
INTRODUCTION
Sunitinib, an oral small molecular multipletarg-eted receptor tyrosine kinase (RTK) inhibitor, has a relatively board spectrum of targets, including the vascular endothelial growth factor receptors (VEGFRs); platelet-derived growth factor receptors (PDGFRs); the stem cell factor receptor (c-KIT) and FMS-like tyrosine kinase 3(FLT3) [1–4], thereby exhibiting both anti-angiogenesis and anti-tumor activities. In the clinic, sunitinib has benefited many patients with advanced renal cell carcinoma (RCC), imatinib-refractory gastrointestinal stromal tumor (GIST) or pancreatic neuroendocrine tumor (pNET) [5–7]. Additionally, clinical data and experimental evidence suggested that sunitinib also has demonstrable efficacy on other solid tumors, such as lung cancer [8, 4, 9, 10].
However, future of sunitinib is facing a major challenge: an emergent resistance to sunitinib will eventually develop. Generally, two groups of underlying mechanisms of resistance have been elucidated: first, tumor cells themselves mediate resistance to sunitinib. For instance, a study uncovered that continued exposure to sunitinib could cause an increased lysosomal capacity in tumor cells, resulting in resistance to sunitinib [11]. Second, the other mechanisms mainly attribute to tumor microenvironment. Finke et al elaborated that myeloid derived suppressor cells (MDSCs) induced sunitinib-resistance via providing enduring angiogenesis and immune suppression [12]. In this study, we focused on the biological behavior changes happened to sunitinib-resistant tumor cells and its possible explanation for sunitinib-resistance.
In the attempt of addressing these questions, we developed a sunitinib-resistant lung cancer cell line in vitro, and then confirmed its drug-resistance to sunitnib in vivo. Moreover, we further investigated the changes in biological behavior and molecular profiles both in vitro and in vivo.
RESULTS
Establishment of acquired resistant LL/2 cell line to sunitinib
LL/2-P cells were treated with sunitinib for more than 6 months at gradually increasing concentrations. Finally, sunitinib-resistant LL/2 cell line, LL/2-R, was established. The values of IC50 for the LL/2 cell sublines were determined by MTT. The IC50 of LL/2-R cell line showed about 5-fold higher concentrations compared to that of LL/2-P cell line: 10.03μM vs.1.94μM (Figure 1A).
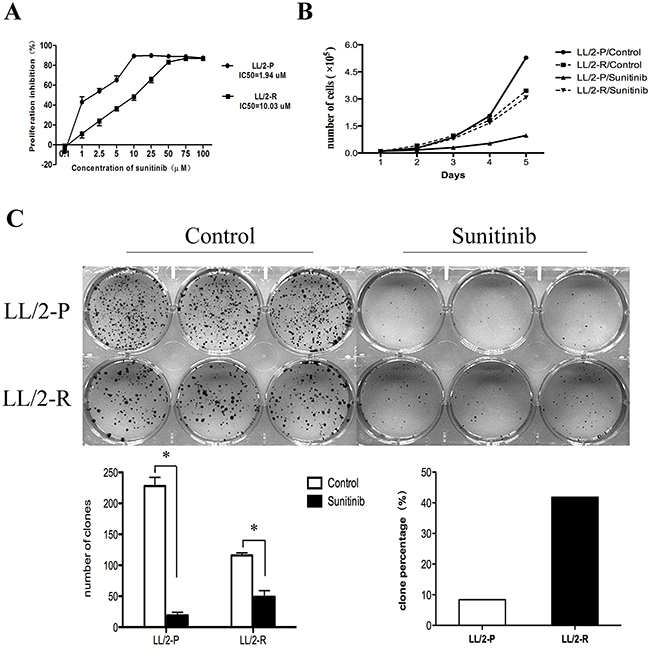
Figure 1: Acquired resistance to sunitinib in LL/2-R cells. To induce resistance, LL/2 cell line was continuously exposed for more than 6 months to gradually increasing concentrations of sunitinib. (A) Value of IC50 to sunitinib increased in LL/2-R cells. Points, mean of three independent experiments; bars, SD. (B) LL/2-R cells had a reduced cell growth inhibition, when treated with sunitnib. (C) LL/2-R cells had an increased clonogenic capacity, when treated with sunitinib. Cells were exposed to 1μm sunitinib for 10 days, and each group is made in triplicate. Columns, mean of three independent experiments; bars, SD; *, P<0.05.
