
INTRODUCTION
The association between thrombosis and cancer has long been recognized and many clinical and experimental studies have indicated the involvement of hypercoagulopathy in cancer progression and unfavorable outcome [1, 2]. Patients with cancer are often at high risk of developing thrombosis, including venous and arterial thrombosis and systemic syndromes, such as disseminated intravascular coagulation [1–3]. Tissue factor (TF) is a 47 kDa transmembrane glycoprotein that is normally present in subendothelial cells away from circulation; upon tissue damage, TF forms a complex with factor VIIa (FVIIa) to initiate the extrinsic coagulation process. However, it is now known that this tightly controlled hemostatic process can be hijacked by tumors. In this setting aberrant expression of TF in several types of aggressive tumor plays a key role in tumor-initiated thrombosis and is associated with metastatic properties and poor disease prognosis [4–6]. It has been suggested that the tumor TF-induced procoagulant state increases fibrin formation and facilitates tumor cell survival, immune escape and metastasis [7–9]. TF/FVIIa complex also binds and stimulates protease activated receptor 2 (PAR2)-mediated intracellular signaling pathway and activates ERK/MAPK and PI3K pathways, which positively regulates integrin signal, production of chemokines, proangiogenic factors and matrix metalloproteinases thereby facilitating inflammation, angiogenesis and tumor growth [10, 11].
Pathological TF expression has been reported in several cancer types [5, 12–21]. The mechanisms for TF dysregulation involve multiple oncogenic drivers, including those of PI3K, KRAS, EGFRvIII and loss of PTEN or p53 [14–18, 22]. TF is also upregulated in response to VEGF, tumor necrosis factor (TNF)-α and hypoxia-inducible factor-1α (HIF-1α) [15, 22]. In addition, post-transcriptional modification of the TF gene via alternative splicing (asTF) and micro RNA (miRNA) also contribute to tumor development and metastasis [13, 23]. A recent report indicates that TF expression is induced by epithelial-mesenchymal transition (EMT) and triggers a procoagulant state and metastasis of circulating tumor cells [24].
TF represents a potential target for cancer treatment. Numerous experimental studies have provided clues that targeting TF in aggressive tumors will likely be beneficial. Specific blockade of TF functions in tumor cells by TF knockdown, monoclonal antibody (mAb) or by tissue factor pathway inhibitor (TFPI) resulted in inhibition of tumor growth, metastasis and angiogenesis [11, 25–32]. A TF mAb (TF-011)-drug conjugate Tisotumab Vedotin showed promising antitumor activity in preclinical model of TF-positive tumors [33] and has entered patient trial (e.g. NCT02001623).
In this report, we found that TF is frequently expressed in highly invasive triple negative breast cancer (TNBC) and in pancreatic adenocarcinoma (PaC). TNBC and PaC are among the most lethal forms of malignancy in the clinic with extremely poor survival and no effective therapy [34, 35]. To explore the potential for TF as a therapeutic target in these challenging tumor types, we have developed a novel anti-TF therapeutic antibody SC1. Abrogation of TF function in TNBC or PaC cells via ShRNA or TF-mAb SC1 blocked tumor-initiated coagulation process, inhibited TF-PAR2-dependent MAPK/ERK phosphorylation cascade and attenuated tumor growth and metastasis. We also show that the antibody-drug conjugate SC1-ADC exerted excellent antitumor activity in tumor models of TNBC and PaC.
RESULTS
TF is aberrantly present in a large percentage of highly invasive Basal-like/TNBC breast cancer and pancreatic cancer
We first analyzed breast cancer cell gene expression profile dataset generated by Neve et al. [36], which included 25 luminal- and 25 basal A/B lines. We found that TF mRNA level was significantly higher in the basal A/B group, many of which are well established TNBC cells (p<0.001) (Figure 1A). Analysis of the Cancer Cell Line Encyclopedia (CCLE) database reached a similar result (not shown). We then examined TF protein in a panel of breast cancer lines by immunoblotting, in which majority of the TNBC lines (MDA-MB-231, HCC1806, Hs578T, HCC1937, HCC38, MDA-MB-468, Bcap-37 and HBL-100) exhibited aberrant TF protein levels compared to that for only one luminal line (Figure 1B). We next examined 522 breast tumor mRNA dataset from TCGA [37]. Tumors were grouped into “Basal-like” or “Luminal-like” based on mRNA expression levels of basal-keratins KRT5/14- vs. luminal-keratins KRT8/18 [38, 39]. We found that TF mRNA level was also higher in the “Basal-like” compared to the “Luminal-like” group of tumors (p<0.001) (Figure 1C). To examine TF levels in PaC, we analyzed GEO dataset with 36 normal- and 36 PaC samples [40]. TF mRNA was much higher in PaC compared to normal pancreas (Figure 1D). This result was verified in PaC cells by immunoblotting (Figure 1E) and was consistent with a previous study employing tissue array [20].
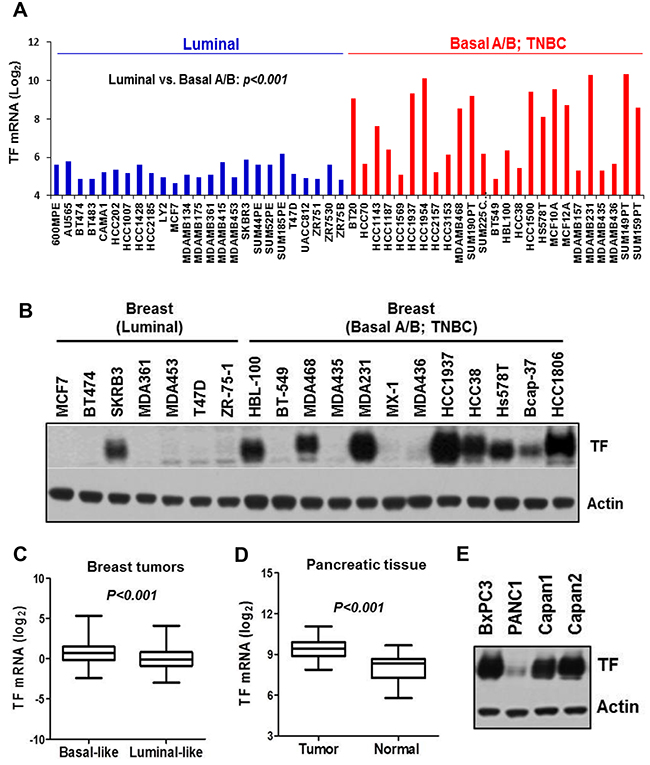
Figure 1: TF is aberrantly expressed in invasive basal-like breast cancer/TNBC and PaC. (A) TF mRNA levels for 25 luminal- and 25 basal A/B breast cell lines classified by Neve et al. [36] were plotted. (B) Total cell lysates prepared from the indicated breast cell lines were immunoblotted for TF protein level. (C) Affymetrix array data for 522 breast tumors [37] were sorted into “Basal-like” vs. “Luminal-like” based on mRNA expression ratio (KRT5 plus KRT14)/(KRT8 plus KRT18) [38] with median value of the dataset served as group cutoff. TF mRNA levels are plotted. (D) TF mRNA levels in pancreatic tumors vs. normal pancreatic tissues were analyzed from Gene Expression Omnibus (GEO) repository GSE15471 [40]. (E) Total cell lysates prepared from the indicated pancreatic cancer cell lines were immunoblotted for TF protein level.
