
Introduction
The phosphoinositide-3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) pathway is commonly activated in cancer by several mechanism including activating mutations of PIK3CA or AKT1 and or loss of phosphatase and tensin homolog (PTEN) [1] . This signaling pathway is critical in the regulation of cell growth, metabolism and survival, angiogenesis, tumor invasion, cell cycle regulation and DNA repair [2, 3]. The activity of different PI3K inhibitors in unselected population in the clinical setting has been limited [1]. Preclinical testing strategies to increase the antitumor activity of these compounds should address effectiveness of combination regimens with chemotherapy or other targeted agents and improvement of patient selection based on biomarkers predictive of an activated PI3K pathway.
Preclinical and clinical evidence supports a role of the PI3K pathway in chemoresistance in different tumor types including ovarian [4], breast [5], and non-small cell lung adenocarcinoma (LADC) [6]. Furthermore, inhibition of the PI3K pathway sensitizes preclinical models to chemotherapy [6-8]. Likewise, resistance to anti-EGFR and anti-HER2 therapies are associated with PI3K pathway activation by PIK3CA mutations in EGFR mutant LADC [9], and HER2-positive breast cancer [10] [11], or by loss of PTEN [12] or HER3 activation (which activates the PI3K pathway) in breast cancer models [13]. In cell lines, the addition of PI3K inhibitors overcomes resistance to anti-EGFR or anti-HER2 agents [10, 11, 14]. This evidence has supported the evaluation of the PI3K inhibitors in combination with anti-EGFR or anti-HER2 therapies in the clinical setting and several combinations are under investigation.
Patient derived tumor xenografts (PDXs) represent promising pre-clinical models as they seem to recapitulate some of the molecular characteristics of the primary tumor [15] as well as clinical tumor response [16-18]. To improve the clinical activity of the PI3K inhibitors, we tested several therapeutic strategies in three different tumor types using PDX selected for deficient PTEN expression as increased sensitivity to PI3K and mTOR inhibitors has been previously described in cancer cell lines [19]. PTEN-deficient tumors seem to signal preferentially through the PI3K beta isoform (p110β) [20, 21] but it remains unclear whether single inhibition of the beta isoform would be sufficient to induce tumor growth inhibition, or whether dual PI3K alpha and beta isoform inhibition would be superior [22].
In the current study, we evaluated two PI3K-mTOR inhibitors in combination with a pan-HER inhibitor dacomitinib, cisplatin or paclitaxel. The two PI3K-mTOR inhibitors (PF-04691502 and PF-05212384), although different in route of administration and pharmacokinetics, both are potent inhibitors of all PI3K isoforms and mTOR [23, 24] and have shown clinical antitumor activity as monotherapies [25, 26]. Dacomitinib is an irreversible tyrosine kinase inhibitor targeting EGFR, HER2 and HER4 [27] with antitumor activity demonstrated pre-clinically in EGFR wild type and mutant LADC models [28], as well as clinically in head and neck squamous cell carcinoma [29] and LADC [30]. Cisplatin is commonly used as the backbone of chemotherapy regimens for many cancers, including LADC [31], ovarian [32], and triple negative breast cancers (TNBC) [33]. Likewise, paclitaxel is widely used in LADC [31], ovarian [32] , and breast cancers [34]. We hypothesize that in tumors deficient in PTEN protein expression, PI3K inhibition might increase the activity of cisplatin, paclitaxel or dacomitinib. We expect that the simultaneous evaluation of these compounds in PDX may expedite the translation of the most promising combinations into the clinical setting.
Results
Molecular characterization
Three PDX models were selected based on their deficient (null or low) PTEN expression. The three models display morphological and molecular characteristics from the original tumor (Supplementary Figure 1). The TNBC model lacks PTEN expression and did not harbor any of the mutations in the Oncocarta panel. PTEN staining was faint by immunohistochemistry in the low-grade serous ovarian cancer (LGSOC) and LADC PDX models. Furthermore, both of the latter models had KRAS mutations (G12R and G12C respectively in the LGSOC and LADC models). The LADC model also had a TP53 co-mutation (R181P) detected by direct sequencing.
PI3K-mTOR inhibitor and Cisplatin
Treatment duration was up to 29 days when experiments were discontinued due to inaccessible tail veins for IV drug administration. Single agent PF-05212384 had no significant activity as single agent in any of the three models (Table 1 and Figure 1). As a single agent, cisplatin showed insignificant anti-tumor activity in the TNBC (Figure 1A) and LGSOC models (Figure 1B), but induced tumor growth inhibition (TGI) > 50% in the LADC model (Figure 1C). The combination of cisplatin and PF-05212384 induced TGI > 50% in all the three models (Table 1). While the combination was synergistic in the TNBC model (p < 0.05), PF-05212384 did not enhance the TGI induced by cisplatin single agent in the LADC model (p = 0.113), nor in the LGSOC model (p = 0.147).
The initial dose selected for PF-05212384 (15 mg/kg) was found to be excessively toxic in the first model evaluated (LADC). Several cases of sudden death occurred after the first administration. Hence, a lower dose (10 mg/kg) was administered in subsequent experiments. After this dose reduction, the treatment was well-tolerated in the three models, with mean weight loss ranging 10 - 15% in the combination arm in the TNBC model and in cisplatin-containing arm in the LGSOC model. No mean weight loss was observed in the LADC model.