In addition, we found that LL/2-R cells had a reduced cell proliferation inhibition and an increased clonogenic capacity compared to LL/2-P cells, when treated with sunitinib. In cell proliferation assay, the doubling time (DT) of LL/2-P cell line was 36.2h and 19.7h in sunitinib- and vehicle- treated cells, respectively, while it was 26.7h and 25.5h for LL/2-R cell line correspondingly. The proliferation inhibition rate was 85.2% in LL/2-P cell line, but only 13.8% in LL/2-R cell line (Figure 1B). In clonogenic assay, the number of clonies in LL/2-R cell line was 49.0±10.0 and 116.0±4.0 in sunitinib- and vehicle-treated wells, respectively, while it was 19±5 and 228±14 in LL/2-P cell line. The percentage of clony formation in LL/2-R cell line was 41.8%, but only 8.3% in LL/2-P cell line (Figure 1C). In conclusion, LL/2-R cells had a higher resistance to sunitinib treatment in contrast to LL/2-P cells in vitro.
Decreased growth-inhibitory effect of sunitinib on LL/2-R cells in vivo
To verify whether LL/2-R cells also had sunitinib-resistance in vivo, LL/2-R cells and LL/2-P cells were injected subcutaneously in the right flank of mice, respectively. Ten days after injection, tumors were established (about 100mm3 in size) and treatment with sunitinib (80mg/kg/day) or vehicle was performed. Sunitinib resulted in a growth inhibition of LL/2-R tumors by 28.2% and LL/2-P tumors by 51.3% (Figure 2A). We confirmed that LL/2-R cells were also more resistant to sunitinib treatment in vivo, compared to LL/2-P cells.
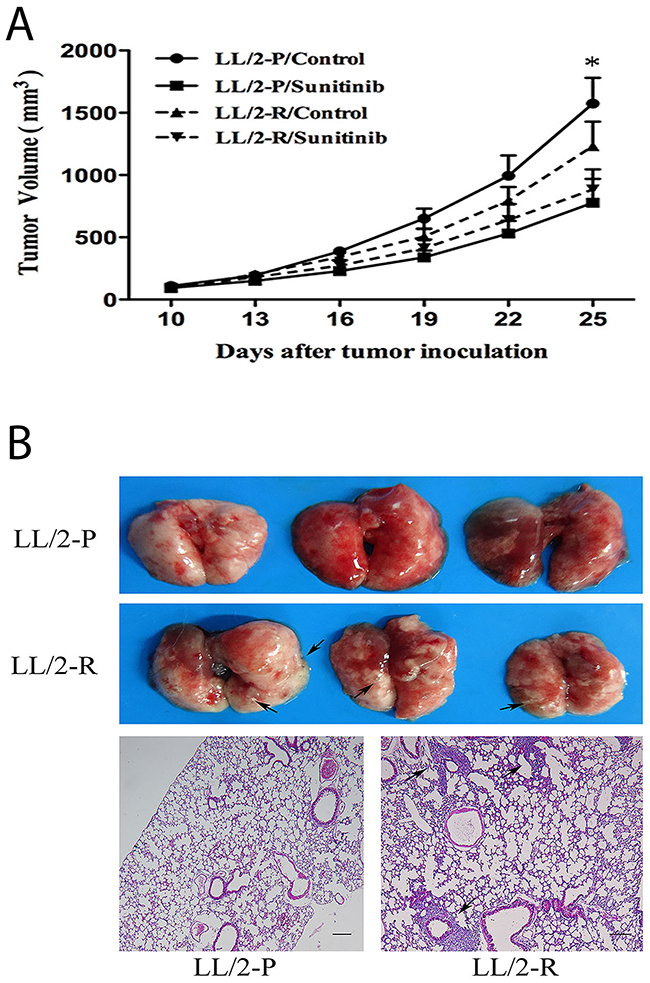
Figure 2: Decreased growth-inhibitory effect of sunitinib and enhanced metastatic potential of LL/2-R cell subline in vivo. (A) Decreased growth-inhibitory effect of sunitinib on LL/2-R cells in vivo. Growth curve of tumors established from LL/2-P and LL/2-R tumors after tumor cells injection (5×105 cells, n=10). Mice received treatment with vehicle or sunitinib (80mg/kg/day) 10 days after tumor cells injection. Points, mean of three independent experiments; bars, SD; *, P<0.05. (B) Increased metastatic potential of LL/2-R cells in vivo. 40 days after LL/2 cell sublines subcutaneous injection (5×105 cells, n=3), lungs in mice were examined and analyzed by histological H&E staining of tissue sections. Metastases were observed, as indicated by the black arrows. Representative images are shown. Scale bars, 200μm(×40).
