation of cell behavior and tissue patterning by bioelectrical signals: challenges and opportunities for biomedical engineering. Annu Rev Biomed Eng. 2012; 14: p. 295-323.
97. Fukumoto T, Blakely R, and Levin M. Serotonin transporter function is an early step in left-right patterning in chick and frog embryos. Dev Neurosci. 2005; 27(6): p. 349-63.
98. Fukumoto T, Kema IP, and Levin M. Serotonin signaling is a very early step in patterning of the left-right axis in chick and frog embryos. Curr Biol. 2005; 15(9): p. 794-803.
99. Adams DS, Robinson KR, Fukumoto T, Yuan S, Albertson RC, Yelick P, Kuo L, McSweeney M, and Levin M. Early, H+-V-ATPase-dependent proton flux is necessary for consistent left-right patterning of non-mammalian vertebrates. Development. 2006; 133: p. 1657-1671.
100. Munaron L, Antoniotti S, Fiorio Pla A, and Lovisolo D. Blocking Ca2+ entry: a way to control cell proliferation. Curr Med Chem. 2004; 11(12): p. 1533-43.
101. Munaron L, Antoniotti S, and Lovisolo D. Intracellular calcium signals and control of cell proliferation: how many mechanisms? J Cell Mol Med. 2004; 8(2): p. 161-8.
102. Carneiro K, Donnet C, Rejtar T, Karger BL, Barisone GA, Diaz E, Kortagere S, Lemire JM, and Levin M. Histone deacetylase activity is necessary for left-right patterning during vertebrate development. BMC Dev Biol. 2011; 11: p. 29.
103. Tseng AS, Carneiro K, Lemire JM, and Levin M. HDAC activity is required during Xenopus tail regeneration. PLoS ONE. 2011; 6(10): p. e26382.
104. Levin M. Reprogramming cells and tissue patterning via bioelectrical pathways: molecular mechanisms and biomedical opportunities. Wiley Interdiscip Rev Syst Biol Med. 2013.
105. Davie JR. Inhibition of histone deacetylase activity by butyrate. J Nutr. 2003; 133(7 Suppl): p. 2485S-2493S.
106. Orchel A, Dzierzewicz Z, Parfiniewicz B, Weglarz L, and Wilczok T. Butyrate-induced differentiation of colon cancer cells is PKC and JNK dependent. Dig Dis Sci. 2005; 50(3): p. 490-8.
107. Hague A, Elder DJ, Hicks DJ, and Paraskeva C. Apoptosis in colorectal tumour cells: induction by the short chain fatty acids butyrate, propionate and acetate and by the bile salt deoxycholate. Int J Cancer. 1995; 60(3): p. 400-6.
108. Rutenburg AM, Shapiro P, and Schweinburg F. Novobiocin in the treatment of surgical infections due to staphylococci and other gram-positive bacteria. N Engl J Med. 1956; 255(7): p. 325-30.
109. Gellert M, O’Dea MH, Itoh T, and Tomizawa J. Novobiocin and coumermycin inhibit DNA supercoiling catalyzed by DNA gyrase. Proc Natl Acad Sci U S A. 1976; 73(12): p. 4474-8.
110. Burlison JA, Neckers L, Smith AB, Maxwell A, and Blagg BS. Novobiocin: redesigning a DNA gyrase inhibitor for selective inhibition of hsp90. J Am Chem Soc. 2006; 128(48): p. 15529-36.
111. Shen LL and Pernet AG. Mechanism of inhibition of DNA gyrase by analogues of nalidixic acid: the target of the drugs is DNA. Proc Natl Acad Sci U S A. 1985; 82(2): p. 307-11.
112. Kunzelmann K. Ion channels and cancer. J Membr Biol. 2005; 205(3): p. 159-73.
113. Becchetti A. Ion channels and transporters in cancer. 1. Ion channels and cell proliferation in cancer. Am J Physiol Cell Physiol. 2011; 301(2): p. C255-65.
114. Borys P. On the biophysics of cathodal galvanotaxis in rat prostate cancer cells: Poisson-Nernst-Planck equation approach. Eur Biophys J. 2012; 41(6): p. 527-34.
115. Tsai HF, Huang CW, Chang HF, Chen JJ, Lee CH, and Cheng JY. Evaluation of EGFR and RTK signaling in the electrotaxis of lung adenocarcinoma cells under direct-current electric field stimulation. PLoS ONE. 2013; 8(8): p. e73418.
