
INTRODUCTION
Hepatocellular carcinoma (HCC) is the fifth most common cancer worldwide [1–3]. The prognosis of HCC has been poor, especially for the patients with advanced and/or metastatic tumors [4–6]. Recent epidemiological data indicates that HCC’s mortality rate could possibly be doubled over the next decades [4–6].
Hepatic stellate cells (HSC) are the main source of extracellular matrix proteins during fibrogenesis [7–9]. Recent studies have proposed that tumor-specific hepatic stellate cells (tHSCs) are critical in the HCC tumorigenesis and progression [7–9]. These cells, characteristically expressing α-smooth muscle actin (α-SMA), could possibly proliferate and differentiate into myofibroblasts [7–9]. tHSCs are tumor-promoting cells, which express intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1) among other factors to promote cancer cell migration and proliferation [7–9].
Recently studies, including ours [10, 11], have also proposed a key function of tHSCs in tumor immunology [7–9]. We have previously shown that tHSCs could induce T cell apoptosis [10]. Further, tHSCs also inhibits T cell response, causing HCC cell migration and invasion [11]. Dendritic cells (DCs) are specialized antigen-presenting cells (APCs), which are responsible for the initiation and regulation of immune response [12, 13]. Activation of DCs is important for anti-tumor activity [12, 13]. Dendritic cell-derived immunoglobulin receptor 2, or DIgR2, is a key inhibitory receptor that inhibits DCs-induced antigen-specific T-cell responses [14]. DC-derived DIgR2 is shown to bind to the receptor in T cells, suppressing T-cell proliferation, cytokine production and cytotoxic T lymphocyte (CTL) activity [14]. In the current study, we show that tHSCs induces DIgR2 expression in DCs, causing T-cell hypo-responsiveness.
RESULTS
tHSCs co-culture induces DIgR2 expression in bone marrow-derived dendritic cells (mDCs)
As described in our previous studies [10], the primary HSCs were derived from livers of control normal Buffalo rats or the xenograft HCC tissues, which were named as quiescent HSCs (qHSCs) and tumor HSCs (tHSCs), respectively [10]. Primary cultured bone marrow-derived dendritic cells (mDCs) were also derived from Buffalo rats [14]. mDCs were then co-cultured with tHSCs or qHSCs (mDCs to HSCs ratio, 20: 1). Expression of DIgR2, the key immune inhibitory receptor of immunoglobulin superfamily [14], was then tested. Quantitative real-time PCR (“qRT-PCR”) assay results in Figure 1A demonstrated that DIgR2 mRNA expression in mDCs was significantly elevated after co-culture of tHSCs. DIgR2 mRNA level increased over 9–10 fold higher in mDCs with tHSCs challenge (Figure 1A). On the other hand, qHSCs co-culture had no significant effect on DIgR2mRNA expression (Figure 1A). Further, DIgR2 protein expression in mDCs was also dramatically induced when co-cultured with tHSCs (but not the qHSCs, three sets of repeated data were quantified in Figure 1B). These results suggest that DIgR2 expression is induced in mDCs after tHSCs co-culture.
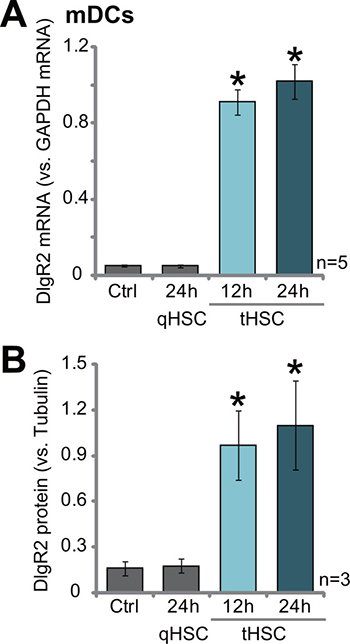
Figure 1: tHSCs co-culture induces DIgR2 expression in bone marrow-derived dendritic cells. Relative mRNA (A) and protein expression (Three sets of repeated blot data were quantified in (B) of DIgR2 in bone marrow-derived dendritic cells (mDCs), co-cultured with/out quiescent HSCs (qHSCs) or tumor HSCs (tHSCs) for applied time, were shown. “Ctrl” stands for mDCs only. “Tubulin” stands for loading control β-Tubulin. *P < 0.05 vs. “Ctrl” group.