Increased migratory, invasive and metastatic potential of LL/2-R cell subline
The cell migration was analyzed by wound healing assay. By 48h post-wounding, LL/2-R cells significantly improved closure of wound compared to LL/2-P cells, showing a faster migration (Figure 3A). The invasive capacity of LL/2 cell sublines was examined by transwell assay. The numbers of LL/2-R and LL/2-P cells that invaded through the basement membrane were 376.0 ± 90.0 and 71.5 ± 16.2, respectively (Figure 3B). To further investigate the metastatic potential of LL/2 cell sublines in vivo, LL/2-R cells and LL/2-P cells were injected subcutaneously in the right flank of mice, respectively. 40 days later, lung metastases could be detected on mice bearing LL/2-R tumors, but not on mice bearing LL/2-P tumors (Figure 2B). In addition, MMPs has been reported to play crucial roles in invasion and metastasis of tumor cells, thus protein levels of two of most important MMPs, MMP-2 and MMP-9, in LL/2-R and LL/2-P cells and tumors were measured by western blotting assay. The expression of MMP-2 and MMP-9 were both higher in LL/2-R than LL/2-P cells (Figure 3C), also higher in tumor tissues from LL/2-R than LL/2-P in mouse models. The data indicated that LL/2-R cells possessed increased migratory, invasive and metastatic potential.
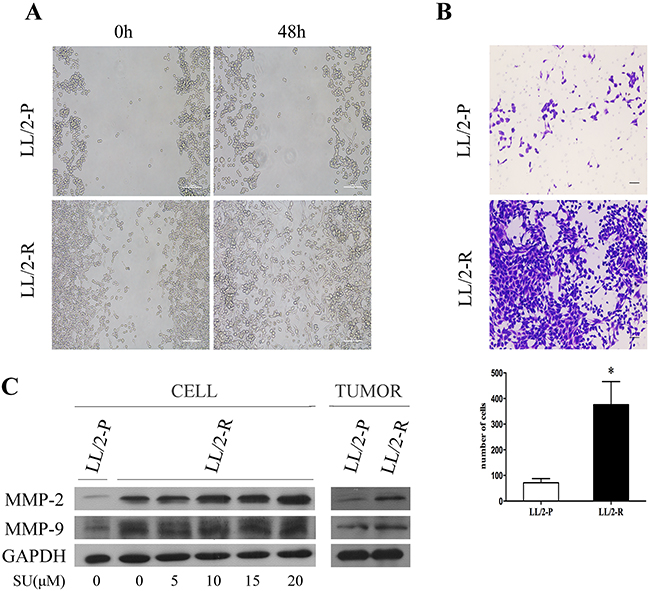
Figure 3: Increased migratory and invasive potential of LL/2-R cell subline. (A) A wound healing assay showed that LL/2-R cells had increased migratory ability. Representative images are shown imediately after a scratch was created (0h) and 48h later. Scale bars, 100μm (×100). (B) LL/2-R cells had increased ability to invade through Matrigel-coated transwell membranes. After 48 hours, the invaded cells were stained, photographed and counted. Representative photographs of transwell membranes showed stained invaded cells. Columns, mean of three independent experiments; bars, SD; *, P<0.05; scale bars, 50μm(×100). (C) Western blotting assay was used to investigate MMP-2 and MMP-9 levels of LL/2 cells and tumors, with GAPDH as a loading control. The MMP2 and MMP9 levels of LL/2-R cells and tumors were upregulated, compared with LL/2-P. The expression of MMP2 and MMP9 in LL/2-R cells seems increased when treated with climbing concentration of sunitinib for 48h from 5μM to 20μM. Three independent experiments were conducted.
EMT characteristics of LL/2-R cell
It has been reported that epithelial-mesenchymal transition (EMT) is associated with increased invasiveness of tumor cells. Firstly, we found that LL/2-R cells changed to flat and spindle-sharped, showing a mesenchymal morphology (Figure 4A). Therefore, we investigate expression of vimentin and snail, and location of β-catenin in LL/2 cell sublines in vitro. The immunofluorescence staining showed that the expression level of vimentin and snail increased in LL/2-R cells relative to LL/2-P cells, while the immunofluorescence staining and western blotting assay both confirmed that β-catenin translocated to the nucleus (Figure 4B and 4C). Additionally, we further tested whether LL/2-R cells also undergone EMT in vivo using western blotting assay. The expression of E-cadherin, an epithelial cell marker, significantly decreased, conversely, the protein level of vimentin, a mesenchymal cell marker, increased in LL/2-R tumors. Next we further analyzed the expression of EMT-related transcriptional factors like snail and twist. They were both upregulated in LL/2-R tumors (Figure 4D). As one of the main player in inducing EMT, the TGFβ1 secretion of LL/2-R cells was also remarkably increased, compared to LL/2-P cells (Figure 4E). Our results demonstrated that LL/2-R cells undergone EMT.
