
INTRODUCTION
B lymphoblastic leukemia (B-ALL), a hematopoietic malignancy derived from B-cell progenitors, is predominantly a childhood disease but can occur in adolescents and adults as well. According to the 2016 United States statistics data of lymphoid neoplasm by World Health Organization classification, the incidence rate of B-ALL is 1.2% in 2011-2012 with an estimated 4930 new cases in 2016 [1]. It accounts for approximately 2% of the lymphoid neoplasms, and the incidence is approximately 11 cases per million persons per year in the United States [2]. Since 1980s, clinical outcomes of B-ALL patients have been steadily improved with an overall complete remission (CR) rate of >95% and 60-85% and overall survival (OS) rate of greater than 80% and less than 50% in children and adults, respectively [3–5]. The current treatment guideline recommends risk stratification based on patient age and BCR-ABL1 translocation status. For example, patients with older ages, higher white blood cell counts, unfavorable cytogenetic changes, and residual disease after induction chemotherapy and comorbidities usually have a greater risk of relapse and shorter OS. The non-pediatric B-ALL patients are subclassified into four different groups based on age and BCR-ABL1 status: 1) Philadelphia positive (Ph+) ALL adolescent and young adult (aged 15-39 years), 2) Ph+ ALL adult (aged ≥ 40 years), 3) Ph negative (Ph-) ALL adolescent and young adult, and 4) Ph- ALL adult [6]. Meanwhile, risk stratification of ALL in childhood is based on clinical and biological factors including age, white blood cell count, cytogenetics, response to initial induction chemotherapy, and involvement in central nervous system and testis [7]. Nevertheless, the current risk stratification system fails to identify a subgroup of refractory patients in low risk groups. The subset of patients with “low risk” behaves aggressively and could be undertreated without appropriate follow-up [8].
There have been major advances in recent years on the underlying pathogenesis of B-ALL, mostly attributed to the recent development of gene expression profiling and genome-wide sequencing analyses. In addition to revealing leukemogenesis of B-ALL in more depth, novel B-ALL subtypes with clinical significance have been proposed based on the newly emerged genetic data. Biomarkers with significant prognostic and predictive values (e.g., IKZF1, CRLF2, JAK, ABL1, ABL2, CSFR, PDGFRB, CREBBP) are identified. These markers would probably be, in part or in whole, integrated into the risk stratification system after validation through large clinical cohorts.
This review will summarize current understanding of B-ALL cytogenetics, and recently identified genetic aberrations, emphasizing on novel subclassification based on genetic changes, prognostic and predictive parameters that are directly related to clinical management of B-ALL patients.
B-ALL CYTOGENTIC ABNORMALITTIES AND SUBCLASSIFICATION
B-ALL is a heterogeneous disease that is associated with a plethora of chromosomal abnormalities, involving both numerical and structural alterations, such as hyperdiploidy, hypodiploidy, translocation, and intrachromosomal amplification. Approximately 75% of B-ALL cases have recurrent chromosomal changes detectable by conventional cytogenetic analysis [9], many of which have impacts on prognosis and are used for risk stratification on some treatment protocols [10] (Table 1, Figure 1).
Table 1: Common recurrent cytogenetic abnormalities in pediatric and adult B-ALL [3, 10, 11]
Risk groups |
Cytogenetic abnormalities |
Clinical significance |
Frequency |
|---|---|---|---|
Good risk |
Hyperdiploidy (>50 chromosomes) |
Favorable prognosis |
25-30% in children; 7-8% in adults |
t(12;21)/ ETV-RUNX1 |
Favorable prognosis in children, undetermined in adults |
25% in children; 0-4% in adults |
|
Intermediate risk |
t(1;19)/E2A-PBX1 |
Intermediate to favorable prognosis |
1-6% in children; 1-3% in adults |
t(5;14)/IL3-IGH |
Intermediate |
Rare |
|
Poor risk |
t (9; 22)/BCR-ABL1 |
Poor prognosis |
1-3% in children; 25-30% in adults |
t(v;11q23)/ KMT2A (MLL) rearrangements |
Poor prognosis |
2/3 in infants; 1-2% in older children; 4-9% in adults |
|
Hypodiploidy (<44 chromosomes) |
Poor prognosis |
6% in children, 7-8% in adults |
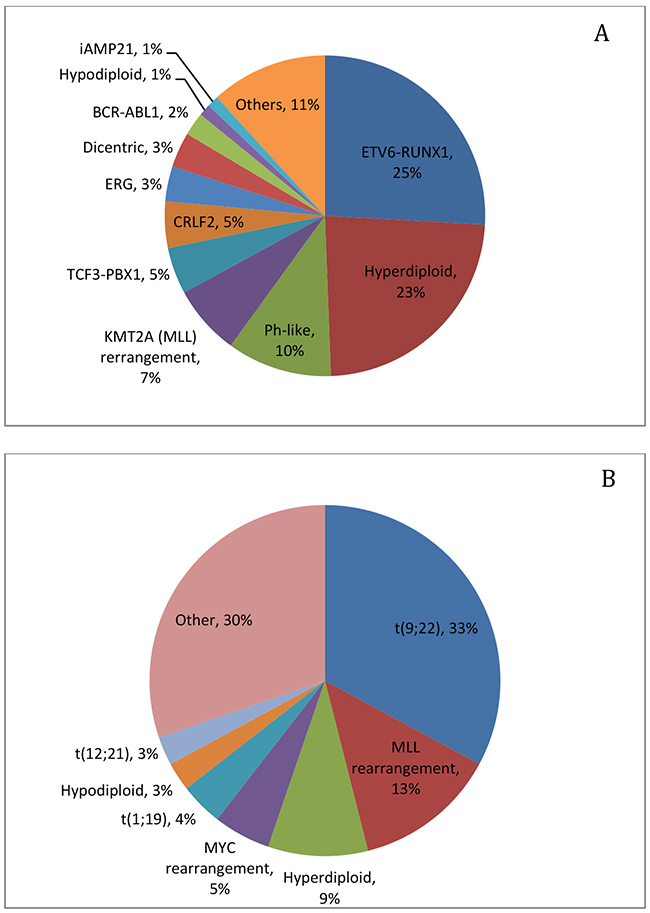
Figure 1: Frequency of cytogenetic and molecular genetic abnormalities in pediatric ALL (A) [9] and adult ALL (B) [12].
Of note, two chromosomal abnormalities, including hyperdiploidy and t(12;21)/ETV6-RUNX1 translocation, are associated with favorable clinical outcome. Cases with hyperdiploidy constitute one of the largest subgroups in B-ALL. The chromosomal gain is most often seen with chromosomes 4, 6, 10, 14, 17, 18, 21 and X [13] and least seen with chromosomes 1, 2 and 3 [14]. The overall prognosis is excellent [15]. However, extra number of specific chromosomes show different prognostic significance. For example, simultaneous trisomies of 4,10 and 17 carry the best prognosis [16]. Gain of chromosomes 4, 6, 10, and 17 indicates good prognosis [17], while gain of chromosome 5 or isochromosome 17 indicates poorer prognosis in this group [18]. Abnormality of t(12;21)/ETV6-RUNX1 is usually cryptic by conventional karyotyping but detectable by fluorescence in situ hybridization (FISH) or polymerase chain reaction (PCR). The fusion protein likely acts in a dominant negative manner, disrupting normal function of the transcription factor RUNX1. Studies show that the translocation is an early event in leukemogenesis but by itself is insufficient for the development of overt leukemia [19]. Further cooperating genetic changes appear to be needed [20, 21].