Table 1: Patient derived xenograft molecular profile and tumor growth inhibition per treatment arm
PDX tumor type |
TNBC |
LGSOC |
LADC |
|||
Molecular profile |
PTEN null |
KRAS G12R PTEN low |
KRAS G12C TP53 R181P PTEN low |
|||
TGI% |
ΔV p-value |
TGI% |
ΔV p-value |
TGI% |
ΔV p-value |
|
Experiment 1 |
||||||
PF-05212384 |
32% |
0.07 |
39% |
0.19 |
-1% |
0.78 |
Cisplatin |
42% |
<0.05** |
43% |
0.03 |
97% |
<0.05** |
PF-05212384 + Cisplatin |
96% |
<0.05** |
68% |
<0.05** |
92% |
<0.05** |
Experiment 2 |
||||||
PF-05212384 |
22% |
0.64 |
40% |
0.21 |
||
PF-04691502 |
33% |
<0.05** |
||||
Paclitaxel |
84% |
<0.05** |
38% |
0.20 |
79% |
<0.05** |
PF-05212384 + Paclitaxel |
110%^ |
<0.05** |
56% |
<0.05** |
||
PF-04691502 + Paclitaxel |
45% |
<0.05** |
||||
Experiment 3 |
||||||
PF-05212384 |
22% |
0.61 |
40% |
0.26 |
||
PF-04691502 |
33% |
<0.05** |
||||
Dacomitinib |
15% |
0.99 |
8% |
0.53 |
23% |
0.35 |
PF-05212384 + Dacomitinib |
55% |
0.09 |
45% |
0.47 |
||
PF-04691502 + Dacomitinib |
32% |
0.055 |
||||
TNBC = triple negative breast cancer; LGSOC = low grade serous ovarian cancer; LADC = non-small cell lung cancer; TGI%: percentages of tumor growth inhibition; ΔV: differences between daily tumor volume change of each treatment arm and the control arm; ^ Tumor regression; ** statistically significant
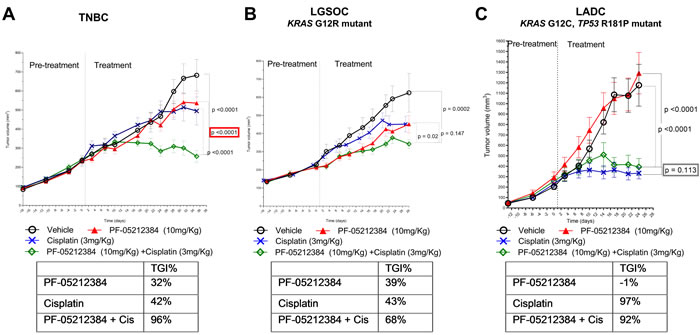
Figure 1: Combined cisplatin and PI3K-mTOR inhibitor in PTEN-deficient gPDX. Tumor growth of A. triple-negative breast cancer (TNBC), B. KRAS mutant (G12R) low-grade ovarian cancer (LGSOC) and C. TP53 (R181P) and KRAS (G12C) mutant lung adenocarcinoma (LADC). The three models were treated with vehicle, PF-05212384 (10 mg/kg, twice weekly, intravenously, cisplatin (3 mg/kg, once weekly, intraperitoneally) or the combination of both agents. Relative tumor volumes are displayed as mean +/- SE. p: p value for daily tumor volume change for each arm in comparison to vehicle arm. TGI%: percentages of tumor growth inhibition in comparison to vehicle arm.
PI3K-mTOR inhibitor and Paclitaxel
Although PF-05212384 and PF-04691502 showed no significant activity as single agents, paclitaxel induced TGI > 50% in the TNBC and LADC models (Table 1). The combination arm only induced TGI > 50% in the LGSOC and TNBC models (Figure 2A, 2B). Although the difference between the paclitaxel single agent and paclitaxel + PF-05212384 arms were not statistically significant, we observed signs of additive activity by adding PF-05212384 to paclitaxel in these two models; In the TNBC model, the combination arm induced tumor regression, which was not achieved in the paclitaxel single agent arm. In the LGSOC model, the combination arm achieved a TGI > 50%, while such TGI was not achieved by any of the single agent arm achieved (Table 1). The results of the LADC model (Figure 2C) should be evaluated with caution, as several mice in the paclitaxel arm suddenly died between weeks 1 and 2 of treatment. These deaths were directly attributed to dose initially selected of paclitaxel (15 mg/kg), as no new events were observed in any of the models once the paclitaxel dose was reduced to 10 mg/kg. No significant weight loss was observed in any of the treatment arms in the three models.
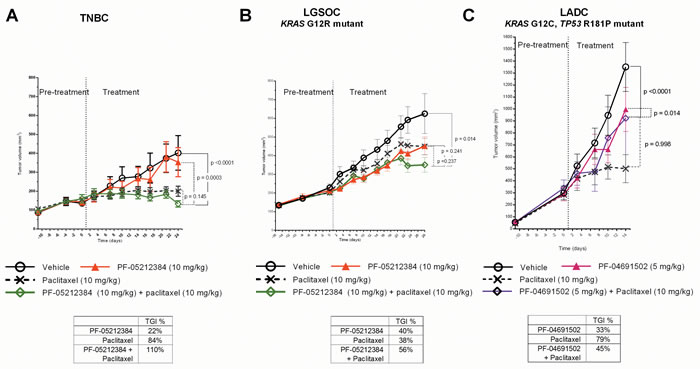
Figure 2: Combined Paclitaxel and PI3K-mTOR inhibitor in PTEN-deficient PDX. Tumor growth of A. triple-negative breast cancer (TNBC); B. KRAS mutant (G12R) low-grade ovarian cancer (LGSOC) and C. TP53 (R181P) and KRAS (G12C) mutant lung adenocarcinoma (LADC). The TNBC and LGSOC models were treated with vehicle, PF-05212384 (10 mg/kg, twice weekly, intravenously, paclitaxel (10 mg/kg, twice weekly, intraperitoneally) or the combination of both agents. The LADC was treated with vehicle, PF-04691502 (5 mg/kg, daily, oral gavage), paclitaxel (10 mg/kg, twice weekly, intraperitoneally) or the combination of both agents. Relative tumor volumes are displayed as mean +/- SE. p: p value for daily tumor volume change for each arm in comparison to vehicle arm. TGI%: percentages of tumor growth inhibition in comparison to vehicle arm.
PI3K-mTOR inhibitor and dacomitinib
As single agents, neither the PI3K-mTOR inhibitors nor dacomitinib achieved a TGI of 50%, (Table 1 and Figure 3). The combination arm only induced a mean TGI > 50% in the TNBC model (TGI = 55 %), but this did not achieve statistical significance according to the mixed effect model (p = 0.09) (Table 1). No relevant weight loss or other toxicity was observed in any of the arms.
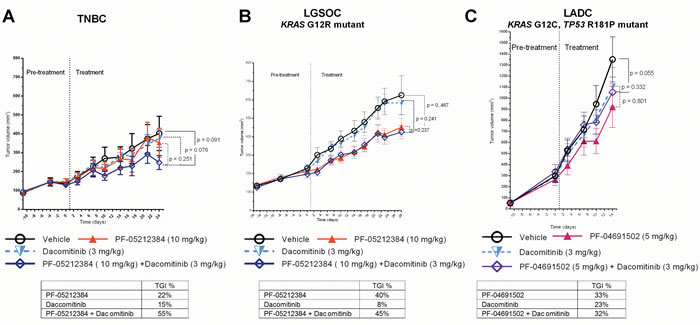
Figure 3: Combined dacomitinib and PI3K-mTOR inhibitor in PTEN-deficient PDX. Tumor growth of A. triple-negative breast cancer PDX (TNBC), KRAS mutant (G12R) low-grade ovarian cancer (LGSOC) and TP53 (R181P) and KRAS (G12C) mutant lung adenocarcinoma (LADC). The TNBC and LGSOC models were treated with vehicle, PF-05212384 (10 mg/kg, twice weekly, intravenously, dacomitinib (3 mg/kg daily, oral gavage) or the combination of both agents. The LADC was treated with vehicle, PF-04691502 (5 mg/kg, daily, oral gavage), dacomitinib (3 mg/kg daily, oral gavage) or the combination of both agents. Relative tumor volumes are displayed as mean +/- SE. p: p value for daily tumor volume change for each arm in comparison to vehicle arm. TGI%: percentages of tumor growth inhibition in comparison to vehicle arm.