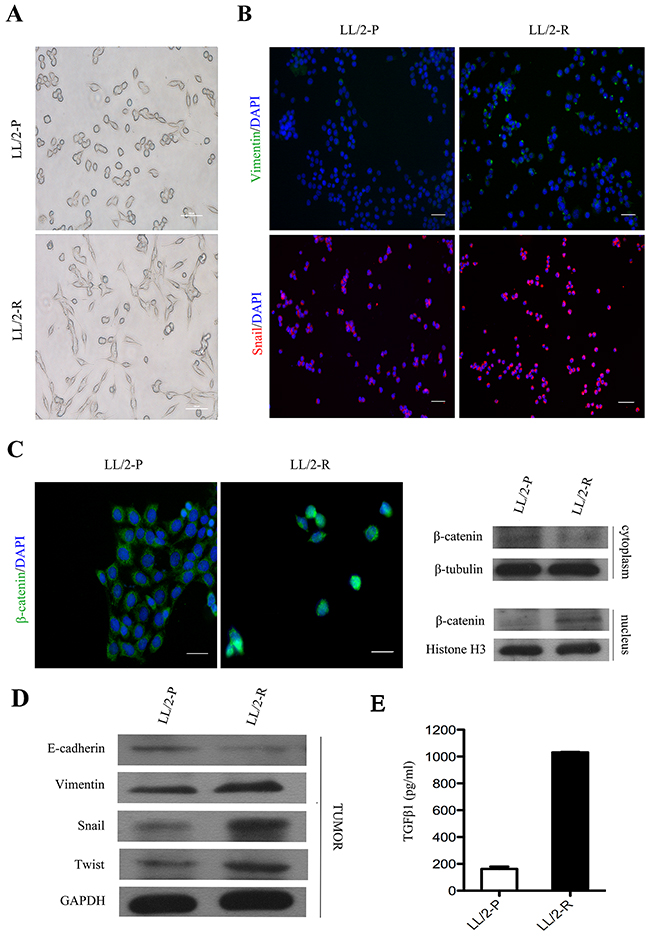
Figure 4: EMT of LL/2-R cell subline both in vitro and in vivo. (A) The morphology of LL/2-P and LL/-R cells. Scale bars, 100μm(×100). (B) Immunofluorescence images of LL/2 cell sublines. First row, vimentin imunolocalisation (green colour) was detectable in LL/2-R cells, but absent from LL/2-P cells. Second row, snail imunolocalisation (red colour) in nucleus was stronger in LL/2-R cells. Cell nuclei are coloured blue after staining with DAPI. Scale bars, 100μm (×100). (C) Nuclear translocation of β-cateninin LL/2-R cells. Immunofluorescence images displayed that β-catenin (green colour) gave nuclear foci pattern of imunolocalisation in LL/2-R cells, but diffuse cytoplasmic pattern in LL/2-P cells. Western blotting also showed that the β-catenin level of nucleus was upregulation, but that of cytoplasm was downregulation in LL/2-R cells. Cell nuclei are coloured blue after staining with DAPI. Scale bars, 50μm(×400). β-tubulin and Histone H3 were used as loading controls respective for cytoplasmic and nuclear protein. (D) Alterations in EMT marker in LL/2-R tumors. Western blotting revealed that E-cadherin was downregulation, and vimentin, snail and twist were all upregulation, with GAPDH as a loading control in LL/2-R tumors. Three independent experiments were conducted. (E) The TGFβ1 level was significantly increased in LL/2-R cells, which is measured by ELISA assays.
DISCUSSION
Tyrosine kinase inhibitors (TKIs), such as sunitinib, are the major targeted therapies for many cancers. Unfortunately, resistance to the TKIs will eventually develop. To study the mechanism, we developed a resistant lung cancer cell and animal model to sunitinib, which have been tested in several clinical trials and preclinical studies in lung cancer [13–15], and subsequently validated the resistance to sunitinib both in vitro and in vivo. Our results that acquired tumor cell resistance to sunitinib causes resistance in vivo support the concept that tumor cells themselves might play a crucial role in resistance to sunitinib.
Thus, we focused our investigation on the changes in biological characteristics of tumor cells, as several studies also pointed that a significant role for tumor cells themselves in development of drug-resistance [16–18]. We observed that sunitinib-resistant LL/2 cancer cells exhibited increased migration and invasion in vitro, and enhanced metastatic potential in vivo, companied by MMP2 and MMP9 overexpression, which play crucial roles in tumor invasion and metastasis [19, 20]. Similarly, there have been previous links reported between drug-resistance and increased invasion [21–23]. Du et al [24] and Paez-Ribes et al [25] also showed that more invasive behavior was observed in cancer cells which are resistant to antiangiogenic therapy. Our data and studies implicate that invading more aggressively into normal tissue is possibly another latent adaption mode of tumor cells.