Genetic abnormalities associated with an increased risk of disease relapse or worse prognosis include t(9;22) translocation (Philadelphia chromosome, or Ph chromosome), KMT2A/MLL translocations, and hypodiploidy. The t(9;22) translocation leads to a 190 kD or 210 kD BCR-ABL fusion protein, which is a dysfunctional tyrosine kinase. The incidence of t(9;22) increases with age and is present in up to 50% in older patients [22]. The clinical outcome with conventional chemotherapy in this patient group is extremely poor. However, tyrosine kinase inhibitors (TKIs) such as imatinib mesylate, in combination with intensive chemotherapy, have been used successfully, although primary or secondary drug resistance and high rates of relapse are problematic [23]. Mutations in ABL1 (frequently T315I, Y253F/H, E255K/V, M351T, G250E, F359C/V, H396R/P, M244V, E355G, F317L, M237I, Q252H/R, D276G, L248V, F486S, etc.) are thought to be the major contributors to the drug resistance [24], for which new TKIs have been developed to bypass the signaling pathways or to bind to alternative sites, such as nilotinib, saracatinib, and ponatinib. They have shown great improvement on the clinical response in certain patients [25]. Nevertheless, clonal evolution, secondary gene aberrations such as deletions or mutations of IKZF1 (discussed below) or other genes are found to be significantly associated with the resistance and relapse [26, 27]. Rearrangements involving the KMT2A/MLL gene located at chromosome 11q23 and one of the ~80 fusion gene partners are most common in infants less than 1 year of age, and are associated with a poor prognosis. There is also a high frequency of central nervous system involvement at diagnosis. The most frequent partner gene is AF4, located at chromosome 4q21, with a fusion protein of KMT2A/MLL-AF4. The fusion proteins have an altered histone methylation pattern of KMT2A/MLL target genes, and subsequently cause leukemic transformation of the hematopoietic cells. Hypodiploidy (<46 chromosomes, with some people suggesting a stricter criteria with <44 chromosomes [28]) is a poor prognostic indicator. It can be further classified into different categories: high hypodiploidy (42-45 chromosomes), low hypodiploidy (33-39 chromosomes) and near haploidy (23-29 chromosomes) [29]. The patient has progressively poor prognosis with decreasing chromosome numbers. Near-haploidy and low hypoploidy B-ALL patients have extremely poor prognosis [30]. B-ALL with rearrangement of IGH locus occurs in less than 5% of the cases and confers poor prognosis [31]. The most common partner gene is cytokine receptor-like factor 2 (CRLF2) located at chromosome X, and other partner genes can be inhibitor of DNA binding 4 (ID4), EPOR, CCAAT/enhancer-binding protein (CEBP) family members, BCL2, the LIM domain homeobox 4 (LHX4) [17]. One of the rare B-ALL subtypes recognized in WHO classification is B-ALL with t(5;14) translocation which involves IL3 and IGH, which is characteristically associated with non-clonal eosinophilia.
Some genetic changes that are associated with poor prognosis include the very rare t(17;19)/E2A-HLF translocation [32], abnormal 17p, and loss of 13q [10], as well as complex karyotype with 5 or more abnormalities in adult B-ALL patients [33]. Certain genetic changes that do not show significant impact on prognosis include t(1;19)/E2A-PBX1 [34], del(6q), del(9p), and del(12p) [10, 11, 35, 36].
NEWLY RECOGNIZED B-ALL SUBTYPES
In the 2016 revision of WHO classification, two new provisional B-ALL subtypes with recurrent genetic abnormalities have been recognized: B-ALL with intrachromosomal amplification of chromosome 21 (iAMP21), and B-ALL with translocations involving tyrosine kinases or cytokine receptors (“BCR-ABL1-like ALL”) [37]. These two entities further identify subgroups of B-ALL patients who have inferior clinical outcome and may benefit from more aggressive therapies or combination regimens with targeted therapy.
B-ALL with intrachromosomal amplification of chromosome 21 (iAMP21)
Intrachromosomal amplification of chromosome 21 (iAMP21) is present in about 2% of pediatric B-ALL, mostly in older children and adolescents (median age 9 years), but is uncommon in adults. The patients harbor amplification of a large but variable region of chromosome 21, which can be detected by FISH with a RUNX1 probe that reveals extra signals (5 or more copies per interphase nucleus, or 3 or more copies on a single abnormal chromosome 21 in metaphase FISH). This aberration manifests instability of chromosome 21 [38].
The patients are characterized by lower white blood cell and blast cell counts, older age, the French-American-British classification (FAB) L1 morphology, and common B-lymphoblast immunophenotype with a subset showing aberrant myeloid-associated antigen expression [39]. These cases can be detected by conventional karyotyping analysis by identification of the absence of a second normal copy of chromosome 21, which may not always be present, and concurrent FISH studies using RUNX1 probe [39].
B-ALL with iAMP21 patients’ presentation of pancytopenia or mildly elevated white blood cell counts at diagnosis (usually ≤10 × 109/L, with most ≤50 × 109/L) may reduce the risk stratification. However, the patients demonstrated a consistently poor prognosis with worse event-free survival and OS when treated with standard-risk chemotherapy regimens [40]. In addition, cytogenetic change of iAMP21 has been confirmed to be a primary genetic event [38]. Therefore, B-ALL with iAMP21 is now considered as a distinct cytogenetic subgroup of B-ALL associated with a poor prognosis in pediatric patient population, and it is justified to assign such patients in the very high-risk group and treat them with more intensive chemotherapy. The clinical outcome has been significantly improved with more aggressive therapy [30, 41]. As iAMP21 is extremely rare in adults, its prognostic effect in adult group is undetermined.
B-ALL with translocations involving tyrosine kinases or cytokine receptors (BCR-ABL1-like ALL, or Ph-like ALL)
BCR-ABL1-like ALL is a subgroup of B-ALL associated with unfavorable prognosis, which was originally described as a subgroup of childhood B-ALL that lacks chromosomal rearrangement of BCR-ABL1 but exhibits similar gene expression profile to that of B-ALL with BCR-ABL1 rearrangement [42, 43], and shares the similar poor prognosis and high risk for relapse [44, 45]. More studies suggest that BCR-ABL1-like ALL occurs in all age groups, accounting for up to 15% of children, 20-25% of adolescents and young adults, and 20-25% of adults with B-ALL, and is associated with event-free and OS rates equal or inferior to high-risk ALL subtypes, including BCR-ABL1 positive and KMT2A/MLL-rearranged B-ALL [8, 46, 47].
BCR-ABL1-like ALL is a heterogeneous subgroup involving many different genetic alterations. There are several common underlying genetic changes: translocations involving tyrosine kinases other than ABL1, translocation involving cytokine receptor genes such as CRLF2, and activating mutations or deletions of critical genes such as tyrosine kinase genes (ABL1, JAK2. etc.) and Ras signaling pathway genes [46] (see below for a more detailed discussion). More importantly, in this group, especially those with translocations involving tyrosine kinases, patients have improved clinical outcome with remarkable responses to TKI therapy [46, 48].
Currently in the clinical setting it is difficult to identify such cases without gene expression profiling assays or genetic approaches such as genome and RNA sequencing, although certain laboratory screening assays have been used to identify some of the cases, such as targeted gene sequencing (RNA sequencing, RNAseq), low-density gene expression arrays, reverse transcription polymerase chain reaction (RT-PCR) and fluorescence in situ hybridization (FISH) for known translocations. Although there has been no standard guideline established for BCR-ABL1-like ALL diagnosis at initial workup of B-ALL, possible workflow was proposed, and is adapted in the following flow chart (Figure 2) [49].