Evaluation of downstream effector phosphoproteins
Despite the limited anti-tumor effect induced by the PI3K-mTOR inhibitors single agents or in combination with dacomitinib, downstream inhibition of the PI3K/AKT/mTOR pathway was observed in all of the arms containing a PI3K-mTOR inhibitor (Figure 4A-4F): all models exhibited pS6 inhibition and the LGSOC and LADC models also exhibited pAKT inhibition. The inhibition of these phospho-proteins was, in general, greater in the combination arm, which might reflect the requirement for dual inhibition of the tyrosine kinase receptor and PI3K to completely abrogate the pathway. Substantial inhibition of AKT and S6 phosphorylation was observed 2 hours after treatment; while their phosphorylation levels partially recovered by 24 hours. The differences in phosphorylation patterns between the LADC, TNBC and LGSOC models might be partially due to the different molecular backgrounds, but also to the different pharmacokinetic profiles of PF-04691502 and PF-05212384.
Although the addition of dacomitinib was expected to induce some degree of inhibition in the MAPK signaling pathway, no effects on pEGFR, nor on pERK levels was observed in the three models (Figure 4A-4F).
Table 2: Treatment arms, dose, frequency and administration route
Cisplatin + PI3K-mTOR inhibitor experiment |
||||
Model |
Drug |
Dose |
Frequency |
Route |
LADC 1 TNBC LGSOC |
Cisplatin |
3 mg / kg |
Weekly |
IP |
PF-05212384 |
10 mg/ Kg |
2 times / week |
IV |
|
Cisplatin PF-05212384 |
3 mg / kg 10 mg/ Kg |
weekly 2 times / week |
IP IV |
|
Vehicle |
weekly 2 times / week |
IP + IV |
||
Paclitaxel + PI3K-mTOR inhibitor experiment |
||||
Model |
Drug |
Dose |
Frequency |
Route |
LADC |
Paclitaxel |
15 mg / kg 2 |
2 times / week |
IP |
PF-04691502 |
5 mg / kg |
daily |
PO |
|
Paclitaxel PF-04691502 |
15 mg / kg 5 mg / kg |
2 times / week daily |
IP PO |
|
Vehicle |
daily 2 times / week |
PO + IP |
||
TNBC LGSOC |
Paclitaxel |
10 mg / kg |
2 times / week |
IP |
PF-05212384 |
10 mg / kg |
2 times / week |
IV |
|
Paclitaxel + PF-05212384 |
10 mg / kg 10 mg / Kg |
2 times / week 2 times / week |
IP IV |
|
Vehicle |
2 times / week |
IP + IV |
||
Dacomitinib + PI3K-mTOR inhibitor |
||||
Model |
Drug |
Dose |
Frequency |
Route |
LADC |
Dacomitinib |
3 mg / kg |
daily |
PO |
PF-04691502 |
5 mg / kg |
daily |
PO |
|
Dacomitinib PF-04691502 |
3 mg / kg 5 mg / kg |
daily daily |
PO PO |
|
Vehicle |
daily |
PO |
||
TNBC LGSOC |
Dacomitinib |
3 mg / Kg |
daily |
PO |
PF-05212384 |
10 mg/ Kg |
2 times / week |
IV |
|
Dacomitinib PF-05212384 |
3 mg / Kg 10 mg/ Kg |
daily 2 times / week |
PO IV |
|
Vehicle |
daily 2 times / week |
PO |
||
IV |
||||
LADC: non-small cell lung cancer; TNBC: triple negative breast cancer; LGSOC: low-grade serous ovarian cancer; IP: intraperitoneal; IV: intravenous; PO: oral gavage; 1 In the LADC model, PF-05212384 at a dose of 15 mg / kg was initially tested. Several sudden deaths occur after the first drug administration; thus, PF-05212384 was reduced to 10 mg / kg in subsequent administrations. The models evaluated afterwards (TNBC and LGOSC) received a dose of 10 mg / kg. 2 Paclitaxel was initially evaluated at a dose of 15 mg / kg in the LADC and subsequently changed to 10 mg/kg based on several unexpected deaths in the paclitaxel-containing arms. Paclitaxel was tested at a dose of 10 mg / kg in the remaining models.
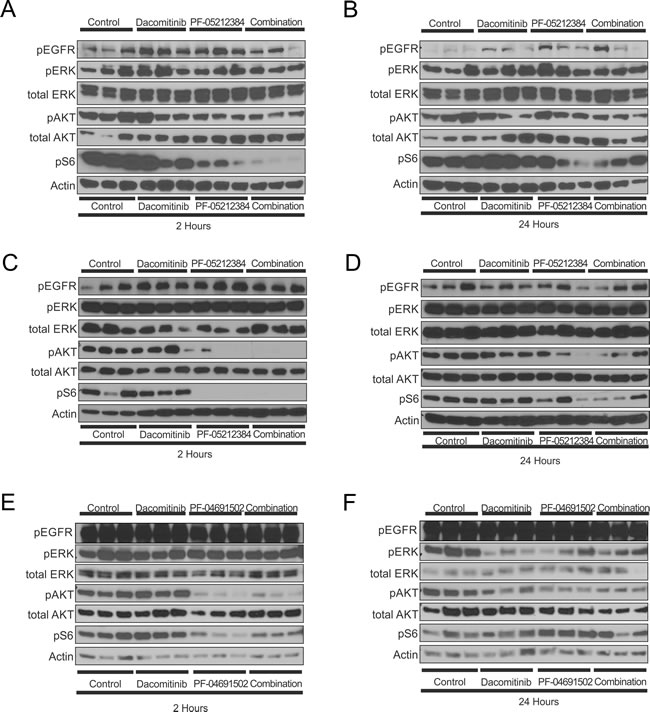
Figure 4: Western blot analysis of the indicated proteins in three independent tumors of the triple-negative breast cancer (TNBC), KRAS mutant (G12R) low-grade ovarian cancer (LGSOC) and TP53 (R181P) and KRAS (G12C) mutant lung adenocarcinoma (LADC). The TNBC A., B. and LGSOC C., D. models were treated with vehicle, PF-05212384 (10 mg/kg, twice weekly, intravenously, dacomitinib (3 mg/kg daily, oral gavage) or the combination of both agents. The LADC model E., F. was treated with vehicle, PF-04691502 (5 mg/kg, daily, oral gavage), dacomitinib (3 mg/kg daily, oral gavage) or the combination of both agents. Tumors were collected 1 hour after treatment (A, C, E) or 24 hours after treatment (B, D, F).