To invade into normal tissue, tumor cells have to dissolve cell-cell junction and cell-ECM adhesion. As a process during which the polarized, stationary epithelial cells break down their cell-cell and cell-ECM contacts and convert intosingle dissociated, non-polarized, motile mesenchymal cells [26], the EMT has been reported that associated with cancer cell invasion and distant metastasis [27, 28]. When cancer cells undergo EMT, some molecular changes occur. For example, E-cadherin, a typical marker of epithelial cells, is lost, on the other hand, vimentin, a typical maker of mesenchymal cells, is induced [29]. Besides, E-cadherin is anchored to β-catenin at the epithelial cell membrane to maintain integrity of cell-cell adhesion junctions (AJs). Therefore, loss of E-cadherin also results in nuclear translocation of β-catenin, inducing activation transcriptional factor LEF/TCF4 which facilitates EMT by activating Wnt signaling pathway [30]. Moreover, EMT is driven by transcriptional factors that repress the expression of E-cadherin, such as snail and twist [31, 32]. In this study, we observed morphology changes, downregulation of E-cadherin, on the contrary, upregulation of vimentin, snail and twist, along with nuclear translocation of β-catenin, in LL/2-R cells. Based on these results, we believe that sunitinib-resistant LL/2 cancer cells may experience EMT, which results in their increased activity to invade. Besides, we detected that the TGFβ1 level of LL/2-R cells was dramatically increased, and previous studies had reported that TGFβ1 could induce EMT. So the EMT process occurred in LL/2-R cells may be TGFβ1-dependent.
In the present study, we described that LL/2-R cancer cells undergone EMT, along with their resistant phenotype. To date, there is growing acceptance that sunitinib-resistance and EMT have strictly connection, as we displayed in our work as well. The EMT phenomena have been observed in some other sunitinib-resistant cancers, such as prostate cancer, hepatocellular carcinoma, renal cell carcinoma [33, 34]. Hammers et al. also described that EMT may be correlated with acquired resistance to sunitinib in patients with clear cell renal carcinoma [35]. Yet, the underlying mechanism between sunitinib-resistance and EMT remains unclear. Previous studies deemed that the EMT is triggered by hypoxic tumor microenvironment, which is induced by anti-angiogenesis therapy, in sunitinib-resistant cancers. However, we found that EMT characteristics also could be observed in sunitinib-resistant cancer cells in vitro in normoxia, meaning that sunitinib has a direct impact on tumor cells, eventually, EMT occurs. A previous study had reported that sunitinib could remarkly induced the expression of TGFβ [36], which is in accordance with our finding. These results suggested that sunitinib could induce the upregulation of TGFβ, which might be associated with EMT. Further research is needed to figure out the underlying mechanisms.
In conclusion, we successfully established LL/2-R cell line, which exhibits decreased sensitivity to sunitinib as opposed to its parental cell line, both in vitro and in vivo. We subsequently showed that resistant cells are possessed of increased invasive capacity and enriched EMT properties, which may be involved in acquisition of a phenotype resistant to sunitinib in LL/2 cells and this EMT maybe TGFβ1-dependent. Thus, our results warrant further studies to investigate the mechanism of resistance and promising therapeutic strategies based on circumvention of EMT during sunitinib treatment.
MATERIALS AND METHODS
Cell culture and reagents
The lewis lung carcinoma cell LL/2 was obtained from ATCC (American Tissue Culture Collection) and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% FBS and maintained in a humidified incubator containing 5% CO2 at 37 °C. Sunitinib malate was purchased from Selleckchem. For the development of sunitinib-resistant subline LL/2-R, the parental cell line LL/2-P was continuously exposed for more than 6 months to gradually increasing concentrations of sunitinib, which is increased by 0.2μm every 48h until the IC50 to 2μm, and then by 0.5μm until to 20μm.
MTT assay
Cells (2×103 per well) were seeded in 96-well plates, and then incubated with different concentrations of sunitinib on the next day. 48hours later, the metabolically active cells were quantified using MTT (5mg/ml, Sigma-Aldrich) by measuring the Optical Density (OD) value at 570nm in ELISA reader. The proliferation inhibition due to different concentration of sunitinib was calculated by the following formula: proliferation inhibition (%)=(ODtreated-ODcontrol)/ (ODcontrol-ODblank)×100. The value of IC50 (the concentration required for a 50% proliferation inhibition) was determined by Graphpad prism 5.0.
Cell proliferation assay
Cells (5×103 per well) were seeded in 24-well plates. After 24 hours, culture medium was replaced with fresh medium or that containing 2μM sunitinib. The cells were counted daily in triplicate by Trypan blue dye exclusion assay. The values of doubling time (DT) and proliferation inhibition were calculated, according to the following formulas: DT(h)=[lg2/(lgNt-lgN0)]×t (Nt = ultimate cell number; N0 = primary cell number; t = termination incubation time) and proliferation inhibition (%)=(N0-N)/N0×100 (N0=number of cells in untreated well; N=number of cells in treated well).