116. Stock C, Ludwig FT, Hanley PJ, and Schwab A. Roles of Ion Transport in Control of Cell Motility. Comprehensive Physiology. 2013; 3(1): p. 59-119.
117. Sun YS, Peng SW, Lin KH, and Cheng JY. Electrotaxis of lung cancer cells in ordered three-dimensional scaffolds. Biomicrofluidics. 2012; 6(1): p. 14102-1410214.
118. Huang CW, Chen HY, Yen MH, Chen JJ, Young TH, and Cheng JY. Gene expression of human lung cancer cell line CL1-5 in response to a direct current electric field. PLoS ONE. 2011; 6(10): p. e25928.
119. Yan X, Han J, Zhang Z, Wang J, Cheng Q, Gao K, Ni Y, and Wang Y. Lung cancer A549 cells migrate directionally in DC electric fields with polarized and activated EGFRs. Bioelectromagnetics. 2009; 30(1): p. 29-35.
120. Pillozzi S, Brizzi MF, Balzi M, Crociani O, Cherubini A, Guasti L, Bartolozzi B, Becchetti A, Wanke E, Bernabei PA, Olivotto M, Pegoraro L, and Arcangeli A. HERG potassium channels are constitutively expressed in primary human acute myeloid leukemias and regulate cell proliferation of normal and leukemic hemopoietic progenitors. Leukemia. 2002; 16(9): p. 1791-8.
121. Kamleiter M, Hanemann CO, Kluwe L, Rosenbaum C, Wosch S, Mautner VF, Muller HW, and Grafe P. Voltage-dependent membrane currents of cultured human neurofibromatosis type 2 Schwann cells. GLIA. 1998; 24(3): p. 313-22.
122. Knutson P, Ghiani CA, Zhou JM, Gallo V, and McBain CJ. K+ channel expression and cell proliferation are regulated by intracellular sodium and membrane depolarization in oligodendrocyte progenitor cells. Journal of Neuroscience. 1997; 17(8): p. 2669-82.
123. Becchetti A. Ion channels and transporters in cancer. 1. Ion channels and cell proliferation in cancer. American journal of physiology. Cell physiology. 2011; 301(2): p. C255-65.
124. Arcangeli A. Ion channels and transporters in cancer. 3. Ion channels in the tumor cell-microenvironment cross talk. American journal of physiology. Cell physiology. 2011; 301(4): p. C762-71.
125. Prevarskaya N, Skryma R, and Shuba Y. Ion channels and the hallmarks of cancer. Trends Mol Med. 2010; 16(3): p. 107-21.
126. Arcangeli A, Crociani O, Lastraioli E, Masi A, Pillozzi S, and Becchetti A. Targeting ion channels in cancer: a novel frontier in antineoplastic therapy. Curr Med Chem. 2009; 16(1): p. 66-93.
127. Pullar CE. The physiology of bioelectricity in development, tissue regeneration, and cancer. Biological effects of electromagnetics series. 2011, Boca Raton: CRC Press.
128. Tsonis PA. Embryogenesis and carcinogenesis: order and disorder. Anticancer Res. 1987; 7(4A): p. 617-23.
129. Pierce GB and Speers WC. Tumors as caricatures of the process of tissue renewal: prospects for therapy by directing differentiation. Cancer Res. 1988; 48(8): p. 1996-2004.
130. Kopelovich L, Henson DE, Gazdar AF, Dunn B, Srivastava S, Kelloff GJ, and Greenwald P. Surrogate anatomic/functional sites for evaluating cancer risk: an extension of the field effect. Clin Cancer Res. 1999; 5(12): p. 3899-905.
131. Burr HS, Strong LC, and Smith GM. Bioelectric correlates of methylcolantherene-induced tumors in mice. Yale J Biol. Med. 1938; 10: p. 539–544.
132. Backman V and Roy HK. Advances in biophotonics detection of field carcinogenesis for colon cancer risk stratification. Journal of Cancer. 2013; 4(3): p. 251-61.
133. Burr HS. Biologic Organization and the Cancer Problem *. The Yale Journal of Biology and Medicine. 1940; 12(3): p. 277-82.
134. Burr HS. Changes in the field properties of mice with transplanted tumors. Yale Journal of Biology & Medicine. 1941; 13: p. 783-788.
135. Nordenstrom BEW. Biologically Closed Electric Circuits: Nordic Medical Publications.
136. Cone CD. Unified theory on the basic mechanism of normal mitotic control and oncogenesis. Journal of Theoretical Biology. 1971; 30(1): p. 151-81.