Discussion
We simultaneously evaluated multiple combinations of novel therapeutic strategies using PDXs as an innovative platform for preclinical evaluation of these combinations. The selected models harbored molecular alterations commonly described in the matched tumor types, such as loss of PTEN expression present in around 30% in patients with TNBC [35] and LADC [36] or KRAS mutation described in around one third of LADC [37] and LGSOC [38]. The response to conventional chemotherapy in the three selected PDXs resembled the response in the matching tumor types reported in clinical trials and retrospective series, from a chemo-sensitive model represented by the LADC to a chemo-resistant model represented by the LGSOC [31, 33, 34, 39]. We observed that the addition of PF-05212384 to cisplatin enhanced the antitumor activity of cisplatin suggesting a chemo-sensitizing effect. There is preclinical evidence supporting the role of the PI3K-AKT-mTOR pathway in DNA repair. The beta isoform of PI3K (p110β) seems to have a role sensing DNA damage and facilitating the binding to DNA of DNA repair proteins from the ATM and ATR pathways [40]. Loss of p110β was found to induce genomic instability, whereas 110β inhibition increased sensitivity to DNA damaging agents [40]. The PI3K-mTOR pathway also regulates the expression of certain DNA repair proteins, such as BRCA1 [41] and FANCD2 [3], as decreased levels of these proteins are observed upon PI3K-mTOR inhibition. PTEN also is involved in chromosomal integrity and DNA repair, specifically the fraction of PTEN localized in the nucleus [42, 43]. In xenografts established from PTEN-deficient cell lines, the addition of a PI3K inhibitor increased the antitumor activity of cisplatin [43]. Our experiments did not assess DNA damage markers or DNA repair proteins, so the specific mechanism by which the addition of PF-05212384 might have induced cisplatin chemo-sensitization was not fully clarified. Furthermore, the use of a pan-isoform PI3K-mTOR inhibitor, does not allow to discern whether the chemo-sensitization induced by PF-05212384 depends on an individual PI3K isoform or whether simultaneous inhibition of different PI3K isoforms is needed. Experiments comparing the activity of pan-isoform PI3K inhibitors and novel beta-isoform PI3K inhibitors combined with chemotherapy would help understanding the mechanism of chemo-sensitization observed in our experiments.
The effects of adding a PI3K-mTOR inhibitors to chemotherapy has varied among the three models tested: from a synergistic effect in the TNBC model to lack of effect in the LADC model; whereas an intermediate effect was achieved in the LGSOC model. These differences between these models may be probably related to their variable molecular backgrounds. The LADC had a TP53 missense mutation causing a complete loss of function of p53 [44]. TP53 mutations have been identified as potential mechanisms of resistance to PI3K inhibition in cell lines and in patients participating in the phase I clinical trial evaluating the alpha-specific PI3K inhibitor BYL719 [45]. Further research is warranted to characterize the role of TP53 status on the activity of the PI3K inhibitors. The LADC and LGSOC models also harbored KRAS mutations (G12C and G12R respectively). Different KRAS mutations seemed to signal preferentially through different downstream pathways [46]; KRAS mutations signaling preferentially through the MAPK pathway, such as G12C, are a well-known resistance factor to PI3K inhibition [47]; whereas KRAS G12R signals through both the ERK and PI3K pathways, which might explain the moderate chemo-sensitization achieved in the LGSOC, disease characterized by its intrinsic relative chemo-resistance. Although the addition of a PI3K-mTOR inhibitor to chemotherapy might be a strategy to increase the antitumor activity of chemotherapy in LGSOC, the combination of MEK and PI3K inhibitors would probably be a more relevant combination based on the high response rate observed in this patient population in the early clinical trials evaluating these combinations [48, 49].
In the experiments evaluating the combination of PI3K-mTOR inhibitors and dacomitinib, no statistically significant difference was observed between the combination arm and the vehicle arm. This lack of activity contrasts with results from the western-blot, in which target inhibition was observed in the arms containing the PI3K-mTOR inhibitors. No significant pEGFR inhibition was observed in the dacomitinib containing arms which might be related to the dose selected, lower than in prior in vivo experiments [27, 28, 50, 51]. The lack of MAPK pathway inhibition in the arm combining PIK3-mTOR inhibitor and dacomitinib may be related to an insufficient pEGFR inhibition or to the presence of activating events in the MAPK pathway, such as KRAS mutations, which may constitute potential mechanisms of resistance to this treatment combination.
The preclinical results generated by our study have supported the translation of these combinations into the clinic and helped define the patient populations for each combination. The combination of cisplatin and PF-05212384 in patients with TNBC is currently under evaluation in a phase I clinical trial (NCT01920061), which has three parallel arms evaluating PF-05212384 in combination with cisplatin, docetaxel, and dacomitinib respectively. Although no molecular selection is required, paired biopsies are being collected for biomarker analysis. This trial should provide further insights into the role of PI3K-mTOR inhibitors as chemosensitizing agents and could help to clarify the effect of PF-05212384 on DNA damage and repair and the role of different molecular biomarkers, such as PTEN deficiency, on such effect. The results provide some evidence that preclinical studies of novel targeted drugs in PDX may reveal the treatment response heterogeneity, which could be informative in refining the design of early stage clinical trials for these drugs.
Materials and Methods
Drugs
The two PI3K-mTOR inhibitors evaluated, PF-05212384 and PF-04691502, were provided by Pfizer Inc. (New York City, NY), as well as the pan-HER inhibitor dacomitinib; cisplatin (Teva Parenteral Medicines, Inc., Irvine, CA) and paclitaxel (Sigma-Aldrich Corporation, St. Louis, MO) were purchased from the hospital pharmacy.