Clonogenic assay
Cells (1×103 per well) were exposed to 1μm sunitinib for 10 days to allow clony formation. Clonies were fixed, stained with crystal violet and counted manually, with a minimal clony cells number of 50 for required counting. The percentage of clony formation is calculated as the number of clonies in treated wells divided by those obtained in untreated wells.
Wound healing assay
Cells (2×106 per well) were seeded in 6-well plates. Scrape the cell monolayer in a straight line to create a scratch with a p200 pipet tip. To obtain the same field during the image acquisition, reference points was made on the outer bottom of the dish. Sequentially, the images were captured with a Nikon Eclipse Ti-U inverted microscope at 0h and 48h, respectively.
Transwell invasion assay
Cells were starved in serum-free DMEM for 48h. Transwell chambers (Corning Costar) consisting of 8μm pore size membrane filter inserts were coated with matrigel (BD Bioscience) overnight in incubator. 1×105 cells resuspended in serum-free medium were placed in the upper chamber, and medium supplemented with 10% FBS were added into the lower chamber. After 48 hours, cells on the upper surface of the membrane were removed with a cotton swab, and cells on the lower surface were stained with crystal violet, photographed and counted using an Olympus BX51 microscope.
Immunofluorescence assay
Cells on coverslips were fixed with 4% paraformaldehyde, permeabilized in 1% (V/V) Triton X-100 in PBS, blocked with 5% BSA and incubated with primary antibodies at 1:100 dilution at 4°C overnight. Secondary FITC-conjugated anti-rabbit or Texas Red-conjugated anti-mouse antibody (Santa Cruz Biotechnology) was used at 1: 50 dilution, and then DAPI (Beyotime) was included in this incubation for last 10 min. Coverslips were photographed with a Nikon Eclipse Ti-U inverted fluorescence microscope. Primary antibodies used are as follows: mouse anti-snail (Merck Millipore), rabbit anti-vimentin and rabbit anti-β-catenin (Cell Signaling Technology).
Western blotting assay
Total protein samples were (30μg) separated by SDS polyacrylamide gel and then transferred to PVDF membrane. After blocking with 5% (W/V) nonfat-dried milk, membranes were incubated with primary antibodies at 1:1000 dilution at 4°C overnight, followed by incubation with the appropriate HRP-conjugated secondary antibodies (ZSGB-BIO) at 1:10000 dilution. The immune complexes were detected by an enhanced chemiluminescence system and exposed on Kodak X-ray films. Primary antibodies used are as follows: mouse anti-GAPDH (Beyotime), rabbit anti-MMP-2, rabbit anti-MMP-9, rabbit anti-E-cadherin, rabbit anti-vimentin, rabbit anti-β-catenin, rabbit anti-Histone H3 (Cell Signaling Technology), mouse anti-β-tubulin (Sigma-Aldrich), rabbit anti-snail and rabbit anti-twist (Zen Bioscience).
Measurement of TGFβ1 level
Cells (2×106 per well) were seeded in 6-well plates. 48 hours later, the TGFβ1 level in cultured medium was quantified using the Mouse TGFβ1 ELISA kit (Neobioscience) in accordance with manufacturer’s protocol.
Tumor growth in vivo
All animal experiments were performed according to procedures approved by the Institutional Animal Use and Care Committee of Sichuan University, China. C57BL/6 mice were purchased from HuaFukang Biological Technology Co. Ltd. (Beijing, China) and housed under SPF (specific pathogen-free) conditions. Tumor cells (5×105 cells per mouse for LL/2-P or LL/2-R) were injected subcutaneously in the right flank of mice. When tumor size reached about 100mm3, the mice were randomly divided 2 groups of 10 mice each and treated daily by gavage with sunitinib at a dose of 80mg/kg or vehicle (control). The tumor size was assessed every 3 days with caliper measurement, and expressed in mm3 using the formula: length×width2×0.52. After 15 days of treatment, mice were sacrificed and tumors were snap-frozen in liquid nitrogen. The growth inhibition was measured by the following formula: growth inhibition (%)=1-V/V0×100 (V0=volume of tumors in control group; V=volume of tumors in sunitinib-treated group).
Tumor metastasis
Three mice were injected subcutaneously in the right flank with LL/2-P or LL/2-R cells, respectively. After 40 days, mice were sacrificed and lungs were examined, embedded in paraffin, stained for hematoxylin and eosin and photographed using an Olympus BX51 microscope.
Statistical analysis
Data are presented as means ± SD. Statistical differences were examined by Student’s t test. A P value less than 0.05 is considered to be statistically significant.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
FUNDING
This work was partly supported by National Natural Science Foundation of China (No.81272457 and No.81572380).
REFERENCES
1. Roskoski R Jr. Sunitinib: a VEGF and PDGF receptor protein kinase and angiogenesis inhibitor. Biochem Biophys Res Commun. 2007;356:323-8. https://doi.org/10.1016/j.bbrc.2007.02.156.