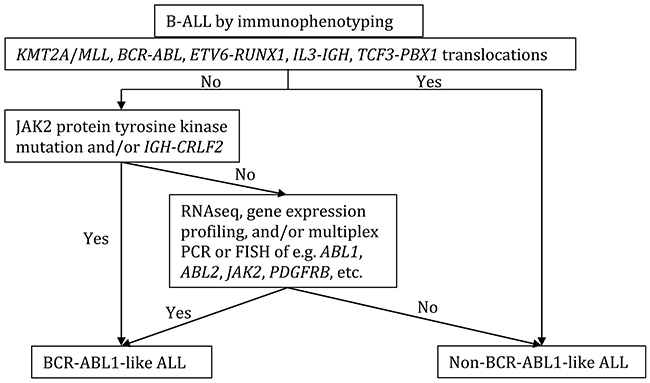
Figure 2: Proposed flow chart for the initial BCR-ABL1-like ALL workup. Modified from [49].
MOLECULAR GENETIC CHANGES
Genome-wide genetic profiling studies on B-ALL have extended our understanding of genetic landscape of B-ALL in children and young adults over the past decade. Mutations involved in various key pathways are found in different subtypes of B-ALL. The genes include transcriptional factors promoting early lymphoid cell development, e.g., PAX5, IKZF1, EBF1, ETV6, LMO2, which were detected in ~40% of B-ALL cases [50], and other genes including tumor suppressor genes and cell cycle regulators (e.g., TP53, RB1, CDKN2A/CDKN2B), cytokine receptor (e.g., CRLF2, RPOR), kinase (e.g., ABL1, ABL2, CSF1R, JAK2, PDGFRB), Ras signaling pathway (e.g., KRAS, NF1, NRAS, PTPN11), lymphoid signaling (e.g., BTLA, CD200), and epigenetic modification (e.g., EZH2, CREBBP, SETD2, MLL2, NSD2) [46, 50]. Among them, a few specific genetic alterations are found to be associated with adverse clinical outcome and increased risk for relapse [51]. Below are the most common molecular genetic changes identified in B-ALL, listed in a rough order of frequency reported in different B-ALL populations, and the most common genetic changes in BCR-ABL1-like B-ALL at the end.
IKZF1 mutations
Mutations of transcription factors involved in early lymphoid development are considered a hallmark of B-ALL genetic changes. The transcription factors include IKZF1, PAX5, EBF1, ETV6, LMO2, etc. Among them, IKZF1 mutation is one of the most frequent genetic aberrations in B-ALL. IKZF1 gene encodes the Ikaros transcription factor that is an important regulator of normal lymphoid development and differentiation [52, 53]. IKZF1 gene mutation is observed in high risk B-ALL, including approximately 80% of BCR-ABL1 positive B-ALL cases and 70% of BCR-ABL1-like B-ALL cases [54, 55]. IKZF1 mutations are often deletions and rarely point mutations [56, 57]. Most deletions are monoallelic and involve exons 3-6, which encode the N-terminal zinc finger DNA-binding domain [56]. The deletions result in dominant negative form of the Ikaros protein that inhibits the function of wild-type Ikaros. It has been shown that induction of mutant, dominant negative Ikaros in early pre-B cells arrest the cell differentiation, suggesting that loss of Ikaros activity contributes to B-ALL leukemogenesis and IKZF1 mutations are likely driver mutations [58]. Multiple studies support that IKZF1 mutation/deletion is an independent indicator of B-ALL unfavorable clinical outcome including chemotherapy resistance and higher risk for relapse [27, 46, 59–61].
CRLF2 alterations
CRLF2 alterations are found in approximately 8% of pediatric B-ALL patients, and more than 50% of patients with Down-syndrome associated B-ALL [62]. CRLF2 alterations are commonly gene rearrangement with immunoglobulin heavy chain locus resulting in IGH-CRLF2 fusion gene, less often interstitial deletions resulting in P2RY8-CRLF2 fusion gene, and rarely can be point mutations [63, 64]. These changes usually result in overexpression of CRLF2 (therefore can be analyzed by flow cytometry). CRLF2 alterations are associated with constitutive activation in the JAK-2 pathway such as JAK-STAT, PI3K/mTOR and BCL-2 transduction [65]. The alterations are often found in high-risk B-ALL [64], although the prognostic significance of CRLF2 deregulation in B-ALL remains controversial [63].
PAX5 alterations
Alterations of PAX5, another key transcription factor involved in normal lymphoid development, have been found in ~30% of B-ALL cases [50]. The alterations include acquired mutations, rearrangements involving various partner genes such as ETV6 and JAK2, and germline mutations [50, 66, 67]. Unlike IKZF1, PAX5 alterations do not appear to impact clinical outcomes, however, the PAX5 mutations may be driver mutations in B-ALL leukemogenesis and play a role in susceptibility of B-ALL [67, 68].
TP53 mutations
TP53 deletions and mutations are initially found in 2-4% of pediatric patients [69] and 8% of adult patients [70] at initial diagnosis of B-ALL. However, next generation sequencing (NGS) data revealed that overall TP53 mutations were present in up to 16% of B-ALL patients and the incidence increased with age and hypodiploid karyotype [71–73]. Notably, half of pediatric low hypodiploid B-ALL with 30-39 chromosomes show constitutional TP53 mutations, indicating a unique association between low hypodiploid B-ALL and Li-Fraumeni syndrome [73]. Multiple studies suggest that TP53 aberrations at diagnosis are independently associated with early relapse and poor OS [74, 75].
CREBBP mutations
Deletions and mutations of CREBBP, which encodes the transcriptional coactivators and acetyltransferase CREB binding protein, are found in 18% of relapsed pediatric B-ALL patients, but less than 1% at diagnosis in those who did not relapse [76], suggesting CREBBP gene mutations are associated with relapse of the disease. The mutations result in loss of function of CREBBP. In one study, CREBBP mutations were associated with hyperdiploid B-ALL relapse. Up to 60% of high-hyperdiploid relapse cases show CREBBP mutation, altering the clinical outcome in the favorable B-ALL group [77]. It might be a marker that can be integrated into risk stratification system after large cohort study.
ERG mutations
Several studies have identified a subgroup of pediatric B-ALL patients, comprising 3-5% of B-ALL cases, with monoallelic deletion of ERG gene, which encodes an ETS-domain-containing transcription factor [78, 79]. The deletions result in an aberrant ERG protein that functions as a competitive inhibitor of wild-type ERG [80]. The ERG deletion and other known classifying genetic lesions are mutually exclusive, suggesting that B-ALL with ERG deletion may be a distinct subtype. Interestingly, these patients generally have excellent prognosis, despite an association with frequent IKZF1 deletions, which is different from BCR-ABL1 positive and BCR-ABL1-like B-ALL cases [79]. Whether or not the ERG mutations function as a negative regulator under IKZF1 mutated status needs to be explored.
Genetic aberrations in BCR-ABL1-like B-ALL
BCR-ABL1-like B-ALL is a unique group that is subcategorized under high risk B-ALL. The genetic abnormalities in this subtype of B-ALL involve a plethora of genes that can be categorized into different subgroups (Figure 3). They usually have a high frequency of IKZF1 deletion (~70%), CRLF2 overexpression (~50%) and JAK mutations (~30%) [46, 64]. Deletions or mutations of IKZF1 are a hallmark of both BCR–ABL1–positive ALL as well as BCR-ABL1-like ALL [43, 50]. There are several types of kinase alteration in BCR-ABL1-like ALL including the rearrangements of CRLF2, rearrangements of ABL tyrosine kinase genes, rearrangements of JAK2 and EPOR, mutations activating Janus kinase and signal transducer and activator of transcription (JAK-STAT) signaling and Ras, and less common kinase alterations (NTRK3 and PTK2B) [81, 82]. Genomic profiling study on 154 B-ALL cases revealed most common rearrangements of kinase and cytokine receptor genes involving JAK2, ABL1 (with partners other than BCR), and other genes controlling tyrosine kinases including ABL2, CRLF2, CSF1R, EPOR, NTRK3, PDGFRB, PTK2B, TSLP, or TYK2, and gene mutations involving FLT3, IL7R, or SH2B3 [46]. Notably, more than 80% of the BCR-ABL1-like cases have one or more genetic abnormalities in genes involved in B lymphoid cell development, including IKZF1, TCF3 (E2A), EBF1, PAX5, and VPREB1 [42]. Kinase activation and signaling via JAK-STAT and ABL-1 pathway are also considered key pathways in B-ALL leukemic transformation. Overexpression of the cytokine receptor CRLF2 was often found to have association with JAK mutations, especially JAK2.