PDX models
The three PDX models were selected from a repository at the University Health Network (UHN) that has been established with patient consent and in accordance to the guidelines of the UHN Human Research Ethics Board. Animal experiments were performed at the Toronto Centre for Phenogenomics in compliance with regulatory guidelines and a protocol approved by the UHN Animal Care Committee. Three models were selected based on their deficient PTEN expression as assessed by immunohistochemistry: a TNBC model established from a liver metastasis biopsy, a LGSOC model established from an ascites sample, and a lung adenocarcinoma (LADC) model established from a surgical specimen. Breeders were purchased from Jackson Laboratories (Bar Harbor, ME) and were used for up to four generations to avoid genetic drift. Cryobanked tumor fragments were revived and expanded as donors in 2-3 serial mouse generations at the subcutaneous flank site of non-obese diabetic severe combined immunodeficiency (NOD-SCID) NOD.CB17-Prkdcscid/J mice.
PDX therapeutic studies
In the experimental phase, groups of 12 tumor-bearing mice were randomized to treatment or vehicle once tumors reached an average volume of 250 mm3 by caliper measurements.
Treatment doses were selected according to previously performed maximum tolerated dose experiments and were refined further based on the observed toxicity profile. Three different drug combinations were evaluated with their matching single agent and vehicle arms. Treatment arm details are summarized in Table 2. In the LADC model, both PI3K-mTOR inhibitors—PF-05212384 and PF-04691502—were tested. However, in the subsequent models, only PF-05212384 was tested as PF-05212384 would be the compound that would be further evaluated in clinical trials based on additional clinical data that became available before starting the TNBC and LGSOC models. Tumor size and mouse weight were evaluated twice per week. Tumor volume was calculated using the formula = length2 x width x 0.52. Mice were sacrificed once tumors reached 1500 mm3 or when other human endpoints were observed in compliance with regulatory guidelines of the Institutional Animal Care Committee. Pieces of extracted tumors were snap-frozen in liquid nitrogen and formalin-fixed and paraffin-embedded for further analysis.
A parallel acute dose experiment was performed to evaluate the pharmacodynamic effect of PF-05212384, PF-04691502, and dacomitinib on downstream effectors in the treatment groups. Three mice replicates were harvested at 2 and 24 hours post-dosing.
Genomic characterization of patient-derived tumor xenografts
PDXs in the repository were characterized using the OncoCarta Panel v 1.0 (238 mutations in 19 key oncogenes) on the MassARRAY System (Agena Bioscience, San Diego, CA) to determine their tumor mutational profile.
Immunoblots of downstream effector proteins
Aliquots of approximately 50 mg of tissue were mixed with 1 ml of lysis RIPA buffer (R 0278, Sigma, Saint Louis, MO) buffer containing a protease inhibitor cocktail (11 836 170 001, Roche Diagnostics GmbH, Mannheim, Germany), 8 mM sodium orthovanadate, and 0.2 mM phenylmethanesulfonyl fluoride. Protein extracts were collected after tissues were homogenized (1 min) and centrifuged (20000 × g, 20 min). Aliquots of 30 µg of protein were mixed with an equal volume of 2 X SDS-PAGE sample loading buffer, and resolved by SDS-PAGE. Resolved samples were transferred to polyvinylidene fluoride (PVDF) membranes by using Trans-Blot® Turbo™ Midi PVDF Transfer Packs, 170-4157 BioRad, Hercules, CA). Membranes were blocked with 5% nonfat dry milk in Tris-Buffered Saline with Tween 20 (1X TBST). Blots were incubated overnight at 4ºC with primary antibodies (EGFR Tyr 1068, ERK, pERK1/2 Thr202/Tyr204, AKT, pAKT Ser 473, pS6 Ser235/236, and β-actin) at 1:1000 dilutions, followed by incubation with a 1: 4000 dilution of HRP-linked anti-rabbit IgG secondary antibodies for one hour . All antibodies used for blotting were from Cell Signaling Technology, Danvers, MA. Immunoreactive protein bands were detected by ECL-Prime Western blotting detection reagent (RPN2236, GE Healthcare, Little Chalfont, UK).
Immunohistochemistry
PTEN immunohistochemistry was performed using the Ventana Benchmark XT autostainer for anti-PTEN (138G6, Cell Signaling Technology, Danvers, MA) with the iVIEW DAB detection system (Ventana Medical Systems, Inc., Tucson, AZ). Absence of staining of tumor cell cytoplasm was classified as loss of expression whereas faint staining was considered as low PTEN expression.
Statistical analysis
Mixed effect model was used to test the differences in tumor growth rates overtime between treatment and control groups within each PDX model. SAS 9.2 (SAS Institute Inc., Cary, NC) was used for the analysis.
Relative mean tumor growth was calculated with the formula
![]()
Tumor volume were plotted as mean +/- standard deviation with GraphPad Prism 6 (GraphPad Software Inc., San Diego, CA).
Author contributions
Experiment design: Irene Brana, Nhu-An Pham, Rafael Sierra, Lillian L Siu and Ming-Sound Tsao.
Model generation: Nhu-An Pham, Ming Li, Rafael Sierra, Benjamin G Neel, Lillian L Siu and Ming-Sound Tsao.
In vivo experiments: Ming Li, Christine Ng, Yuhui Wang, Peter Loparco, Rafael Sierra, Irene Brana, Nhu-An Pham, Ming-Sound Tsao.
Western-blot: Christine Ng, Irene Brana, Nhu-An Pham.
Pathology review and sample quality assessment: Lucia Kim, Shingo Sakashita, Blaise A. Clarke, Ming-Sound Tsao.
Statistical analysis: Lisa Wang.
Data interpretation: Irene Brana, Nhu-An Pham, Benjamin G. Neel, Lillian L Siu, Ming-Sound Tsao.
Manuscript preparation: Irene Brana, Nhu-An Pham, Lillian L Siu, Ming-Sound Tsao.
Manuscript review: all authors.
Acknowledgments
Irene Brana is a Conquer Cancer Foundation Young Investigator Award 2012 recipient, a SEOM translational Research Grant recipient and a “La Caixa” fellowship award recipient.
This work has been performed within the Department of Medicine from the Universitat Autònoma de Barcelona Doctoral Program framework.
Conflicts of Interest
Lillian L. Siu: funding from Pfizer to support clinical trials and to conduct of this preclinical study.
Ming S. Tsao: funding from Pfizer to conduct this preclinical study.
Rest of the authors: no conflict of interest to disclose.