2. Faivre S, Demetri G, Sargent W, Raymond E. Molecular basis for sunitinib efficacy and future clinical development. Nat Rev Drug Discov. 2007;6:734-45. https://doi.org/10.1038/nrd2380.
3. O’Farrell AM, Abrams TJ, Yuen HA, Ngai TJ, Louie SG, Yee KW, Wong LM, Hong W, Lee LB, Town A, Smolich BD, Manning WC, Murray LJ, et al. SU11248 is a novel FLT3 tyrosine kinase inhibitor with potent activity in vitro and in vivo. Blood. 2003;101:3597-605. https://doi.org/10.1182/blood-2002-07-2307.
4. Abrams TJ, Lee LB, Murray LJ, Pryer NK, Cherrington JM. SU11248 inhibits KIT and platelet-derived growth factor receptor beta in preclinical models of human small cell lung cancer. Mol Cancer Ther. 2003;2:471-8.
5. Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, Oudard S, Negrier S, Szczylik C, Kim ST, Chen I, Bycott PW, Baum CM, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356:115-24. https://doi.org/10.1056/NEJMoa065044.
6. Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, McArthur G, Judson IR, Heinrich MC, Morgan JA, Desai J, Fletcher CD, George S, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368:1329-38. https://doi.org/10.1016/S0140-6736(06)69446-4.
7. Raymond E, Dahan L, Raoul JL, Bang YJ, Borbath I, Lombard-Bohas C, Valle J, Metrakos P, Smith D, Vinik A, Chen JS, Hörsch D, Hammel P, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med. 2011;364:501-13. https://doi.org/10.1056/NEJMoa1003825.
8. Ready NE, Pang HH, Gu L, Otterson GA, Thomas SP, Miller AA, Baggstrom M, Masters GA, Graziano SL, Crawford J, Bogart J, Vokes EE. Chemotherapy with or without maintenance sunitinib for untreated extensive-stage small-cell lung cancer: a randomized, double-blind, placebo-controlled phase II study-CALGB 30504 (alliance). J Clin Oncol. 2015;33:1660-5. https://doi.org/10.1200/JCO.2014.57.3105.
9. Blumenschein GR Jr, Ciuleanu T, Robert F, Groen HJ, Usari T, Ruiz-Garcia A, Tye L, Chao RC, Juhasz E. Sunitinib plus erlotinib for the treatment of advanced/metastatic non-small-cell lung cancer: a lead-in study. J Thorac Oncol. 2012;7:1406-16. https://doi.org/10.1097/JTO.0b013e31825cca1c.
10. James C. Antitumor efficacy of sunitinib malate in concurrent and sequential combinations with standard chemotherapeutic agents in non-small cell lung cancer (NSCLC) nonclinical models. Proceedings of the 99th Annual Meeting of the American Association for Cancer Research. 2008.
11. Gotink KJ, Broxterman HJ, Labots M, de Haas RR, Dekker H, Honeywell RJ, Rudek MA, Beerepoot LV, Musters RJ, Jansen G, Griffioen AW, Assaraf YG, Pili R, et al. Lysosomal sequestration of sunitinib: a novel mechanism of drug resistance. Clin Cancer Res. 2011;17:7337-46. https://doi.org/10.1158/1078-0432.CCR-11-1667.
12. Finke J, Ko J, Rini B, Rayman P, Ireland J, Cohen P. MDSC as a mechanism of tumor escape from sunitinib mediated anti-angiogenic therapy. Int Iimmunopharmacol. 2011;11:856-61. https://doi.org/10.1016/j.intimp.2011.01.030.
13. Socinski MA, Novello S, Brahmer JR, Rosell R, Sanchez JM, Belani CP, Govindan R, Atkins JN, Gillenwater HH, Pallares C, Tye L, Selaru P, Chao RC, et al. Multicenter, phase II trial of sunitinib in previously treated, advanced non-small-cell lung cancer. J Clin Oncol. 2008;26:650-6. https://doi.org/10.1200/JCO.2007.13.9303.
14. Novello S, Scagliotti GV, Rosell R, Socinski MA, Brahmer J, Atkins J, Pallares C, Burgess R, Tye L, Selaru P, Wang E, Chao R, Govindan R. Phase II study of continuous daily sunitinib dosing in patients with previously treated advanced non-small cell lung cancer. Br J Cancer. 2009;101:1543-8. https://doi.org/10.1038/sj.bjc.6605346.
15. Mendel DB, Laird AD, Xin X, Louie SG, Christensen JG, Li G, Schreck RE, Abrams TJ, Ngai TJ, Lee LB, Murray LJ, Carver J, Chan E, et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003;9:327-37.