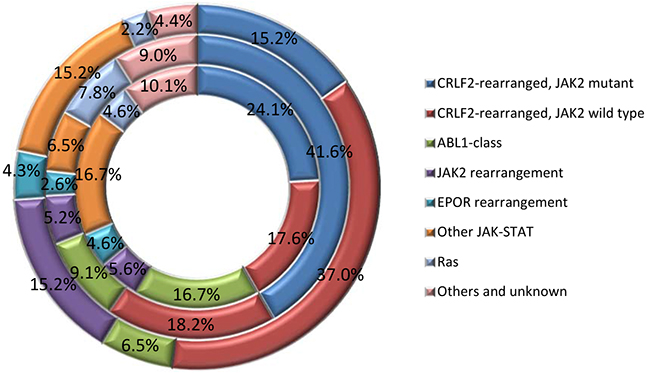
Figure 3: Breakdown of kinase alterations in children (inner doughnut), adolescents (middle doughnut) and young adults (outer doughnut) BCR-ABL1-like ALL [46, 84].
In a brief summary, genetic changes underlying kinase-signaling pathways dysregulation include 1) ABL1-like rearrangements involving ABL1, ABL2, CSF1R and PDGFRB; 2) JAK2 or EPOR rearrangements; 3) CRLF2 rearrangements (often with JAK gene mutations and activation of JAK-STAT signaling); 4) Ras signaling pathway gene mutations; and 5) uncommon kinase alterations including NTRK3, PTK2B TYK2, etc. [46]. The clinical significance of the activation of kinase-signaling pathways is that the patients can be benefited from tyrosine kinase inhibitor therapy [46, 83].
CLINICAL IMPLICATIONS OF GENETIC ABNORMALITIES
The current treatment regimens recommended by National Comprehensive Cancer Network guideline has mainly been based on risk stratification with appropriate chemotherapy intensification for high-risk patients [6]. Over the last several decades, advances in the treatment of B-ALL have significantly improved the clinical outcomes. Besides the successful utilization of TKIs in the treatment of BCR-ABL1-positive B-ALL, collaborative studies such as the project Therapeutically Applicable Research to Generate Effective Treatments (TARGET) (https://ocg.cancer.gov/programs/target/) use comprehensive molecular characterization to determine the genetic changes and identify therapeutic targets and prognostic markers. The studies identified novel targets for therapy in high risk and relapsed B-ALL, as summarized in Table 2 [85].
Table 2: Potential targeted therapy in B-ALL ([46, 85], if not otherwise specified)
Altered singling pathways |
Inhibitor |
FDA approved medication |
Potential agents |
|---|---|---|---|
BCR-ABL1-like ALL |
|||
ABL1, ABL2, CSFR, PDGFRB |
TKIs |
Imatinib * |
|
CRLF2, JAK2, EPOR, TSLP |
JAK2 inhibitor |
Ruxolitinib # |
|
IL2RB |
JAK1/JAK3 inhibitor |
Tofacitinib # |
|
NTRK3 |
NTRK3 inhibitor |
Crizotinib # |
|
TYK2 |
TYK2 inhibitor |
Ndi-031301 [92] |
|
PTK2B |
FAK inhibitor |
VS-4718 [93] |
|
CREBBP (CREB-binding protein- CBP) |
Histone deacetylase (HiDAC) inhibitors |
ICG-001 (bind to CBP) [94] |
|
Mutations in Ras/RTK pathway and PI3K pathway genes |
PI3K/mTOR inhibitors |
Rapamycin # [95] |
|
MLL/KTM2A rearrangement |
Inhibitor of histone methyltransferase: DOLT1, FLT3 inhibitors |
||
Hypodiploidy (TP53, RAS/RTK/PI3K pathways) |
MEK inhibitors PI3K inhibitors |
Trametinib # [91] |
Selumetinib [90] |
Hyperdiploidy (RAS pathway) |
MEK inhibitors |
Selumetinib [90] |
*. FDA approved for lymphoblastic leukemia treatment
#. FDA approved for other diseases but not for acute lymphoblastic leukemia
TKIs addition to cytotoxic chemotherapy in patients with BCR-ABL1-like B-ALL has significantly improved even-free survival and OS [46, 48, 83, 86]. The group of TKIs has been widely implicated in B-ALL patients who harbored ABL1, CSF1R and PDGFRB aberrations. Studies have shown that BCR-ABL1-like B-ALL cases, including cases with gene rearrangement involving ABL1, JAK2, PDGFRB and IL7R and other tyrosine kinase genes, had a poor response to conventional induction chemotherapy but showed sustained responses to TKIs [46, 48, 83, 86].
Preclinical studies demonstrated that JAK kinase inhibitors can be used in B-ALL with activated JAK-STAT signaling such as in B-ALL with CRLF2 rearrangements and JAK gene mutations [87]. JAK2 inhibitors are particularly used in the subgroup of B-ALL with JAK2, EPOR, CRLF2 and TSLP aberrations, while JAK1 and JAK3 inhibitors selectively inhibit patients with IL2RB gene rearrangements. Likewise, PI3K/mTOR pathway inhibitors and Ras signaling pathway inhibitors are exploited and may become new therapeutic targets for high risk and relapsed B-ALL subgroups [88, 89]. Multiple small molecules developed to target specific pathways in the preclinical studies have shown potential treatment effects as well. MEK inhibitors (e.g. selumetinib) can overcome glucocorticosteroid resistance in B-ALL [90]. Korfi K. et al. used anti-MEK molecule, MEKi/Trametinib, and BCL-2/BCL-XL family inhibitors to increase inhibitory functions and to induce apoptosis in B-ALL cells [91].
Besides the aforementioned targeted therapies for newly diagnosed or refractory B-ALL, several monoclonal antibodies (anti-CD20, rituximab; anti-CD52, alemtuzumab; and anti-CD22, epratuzumab) are developed and added to conventional chemotherapy to achieve optimal clinical outcomes. Novel agents such as inotuzumab ozogamicin (anti-CD22, immunoconjugate) and blinatumomab (anti-CD19 BiTE antibody) have also been adopted as single agent therapy for those with relapsed or refractory B-ALL [99]. Clinically a combination of monoclonal antibody and targeted pathway inhibitor might be the option to achieve synergistic effects. In addition, targeted immunotherapty using chimeric antigen receptor (CAR) modified T cells targeting CD19 has emerged as a powerful targeted immunotherapy, particularly to relapsed and refractory B-ALL with high response rates and durable remissions reported [100].
However, it is still in early stage for B-ALL target therapy, and it may take a lengthy time to translate the promising therapeutic agents in preclinical studies to clinical implementation. Development of more inhibitory small molecules, study of their efficacy and side effects clinically and careful clinical evaluation of the long term outcome of the targeted agents, including combination use of two or more agents, are the future directions. It will be necessary to put more priority on treatment of high-risk and relapsed B-ALL cases, and to minimize development of drug resistance.