TF mRNA level in breast cancer correlates with “Basal-like” and “EMT-high” gene signature
In the dataset of 50 breast cancer lines TF expression clustered with higher levels of basal-marker vimentin, keratin KRT5/14 and caveolin-1 (CAV1) but lower levels of luminal-marker keratin KRT8/18, ERBB3 and ESR1 (Figure 2A). Comparative analysis of 522 tumors revealed a similar association of TF with higher expression of basal markers but lower expression of luminal markers (Figure 2B top panel). We further grouped tumors by “EMT-high” or “EMT-low” based on levels of vimentin- vs. E-cadherin expression [41]. TF mRNA level was higher in the “EMT-high” tumors (p<0.001), which also clustered with higher basal markers and lower luminal markers (Figure 2B bottom panel). Together, these results indicate that TF is aberrantly present in a large percentage of aggressive tumors with highly invasive and metastatic properties and may present as a promising target for treatment of TNBC.
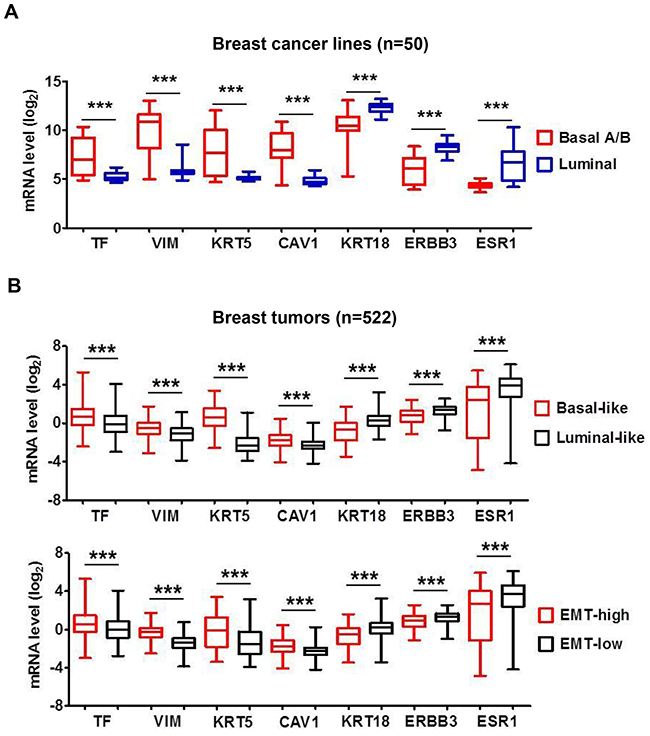
Figure 2: Breast cancer TF mRNA level correlates with “Basal-like” and “EMT-high” signature. (A) The panel of 50 breast cancer cell lines as in Figure 1A was analyzed employing the Affymetrix array data file (36). Representative mRNA levels of the indicated genes in the 50 cell lines were analyzed and plotted. Vimentin (VIM), keratin-5 (KRT5) and caveolin-1 (CAV1) are representative Basal-markers, while keratin-18 (KRT18), ERBB3 and ESR1 are representative Luminal-markers. (B) Affymetrix array data for 522 breast tumors [37] were sorted into “Basal-like” vs. “Luminal-like” or “EMT-high” vs. “EMT-low” based on mRNA expression ratio (KRT5 plus KRT14)/(KRT8 plus KRT18) [38] or vimentin/E-cadherin [39], respectively. Median values of the datasets served as group cutoff. Representative mRNA levels for TF, VIM, KRT5, CAV1, KRT18, ERBB3 and ESR1 were analyzed and compared between “Basal-like” vs. “Luminal-like” tumors (top panel) or “EMT-high” vs. “EMT-low” tumors (bottom panel), respectively. ***, P<0.001.
SC1 is a novel TF-mAb with high affinity in blocking cell surface TF, TF:FVIIa-PAR2 signaling and tumor-initiated coagulation in TNBC and PaC
We immunized mice with the extracellular domain of human TF (TF-ECD) and obtained a panel of original human TF-specific monoclonal antibodies (Supplementary Figure 1A). Biochemical affinity assays and cellular pharmacology results identified SC1 (mouse IgG2bκ) as a candidate for further development. IgG variable regions of the SC1 heavy (VH) and light (VL) chains were cloned and sequenced, and sufficient quantity of purified SC1 was obtained (Supplementary Figure 1B). As expected, SC1 immunoblotted well with the cell lysates of high-TF TNBC and PaC (Supplementary Figure 1C) and showed similar immunostaining as the previously established TF-mAb TF-011 [33] in the BxPC3 tumors with the strongest staining on surface of the tumor cells (Supplementary Figure 1D). In ELISA measurement, SC1 binds to TF-ECD with a mean EC50 value 0.019 nM (Figure 3A). SC1 elicited a dose-dependent binding to cell surface TF in the TNBC MDA-MB-231 and PaC BxPC3 cells with mean EC50 values 2.5 nM and 2.6 nM, respectively (Figure 3B, 3C). In BxPC3 cells, the TF:FVIIa-PAR2-induced ERK/MAPK phosphorylation was blocked by SC1 at 2-3 nM (Figure 3D). Lastly SC1 and TF-011 inhibited both the MDA-MB-231- and BxPC3-initiated coagulation as assessed by clotting time (Figure 3E, Supplementary Figure 2A) or FXa generation assay (Figure 3F, Supplementary Figure 2B) exhibiting the low nM IC50 values. These results indicate that SC1 recognizes the functional epitope(s) in TF extracellular domain and perturbs at least two major functions of TF, namely the coagulation function and the TF:FVII-PAR2 intracellular signaling activity.
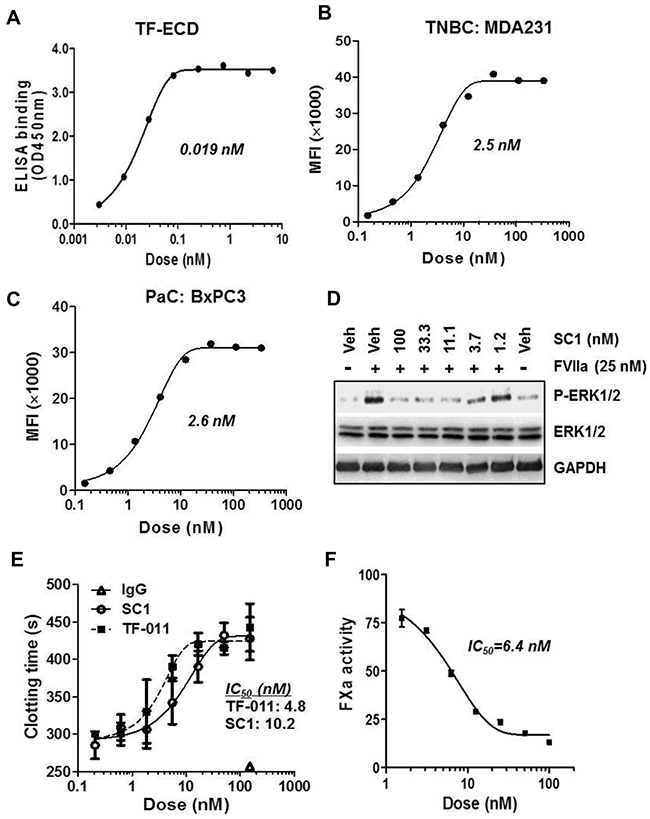
Figure 3: Biochemical and cellular activity profile of SC1. (A) Binding affinity of SC1 to TF-ECD as measured by ELISA. SC1 dose response curve and EC50 value are shown. (B and C) Binding affinity of SC1 to cell surface TF in MDA-MB-231 (B) and BxPC3 (C) was assessed by FACS analysis. SC1 dose response curves and EC50 values are shown. (D) Serum-starved BxPC3 cells were pre-treated with SC1 for 1 h, stimulated with FVIIa for 15 min and subjected to immunoblotting. (E and F) MDA-MB-231 cell-initiated coagulation was inhibited by SC1. Assays for clotting with SC1 and TF-011 (E) and FXa generation with SC1 (F) were performed as described in Methods. Representative assay results are shown. The TF-011 VL/VH regions were prepared as described [49]. MFI, mean fluorescence intensity.