MEK-ERK activation is required for DIgR2 expression in tHSCs-stimulated mDCs
Next, we studied the potential mechanism of DIgR2 expression in mDCs with tHSCs co-culture. Western blotting assay results showed that co-culture with tHSCs induced significant activation of MAPK/ERK kinase (MEK)-extracellular signal-regulated kinase (ERK) cascade in mDCs (Figure 2A). Phosphorylated (p-) MEK1/2 and p-ERK1/2 in mDCs were significantly increased following co-culture of tHSCs (Figure 2A). To study the link between MEK-ERK activation and DIgR2 expression, pharmacological MEK-ERK inhibitors were first applied, including PD98059, U0126 and MEK-162 [15, 16]. As shown in Figure 2B, treatment with these inhibitors almost completely blocked MEK-ERK cascade activation in mDCs with tHSCs co-culture. Consequently, DIgR2 mRNA (Figure 2C) and protein (Figure 2D) expressions were also largely inhibited. The pharmacological evidences suggest that activation of MEK-ERK signaling is required for DIgR2 expression in tHSCs-stimulated mDCs.
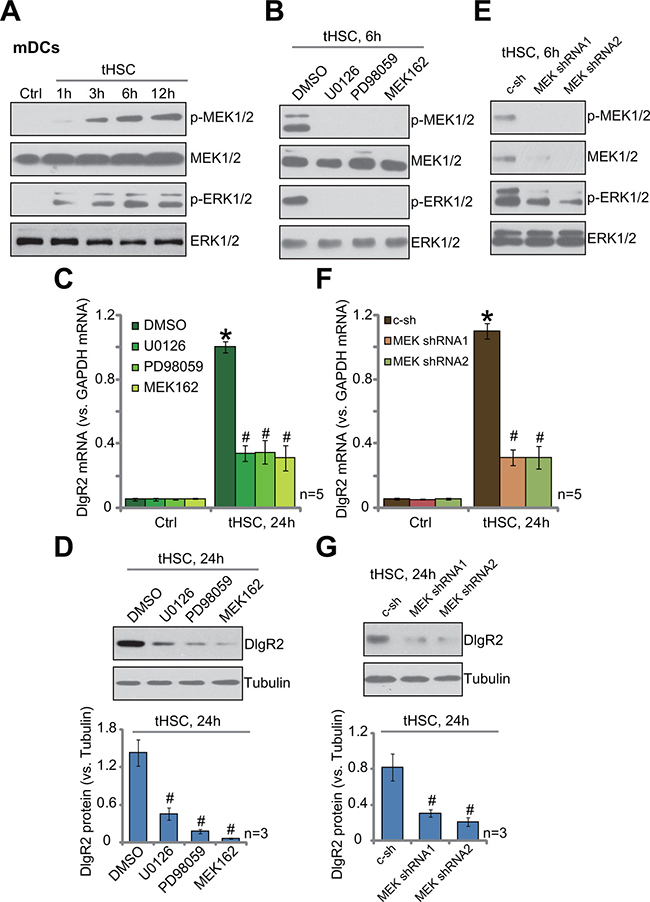
Figure 2: MEK-ERK activation is required for DIgR2 expression in tHSCs-stimulated mDCs. Bone marrow-derived dendritic cells (mDCs) were co-cultured with tumor HSCs (tHSCs) for indicated time; MEK-ERK signaling activation was tested by Western blotting assay (A). mDCs were pretreated with PD98059 (100 nM), U0126 (100 nM), MEK162 (1 μM) or vehicle (0.1% “DMSO”) for 1 hour, followed by tHSCs co-culture for applied time; Signaling was tested by Western blotting assay (B); DIgR2 mRNA (C) and protein (D), three sets of repeated blot data were quantified) expressions were also tested. mDCs were infected with applied MEK1/2-shRNA (“MEK shRNA1/2”) or non-sense scramble control shRNA (“c-sh”), cells were further co-cultured with tumor HSCs (tHSCs) for indicated time; Signaling was tested by Western blotting assay (E); Relative mRNA (F) and protein expression (G, three sets of repeated blot data were quantified) of DIgR2 were also tested. “Ctrl” stands for mDCs only. *P < 0.05 vs. “Ctrl”. #P < 0.05 vs. “DMSO” group (C) or “c-sh” group (D, F and G).