137. Cone CD, Jr. Variation of the transmembrane potential level as a basic mechanism of mitosis control. Oncology. 1970; 24(6): p. 438-70.
138. Binggeli R and Weinstein R. Membrane potentials and sodium channels: hypotheses for growth regulation and cancer formation based on changes in sodium channels and gap junctions. Journal of Theoretical Biology. 1986; 123: p. 377-401.
139. Higashimori H and Sontheimer H. Role of Kir4.1 channels in growth control of glia. GLIA. 2007; 55(16): p. 1668-79.
140. Arcangeli A, Bianchi L, Becchetti A, Faravelli L, Coronnello M, Mini E, Olivotto M, and Wanke E. A novel inward-rectifying K+ current with a cell-cycle dependence governs the resting potential of mammalian neuroblastoma cells. Journal of Physiology. 1995; 489(Pt 2): p. 455-71.
141. Nilius B, Schwarz G, and Droogmans G. Control of intracellular calcium by membrane potential in human melanoma cells. Am J Physiol. 1993; 265(6 Pt 1): p. C1501-10.
142. Yamashita M. Ion channel activities in neural stem cells of the neuroepithelium. Stem cells international. 2012; 2012: p. 247670.
143. Fenno L, Yizhar O, and Deisseroth K. The development and application of optogenetics. Annual Review of Neuroscience. 2011; 34: p. 389-412.
144. Adams DS, Tseng AS, and Levin M. Light-activation of the Archaerhodopsin H(+)-pump reverses age-dependent loss of vertebrate regeneration: sparking system-level controls in vivo. Biology Open. 2013; 2(3): p. 306-13.
145. Stuhmer W, Alves F, Hartung F, Zientkowska M, and Pardo LA. Potassium channels as tumour markers. FEBS Lett. 2006; 580(12): p. 2850-2.
146. Killion JJ. Electrical properties of normal and transformed mammalian cells. Biophysical Journal. 1984; 45(3): p. 523-8.
147. Bianchi L, Wible B, Arcangeli A, Taglialatela M, Morra F, Castaldo P, Crociani O, Rosati B, Faravelli L, Olivotto M, and Wanke E. herg encodes a K+ current highly conserved in tumors of different histogenesis: a selective advantage for cancer cells? Cancer Research. 1998; 58(4): p. 815-22.
148. Wissenbach U, Niemeyer B, Himmerkus N, Fixemer T, Bonkhoff H, and Flockerzi V. TRPV6 and prostate cancer: cancer growth beyond the prostate correlates with increased TRPV6 Ca2+ channel expression. Biochem Biophys Res Commun. 2004; 322(4): p. 1359-63.
149. Marino A, Morris D, Schwalke M, Iliev I, and Rogers S. Electrical potential measurements in human breast cancer and benign lesions. Tumour Biology. 1994; 15(3): p. 147-52.
150. Fraser SP, Grimes JA, and Djamgoz MB. Effects of voltage-gated ion channel modulators on rat prostatic cancer cell proliferation: comparison of strongly and weakly metastatic cell lines. Prostate. 2000; 44(1): p. 61-76.
151. Prevarskaya N, Zhang L, and Barritt G. TRP channels in cancer. Biochim Biophys Acta. 2007; 1772(8): p. 937-46.
152. Fraser SP, Diss JK, Chioni AM, Mycielska ME, Pan H, Yamaci RF, Pani F, Siwy Z, Krasowska M, Grzywna Z, Brackenbury WJ, Theodorou D, Koyuturk M, Kaya H, Battaloglu E, De Bella MT, et al. Voltage-gated sodium channel expression and potentiation of human breast cancer metastasis. Clin Cancer Res. 2005; 11(15): p. 5381-9.
153. Onkal R and Djamgoz MB. Molecular pharmacology of voltage-gated sodium channel expression in metastatic disease: clinical potential of neonatal Nav1.5 in breast cancer. Eur J Pharmacol. 2009; 625(1-3): p. 206-19.
154. Ouwerkerk R, Jacobs MA, Macura KJ, Wolff AC, Stearns V, Mezban SD, Khouri NF, Bluemke DA, and Bottomley PA. Elevated tissue sodium concentration in malignant breast lesions detected with non-invasive 23Na MRI. Breast Cancer Res Treat. 2007; 106(2): p. 151-60.
155. Jansson B. Potassium, sodium, and cancer: a review. J Environ Pathol Toxicol Oncol. 1996; 15(2-4): p. 65-73.
156. Cameron IL, Smith NK, Pool TB, and Sparks RL. Intracellular concentration of sodium and other elements as related to mitogenesis and oncogenesis in vivo. Cancer Res. 1980; 40(5): p. 1493-500.