SC1 inhibits TF-dependent tumor cell migration in vitro and hematogenous metastasis in vivo
Given that both the TF:FVIIa-PAR2 signaling [42] and TF-mediated coagulation [43, 44] can contribute to tumor metastasis, we examined the effect of SC1 in anti-tumor cell migration and hematogenous metastasis in vivo. We first established the doxycycline (Dox)-inducible TF knockdown cell lines of TNBC MDA-MB-231, HCC1806 and PaC BxPC3, in which efficient depletions of TF were achieved by Dox treatment (Figure 4A, Supplementary Figure 3A, 3C). When TF was depleted, both MDA-MB-231 and BxPC3 cells displayed a significant reduction in cell migration (Figure 4B, 4C). Next, consistent with TF knockdown, treatment with 100, 33.3 and 11.1 nM SC1 resulted in a dose-dependent inhibition of cell migration in the TF-high MDA-MB-231, BxPC3, HCC1806 and Hs578T cells but not in the TF-low PANC1 and A549 cells (Figure 4D, 4E, 4F). For in vivo studies, we tagged these cell lines with firefly luciferase gene. Depletion of TF in MDA-MB-231-Luc (Figure 5A, 5B), HCC1806-Luc (Supplementary Figure 3A, 3B) and BxPC3-Luc (Supplementary Figure 3C, 3D) cells all resulted in a profound inhibition in lung invasion as assessed by bioluminescence imaging at 4 h following tail-vein injection of tumor cells. Similarly, co-injection of SC1 with tumor cells also dramatically reduced MDA-MB-231 lung colonization as detected by bioluminescence at 4 h (Figure 5C, 5D) or tumor foci quantified 6 weeks following injection (Figure 5E, 5F). These results strongly indicate that TF plays an important role in tumor cell early metastasis. Blockade of TF function by SC1 and, in addition, the SC1 antibody-dependent cell-mediated cytotoxicity (ADCC) can effectively inhibit this process.
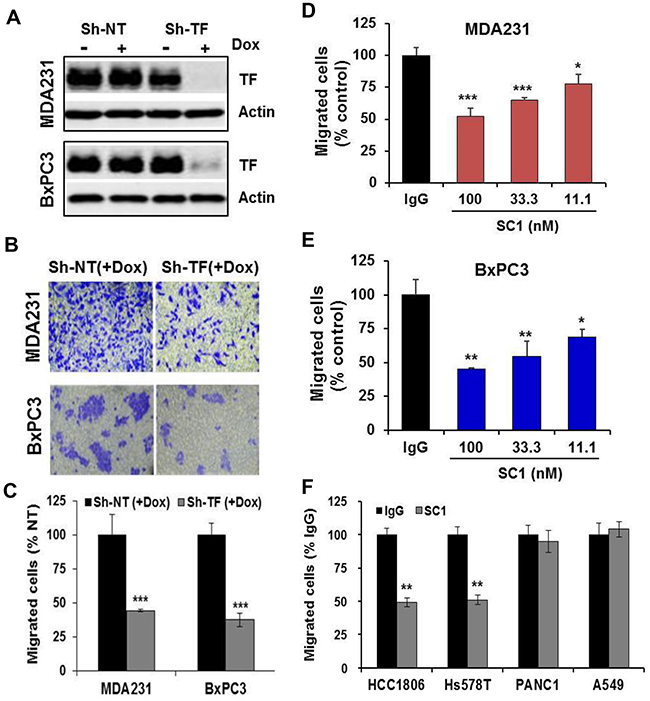
Figure 4: SC1 inhibits TF-dependent tumor cell migration. (A-C) MDA-MB-231 and BxPC3 cells transfected with TRIPZ non-targeting ShRNA (Sh-NT) or TF ShRNA (Sh-TF) were induced with doxycycline (Dox) for 5-7 days to deplete TF. The cells were subjected to immunoblotting (A) or cell migration assay. Migrated cells were stained with crystal violet (B) and the quantified results are plotted (C). (D-F) The indicated cell lines were assayed similarly for cell migration with various doses of SC1 or IgG (D, E) or 100 nM SC1 (F). Quantified results are plotted. *, P<0.05; **, P<0.01; ***, P<0.001.
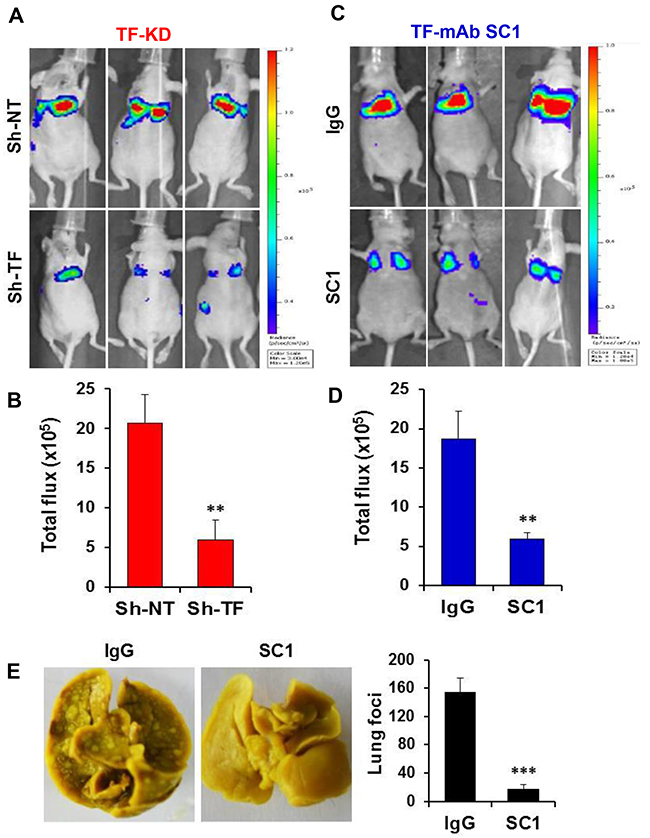
Figure 5: SC1 inhibits TNBC MDA-MB-231 cell lung metastasis. (A and B) Luciferase-tagged MDA-MB-231 cells expressing TRIPZ Sh-NT or Sh-TF were induced with doxycycline for 7 days and injected into the tail vein of Balb/c nude mice (n=6). Bioluminescence was measured 4 h later as described in Methods (A) and the results quantified based on total photon flux are plotted (B). (C and D) MDA-MB-231-Luc cells were mixed with 0.1 mg SC1 or IgG and injected into the tail vein of nude mice (n=4). Bioluminescence was similarly measured and analyzed. (E) MDA-MB-231 cells were injected into the tail vein of SCID mice (n=7) and tumor nodules in the lung were examined 6 weeks later (left panel) and quantified (right panel). **, P<0.01; ***, P<0.001.
SC1 attenuates tumor growth in vivo
Although treatment of TF-high tumor cells with SC1 did not acutely block cell proliferation in vitro (not shown), TF may be required for optimal tumor growth in vivo. Depletion of TF attenuated, albeit moderately, the growth of HCC1806 tumors (p<0.001) (Figure 6A). Co-injection of 10-100 μg SC1 with HCC1806 cells (Figure 6B) or BxPC3 cells (Figure 6C) caused a dose-dependent reduction in tumor growth (p<0.001). Finally, the weekly intravenous administration of SC1 also moderately reduced growth of established BxPC3 tumors (Figure 6D). Next, we examined stromal fibrosis, an important characteristic for PaC progression and therapy impediment [45]. The level of collagen deposition as measured by Masson’s trichrome staining was substantially reduced in the SC1-treated BxPC3 tumors compared to that of IgG control (p<0.001) (Figure 6E), indicating an involvement of TF in PaC stromal remodeling. We then analyzed blood vessel distribution by CD31 staining. While SC1 treatment did not reduce the overall level of CD31 staining, it caused a significant reduction in vessel lumen area, indicating a potential role for TF in vessel performance in these tumors (Figure 6F). Together, these results highlight a role for TF in tumor growth, which involves in part its function in tumor stromal remodeling and angiogenesis.