To further support our hypothesis, shRNA method was applied to knockdown MEK1/2 in mDCs. Two MEK1/2 shRNAs with non-overlapping sequences, named as “MEK shRNA1/2”, were applied. MEK1/2 expression was indeed dramatically downregulated after shRNA infection (Figure 2E). MEK-ERK activation in mDCs, tested against by p-MEK1/2 and p-ERK1/2, was also largely attenuated (Figure 2E). tHSCs-stimulated DIgR2 mRNA(Figure 2F) and protein (Figure 2G) expression was largely inhibited in MEK-silenced mDCs. Collectively, these results suggest that activation of MEK-ERK cascade is required for DIgR2 expression in tHSCs-stimulated mDCs.
shRNA knockdown of DIgR2 in mDCs
The aim of this study is to test the potential effect of tHSCs on the activity of DCs. More specifically, we wanted to know if tHSCs-induced DIgR2 expression could influence the functions of mDCs. Thus, lentiviral shRNA method was applied to selectively knockdown DIgR2 in mDCs. A set of three lentiviral DIgR2 shRNAs, targeting the non-overlapping sequence of DIgR2, were established. The three shRNAs were named as “DlgR2-shRNA Sq1/2/3”. qRT-PCR assay results in Figure 3A confirmed that adding each of the three shRNAs significantly inhibited DIgR2 mRNA expression in mDCs with co-culture of tHSCs. Consequently, DIgR2 protein expression was also silenced (Three sets of repeated data were quantified in Figure 3B). Among three tested DIgR2 shRNAs, the DIgR2 shRNA “Sq3” showed highest efficiency in knocking down DIgR2 (Figure 3A and 3B). This DlgR2-shRNA (“Sq3”) was then selected for further functional studies. Notably, the non-sense scramble control shRNA (“c-sh”) failed to decrease DIgR2 expression in mDCs (Figure 3A and 3B). Notably, DIgR2 shRNA failed to inhibit survival of mDCs (tested by the trypan blue assay).
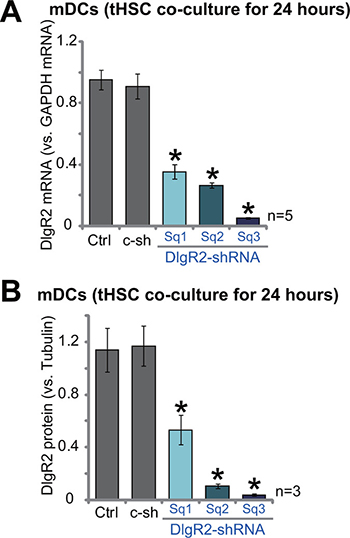
Figure 3: shRNA knockdown of DIgR2 in bone marrow-derived dendritic cells (mDCs). Bone marrow-derived dendritic cells (mDCs) were infected with DlgR2-shRNA (“Sq1/2/3”) or non-sense scramble control shRNA (“c-sh”), cells were further co-cultured with tumor HSCs (tHSCs) for 24 hours, relative mRNA (A) and protein expression (Three sets of repeated blot data were quantified in (B) of DIgR2 were shown. “Ctrl” stands for no shRNA group. *P < 0.05 vs. “Ctrl”.