157. Barth LJ and Barth LG. Effect of the potassium ion on induction of notochord from gastrula ectoderm of Rana pipiens. Biol Bull. 1974; 146(3): p. 313-25.
158. Barth LG and Barth LJ. Ionic regulation of embryonic induction and cell differentiation in Rana pipiens. Dev Biol. 1974; 39(1): p. 1-22.
159. Barth LG and Barth LJ. The sodium dependence of embryonic induction. Developmental biology. 1969; 20(3): p. 236-62.
160. Uzman JA, Patil S, Uzgare AR, and Sater AK. The role of intracellular alkalinization in the establishment of anterior neural fate in Xenopus. Developmental Biology. 1998; 193(1): p. 10-20.
161. Obokata H, Wakayama T, Sasai Y, Kojima K, Vacanti MP, Niwa H, Yamato M, and Vacanti CA. Stimulus-triggered fate conversion of somatic cells into pluripotency. Nature. 2014; 505(7485): p. 641-7.
162. Raggi C, Factor VM, Seo D, Holczbauer A, Gillen MC, Marquardt JU, Andersen JB, Durkin M, and Thorgeirsson SS. Epigenetic reprogramming modulates malignant properties of human liver cancer. Hepatology. 2014.
163. Ohnishi K, Semi K, Yamamoto T, Shimizu M, Tanaka A, Mitsunaga K, Okita K, Osafune K, Arioka Y, Maeda T, Soejima H, Moriwaki H, Yamanaka S, Woltjen K, and Yamada Y. Premature termination of reprogramming in vivo leads to cancer development through altered epigenetic regulation. Cell. 2014; 156(4): p. 663-77.
164. Friedmann-Morvinski D and Verma IM. Dedifferentiation and reprogramming: origins of cancer stem cells. EMBO reports. 2014.
165. Halley-Stott RP and Gurdon JB. Epigenetic memory in the context of nuclear reprogramming and cancer. Briefings in functional genomics. 2013; 12(3): p. 164-73.
166. Wang P, Zhang C, Yu P, Tang B, Liu T, Cui H, and Xu J. Regulation of colon cancer cell migration and invasion by CLIC1-mediated RVD. Mol Cell Biochem. 2012; 365(1-2): p. 313-21.
167. Setti M, Savalli N, Osti D, Richichi C, Angelini M, Brescia P, Fornasari L, Carro MS, Mazzanti M, and Pelicci G. Functional role of CLIC1 ion channel in glioblastoma-derived stem/progenitor cells. J Natl Cancer Inst. 2013; 105(21): p. 1644-55.
168. Bowman EJ, Gustafson KR, Bowman BJ, and Boyd MR. Identification of a new chondropsin class of antitumor compound that selectively inhibits V-ATPases. J Biol Chem. 2003.
169. Sennoune SR, Luo D, and Martinez-Zaguilan R. Plasmalemmal vacuolar-type H+-ATPase in cancer biology. Cell Biochem Biophys. 2004; 40(2): p. 185-206.
170. Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, and Lander ES. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009; 138(4): p. 645-59.
171. Levin M. Large-scale biophysics: ion flows and regeneration. Trends Cell Biol. 2007; 17(6): p. 261-70.
172. Lagger G, O’Carroll D, Rembold M, Khier H, Tischler J, Weitzer G, Schuettengruber B, Hauser C, Brunmeir R, Jenuwein T, and Seiser C. Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO J. 2002; 21(11): p. 2672-81.
173. Marks PA, Richon VM, and Rifkind RA. Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. J Natl Cancer Inst. 2000; 92(15): p. 1210-6.
174. Tan J, Cang S, Ma Y, Petrillo RL, and Liu D. Novel histone deacetylase inhibitors in clinical trials as anti-cancer agents. J Hematol Oncol. 2010; 3: p. 5.
175. Wagner JM, Hackanson B, Lubbert M, and Jung M. Histone deacetylase (HDAC) inhibitors in recent clinical trials for cancer therapy. Clin Epigenetics. 2010; 1(3-4): p. 117-136.
176. Marks PA and Xu WS. Histone deacetylase inhibitors: Potential in cancer therapy. J Cell Biochem. 2009; 107(4): p. 600-8.
177. Qiu T, Zhou L, Zhu W, Wang T, Wang J, Shu Y, and Liu P. Effects of treatment with histone deacetylase inhibitors in solid tumors: a review based on 30 clinical trials. Future Oncol. 2013; 9(2): p. 255-69.