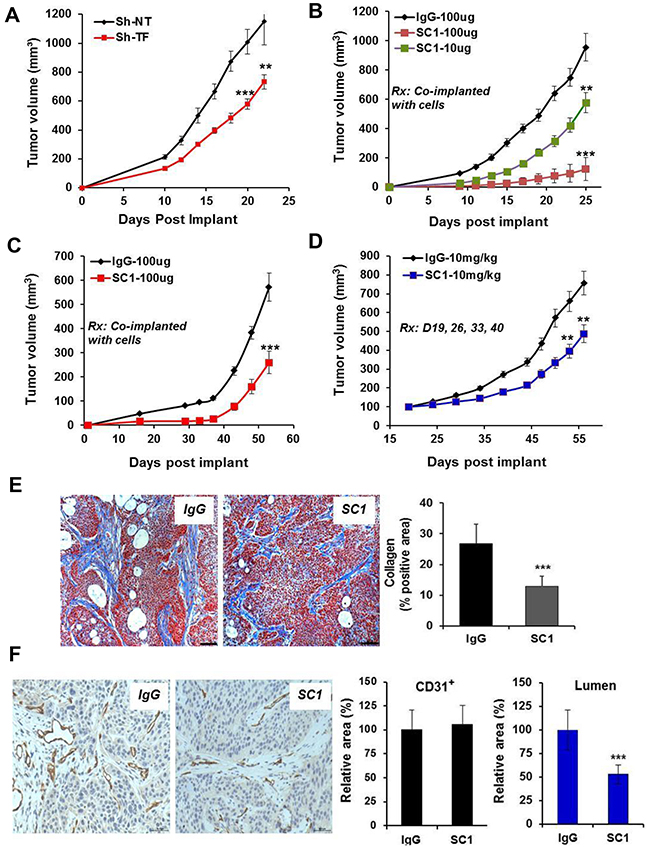
Figure 6: SC1 attenuates tumor growth in vivo. (A) HCC1806 cells expressing TRIPZ Sh-NT or Sh-TF were pre-induced with doxycycline for 7 days and were implanted into the mammary fat pad (MFP) of nude mice (n=10) and maintained with 0.5 mg/mL doxycycline in drinking water. (B and C) HCC1806 cells (B) and BxPC3 cells (C) were pre-mixed with the indicated doses of SC1 or IgG and were implanted to MFP (n=10) or flank (n=10), respectively, in nude mice. (D) Nude mice bearing established BxPC3 Tumors (n=8) were dosed with IgG or SC1 i.v. 1x weekly as indicated. Tumor growth curves are shown. (E and F) On final day of study in C, 3 tumors from each group were collected and subjected to Masson’s trichrome staining (E) and CD31 staining (F). Eight view fields per tumor (n=3 tumors) were assessed for quantification analysis. Representative views are shown. Rx, treatment; **, P<0.01; ***, P<0.001.
SC1 antibody-drug conjugates elicit potent cytotoxicity in TF-expressing cancer cells
To explore the potential utility of SC1 in antibody-drug conjugate (ADC) targeted therapy, we first analyzed its internalization characteristics in MDA-MB-231 cells by confocal microscopy. SC1 was rapidly and extensively internalized into the cytoplasm and co-localized with the lysosomal marker LAMP-2 (Figure 7A) rendering it a suitable candidate for ADC development. We then generated SC1 conjugates with emtansine (SC1-DM1) or monomethyl auristatin E (SC1-MMAE). In a panel of 10 cell lines with high (BxPC3, MDA-MB-231, HCC1806, Hs578T), low (A549, MDA-MB-453, T47D, MCF7) or moderate (H1975 and U87MG) TF expression, both SC1-DM1 and SC1-MMAE elicited exquisite cytotoxicity toward the high-TF cells (IC50 ranging 0.02-0.1 nM) but not in the low-TF cells (IC50 >100 nM) and were modestly active in the medium-TF cells (IC50 0.55-32 nM) (Figure 7B, 7C, Supplementary Figure 4A, 4C). The IC50 values were inversely proportional to TF-mRNA levels, achieving 1000 to >5000 fold target selective cytotoxicity in high-TF TNBC and PaC tumor cells (Figure 7D, Supplementary Figure 4B, 4D) thus validating the potential utility of SC1-ADCs in TF-targeted treatment. Cell cycle analysis showed that treatment of HCC1806 cells with 10 nM SC1-MMAE or 100 nM docetaxel for 48 h resulted in an expected G2/M blockade and cell death in both treatments (Figure 7E). Taken together, SC1-ADCs along with the previously established TF-ADCs [33, 46] represent a new class of highly potent cytotoxic agents for specific targeting of TF-dysregulated cancer cells.
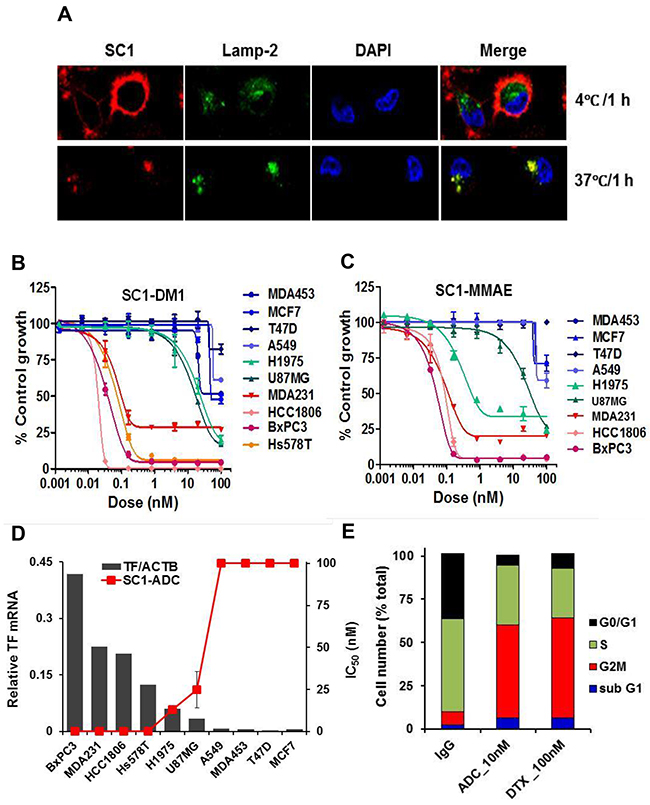
Figure 7: TF-ADCs potently inhibit proliferation of TF-overexpressing tumor cells. (A) MDA-MB-231 cells were incubated with 10 μg/mL SC1 at 4°C or 37°C for 1 h. Cells were fixed and subjected to immunofluorescence (IF). (B and C) A panel of cancer cell lines were treated with various doses of SC1-DM1 (B) or SC1-MMAE (C) for 4 days and analyzed for cell viability. Growth inhibition curves are plotted. (D) Growth inhibition IC50 values for the cell lines (mean of SC1-DM1 and SC1-MMAE designated as SC1-ADC) were plotted against the TF-mRNA expression levels [36]. (E) HCC1806 cells were treated with 10 nM SC1-MMAE or 100 nM docetaxel (DTX) for 48 h. Cell cycle profile as analyzed by FACS are shown. ACTB, β-actin.