tHSCs co-culture inhibits production of multiple cytokines in mDCs, abolished after DlgR2 silence
Next, we tested the function of mDCs after co-culture of tHSCs. Several DC-associated cytokines or activation markers, including CD80, CD86 and interleukin (IL)-12 [17–20], were tested. qRT-PCR assay was performed, and results showed that mRNA expressions of CD80 (surface co-stimulatory molecule, Figure 4A), CD86 (another surface co-stimulatory molecule, Figure 4B) and IL-12 (DC activation marker cytokine, Figure 4C) in mDCs were decreased significantly after co-culture of tHSCs. On the other hand, qHSCs co-culture failed to change mRNA expression of the above-mentioned cytokines (Figure 4A–4C). Remarkably, DlgR2 silence, by the DlgR2-shRNA (“Sq3”) in mDCs, almost restored mRNA expression of the cytokines (Figure 4A–4C). These results indicate that DlgR2 induction by tHSCs is important for subsequent inhibition of above cytokine expression. Further ELISA assay results showed that productions of CD80 (Figure 4D), CD86 (Figure 4E) and IL-12 (Figure 4F) were also decreased in tHSCs-stimulated mDCs. Such effects were again almost abolished with shRNA knockdown of DlgR2 (Figure 4D–4E). Notably, the non-sense scramble control shRNA (“c-sh”) was in-effective (Figure 4A–4F).
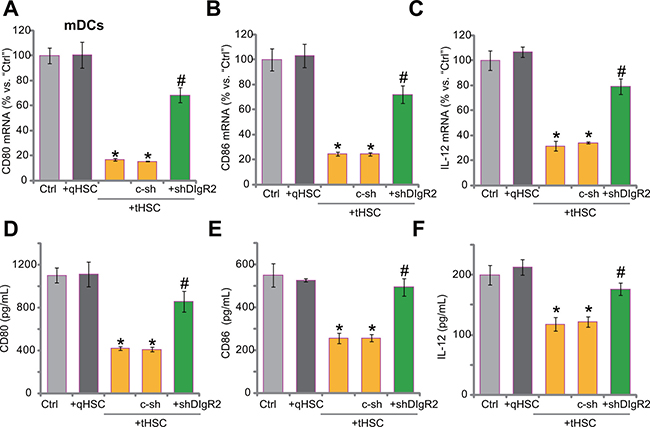
Figure 4: tHSCs co-culture inhibits production of multiple cytokines in mDCs, abolished after DlgR2 silence. mDCs were infected with DlgR2-shRNA (“shDlgR2, Sq/3”) or non-sense scramble control shRNA (“c-sh”), cells were further co-cultured with tumor HSCs (tHSCs) for 72 hours, mRNA expression (in mDCs, qRT-PCR assay, (A–C) and protein content (in conditional medium, ELISA assay, (D–F) of listed cytokines were analyzed. “Ctrl” stands for mDCs only. *P < 0.05 vs. “Ctrl”. #P < 0.05 vs. “c-sh” group
tHSCs-stimulated DlgR2 expression in mDCs inhibits splenic T cells
It has been previously shown that DC-derived DIgR2 binds to the receptor in T cells, which shall inhibit normal T cell functions. Thus, we then co-cultured splenic T cells with mDCs (T cells/mDCs ration, 20:1). OVA-II peptide CTL assay results in Figure 5A confirmed that co-culture with tHSCs-stimulated mDCs significantly inhibited the CTL activity of splenic T cells. Further, lipopolysaccharide (LPS)-induced interferon γ (IFN-γ) production in splenic T cells was also largely inhibited with the existence of tHSCs-stimulated mDCs (Figure 5B). Remarkably, such effects in splenic T cells were almost completely reversed with silence of DlgR2 in tHSCs-stimulated mDCs (Figure 5A and 5B). These results indicate that tHSCs-stimulated DlgR2 expression/production in mDCs should mediate inhibition of splenic T cells. Further studies confirmed that co-culture of tHSCs-stimulated mDCs induced proliferation inhibition (Figure 5C and 5D) and apoptosis (Figure 5E) in splenic T cells, which were again largely attenuated with DlgR2 shRNA in mDCs (Figure 5C–5E). Notably, qHSCs-stimulated mDCs had no significant inhibition on splenic T cells (Figure 5A–5E). Also, the non-sense scramble control shRNA (“c-sh”) failed to rescue splenic T cells from tHSCs-treated mDCs (Figure 5A–5E).