CONCLUSIONS
In conclusion, gene expression profiling and genome-wide sequencing analyses have made great advancement over the past few years in understanding B-ALL biology and genetics. The development is very helpful in subclassifying B-ALL patients with different risks, identifying novel therapeutic targets, and improving the overall clinical outcomes. Biomarkers with prognostic and predictive values, as well as targeted therapeutic agents, have been emerged as promising approaches in clinical care of B-ALL in the era of personalized medicine.
Still, current challenges include fully understanding the genetic basis of B-ALL, discovering more efficacious therapeutic regimens, and importantly, identifying B-ALL subgroups with characteristic molecular features that can be used in targeted therapy, such as B-ALL with other kinase-activating aberrancies. There is a need to implement molecular diagnosis and subclassification in our practice, in order to utilize the potentially more efficacious therapeutic agents for this malignancy in the era of personalized medicine.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Podyma-Inoue KA, Moriwaki T, Rajapakshe AR, Terasawa K, Hara-Yokoyama M. Characterization of heparan sulfate proteoglycan-positive recycling endosomes isolated from glioma cells. Cancer Genomics Proteomics. 2016; 13: 443-52.
2. Dores GM, Devesa SS, Curtis RE, Linet MS, Morton LM. Acute leukemia incidence and patient survival among children and adults in the United States, 2001-2007. Blood. 2012; 119: 34-43.
3. Swerdlow SH, International Agency for Research on Cancer, World Health Organization. (2008). WHO classification of tumours of haematopoietic and lymphoid tissues. (Lyon, France: International Agency for Research on Cancer).
4. Trigg ME, Sather HN, Reaman GH, Tubergen DG, Steinherz PG, Gaynon PS, Uckun FM, Hammond GD. Ten-year survival of children with acute lymphoblastic leukemia: a report from the Children’s Oncology Group. Leuk Lymphoma. 2008; 49: 1142-54.
5. Hunger SP, Lu X, Devidas M, Camitta BM, Gaynon PS, Winick NJ, Reaman GH, Carroll WL. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the children’s oncology group. J Clin Oncol. 2012; 30: 1663-9.
6. National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology (NCCN guidelines) acute lymphoblatic leukemia (Version 1.2017). https://www.nccn.org/professionals/physician_gls/pdf/all.pdf. Accessed June 27, 2017.
7. Schultz KR, Pullen DJ, Sather HN, Shuster JJ, Devidas M, Borowitz MJ, Carroll AJ, Heerema NA, Rubnitz JE, Loh ML, Raetz EA, Winick NJ, Hunger SP, et al. Risk- and response-based classification of childhood B-precursor acute lymphoblastic leukemia: a combined analysis of prognostic markers from the Pediatric Oncology Group (POG) and Children’s Cancer Group (CCG). Blood. 2007; 109: 926-35.
8. Jain N, Roberts KG, Jabbour E, Patel K, Eterovic AK, Chen K, Zweidler-McKay P, Lu X, Fawcett G, Wang SA, Konoplev S, Harvey RC, Chen IM, et al. Ph-like acute lymphoblastic leukemia: a high-risk subtype in adults. Blood. 2017; 129: 572-81.
9. Mullighan CG. Molecular genetics of B-precursor acute lymphoblastic leukemia. J Clin Invest. 2012; 122: 3407-15.
10. Moorman AV, Ensor HM, Richards SM, Chilton L, Schwab C, Kinsey SE, Vora A, Mitchell CD, Harrison CJ. Prognostic effect of chromosomal abnormalities in childhood B-cell precursor acute lymphoblastic leukaemia: results from the UK Medical Research Council ALL97/99 randomised trial. Lancet Oncol. 2010; 11: 429-38.
11. Mrozek K, Harper DP, Aplan PD. Cytogenetics and molecular genetics of acute lymphoblastic leukemia. Hematol Oncol Clin North Am. 2009; 23: 991-1010.
12. Pui CH, Jeha S. New therapeutic strategies for the treatment of acute lymphoblastic leukaemia. Nat Rev Drug Discov. 2007; 6: 149-65.
13. Paulsson K, Johansson B. High hyperdiploid childhood acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2009; 48: 637-60.
14. Heerema NA, Raimondi SC, Anderson JR, Biegel J, Camitta BM, Cooley LD, Gaynon PS, Hirsch B, Magenis RE, McGavran L, Patil S, Pettenati MJ, Pullen J, et al. Specific extra chromosomes occur in a modal number dependent pattern in pediatric acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2007; 46: 684-93.
15. Jarosova M, Volejnikova J, Porizkova I, Holzerova M, Pospisilova D, Novak Z, Vrbkova J, Mihal V. Chromosomal aberrations in childhood acute lymphoblastic leukemia: 15-year single center experience. Cancer Genet. 2016; 209: 340-7.
16. Sutcliffe MJ, Shuster JJ, Sather HN, Camitta BM, Pullen J, Schultz KR, Borowitz MJ, Gaynon PS, Carroll AJ, Heerema NA. High concordance from independent studies by the Children’s Cancer Group (CCG) and Pediatric Oncology Group (POG) associating favorable prognosis with combined trisomies 4, 10, and 17 in children with NCI Standard-Risk B-precursor Acute Lymphoblastic Leukemia: a Children’s Oncology Group (COG) initiative. Leukemia. 2005; 19: 734-40.
17. Ghazavi F, Lammens T, Van Roy N, Poppe B, Speleman F, Benoit Y, Van Vlierberghe P, De Moerloose B. Molecular basis and clinical significance of genetic aberrations in B-cell precursor acute lymphoblastic leukemia. Exp Hematol. 2015; 43: 640-53.
18. Heerema NA, Sather HN, Sensel MG, Zhang T, Hutchinson RJ, Nachman JB, Lange BJ, Steinherz PG, Bostrom BC, Reaman GH, Gaynon PS, Uckun FM. Prognostic impact of trisomies of chromosomes 10, 17, and 5 among children with acute lymphoblastic leukemia and high hyperdiploidy (> 50 chromosomes). J Clin Oncol. 2000; 18: 1876-87.
19. Wiemels JL, Cazzaniga G, Daniotti M, Eden OB, Addison GM, Masera G, Saha V, Biondi A, Greaves MF. Prenatal origin of acute lymphoblastic leukaemia in children. Lancet. 1999; 354: 1499-503.
20. Zuna J, Madzo J, Krejci O, Zemanova Z, Kalinova M, Muzikova K, Zapotocky M, Starkova J, Hrusak O, Horak J, Trka J. ETV6/RUNX1 (TEL/AML1) is a frequent prenatal first hit in childhood leukemia. Blood. 2011; 117: 368-9; author reply 70-1.
21. van der Weyden L, Giotopoulos G, Wong K, Rust AG, Robles-Espinoza CD, Osaki H, Huntly BJ, Adams DJ. Somatic drivers of B-ALL in a model of ETV6-RUNX1; Pax5(+/−) leukemia. BMC Cancer. 2015; 15: 585.
22. Rambaldi A, Attuati V, Bassan R, Neonato MG, Viero P, Battista R, Di Bona E, Rossi G, Pogliani E, Ruggeri M, Amaru R, Rivolta A, Giudici G, et al. Molecular diagnosis and clinical relevance of t(9;22), t(4;11) and t(1;19) chromosome abnormalities in a consecutive group of 141 adult patients with acute lymphoblastic leukemia. Leuk Lymphoma. 1996; 21: 457-66.
23. Liu-Dumlao T, Kantarjian H, Thomas DA, O’Brien S, Ravandi F. Philadelphia-positive acute lymphoblastic leukemia: current treatment options. Curr Oncol Rep. 2012; 14: 387-94.
24. Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J, Sawyers CL. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002; 2: 117-25.