SC1-MMAE and a humanized hSC1-MMAE demonstrate promising efficacy in preclinical in vivo models of TNBC and PaC
Nude mice bearing established BxPC3 tumors were treated 1x weekly with IgG-MMAE, SC1-MMAE or docetaxel for 4 weeks. SC1-MMAE elicited a substantial (3.75 mg/kg) or complete tumor regression (15 mg/kg) (Figure 8A). While 15 mg/kg docetaxel showed a similar tumor inhibition as 3.75 mg/kg SC1-MMAE, it caused a severe body weight loss (Figure 8B). Similar treatment of HCC1806-bearing mice with SC1-MMAE for 2 weeks documented a partial to complete tumor inhibition at 0.7 to 2 mg/kg and a nearly complete tumor regression at 7 mg/kg (Figure 8C). TUNEL staining showed that there was a significant increase in apoptosis in HCC1806 tumor tissue in the mice treated with 2 and 7 mg/kg SC1-MMAE (Figure 8D). We developed a humanized SC1 (hSC1; IgG1κ) and its MMAE conjugate (hSC1-MMAE). The unconjugated hSC1 retained full TF-binding affinity in vitro (Figure 3A, Supplementary Figure 5A) and in vivo antitumor activity compared to SC1 (Figure 6B, Supplementary Figure 5B). 1x weekly treatment with hSC1-MMAE resulted in a substantial inhibition (0.3 mg/kg) or a nearly complete regression (1 mg/kg) of established BxPC3 tumors (Figure 8E). Together these results indicate that SC1/hSC1-MMAE is a potent antitumor agent in vivo and can be potentially developed as targeted therapy for treating TF-positive TNBC and PaC.
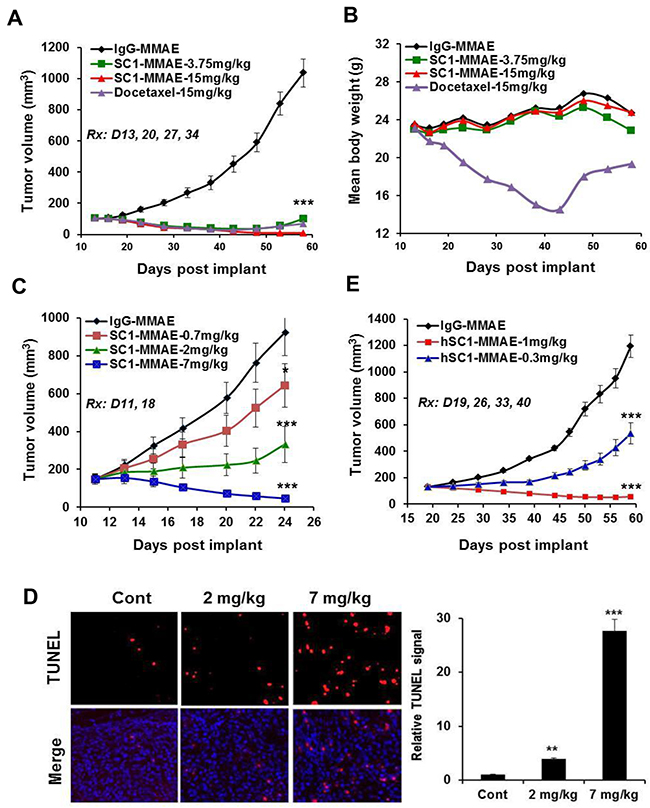
Figure 8: SC1/hSC1-MMAE inhibits TF-expressing TNBC and PaC tumor growth in vivo. (A and B) Mice bearing established BxPC3 tumors (n=8) were treated with SC1-MMAE or docetaxel i.v. 1x weekly as indicated. Tumor growth curves (A) and mouse body weight changes (B) are plotted. (C) Mice bearing established HCC1806 tumors (n=8) were treated with the indicated doses of SC1-MMAE. Tumor growth curves are plotted. (D) On final day of study in C, HCC1806 tumors were collected 24 h after the last dosing and subjected to TUNEL assay. TUNEL images (n=3) were acquired (left panel) and quantified (right panel). (E) Mice bearing established BxPC3 tumors (n=8) were treated with humanized hSC1-MMAE 1x weekly as indicated. Tumor growth curves are shown. *, P<0.05; **, P<0.01; ***, P<0.001.
DISCUSSION
In analysis of several gene expression profiling database, we found that aberrant TF expression occurs frequently in invasive cancers particularly in “Basal-like” breast cancer, an intrinsic subtype often synonymous with the clinical cohort of TNBC. We also observed TF expression in large percentage of PaC. Because TNBC and PaC are among the most challenging tumors to treat [34, 35], we explored TF-targeting as potential treatment strategy. We have thus far generated a novel and specific TF monoclonal antibody SC1, which displayed exceedingly high inhibition potency against TF extracellular domain, TF-overexpressing TNBC or PaC cells, PAR2 signaling and tumor TF-initiated coagulation. Therefore, SC1 can serve as an excellent pharmacological tool for exploring the therapeutic potential of TF-targeted therapy as cancer treatment.
Previous reports have indicated an association between TF and metastatic property in invasive tumors and have highlighted a critical role for TF-dependent coagulation function in protecting tumor cells in the circulation against anoikis or natural killer cells [8, 24, 43, 44]. We found that TF expression in breast cancer lines and tumors closely clustered with higher levels of EMT (high vimentin/low E-cadherin) and classical basal-type biomarkers KRT5, KRT14 and caveolin-1, while it clustered with lower levels luminal-type biomarkers KRT8, KRT18, ERBB3 and ESR1. In a recent report, EMT was shown to trigger TF expression and metastasis, and co-expression of TF and vimentin was found in subpopulation of circulating tumor cells (CTC) in metastatic breast cancer patients [24]. We have shown that depletion of TF protein or application of SC1 each inhibited TF/FVIIa-induced cell migration in TF-positive TNBC and PaC cells but had no effect in TF-negative cells. These TF-targeted treatments dramatically reduced their ability in metastatic colonization to the lung in mice. Our results are therefore further support a close relationship between TF-triggered procoagulant state and metastatic mechanism in highly invasive tumors including TNBC and PaC.
While depletion of TF or application of SC1 did not directly impact cell proliferation in vitro, these treatments significantly attenuated growth of TNBC HCC1806 tumors in vivo, indicating that TF is required for optimal in vivo growth in these settings. These observations are consistent with the previous TF-depletion study for colon cancer cells [18] and the TF-mAb study for breast cancer cells [11] and collectively highlight the importance of TF in tumor environment. Furthermore, the fact that co-injection of SC1 or hSC1 with TF-positive tumor cells produced better antitumor efficacy compared to that of TF-depletion strongly implies an involvement of the antibody-dependent cell-mediated cytotoxicity (ADCC). Indeed, both the IgG2b (SC1) and IgG1 (hSC1) are well documented for rendering ADCC effect [47], which could present a significant component of the TF-targeted mAb treatment strategy.
In PaC BxPC3 tumor model, treatment with SC1 reduced tumor growth, which was associated with a diminished tumor vessel lumen area leading potentially to a compromised tumor angiogenesis. SC1 may exert such an effect through both the TF-coagulant function and TF-PAR2 signaling [6]. In this context a reduced FXa/thrombin generation indirectly dampens angiogenesis, and the blockade of TF-PAR2 signaling inhibits gene transcription of several key angiogenesis factors such as VEGF, IL-8 and CXCL-1 [10, 11, 28, 29]. The SC1 treatment also reduced tumor stromal fibrosis. Because stromal fibrotic remodeling is a hall mark of PaC and impedes drug penetration [45], targeting TF by SC1 may further improve antitumor efficacy when used in combination with other suitable therapeutic agents. Future studies are required to elucidate the mechanistic detail on how SC1 perturbs the stromal fibrotic response in PaC.