Funding
The work was supported by research grants from Pfizer Canada, Inc., Ontario Research Fund - Research Excellence RE-03-020, Ontario Institute of Cancer Research, Princess Margaret Cancer Foundation and the Ontario Ministry of Health and Long Term Care. Dr. Brana was supported by the Conquer Cancer Foundation Young Investigator Award 2012, the SEOM translational Research Grant, and the “La Caixa” Fellowship award. Drs. Sakashita and Dr. Kim were supported by the Terry Fox Foundation STIHR in Clinician Scientists in Molecular Oncologic Pathology at CIHR (STP 53912). Dr. Tsao holds the M. Qasim Choksi Chair in Lung Cancer Translational Research. Dr. Neel was partially supported by a grant from the Ontario Ministry of Health and Long Term Care, The Princess Margaret Cancer Foundation, and a Terrry Fox Foundation Program Project Grant.
References
1. Brana I, Siu LL. Clinical development of phosphatidylinositol 3-kinase inhibitors for cancer treatment. BMC Med. 2012; 10: 161. doi: 10.1186/1741-7015-10-161.
2. Courtney KD, Corcoran RB, Engelman JA. The PI3K Pathway As Drug Target in Human Cancer. Journal of Clinical Oncology. 2010; 28: 1075-83. doi: 10.1200/jco.2009.25.3641.
3. Shen C, Oswald D, Phelps D, Cam H, Pelloski CE, Pang Q, Houghton PJ. Regulation of FANCD2 by the mTOR Pathway Contributes to the Resistance of Cancer Cells to DNA Double-Strand Breaks. Cancer Research. 2013; 73: 3393-401. doi: 10.1158/0008-5472.can-12-4282.
4. Yang X, Fraser M, Moll UM, Basak A, Tsang BK. Akt-Mediated Cisplatin Resistance in Ovarian Cancer: Modulation of p53 Action on Caspase-Dependent Mitochondrial Death Pathway. Cancer Research. 2006; 66: 3126-36. doi: 10.1158/0008-5472.can-05-0425.
5. Knuefermann C, Lu Y, Liu B, Jin W, Liang K, Wu L, Schmidt M, Mills GB, Mendelsohn J, Fan Z. HER2/PI-3K/Akt activation leads to a multidrug resistance in human breast adenocarcinoma cells. Oncogene. 2003; 22: 3205-12. doi: doi:10.1038/sj.onc.1206394.
6. Brognard J, Clark AS, Ni Y, Dennis PA. Akt/protein kinase B is constitutively active in non-small cell lung cancer cells and promotes cellular survival and resistance to chemotherapy and radiation. Cancer Research. 2001; 61: 3986-97.
7. Wallin JJ, Guan J, Prior WW, Lee LB, Berry L, Belmont LD, Koeppen H, Belvin M, Friedman LS, Sampath D. GDC-0941, a Novel Class I Selective PI3K Inhibitor, Enhances the Efficacy of Docetaxel in Human Breast Cancer Models by Increasing Cell Death In Vitro and In Vivo. Clinical Cancer Research. 2012; 18: 3901-11. doi: 10.1158/1078-0432.ccr-11-2088.
8. Hu L, Hofmann J, Lu Y, Mills GB, Jaffe RB. Inhibition of phosphatidylinositol 3’-kinase increases efficacy of paclitaxel in in vitro and in vivo ovarian cancer models. Cancer Research. 2002; 62: 1087-92.
9. Engelman JA, Mukohara T, Zejnullahu K, Lifshits E, Borras AM, Gale CM, Naumov GN, Yeap BY, Jarrell E, Sun J, Tracy S, Zhao X, Heymach JV, et al. Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. J Clin Invest. 2006; 116: 2695-706. doi: 10.1172/jci28656.
10. Junttila TT, Akita RW, Parsons K, Fields C, Lewis Phillips GD, Friedman LS, Sampath D, Sliwkowski MX. Ligand-Independent HER2/HER3/PI3K Complex Is Disrupted by Trastuzumab and Is Effectively Inhibited by the PI3K Inhibitor GDC-0941. Cancer Cell. 2009; 15: 429-40. doi: 10.1016/j.ccr.2009.03.020.
11. Eichhorn PJA, Gili M, Scaltriti M, Serra V, Guzman M, Nijkamp W, Beijersbergen RL, Valero V, Seoane J, Bernards R, Baselga J. Phosphatidylinositol 3-kinase hyperactivation results in lapatinib resistance that is reversed by the mTOR/phosphatidylinositol 3-kinase inhibitor NVP-BEZ235. Cancer Research. 2008; 68: 9221-30. doi: 10.1158/0008-5472.CAN-08-1740.
12. Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA, Klos KS, Li P, Monia BP, Nguyen NT, Hortobagyi GN, Hung MC, Yu D. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004; 6: 117-27. doi: 10.1016/j.ccr.2004.06.022.
13. Sergina NV, Rausch M, Wang D, Blair J, Hann B, Shokat KM, Moasser MM. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature. 2007; 445: 437-41. doi: 10.1038/nature05474.
14. Wang MY, Lu KV, Zhu S, Dia EQ, Vivanco I, Shackleford GM, Cavenee WK, Mellinghoff IK, Cloughesy TF, Sawyers CL, Mischel PS. Mammalian target of rapamycin inhibition promotes response to epidermal growth factor receptor kinase inhibitors in PTEN-deficient and PTEN-intact glioblastoma cells. Cancer Res. 2006; 66: 7864-9. doi: 10.1158/0008-5472.can-04-4392.
15. Hidalgo M, Amant F, Biankin AV, Budinska E, Byrne AT, Caldas C, Clarke RB, de Jong S, Jonkers J, Maelandsmo GM, Roman-Roman S, Seoane J, Trusolino L, et al. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov. 2014; 4: 998-1013. doi: 10.1158/2159-8290.cd-14-0001.
16. Garralda E, Paz K, Lopez-Casas PP, Jones S, Katz A, Kann LM, Lopez-Rios F, Sarno F, Al-Shahrour F, Vasquez D, Bruckheimer E, Angiuoli SV, Calles A, et al. Integrated next-generation sequencing and avatar mouse models for personalized cancer treatment. Clin Cancer Res. 2014; 20: 2476-84. doi: 10.1158/1078-0432.ccr-13-3047.
17. Stewart EL, Mascaux C, Pham NA, Sakashita S, Sykes J, Kim L, Yanagawa N, Allo G, Ishizawa K, Wang D, Zhu CQ, Li M, Ng C, et al. Clinical Utility of Patient-Derived Xenografts to Determine Biomarkers of Prognosis and Map Resistance Pathways in EGFR-Mutant Lung Adenocarcinoma. Journal of Clinical Oncology. 2015. doi: 10.1200/jco.2014.60.1492.
18. Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, Singh M, Zhang C, Schnell C, Yang G, Zhang Y, Balbin OA, Barbe S, Cai H, et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med. 2015; 21: 1318-25. doi: 10.1038/nm.3954.
19. deGraffenried LA, Fulcher L, Friedrichs WE, Grunwald V, Ray RB, Hidalgo M. Reduced PTEN expression in breast cancer cells confers susceptibility to inhibitors of the PI3 kinase/Akt pathway. Annals of Oncology. 2004; 15: 1510-6. doi: 10.1093/annonc/mdh388.
20. Jia S, Liu Z, Zhang S, Liu P, Zhang L, Lee SH, Zhang J, Signoretti S, Loda M, Roberts TM, Zhao JJ. Essential roles of PI(3)K-p110[bgr] in cell growth, metabolism and tumorigenesis. Nature. 2008; 454: 776-9. doi: 10.1038/nature07091.
21. Wee S, Wiederschain D, Maira SM, Loo A, Miller C, deBeaumont R, Stegmeier F, Yao YM, Lengauer C. PTEN-deficient cancers depend on PIK3CB. Proceedings of the National Academy of Sciences. 2008; 105: 13057-62. doi: 10.1073/pnas.0802655105.
22. Schwartz S, Wongvipat J, Trigwell Cath B, Hancox U, Carver Brett S, Rodrik-Outmezguine V, Will M, Yellen P, de Stanchina E, Baselga J, Scher Howard I, Barry Simon T, Sawyers Charles L, et al. Feedback Suppression of PI3Kα Signaling in PTEN-Mutated Tumors Is Relieved by Selective Inhibition of PI3Kβ. Cancer Cell. 2015. doi: 10.1016/j.ccell.2014.11.008.
23. Mallon R, Feldberg LR, Lucas J, Chaudhary I, Dehnhardt C, Santos ED, Chen Z, dos Santos O, Ayral-Kaloustian S, Venkatesan A, Hollander I. Antitumor Efficacy of PKI-587, a Highly Potent Dual PI3K/mTOR Kinase Inhibitor. Clinical Cancer Research. 2011; 17: 3193-203. doi: 10.1158/1078-0432.ccr-10-1694.
24. Yuan J, Mehta PP, Yin MJ, Sun S, Zou A, Chen J, Rafidi K, Feng Z, Nickel J, Engebretsen J, Hallin J, Blasina A, Zhang E, et al. PF-04691502, a Potent and Selective Oral Inhibitor of PI3K and mTOR Kinases with Antitumor Activity. Molecular Cancer Therapeutics. 2011; 10: 2189-99. doi: 10.1158/1535-7163.mct-11-0185.
25. Shapiro GI, Bell-McGuinn KM, Molina JR, Bendell J, Spicer J, Kwak EL, Pandya SS, Millham R, Borzillo G, Pierce KJ, Han L, Houk BE, Gallo JD, et al. First-in-Human Study of PF-05212384 (PKI-587), a Small-Molecule, Intravenous, Dual Inhibitor of PI3K and mTOR in Patients with Advanced Cancer. Clin Cancer Res. 2015; 21: 1888-95. doi: 10.1158/1078-0432.ccr-14-1306.
26. Britten CD, Adjei AA, Millham R, Houk BE, Borzillo G, Pierce K, Wainberg ZA, LoRusso PM. Phase I study of PF-04691502, a small-molecule, oral, dual inhibitor of PI3K and mTOR, in patients with advanced cancer. Invest New Drugs. 2014; 32: 510-7. doi: 10.1007/s10637-013-0062-5.
27. Gonzales AJ, Hook KE, Althaus IW, Ellis PA, Trachet E, Delaney AM, Harvey PJ, Ellis TA, Amato DM, Nelson JM, Fry DW, Zhu T, Loi CM, et al. Antitumor activity and pharmacokinetic properties of PF-00299804, a second-generation irreversible pan-erbB receptor tyrosine kinase inhibitor. Molecular Cancer Therapeutics. 2008; 7: 1880-9. doi: 10.1158/1535-7163.mct-07-2232.
28. Engelman JA, Zejnullahu K, Gale CM, Lifshits E, Gonzales AJ, Shimamura T, Zhao F, Vincent PW, Naumov GN, Bradner JE, Althaus IW, Gandhi L, Shapiro GI, et al. PF00299804, an Irreversible Pan-ERBB Inhibitor, Is Effective in Lung Cancer Models with EGFR and ERBB2 Mutations that Are Resistant to Gefitinib. Cancer Research. 2007; 67: 11924-32. doi: 10.1158/0008-5472.can-07-1885.
29. Abdul Razak AR, Soulieres D, Laurie SA, Hotte SJ, Singh S, Winquist E, Chia S, Le Tourneau C, Nguyen-Tan PF, Chen EX, Chan KK, Wang T, Giri N, et al. A phase II trial of dacomitinib, an oral pan-human EGF receptor (HER) inhibitor, as first-line treatment in recurrent and/or metastatic squamous-cell carcinoma of the head and neck. Ann Oncol. 2013; 24: 761-9. doi: 10.1093/annonc/mds503.
30. Janne PA, Ou SH, Kim DW, Oxnard GR, Martins R, Kris MG, Dunphy F, Nishio M, O’Connell J, Paweletz C, Taylor I, Zhang H, Goldberg Z, et al. Dacomitinib as first-line treatment in patients with clinically or molecularly selected advanced non-small-cell lung cancer: a multicentre, open-label, phase 2 trial. Lancet Oncol. 2014; 15: 1433-41. doi: 10.1016/s1470-2045(14)70461-9.
31. Schiller JH, Harrington D, Belani CP, Langer C, Sandler A, Krook J, Zhu J, Johnson DH. Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N Engl J Med. 2002; 346: 92-8. doi: 10.1056/NEJMoa011954.
32. Parmar M, Ledermann J, Colombo N, du Bois A, Delaloye J, Kristensen G, Wheeler S, Swart A, Qian W, Torri V, Floriani I, Jayson G, Lamont A, et al. Paclitaxel plus platinum-based chemotherapy versus conventional platinum-based chemotherapy in women with relapsed ovarian cancer: the ICON4/AGO-OVAR-2.2 trial. The Lancet. 2003; 361: 2099-106. doi: 10.1016/S0140-6736(03)13718-X.