178. Marks PA. The clinical development of histone deacetylase inhibitors as targeted anticancer drugs. Expert Opin Investig Drugs. 2010; 19(9): p. 1049-66.
179. Khan O and La Thangue NB. HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunol Cell Biol. 2012; 90(1): p. 85-94.
180. Santoro F, Botrugno OA, Dal Zuffo R, Pallavicini I, Matthews GM, Cluse L, Barozzi I, Senese S, Fornasari L, Moretti S, Altucci L, Pelicci PG, Chiocca S, Johnstone RW, and Minucci S. A dual role for Hdac1: oncosuppressor in tumorigenesis, oncogene in tumor maintenance. Blood. 2013; 121(17): p. 3459-68.
181. Ermolaeva MA, Segref A, Dakhovnik A, Ou HL, Schneider JI, Utermohlen O, Hoppe T, and Schumacher B. DNA damage in germ cells induces an innate immune response that triggers systemic stress resistance. Nature. 2013; 501(7467): p. 416-20.
182. Martin OA, Redon CE, Nakamura AJ, Dickey JS, Georgakilas AG, and Bonner WM. Systemic DNA damage related to cancer. Cancer Res. 2011; 71(10): p. 3437-41.
183. Jernberg C, Lofmark S, Edlund C, and Jansson JK. Long-term impacts of antibiotic exposure on the human intestinal microbiota. Microbiology. 2010; 156(Pt 11): p. 3216-23.
184. Fukuda M, Kanauchi O, Araki Y, Andoh A, Mitsuyama K, Takagi K, Toyonaga A, Sata M, Fujiyama Y, Fukuoka M, Matsumoto Y, and Bamba T. Prebiotic treatment of experimental colitis with germinated barley foodstuff: a comparison with probiotic or antibiotic treatment. Int J Mol Med. 2002; 9(1): p. 65-70.
185. Young VB and Schmidt TM. Antibiotic-associated diarrhea accompanied by large-scale alterations in the composition of the fecal microbiota. J Clin Microbiol. 2004; 42(3): p. 1203-6.
186. Dethlefsen L, Huse S, Sogin ML, and Relman DA. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008; 6(11): p. e280.
187. Pryde SE, Duncan SH, Hold GL, Stewart CS, and Flint HJ. The microbiology of butyrate formation in the human colon. FEMS Microbiol Lett. 2002; 217(2): p. 133-9.
188. Gottenbos B, Grijpma DW, van der Mei HC, Feijen J, and Busscher HJ. Antimicrobial effects of positively charged surfaces on adhering Gram-positive and Gram-negative bacteria. J Antimicrob Chemother. 2001; 48(1): p. 7-13.
189. Terada A, Yuasa A, Kushimoto T, Tsuneda S, Katakai A, and Tamada M. Bacterial adhesion to and viability on positively charged polymer surfaces. Microbiology. 2006; 152(Pt 12): p. 3575-83.
190. Sinha G. Charged by GSK investment, battery of electroceuticals advance. Nature medicine. 2013; 19(6): p. 654.
191. Famm K, Litt B, Tracey KJ, Boyden ES, and Slaoui M. Drug discovery: a jump-start for electroceuticals. Nature. 2013; 496(7444): p. 159-61.
192. Sive HL, Grainger RM, and Harland RM. Early development of Xenopus laevis : a laboratory manual. 2000, Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press. ix, 338 p., 7 p. of plates.
193. Nieuwkoop PD and Faber J. Normal Table of Xenopus Laevis (Daudin). 1967, Amesterdam: North-Holland Publishing Company
194. Yang S, Lockwood A, Hollett P, Ford R, and Kao K. Overexpression of a novel Xenopus rel mRNA gene induces tumors in early embryos. J Biol Chem. 1998; 273(22): p. 13746-52.
195. Miyanaga Y, Shiurba R, Nagata S, Pfeiffer CJ, and Asashima M. Induction of blood cells in Xenopus embryo explants. Dev Genes Evol. 1998; 207(7): p. 417-26.
196. James-Kracke MR. Quick and accurate method to convert BCECF fluorescence to pHi: calibration in three different types of cell preparations. J Cell Physiol. 1992; 151(3): p. 596-603.
197. Vega AV, Avila G, and Matthews G. Interaction between the transcriptional corepressor Sin3B and voltage-gated sodium channels modulates functional channel expression. Sci Rep. 2013; 3: p. 2809.