16. Cillo C, Dick JE, Ling V, Hill RP. Generation of drug-resistant variants in metastatic B16 mouse melanoma cell lines. Cancer Res. 1987;47:2604-8.
17. Sakai I, Miyake H, Fujisawa M. Acquired resistance to sunitinib in human renal cell carcinoma cells is mediated by constitutive activation of signal transduction pathways associated with tumour cell proliferation. BJU Int. 2013;112:E211-20. https://doi.org/10.1111/j.1464-410X.2012.11655.x.
18. O’Loughlin C, Heenan M, Coyle S, Clynes M. Altered cell cycle response of drug-resistant lung carcinoma cells to doxorubicin. Eur J Cancer. 2000;36:1149-60.
19. Fishman DA, Liu Y, Ellerbroek SM, Stack MS. Lysophosphatidic acid promotes matrix metalloproteinase (MMP) activation and MMP-dependent invasion in ovarian cancer cells. Cancer Res. 2001;61:3194-9.
20. Wang X, Lu H, Urvalek AM, Li T, Yu L, Lamar J, DiPersio CM, Feustel PJ, Zhao J. KLF8 promotes human breast cancer cell invasion and metastasis by transcriptional activation of MMP9. Oncogene. 2011;30:1901-11. https://doi.org/10.1038/onc.2010.563.
21. Damiano JS, Cress AE, Hazlehurst LA, Shtil AA, Dalton WS. Cell adhesion mediated drug resistance (CAM-DR): role of integrins and resistance to apoptosis in human myeloma cell lines. Blood. 1999;93:1658-67.
22. Sandri S, Faiao-Flores F, Tiago M, Pennacchi PC, Massaro RR, Alves-Fernandes DK, Berardinelli GN, Evangelista AF, de Lima Vazquez V, Reis RM, Maria-Engler SS. Vemurafenib resistance increases melanoma invasiveness and modulates the tumor microenvironment by MMP-2 upregulation. Pharmacol Res. 2016;111:523-33. https://doi.org/10.1016/j.phrs.2016.07.017.
23. DeLay M, Jahangiri A, Carbonell WS, Hu YL, Tsao S, Tom MW, Paquette J, Tokuyasu TA, Aghi MK. Microarray analysis verifies two distinct phenotypes of glioblastomas resistant to antiangiogenic therapy. Clin Cancer Res. 2012;18:2930-42. https://doi.org/10.1158/1078-0432.CCR-11-2390.
24. Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegue E, Song H, Vandenberg S, Johnson RS, Werb Z, Bergers G. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008;13:206-20. https://doi.org/10.1016/j.ccr.2008.01.034.
25. Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, Inoue M, Bergers G, Hanahan D, Casanovas O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15:220-31. https://doi.org/10.1016/j.ccr.2009.01.027.
26. Hay ED. An overview of epithelio-mesenchymal transformation. Acta Anat (Basel). 1995;154:8-20.
27. Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14:818-29. https://doi.org/10.1016/j.devcel.2008.05.009.
28. Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131-42. https://doi.org/10.1038/nrm1835.
29. Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest. 2009;119:1429-37. https://doi.org/10.1172/JCI36183.
30. Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469-80. https://doi.org/10.1016/j.cell.2006.10.018.
31. Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415-28. https://doi.org/10.1038/nrc2131.
32. Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927-39. https://doi.org/10.1016/j.cell.2004.06.006.
33. Marijon H, Dokmak S, Paradis V, Zappa M, Bieche I, Bouattour M, Raymond E, Faivre S. Epithelial-to-mesenchymal transition and acquired resistance to sunitinib in a patient with hepatocellular carcinoma. J Hepatol. 2011;54:1073-8. https://doi.org/10.1016/j.jhep.2010.11.011.
34. Dos Santos C, Tijeras-Raballand A, Serova M, Sebbagh S, Slimane K, Faivre S, de Gramont A, Raymond E. Effects of preset sequential administrations of sunitinib and everolimus on tumour differentiation in Caki-1 renal cell carcinoma. Br J Cancer. 2015;112:86-94. https://doi.org/10.1038/bjc.2014.578.
35. Hammers HJ, Verheul HM, Salumbides B, Sharma R, Rudek M, Jaspers J, Shah P, Ellis L, Shen L, Paesante S, Dykema K, Furge K, Teh BT, et al. Reversible epithelial to mesenchymal transition and acquired resistance to sunitinib in patients with renal cell carcinoma: evidence from a xenograft study. Mol Cancer Ther. 2010;9:1525-35. https://doi.org/10.1158/1535-7163.MCT-09-1106.
36. Yin T, He S, Ye T, Shen G, Wan Y, Wang Y. Antiangiogenic therapy using sunitinib combined with rapamycin retards tumor growth but promotes metastasis. Transl Oncol. 2014; 7: 221-229. https://doi.org/10.1016/j.tranon.2014.02.007