With the FDA approval of Adcetris and Kadcyla, development of ADC as targeted cancer therapy has become a reality and numerous novel ADC candidates have entered clinical trials in cancer patients [48]. Our results support the idea that TF could be a promising target for ADC development. We showed that high levels of TF are expressed in large percentage of invasive tumors particularly in TNBC and PaC, two most lethal forms of malignancy for which new and effective therapies are urgently needed [34, 35]. We observed a rapid (1 h) and extensive SC1 internalization when it binds to TF-positive cancer cells. Both SC1-MMAE and SC1-DM1 elicited a potent cytotoxicity in the TF-positive TNBC and PaC cells in vitro but not in TF-negative cells with a selectivity ratio >5000 fold. SC1-MMAE and a humanized hSC1-MMAE were both highly efficacious in in vivo models of TNBC and PaC achieving a favorable therapeutic window. It should be noted that because SC1 has poor affinity to murine TF, its tolerability in mice may not translate to humans and requires further evaluation in non-human primates (NHP). Verploegen et al. showed that although TF-011 potently inhibited TF-induced clotting in vitro, it did not induce a serious bleeding issue in NHP [49]. TF-011-MMAE Tisotumab Vedotin is well tolerated in humans without major bleeding events and has demonstrated early encouraging antitumor efficacy [50]. Therefore, these results collectively provide both the scientific rationale and clinical evidence in support for TF-targeted ADC as new anticancer treatment.
In summary, our studies have identified a novel and potent anti-TF therapeutic antibody SC1. The prevalence of aberrant TF expression in TNBC and PaC render these two most deadly cancers susceptible to SC1 in preclinical setting and may offer new opportunity in cancer patients.
MATERIALS AND METHODS
Chemicals
All general chemicals used in buffers and assays were purchased from Sigma-Aldrich Corporation unless otherwise specified. Specific chemicals are indicated with methods.
Generation of anti-human TF monoclonal antibody
A histidine-tagged extracellular domain of human TF (TF-ECD) (Sino Biological, Cat#13133-H08H-20) was used to immunize Balb/c mice. Hybridomas were generated by fusion of splenocytes with SP2/0 murine myeloma cells (Sigma-Aldrich). TF-specific hybridomas were identified by screening with TF-specific ELISA as described below. Antibodies with high binding affinity to human TF were scaled up by intraperitoneal injection of hybridomas into Balb/c mice then purified from ascites using protein G affinity chromatography (GE Healthcare). Antibody concentration was determined by A280 measurement and extinction coefficient (A280/E). The variable region sequences of monoclonal antibodies were obtained by GeneRacer kit (Invitrogen, Cat#L1502-02) and DNA sequencing, and were subsequently humanized.
Cell culture, gene depletion and growth assays
Cell lines of MCF-7, BT-474, SKRB3, MDA-MB-453, MDA-MB-361, MDA-MB-231, HCC1806, BxPC3, Capan-2, A549, H1975 and U87MG were obtained from American Type Culture Collection (ATCC). T47D, ZR-75-1, HBL-100, BT-549, MDA-MB-468, MDA-MB-435, MDA-MB-436, HCC1937, HCC38, Hs578T, MX-1, Bcap-37, PANC-1 and Capan-2 were obtained from the Cell Bank of Chinese Academy of Sciences (CAS, Shanghai). Cells were cultured in a 37°C incubator with 5% CO2 using standard cell culture methods and reagents (Invitrogen). To create gene depletion, validated pTRIPZ lentiviral inducible shRNA for TF (Cat#V3LHS_371301, V3LHS_371304) and non-targeting (Cat#RHS4346) were obtained from Open Biosystems. These constructs were packaged in 293T cells and validated according to manufacturer’s instruction. Virus-infected tumor cells were selected by a pre-determined concentration of puromycin. TF depletion was induced by incubating the cells with 1 μg/mL of doxycycline (Dox) for 5-7 days. Cell growth assays were conducted in 96-well plate at 8-10% starting confluence with treatment for 4 days. Cell viability was assessed using MTS reagent (Promega) as reported previously [51].
Assays of TF antibody binding and internalization
Antigen binding was assayed via ELISA. ELISA plate was coated with 0.5 μg/mL histidine-tagged TF-ECD for overnight, sequentially washed, blocked with 2% BSA in PBS and incubated with various doses of TF antibody, then detected with HRP-labeled goat anti-mouse IgG (Jackson ImmunoResearch, Cat#115-035-146). To measure binding to cell surface TF, MDA-MB-231 or BxPC3 cells (3x105) were incubated with various doses of TF antibody in 200 μL serum-free medium at 4°C for 1 h, washed with PBS and bound antibody was detected with R-PE-conjugated goat anti-mouse IgG (Jackson ImmunoResearch, Cat#115-116-071) by FACS analysis (BD FACSArialTM IIU).
To analyze the internalization of antibody into cancer cells, MDA-MB-231 cells were plated at 50% confluence in glass bottom cell culture dish (NEST, Cat#801002) and incubated with 10 μg/mL of TF antibody at 37°C or 4°C for the indicated time. The cells were fixed with 4% formaldehyde, permeablized and detected with Alexa Fluor 647-conjugated AffiniPure donkey anti-mouse IgG (Jackson ImmunoResearch, Cat#715-605-150). The lysosomes were visualized via incubating cells with a rabbit anti-human LAMP-2 (Abcam, Cat#ab125068) and detected with Alexa Fluor 488-conjugated AffiniPure donkey anti-Rabbit IgG (Jackson ImmunoResearch, Cat#711-545-152). Images were acquired under confocal microscope (Zeiss, LSM710).
Coagulation assays
Coagulation assay was performed as described [26]. Briefly, MDA-MB-231 or BxPC3 cells (30,000 in 50 μL) prepared in HBSS buffer with 5 mM CaCl2 were mixed with various doses of TF antibody (50 μL) at 25°C for 15 min on a Titer Plate Shaker. The clotting was initiated by the addition of 50 μL citrated human plasma and read immediately for opacity at 405 nm on a kinetic plate reader every 15 sec up to 2 h. Clotting time was plotted as a function of antibody concentration. For FXa assay, 15,000 MDA-MB-231 or BxPC3 cells were incubated with 3 nM FVIIa (Novo Nordisk) and TF antibody (100 μL) at 25°C for 20 min. Reaction was initiated by adding 50 μL FX (final 50 nM) (Haematologic Technologies, Cat#HCX-0050) for 10 min and terminated with 25 μL of 1 M EDTA. FXa was measured by adding 25 μL of 3 mM S2765 (Aglyco, Cat#AG00-0102) and read immediately on a kinetic plate reader every 15 sec up to 1 h.
Protein lysates and immunoblotting
After various treatments cells were lysed using NuPAGE-LDS sample buffer (Invitrogen). For profile of cancer cell line panel, cells were lyased as described [52]. Total cell lysates were immunoblotted with antibodies for human TF (R&D Systems, Cat#MAB2339), P-ERK and ERK (Cell Signaling), GAPDH and actin (Bioworld).
Cell migration and experimental metastasis
For cell migration, appropriate number of cells in 200 μL serum-free medium were added to the upper chamber of a Transwell system (Corning, Cat#3422) with TF antibody or control IgG and allowed to migrate for 8 h at 37°C, 5% CO2 toward serum-containing medium in the lower chamber. Cells on the underside of the membrane were stained with crystal violet and counted under a microscope in 5 representative viewing fields at 200x magnification. For lung metastasis, 6-8 weeks old female Balb/c nude mice or SCID mice were purchased from B&K Laboratory Animal Corporation. Luciferase-tagged MDA-MB-231 cells (2×106 cells in 200 μL PBS) were mixed with TF antibody or control IgG and injected into the lateral tail vein of mice. For bioluminescence imaging 4 h after cell injection, mice were given D-luciferin potassium salt (150 mg/kg) intraperitoneally and imaged 6 min later for 1 min exposure in an IVIS Spectrum in vivo imaging system. Mice were sacrificed 6 weeks after cell injection and lung tumor nodules were counted.