25. Bitencourt R, Zalcberg I, Louro ID. Imatinib resistance: a review of alternative inhibitors in chronic myeloid leukemia. Rev Bras Hematol Hemoter. 2011; 33: 470-5.
26. Wetzler M, Dodge RK, Mrozek K, Stewart CC, Carroll AJ, Tantravahi R, Vardiman JW, Larson RA, Bloomfield CD. Additional cytogenetic abnormalities in adults with Philadelphia chromosome-positive acute lymphoblastic leukaemia: a study of the Cancer and Leukaemia Group B. Br J Haematol. 2004; 124: 275-88.
27. Martinelli G, Iacobucci I, Storlazzi CT, Vignetti M, Paoloni F, Cilloni D, Soverini S, Vitale A, Chiaretti S, Cimino G, Papayannidis C, Paolini S, Elia L, et al. IKZF1 (Ikaros) deletions in BCR-ABL1-positive acute lymphoblastic leukemia are associated with short disease-free survival and high rate of cumulative incidence of relapse: a GIMEMA AL WP report. J Clin Oncol. 2009; 27: 5202-7.
28. Nachman JB, Heerema NA, Sather H, Camitta B, Forestier E, Harrison CJ, Dastugue N, Schrappe M, Pui CH, Basso G, Silverman LB, Janka-Schaub GE. Outcome of treatment in children with hypodiploid acute lymphoblastic leukemia. Blood. 2007; 110: 1112-5.
29. Harrison CJ, Moorman AV, Broadfield ZJ, Cheung KL, Harris RL, Reza Jalali G, Robinson HM, Barber KE, Richards SM, Mitchell CD, Eden TO, Hann IM, Hill FG, et al. Three distinct subgroups of hypodiploidy in acute lymphoblastic leukaemia. Br J Haematol. 2004; 125: 552-9.
30. Safavi S, Forestier E, Golovleva I, Barbany G, Nord KH, Moorman AV, Harrison CJ, Johansson B, Paulsson K. Loss of chromosomes is the primary event in near-haploid and low-hypodiploid acute lymphoblastic leukemia. Leukemia. 2013; 27: 248-50.
31. Dyer MJ, Akasaka T, Capasso M, Dusanjh P, Lee YF, Karran EL, Nagel I, Vater I, Cario G, Siebert R. Immunoglobulin heavy chain locus chromosomal translocations in B-cell precursor acute lymphoblastic leukemia: rare clinical curios or potent genetic drivers? Blood. 2010; 115: 1490-9.
32. de Boer J, Yeung J, Ellu J, Ramanujachar R, Bornhauser B, Solarska O, Hubank M, Williams O, Brady HJ. The E2A-HLF oncogenic fusion protein acts through Lmo2 and Bcl-2 to immortalize hematopoietic progenitors. Leukemia. 2011; 25: 321-30.
33. Moorman AV. The clinical relevance of chromosomal and genomic abnormalities in B-cell precursor acute lymphoblastic leukaemia. Blood Rev. 2012; 26: 123-35.
34. Uckun FM, Sensel MG, Sather HN, Gaynon PS, Arthur DC, Lange BJ, Steinherz PG, Kraft P, Hutchinson R, Nachman JB, Reaman GH, Heerema NA. Clinical significance of translocation t(1;19) in childhood acute lymphoblastic leukemia in the context of contemporary therapies: a report from the Children’s Cancer Group. J Clin Oncol. 1998; 16: 527-35.
35. Cytogenetic abnormalities in adult acute lymphoblastic leukemia: correlations with hematologic findings outcome. A Collaborative Study of the Group Francais de Cytogenetique Hematologique. Blood. 1996; 87: 3135-42.
36. Secker-Walker LM, Prentice HG, Durrant J, Richards S, Hall E, Harrison G. Cytogenetics adds independent prognostic information in adults with acute lymphoblastic leukaemia on MRC trial UKALL XA. MRC Adult Leukaemia Working Party. Br J Haematol. 1997; 96: 601-10.
37. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M, Vardiman JW. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016; 127: 2391-405.
38. Rand V, Parker H, Russell LJ, Schwab C, Ensor H, Irving J, Jones L, Masic D, Minto L, Morrison H, Ryan S, Robinson H, Sinclair P, et al. Genomic characterization implicates iAMP21 as a likely primary genetic event in childhood B-cell precursor acute lymphoblastic leukemia. Blood. 2011; 117: 6848-55.
39. Johnson RC, Weinberg OK, Cascio MJ, Dahl GV, Mitton BA, Silverman LB, Cherry AM, Arber DA, Ohgami RS. Cytogenetic variation of B-lymphoblastic leukemia with intrachromosomal amplification of chromosome 21 (iAMP21): a multi-institutional series review. Am J Clin Pathol. 2015; 144: 103-12.
40. Moorman AV, Richards SM, Robinson HM, Strefford JC, Gibson BE, Kinsey SE, Eden TO, Vora AJ, Mitchell CD, Harrison CJ, UK Medical Research Council (MRC)/National Cancer Research Institute (NCRI) Childhood Leukaemia Working Party (CLWP). Prognosis of children with acute lymphoblastic leukemia (ALL) and intrachromosomal amplification of chromosome 21 (iAMP21). Blood. 2007; 109: 2327-30.
41. Harrison CJ. Blood Spotlight on iAMP21 acute lymphoblastic leukemia (ALL), a high-risk pediatric disease. Blood. 2015; 125: 1383-6.
42. Den Boer ML, van Slegtenhorst M, De Menezes RX, Cheok MH, Buijs-Gladdines JG, Peters ST, Van Zutven LJ, Beverloo HB, Van der Spek PJ, Escherich G, Horstmann MA, Janka-Schaub GE, Kamps WA, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol. 2009; 10: 125-34.
43. Mullighan CG, Su X, Zhang J, Radtke I, Phillips LA, Miller CB, Ma J, Liu W, Cheng C, Schulman BA, Harvey RC, Chen IM, Clifford RJ, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009; 360: 470-80.
44. Boer JM, Koenders JE, van der Holt B, Exalto C, Sanders MA, Cornelissen JJ, Valk PJ, den Boer ML, Rijneveld AW. Expression profiling of adult acute lymphoblastic leukemia identifies a BCR-ABL1-like subgroup characterized by high non-response and relapse rates. Haematologica. 2015; 100: e261-4.
45. Moorman AV. New and emerging prognostic and predictive genetic biomarkers in B-cell precursor acute lymphoblastic leukemia. Haematologica. 2016; 101: 407-16.
46. Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, McCastlain K, Ding L, Lu C, Song G, Ma J, Becksfort J, Rusch M, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014; 371: 1005-15.
47. Roberts KG, Gu Z, Payne-Turner D, McCastlain K, Harvey RC, Chen IM, Pei D, Iacobucci I, Valentine M, Pounds SB, Shi L, Li Y, Zhang J, et al. High frequency and poor outcome of Philadelphia chromosome-like acute lymphoblastic leukemia in adults. J Clin Oncol. 2017; 35: 394-401.
48. Weston BW, Hayden MA, Roberts KG, Bowyer S, Hsu J, Fedoriw G, Rao KW, Mullighan CG. Tyrosine kinase inhibitor therapy induces remission in a patient with refractory EBF1-PDGFRB-positive acute lymphoblastic leukemia. J Clin Oncol. 2013; 31: e413-6.
49. Herold T, Gokbuget N. Philadelphia-like acute lymphoblastic leukemia in adults. Curr Oncol Rep. 2017; 19: 31.
50. Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, Girtman K, Mathew S, Ma J, Pounds SB, Su X, Pui CH, Relling MV, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007; 446: 758-64.