33. Isakoff S, Goss P, Mayer E, Traina T, Carey L, Krag K, Rugo H, Liu M, Stearns V, Come S. (2011). TBCRC009: A multicenter phase II study of cisplatin or carboplatin for metastatic triple-negative breast cancer and evaluation of p63/p73 as a biomarker of response. 2011 ASCO Annual Meeting. (Chicaco, IL: Journal of Clinical Oncology), pp. Abstract 1025.
34. Perez EA, Vogel CL, Irwin DH, Kirshner JJ, Patel R. Multicenter Phase II Trial of Weekly Paclitaxel in Women With Metastatic Breast Cancer. Journal of Clinical Oncology. 2001; 19: 4216-23. doi: 10.1200/JCO.2001.19.22.4216.
35. Gonzalez-Angulo AM, Ferrer-Lozano J, Stemke-Hale K, Sahin A, Liu S, Barrera JA, Burgues O, Lluch AM, Chen H, Hortobagyi GN, Mills GB, Meric-Bernstam F. PI3K pathway mutations and PTEN levels in primary and metastatic breast cancer. Mol Cancer Ther. 2011; 10: 1093-101. doi: 10.1158/1535-7163.mct-10-1089.
36. Yanagawa N, Leduc C, Kohler D, Saieg MA, John T, Sykes J, Yoshimoto M, Pintilie M, Squire J, Shepherd FA, Tsao MS. Loss of phosphatase and tensin homolog protein expression is an independent poor prognostic marker in lung adenocarcinoma. J Thorac Oncol. 2012; 7: 1513-21. doi: 10.1097/JTO.0b013e3182641d4f.
37. Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014; 511: 543-50. doi: 10.1038/nature13385.
38. Singer G, Oldt R 3rd, Cohen Y, Wang BG, Sidransky D, Kurman RJ, Shih Ie M. Mutations in BRAF and KRAS characterize the development of low-grade ovarian serous carcinoma. J Natl Cancer Inst. 2003; 95: 484-6.
39. Gershenson DM, Sun CC, Bodurka D, Coleman RL, Lu KH, Sood AK, Deavers M, Malpica AL, Kavanagh JJ. Recurrent low-grade serous ovarian carcinoma is relatively chemoresistant. Gynecol Oncol. 2009; 114: 48-52. doi: 10.1016/j.ygyno.2009.03.001.
40. Kumar A, Fernandez-Capetillo O, Carrera AC. Nuclear phosphoinositide 3-kinase β controls double-strand break DNA repair. Proceedings of the National Academy of Sciences. 2010; 107: 7491-6. doi: 10.1073/pnas.0914242107.
41. Ibrahim YH, Garcia-Garcia C, Serra V, He L, Torres-Lockhart K, Prat A, Anton P, Cozar P, Guzman M, Grueso J, Rodriguez O, Calvo MT, Aura C, et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov. 2012; 2: 1036-47. doi: 10.1158/2159-8290.cd-11-0348.
42. Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP, Yin Y. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 2007; 128: 157-70. doi: 10.1016/j.cell.2006.11.042.
43. Bassi C, Ho J, Srikumar T, Dowling RJO, Gorrini C, Miller SJ, Mak TW, Neel BG, Raught B, Stambolic V. Nuclear PTEN controls DNA repair and sensitivity to genotoxic stress. Science. 2013; 341: 395-9. doi: 10.1126/science.1236188.
44. Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, Olivier M. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Database version (R17, November 2013 is the latest). Hum Mutat. 2007; 28: 622-9. doi: 10.1002/humu.20495.
45. Rodon J, Juric D, Gonzalez-Angulo A, Bendell J, Berlin J, Bootle D, Gravelin K, Huang A, Derti A, Lehar J. (2013). Towards defining the genetic framework for clinical response to treatment with BYL719, a PI3Kalpha-specific inhibitor. 2013 American Association for Cancer Research Annual Meeting. (Washington D.C., US: Cancer Research).
46. Ihle NT, Byers LA, Kim ES, Saintigny P, Lee JJ, Blumenschein GR, Tsao A, Liu S, Larsen JE, Wang J, Diao L, Coombes KR, Chen L, et al. Effect of KRAS oncogene substitutions on protein behavior: implications for signaling and clinical outcome. J Natl Cancer Inst. 2012; 104: 228-39. doi: 10.1093/jnci/djr523.
47. Ihle NT, Lemos R, Wipf P, Yacoub A, Mitchell C, Siwak D, Mills GB, Dent P, Kirkpatrick DL, Powis G. Mutations in the Phosphatidylinositol-3-Kinase Pathway Predict for Antitumor Activity of the Inhibitor PX-866 whereas Oncogenic Ras Is a Dominant Predictor for Resistance. Cancer Research. 2009; 69: 143-50. doi: 10.1158/0008-5472.can-07-6656.
48. Bedard PL, Tabernero J, Janku F, Wainberg ZA, Paz-Ares L, Vansteenkiste J, Van Cutsem E, Pérez-García J, Stathis A, Britten CD, Le N, Carter K, Demanse D, et al. A Phase Ib Dose-Escalation Study of the Oral Pan-PI3K Inhibitor Buparlisib (BKM120) in Combination with the Oral MEK1/2 Inhibitor Trametinib (GSK1120212) in Patients with Selected Advanced Solid Tumors. Clinical Cancer Research. 2015; 21: 730-8. doi: 10.1158/1078-0432.ccr-14-1814.
49. Spreafico A, Oza AM, Clarke BA, Mackay HJ, Shaw P, Butler M, Dhani NC, Lheureux S, Wilson MK, Welch S, Zhang T, Yu C, Stockley T, et al. Genotype-matched treatment for patients with advanced type I epithelial ovarian cancer (EOC). Gynecol Oncol. 2017; 144: 250-5. doi: 10.1016/j.ygyno.2016.12.002.
50. Ercan D, Zejnullahu K, Yonesaka K, Xiao Y, Capelletti M, Rogers A, Lifshits E, Brown A, Lee C, Christensen JG, Kwiatkowski DJ, Engelman JA, Janne PA. Amplification of EGFR T790M causes resistance to an irreversible EGFR inhibitor. Oncogene. 2010; 29: 2346-56. doi: 10.1038/onc.2009.526.
51. Williams JP, Kim I, Ito E, Shi W, Yue S, Siu LL, Waldron J, O’Sullivan B, Yip KW, Liu FF. Pre-clinical characterization of Dacomitinib (PF-00299804), an irreversible pan-ErbB inhibitor, combined with ionizing radiation for head and neck squamous cell carcinoma. PLoS One. 2014; 9: e98557. doi: 10.1371/journal.pone.0098557.