Antibody-drug conjugates
Anti-TF IgG was conjugated with monomethyl auristatin E through a protease-cleavable valinecitrulline (VC) dipeptide or with maytansine by N-succinimidyl 4-(N-maleimidomethyl) cyclohexane-1-carboxylate (SMCC) [53]. The average drug-antibody ratio (DAR) was 3.6.
In vivo tumor growth, immunohistochemistry (IHC) and apoptosis
In vivo efficacy studies were performed under protocols approved by institutional IACUC of Fudan University. Xenograft tumor models were established in female Balb/c nude mice by mammary fat pat (MFP)- or subcutaneous implantation with 3x106 cells (HCC1806) or 10x106 cells (BxPC3), respectively. For co-injection experiments, cells were mixed with TF-mAb or control IgG (n=10) immediately before injection. For therapeutic treatments, established tumors were staged at 100-200 mm3 (n=8), various doses of mAb or ADC were administered i.v. 1x weekly following the indicated schedules. Tumor growth was monitored twice weekly and calculated using the formula V=LW2/2 (where V=volume, L=length and W=width). For IHC, tumors were snap-frozen in liquid nitrogen or formalin fixed for 24 h and embedded in paraffin. Slides were processed for staining with anti-CD31 (Cell Signaling) or Masson’s trichrome (Sigma, Cat#HT15-1KT). For apoptosis assay, tumor slides were processed for terminal deoxynuleotidyl transferase dUTP nick end labeling (TUNEL) using an assay kit (KeyGen Biotech, KGA7061) following vendor’s instruction manual. Images were acquired using Leica microscope (model DMI4000D). Staining levels were analyzed on the basis of mean optical density quantified by software image pro plus 6.0.
Statistical analysis
All numerical data were presented as mean ± standard deviation (SD) except for mouse xenograft studies data which were presented as mean ± standard error (SE). Numerical data processing and statistical analysis were performed with Microsoft Excel and GraphPad Prism 5 software. P values were calculated using unpaired two-tailed Student-t test. In all tests, differences with P values < 0.05 were considered to be statistically significant.
ACKNOWLEDGMENTS
This work was supported by a startup grant from Fudan University (EZF301002), National Natural Science Foundation of China (81273367) and National Basic Research 973 Program of China (2013CB932500). We thank Dr. Qing Lin, Dr. Liyuan Zhu, Dr. Qun He and Mengjun Shu for technical assistance.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest
REFERENCES
1. Heit JA. Cancer and venous thromboembolism: scope of the problem. Cancer Control. 2005; 12 Suppl 1:5-10.
2. Hisada Y, Geddings J, Ay C, Mackman N. Venous thrombosis and cancer: from mouse models to clinical trials. J Thromb Haemost. 2015; 13:1372-82.
3. Manly DA, Boles J, Mackman N. Role of tissue factor in venous thrombosis. Annu Rev Physiol. 2011; 73:515-25.
4. Kasthuri RS, Taubman MB, Mackman N. Role of tissue factor in cancer. J Clin Oncol. 2009; 27:4834-8.
5. Ruf W, Disse J, Carneiro-Lobo TC, Yokota N, Schaffner F. Tissue factor and cell signalling in cancer progression and thrombosis. J Thromb Haemost. 2011; 9:306-15.
6. Versteeg HH. Tissue factor: old and new links with cancer biology. Semin Thromb Hemost. 2015; 41:747-55.
7. Nierodzik ML, Karpatkin S. Thrombin induces tumor growth, metastasis, and angiogenesis: evidence for a thrombin-regulated dormant tumor phenotype. Cancer Cell. 2006; 10:355-62.
8. Palumbo JS, Talmage KE, Massari JV, La Jeunesse CM, Flick MJ, Kombrinck KW, Hu Z, Barney KA, Degen JL. Tumor cell-associated tissue factor and circulating hemostatic factors cooperate to increase metastatic potential through natural killer cell–dependent and–independent mechanisms. Blood. 2007; 110:133-41.
9. Wojtukiewicz MZ, Hempel D, Sierko E, Tucker SC, Honn KV. Thrombin-unique coagulation system protein with multifaceted impacts on cancer and metastasis. Cancer Metastasis Rev. 2016; 35:213-33.
10. Schaffner F, Ruf W. Tissue factor and PAR2 signaling in the tumor microenvironment. Arterioscler Thromb Vasc Biol. 2009; 29:1999-2004.
11. Versteeg HH, Schaffner F, Kerver M, Petersen HH, Ahamed J, Felding-Habermann B, Takada Y, Mueller BM, Ruf W. Inhibition of tissue factor signaling suppresses tumor growth. Blood. 2008; 111:190-9.
12. Ueno T, Toi M, Koike M, Nakamura S, Tominaga T. Tissue factor expression in breast cancer tissues: its correlation with prognosis and plasma concentration. Br J Cancer. 2000; 83:164-70.
13. Kocatürk B, Van den Berg YW, Tieken C, Mieog JS, de Kruijf EM, Engels CC, van der Ent MA, Kuppen PJ, Van de Velde CJ, Ruf W, Reitsma PH, Osanto S, Liefers GJ, et al. Alternatively spliced tissue factor promotes breast cancer growth in a β1 integrin-dependent manner. Proc Natl Acad Sci U S A. 2013; 110:11517-22.
14. Hu C, Huang L, Gest C, Xi X, Janin A, Soria C, Li H, Lu H. Opposite regulation by PI3K/Akt and MAPK/ERK pathways of tissue factor expression, cell-associated procoagulant activity and invasiveness in MDA-MB-231 cells. J Hematol Oncol. 2012; 5:16.
15. Magnus N, Garnier D, Rak J. Oncogenic epidermal growth factor receptor up-regulates multiple elements of the tissue factor signaling pathway in human glioma cells. Blood. 2010; 116:815-8.
16. Rong Y, Belozerov VE, Tucker-Burden C, Chen G, Durden DL, Olson JJ, Van Meir EG, Mackman N, Brat DJ. Epidermal growth factor receptor and PTEN modulate tissue factor expression in glioblastoma through JunD/activator protein-1 transcriptional activity. Cancer Res. 2009; 69:2540-9.
17. Shigemori C, Wada H, Matsumoto K, Shiku H, Nakamura S, Suzuki H. Tissue factor expression and metastatic potential of colorectal cancer. Thromb Haemost. 1998; 80:894-8.
18. Yu JL, May L, Lhotak V, Shahrzad S, Shirasawa S, Weitz JI, Coomber BL, Mackman N, Rak JW. Oncogenic events regulate tissue factor expression in colorectal cancer cells: implications for tumor progression and angiogenesis. Blood. 2005; 105:1734-41.
19. Nitori N, Ino Y, Nakanishi Y, Yamada T, Honda K, Yanagihara K, Kosuge T, Kanai Y, Kitajima M, Hirohashi S. Prognostic significance of tissue factor in pancreatic ductal adenocarcinoma. Clin Cancer Res. 2005; 11:2531-9.
20. Khorana AA, Ahrendt SA, Ryan CK, Francis CW, Hruban RH, Hu YC, Hostetter G, Harvey J, Taubman MB. Tissue factor expression, angiogenesis, and thrombosis in pancreatic cancer. Clin Cancer Res. 2007; 13:2870-75.
21. Tieken C, Verboom MC, Ruf W, Gelderblom H, Bovée JV, Reitsma PH, Cleton-Jansen AM, Versteeg HH. Tissue factor associates with survival and regulates tumour progression in osteosarcoma. Thromb Haemost. 2016; 115:1025-33.
22. Leppert U, Eisenreich A. The role of tissue factor isoforms in cancer biology. Int J Cancer. 2015; 137:497-503.
23. Eisenreich A, Leppert U. The impact of microRNAs on the regulation of tissue factor biology. Trends Cardiovasc Med. 2014; 24:128-32.