51. Liu YF, Wang BY, Zhang WN, Huang JY, Li BS, Zhang M, Jiang L, Li JF, Wang MJ, Dai YJ, Zhang ZG, Wang Q, Kong J, et al. Genomic profiling of adult and pediatric B-cell acute lymphoblastic leukemia. EBioMedicine. 2016; 8: 173-83.
52. Rebollo A, Schmitt C. Ikaros, Aiolos and Helios: transcription regulators and lymphoid malignancies. Immunol Cell Biol. 2003; 81: 171-5.
53. Georgopoulos K, Bigby M, Wang JH, Molnar A, Wu P, Winandy S, Sharpe A. The Ikaros gene is required for the development of all lymphoid lineages. Cell. 1994; 79: 143-56.
54. Ensor HM, Schwab C, Russell LJ, Richards SM, Morrison H, Masic D, Jones L, Kinsey SE, Vora AJ, Mitchell CD, Harrison CJ, Moorman AV. Demographic, clinical, and outcome features of children with acute lymphoblastic leukemia and CRLF2 deregulation: results from the MRC ALL97 clinical trial. Blood. 2011; 117: 2129-36.
55. Hertzberg L, Vendramini E, Ganmore I, Cazzaniga G, Schmitz M, Chalker J, Shiloh R, Iacobucci I, Shochat C, Zeligson S, Cario G, Stanulla M, Strehl S, et al. Down syndrome acute lymphoblastic leukemia, a highly heterogeneous disease in which aberrant expression of CRLF2 is associated with mutated JAK2: a report from the International BFM Study Group. Blood. 2010; 115: 1006-17.
56. Mullighan CG, Miller CB, Radtke I, Phillips LA, Dalton J, Ma J, White D, Hughes TP, Le Beau MM, Pui CH, Relling MV, Shurtleff SA, Downing JR. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature. 2008; 453: 110-4.
57. Olsson L, Johansson B. Ikaros and leukaemia. Br J Haematol. 2015; 169: 479-91.
58. Joshi I, Yoshida T, Jena N, Qi X, Zhang J, Van Etten RA, Georgopoulos K. Loss of Ikaros DNA-binding function confers integrin-dependent survival on pre-B cells and progression to acute lymphoblastic leukemia. Nat Immunol. 2014; 15: 294-304.
59. Yao QM, Liu KY, Gale RP, Jiang B, Liu YR, Jiang Q, Jiang H, Zhang XH, Zhang MJ, Chen SS, Huang XJ, Xu LP, Ruan GR. Prognostic impact of IKZF1 deletion in adults with common B-cell acute lymphoblastic leukemia. BMC Cancer. 2016; 16: 269.
60. Boer JM, van der Veer A, Rizopoulos D, Fiocco M, Sonneveld E, de Groot-Kruseman HA, Kuiper RP, Hoogerbrugge P, Horstmann M, Zaliova M, Palmi C, Trka J, Fronkova E, et al. Prognostic value of rare IKZF1 deletion in childhood B-cell precursor acute lymphoblastic leukemia: an international collaborative study. Leukemia. 2016; 30: 32-8.
61. Clappier E, Grardel N, Bakkus M, Rapion J, De Moerloose B, Kastner P, Caye A, Vivent J, Costa V, Ferster A, Lutz P, Mazingue F, Millot F, et al. IKZF1 deletion is an independent prognostic marker in childhood B-cell precursor acute lymphoblastic leukemia, and distinguishes patients benefiting from pulses during maintenance therapy: results of the EORTC Children’s Leukemia Group study 58951. Leukemia. 2015; 29: 2154-61.
62. Buitenkamp TD, Pieters R, Gallimore NE, van der Veer A, Meijerink JP, Beverloo HB, Zimmermann M, de Haas V, Richards SM, Vora AJ, Mitchell CD, Russell LJ, Schwab C, et al. Outcome in children with Down’s syndrome and acute lymphoblastic leukemia: role of IKZF1 deletions and CRLF2 aberrations. Leukemia. 2012; 26: 2204-11.
63. Zhou Y, Kanagal-Shamanna R, Zuo Z, Tang G, Medeiros LJ, Bueso-Ramos CE. Advances in B-lymphoblastic leukemia: cytogenetic and genomic lesions. Ann Diagn Pathol. 2016; 23: 43-50.
64. Harvey RC, Mullighan CG, Wang X, Dobbin KK, Davidson GS, Bedrick EJ, Chen IM, Atlas SR, Kang H, Ar K, Wilson CS, Wharton W, Murphy M, et al. Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling: correlation with genome-wide DNA copy number alterations, clinical characteristics, and outcome. Blood. 2010; 116: 4874-84.
65. Tasian SK, Doral MY, Borowitz MJ, Wood BL, Chen IM, Harvey RC, Gastier-Foster JM, Willman CL, Hunger SP, Mullighan CG, Loh ML. Aberrant STAT5 and PI3K/mTOR pathway signaling occurs in human CRLF2-rearranged B-precursor acute lymphoblastic leukemia. Blood. 2012; 120: 833-42.
66. Nebral K, Denk D, Attarbaschi A, Konig M, Mann G, Haas OA, Strehl S. Incidence and diversity of PAX5 fusion genes in childhood acute lymphoblastic leukemia. Leukemia. 2009; 23: 134-43.
67. Shah S, Schrader KA, Waanders E, Timms AE, Vijai J, Miething C, Wechsler J, Yang J, Hayes J, Klein RJ, Zhang J, Wei L, Wu G, et al. A recurrent germline PAX5 mutation confers susceptibility to pre-B cell acute lymphoblastic leukemia. Nat Genet. 2013; 45: 1226-31.
68. Auer F, Ruschendorf F, Gombert M, Husemann P, Ginzel S, Izraeli S, Harit M, Weintraub M, Weinstein OY, Lerer I, Stepensky P, Borkhardt A, Hauer J. Inherited susceptibility to pre B-ALL caused by germline transmission of PAX5 c.547G>A. Leukemia. 2014; 28: 1136-8.
69. Zhang J, Mullighan CG, Harvey RC, Wu G, Chen X, Edmonson M, Buetow KH, Carroll WL, Chen IM, Devidas M, Gerhard DS, Loh ML, Reaman GH, et al. Key pathways are frequently mutated in high-risk childhood acute lymphoblastic leukemia: a report from the Children’s Oncology Group. Blood. 2011; 118: 3080-7.
70. Chiaretti S, Brugnoletti F, Tavolaro S, Bonina S, Paoloni F, Marinelli M, Patten N, Bonifacio M, Kropp MG, Sica S, Guarini A, Foa R. TP53 mutations are frequent in adult acute lymphoblastic leukemia cases negative for recurrent fusion genes and correlate with poor response to induction therapy. Haematologica. 2013; 98: e59-61.
71. Stengel A, Schnittger S, Weissmann S, Kuznia S, Kern W, Kohlmann A, Haferlach T, Haferlach C. TP53 mutations occur in 15.7% of ALL and are associated with MYC-rearrangement, low hypodiploidy, and a poor prognosis. Blood. 2014; 124: 251-8.
72. Muhlbacher V, Zenger M, Schnittger S, Weissmann S, Kunze F, Kohlmann A, Bellos F, Kern W, Haferlach T, Haferlach C. Acute lymphoblastic leukemia with low hypodiploid/near triploid karyotype is a specific clinical entity and exhibits a very high TP53 mutation frequency of 93%. Genes Chromosomes Cancer. 2014; 53: 524-36.
73. Holmfeldt L, Wei L, Diaz-Flores E, Walsh M, Zhang J, Ding L, Payne-Turner D, Churchman M, Andersson A, Chen SC, McCastlain K, Becksfort J, Ma J, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet. 2013; 45: 242-52.
74. Salmoiraghi S, Montalvo ML, Ubiali G, Tosi M, Peruta B, Zanghi P, Oldani E, Boschini C, Kohlmann A, Bungaro S, Intermesoli T, Terruzzi E, Angelucci E, et al. Mutations of TP53 gene in adult acute lymphoblastic leukemia at diagnosis do not affect the achievement of hematologic response but correlate with early relapse and very poor survival. Haematologica. 2016; 101: e245-8.
75. Hof J, Krentz S, van Schewick C, Korner G, Shalapour S, Rhein P, Karawajew L, Ludwig WD, Seeger K, Henze G, von Stackelberg A, Hagemeier C, Eckert C, et al. Mutations and deletions of the TP53 gene predict nonresponse to treatment and poor outcome in first relapse of childhood acute lymphoblastic leukemia. J Clin Oncol. 2011; 29: 3185-93.
76. Mullighan CG, Zhang J, Kasper LH, Lerach S, Payne-Turner D, Phillips LA, Heatley SL, Holmfeldt L, Collins-Underwood JR, Ma J, Buetow KH, Pui CH, Baker SD, et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature. 2011; 471: 235-9.
77. Inthal A, Zeitlhofer P, Zeginigg M, Morak M, Grausenburger R, Fronkova E, Fahrner B, Mann G, Haas OA, Panzer-Grumayer R. CREBBP HAT domain mutations prevail in relapse cases of high hyperdiploid childhood acute lymphoblastic leukemia. Leukemia. 2012; 26: 1797-803.
78. Zaliova M, Zimmermannova O, Dorge P, Eckert C, Moricke A, Zimmermann M, Stuchly J, Teigler-Schlegel A, Meissner B, Koehler R, Bartram CR, Karawajew L, Rhein P, et al. ERG deletion is associated with CD2 and attenuates the negative impact of IKZF1 deletion in childhood acute lymphoblastic leukemia. Leukemia. 2014; 28: 182-5.
79. Clappier E, Auclerc MF, Rapion J, Bakkus M, Caye A, Khemiri A, Giroux C, Hernandez L, Kabongo E, Savola S, Leblanc T, Yakouben K, Plat G, et al. An intragenic ERG deletion is a marker of an oncogenic subtype of B-cell precursor acute lymphoblastic leukemia with a favorable outcome despite frequent IKZF1 deletions. Leukemia. 2014; 28: 70-7.
80. Mullighan CG. The molecular genetic makeup of acute lymphoblastic leukemia. Hematology Am Soc Hematol Educ Program. 2012; 2012: 389-96.
81. Hunger SP, Mullighan CG. Redefining ALL classification: toward detecting high-risk ALL and implementing precision medicine. Blood. 2015; 125: 3977-87.
82. Mullighan CG. The genomic landscape of acute lymphoblastic leukemia in children and young adults. Hematology Am Soc Hematol Educ Program. 2014; 2014: 174-80.
83. Lengline E, Beldjord K, Dombret H, Soulier J, Boissel N, Clappier E. Successful tyrosine kinase inhibitor therapy in a refractory B-cell precursor acute lymphoblastic leukemia with EBF1-PDGFRB fusion. Haematologica. 2013; 98: e146-8.
84. Roberts KG, Mullighan CG. Genomics in acute lymphoblastic leukaemia: insights and treatment implications. Nat Rev Clin Oncol. 2015; 12: 344-57.
85. Harrison CJ. Targeting signaling pathways in acute lymphoblastic leukemia: new insights. Hematology Am Soc Hematol Educ Program. 2013; 2013: 118-25.
86. Roberts KG, Morin RD, Zhang J, Hirst M, Zhao Y, Su X, Chen SC, Payne-Turner D, Churchman ML, Harvey RC, Chen X, Kasap C, Yan C, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell. 2012; 22: 153-66.
87. Wu SC, Li LS, Kopp N, Montero J, Chapuy B, Yoda A, Christie AL, Liu H, Christodoulou A, van Bodegom D, van der Zwet J, Layer JV, Tivey T, et al. Activity of the type II JAK2 inhibitor CHZ868 in B cell acute lymphoblastic leukemia. Cancer Cell. 2015; 28: 29-41.
88. Tasian SK, Teachey DT, Rheingold SR. Targeting the PI3K/mTOR pathway in pediatric hematologic malignancies. Front Oncol. 2014; 4: 108.
89. Knight T, Irving JA. Ras/Raf/MEK/ERK pathway activation in childhood acute lymphoblastic leukemia and its therapeutic targeting. Front Oncol. 2014; 4: 160.
90. Polak A, Kiliszek P, Sewastianik T, Szydlowski M, Jablonska E, Bialopiotrowicz E, Gorniak P, Markowicz S, Nowak E, Grygorowicz MA, Prochorec-Sobieszek M, Nowis D, Golab J, et al. MEK inhibition sensitizes precursor B-cell acute lymphoblastic leukemia (B-ALL) cells to dexamethasone through modulation of mTOR activity and stimulation of autophagy. PLoS One. 2016; 11: e0155893.
91. Korfi K, Smith M, Swan J, Somervaille TC, Dhomen N, Marais R. BIM mediates synergistic killing of B-cell acute lymphoblastic leukemia cells by BCL-2 and MEK inhibitors. Cell Death Dis. 2016; 7: e2177.
92. Akahane K, Li Z, Etchin J, Berezovskaya A, Gjini E, Masse CE, Miao W, Rocnik J, Kapeller R, Greenwood JR, Tiv H, Sanda T, Weinstock DM, et al. Anti-leukaemic activity of the TYK2 selective inhibitor NDI-031301 in T-cell acute lymphoblastic leukaemia. Br J Haematol. 2017; 177: 271-82.
93. Wang X, Mak PY, Mu H, Jiang X, Mak D, Pachter JA, Weaver DT, Xu B, Andreeff M, Carter BZ. Inhibition of FAK exerts anti-leukemic activity and potentiates ABT-199-induced apoptosis in AML. Blood. 2016; 128: 1574.
94. Gang EJ, Hsieh YT, Pham J, Zhao Y, Nguyen C, Huantes S, Park E, Naing K, Klemm L, Swaminathan S, Conway EM, Pelus LM, Crispino J, et al. Small-molecule inhibition of CBP/catenin interactions eliminates drug-resistant clones in acute lymphoblastic leukemia. Oncogene. 2014; 33: 2169-78.
95. Dinner S, Platanias LC. Targeting the mTOR pathway in leukemia. J Cell Biochem. 2016; 117: 1745-52.
96. Knapper S, Russell N, Gilkes A, Hills RK, Gale RE, Cavenagh JD, Jones G, Kjeldsen L, Grunwald MR, Thomas I, Konig H, Levis MJ, Burnett AK. A randomised assessment of adding the kinase inhibitor lestaurtinib to 1st-line chemotherapy for FLT3-mutated AML. Blood. 2016.
97. Brown P, Levis M, McIntyre E, Griesemer M, Small D. Combinations of the FLT3 inhibitor CEP-701 and chemotherapy synergistically kill infant and childhood MLL-rearranged ALL cells in a sequence-dependent manner. Leukemia. 2006; 20: 1368-76.
98. Kerstjens M, Driessen EM, Willekes M, Pinhancos SS, Schneider P, Pieters R, Stam RW. MEK inhibition is a promising therapeutic strategy for MLL-rearranged infant acute lymphoblastic leukemia patients carrying RAS mutations. Oncotarget. 2017; 8: 14835–46. https://doi.org/10.18632/oncotarget.11730.
99. Portell CA, Advani AS. Novel targeted therapies in acute lymphoblastic leukemia. Leuk Lymphoma. 2014; 55: 737-48.
100. Maude SL, Teachey DT, Porter DL, Grupp SA. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood. 2015; 125: 4017-23.