24. Bourcy M, Suarez-Carmona M, Lambert J, Francart ME, Schroeder H, Delierneux C, Skrypek N, Thompson EW, Jérusalem G, Berx G, Thiry M, Blacher S, Hollier BG, et al. Tissue factor induced by epithelial-mesenchymal transition triggers a procoagulant state that drives metastasis of circulating tumor cells. Cancer Res. 2016; 76:4270-82.
25. Liu Y, Jiang P, Capkova K, Xue D, Ye L, Sinha SC, Mackman N, Janda KD, Liu C. Tissue factor–activated coagulation cascade in the tumor microenvironment is critical for tumor progression and an effective target for therapy. Cancer Res. 2011; 71:6492-502.
26. Ngo CV, Picha K, McCabe F, Millar H, Tawadros R, Tam SH, Nakada MT, Anderson GM. CNTO 859, a humanized anti-tissue factor monoclonal antibody, is a potent inhibitor of breast cancer metastasis and tumor growth in xenograft models. Int J Cancer. 2007; 120:1261-67.
27. ClinicalTrials.gov. A study of ALT-836 in combination with gemcitabine for locally advanced or metastatic solid tumors. clinicaltrials.gov identifier: NCT01325558.
28. Belting M, Dorrell MI, Sandgren S, Aguilar E, Ahamed J, Dorfleutner A, Carmeliet P, Mueller BM, Friedlander M, Ruf W. Regulation of angiogenesis by tissue factor cytoplasmic domain signaling. Nat Med. 2004; 10:502-9.
29. Schaffner F, Versteeg HH, Schillert A, Yokota N, Petersen LC, Mueller BM, Ruf W. Cooperation of tissue factor cytoplasmic domain and PAR2 signaling in breast cancer development. Blood. 2010; 116:6106-13.
30. Hembrough TA, Swartz GM, Papathanassiu A, Vlasuk GP, Rote WE, Green SJ, Pribluda VS. Tissue factor/factor VIIa inhibitors block angiogenesis and tumor growth through a nonhemostatic mechanism. Cancer Res. 2003; 63:2997-3000.
31. Rak AS, Mackman N, Versteeg HH, Bogdanov VY. Antibody-based targeting of alternatively spliced tissue factor: a new approach to impede the primary growth and spread of pancreatic ductal adenocarcinoma. Oncotarget. 2016; 7:25264-75. doi: 10.18632/oncotarget.7955.
32. Saito Y, Hashimoto Y, Kuroda J, Yasunaga M, Koga Y, Takahashi A, Matsumura Y. The inhibition of pancreatic cancer invasion-metastasis cascade in both cellular signal and blood coagulation cascade of tissue factor by its neutralization antibody. Eur J Cancer. 2011; 47:2230-9.
33. Breij EC, de Goeij BE, Verploegen S, Schuurhuis DH, Amirkhosravi A, Francis J, Miller VB, Houtkamp M, Bleeker WK, Satijn D, Parren PW. An antibody-drug conjugate that targets tissue factor exhibits potent therapeutic activity against a broad range of solid tumors. Cancer Res. 2014; 74:1214-26.
34. Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010; 363:1938-48.
35. Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med. 2014; 371:1039-49.
36. Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, Speed T, Spellman PT, DeVries S, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006; 10:515-27.
37. Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumors. Nature. 2012; 490:61-70.
38. Abd El-Rehim DM, Pinder SE, Paish CE, Bell J, Blamey RW, Robertson JF, Nicholson RI, Ellis IO. Expression of luminal and basal cytokeratins in human breast carcinoma. J Pathol. 2004; 203:661-71.
39. Kreike B, van Kouwenhove M, Horlings H, Weigelt B, Peterse H, Bartelink H, van de Vijver MJ. Gene expression profiling and histopathological characterization of triple-negative/basal-like breast carcinomas. Breast Cancer Res. 2007; 9:R65.
40. Badea L, Herlea V, Dima SO, Dumitrascu T, Popescu I. Combined gene expression analysis of whole-tissue and microdissected pancreatic ductal adenocarcinoma identifies genes specifically overexpressed in tumor epithelia. Hepatogastroenterology. 2008; 55:2016-27.
41. Jeong H, Ryu YJ, An J, Lee Y, Kim A. Epithelial-mesenchymal transition in breast cancer correlates with high histological grade and triple-negative phenotype. Histopathology. 2012; 60:E87-95.
42. Hjortoe GM, Petersen LC, Albrektsen T, Sorensen BB, Norby PL, Mandal SK, Pendurthi UR, Rao LV. Tissue factor-factor VIIa–specific up-regulation of IL-8 expression in MDA-MB-231 cells is mediated by PAR-2 and results in increased cell migration. Blood. 2004; 103:3029-37.
43. Yokota N, Zarpellon A, Chakrabarty S, Bogdanov VY, Gruber A, Castellino FJ, Mackman N, Ellies LG, Weiler H, Ruggeri ZM. Contributions of thrombin targets to tissue factor-dependent metastasis in hyperthrombotic mice. J Thromb Haemost. 2014; 12:71-81.
44. Camerer E, Qazi AA, Duong DN, Cornelissen I, Advincula R, Coughlin SR. Platelets, protease-activated receptors, and fibrinogen in hematogenous metastasis. Blood. 2004; 104:397-401.
45. Mahadevan D, Von Hoff DD. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol Cancer Ther. 2007; 6:1186-97.
46. Koga Y, Manabe S, Aihara Y, Sato R, Tsumura R, Iwafuji H, Furuya F, Fuchigami H, Fujiwara Y, Hisada Y, Yamamoto Y, Yasunaga M, Matsumura Y. Antitumor effect of antitissue factor antibody-MMAE conjugate in human pancreatic tumor xenografts. Int J Cancer. 2015; 137:1457-66.
47. Nimmerjahn F, Lux A, Albert H, Woigk M, Lehmann C, Dudziak D, Smith P, Ravetch JV. FcγRIV deletion reveals its central role for IgG2a and IgG2b activity in vivo. Proc Natl Acad Sci U S A. 2010; 107:19396-401.
48. de Goeij BE, Lambert JM. New developments for antibody-drug conjugate-based therapeutic approaches. Curr Opin Immunol. 2016; 40:14-23.
49. Verploegen S, Satijn D, Hoet R, Parren P, Winkel J, Breinholt VM, Ehrnrooth E, Baadsgaard O, Vink T, Bleeker WK, Houtkamp M, Oudshoorn M, De Jong RN. Human antibodies against tissue factor and methods of use thereof, 2016; Patent Application US2016053020A1.
50. Lassen UN, Hong DS, Diamantis N, Subbiah V, Kumar R, Sorensen M, Lisby S, Coleman RL, De Bono JS. A phase I, first-in-human study to evaluate the tolerability, pharmacokinetics and preliminary efficacy of HuMax-tissue factor-ADC (TF-ADC) in patients with solid tumors. J Clin Oncol. 2015; 33:2570.
51. Yu K, Toral-Barza L, Shi C, Zhang WG, Lucas J, Shor B, Kim J, Verheijen J, Curran K, Malwitz DJ. Biochemical, cellular, and in vivo activity of novel ATP-competitive and selective inhibitors of the mammalian target of rapamycin. Cancer Res. 2009; 69:6232-40.
52. Yu K, Toral-Barza L, Shi C, Zhang WG, Zask A. Response and determinants of cancer cell susceptibility to PI3K inhibitors: combined targeting of PI3K and Mek1 as an effective anticancer strategy. Cancer Biol Ther. 2008; 7:307-15.
53. Ducry L, Stump B. Antibody-drug conjugates: linking cytotoxic payloads to monoclonal antibodies. Bioconjug Chem. 2010; 21:5-13.