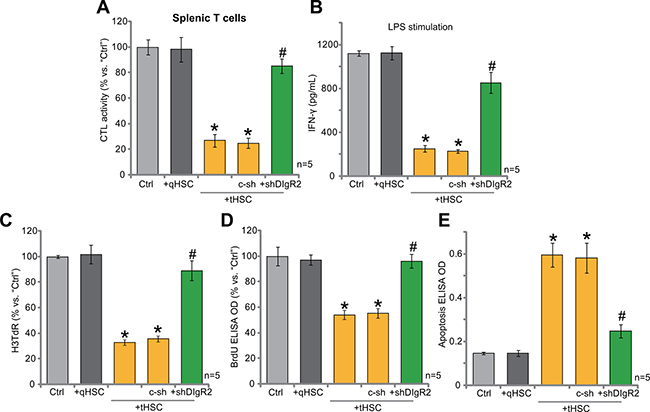
Figure 5: tHSCs-stimulated DlgR2 expression in mDCs inhibits splenic T cells. mDCs were infected with DlgR2-shRNA (“shDlgR2, Sq/3”) or non-sense scramble control shRNA (“c-sh”), cells were further co-cultured with/out tumor HSCs (tHSCs) or quiescent HSCs (qHSCs) for 24 hours; Stimulated mDCs were then co-cultured with splenic T cells; OVA-II peptide CTL assay activity (after 24 hours, (A), LPS (100 ng/mL)-induced IFN-γ production (after 24 hours, (B), cell proliferation (C and D), after 96 hours) and cell apoptosis (E), Histone DNA ELISA assay, after 96 hours) in the T cells were analyzed. “Ctrl” stands for co-culture of regular mDCs. *P < 0.05 vs. “Ctrl”. *P < 0.05 vs. “c-sh”.
DISCUSSION
tHSCs are the major source of extracellular matrix proteins during fibrogenesis [21–23], and are vital in the tumorigenesis and progression of human HCC [7, 8]. tHSCs could infiltrate HCC stroma and peri-tumor tissues, and could also be localized around tumor sinusoids, fibrous septae and the tumor capsule [7–9]. tHSCs promote HCC progression via regulating diverse biological processes, i.e. facilitating extracellular matrix (ECM) turnover, growth factor and cytokine signalling, as well as promoting tumor angiogenesis [7–9]. Recent studies have also proposed a pivotal function of tHSCs in tumor immunity [7–9].
Immune evasion of HCC cells [24–26] and other tumor cells is critical for cancer progression [26, 27]. One underling mechanism is to decrease the number and activity of anti-tumor immune cells, at both the tumor site and in the lymphoid organs [26, 27]. tHSCs could induce T-cell hypo-responsiveness, an effect that is not observed with the qHSCs. Previous studies have shown that B7-H1 (a key member of the B7 family of co-stimulatory molecules) upregulation in tHSCs could result in increased ligation of PD-1 receptor on activated T-cells, causing T cell apoptosis and inhibition of T-cell-mediated tumor cell apoptosis [28–30]. Meanwhile, activated tHSCs could also increase the number of in T-reg cells to exert immunosuppressive functions [31, 32].
We here discovered another key mechanism of tHSCs in immuno-regulation: via direct communicating with DCs. tHSCs stimulation induced upregulation of DIgR2 in mDCs, which is a key negative regulator of DC-initiated T-cell responses [14]. Further, tHSCs-stimulated mDCs induced T-cell hypo-responsiveness, causing decreased CTL activity and reduced IFN-γ production as well as T cell proliferation inhibition and apoptosis. Remarkably, such effects were largely attenuated with DIgR2 shRNA in mDCs. Based on these results, we propose that tHSCs stimulation directly induces DIgR2 expression and production in DCs, which binds to T-cells, causing T-cell hypo-responsiveness and apoptosis.
Intriguingly, we showed that activation of MEK-ERK cascade is required for tHSCs-stimulated DIgR2 expression in mDCs. MEK-ERK inhibitors (PD98059, U0126 and MEK-162) or shRNA-mediated knockdown of MEK1/2 largely attenuated tHSCs-induced DIgR2 expression in mDCs. Further studies will be needed to further characterize the underlying signaling mechanisms of DIgR2 expression in mDCs.
MATERIALS AND METHODS
Chemicals, reagents and antibodies
All the antibodies utilized in this study were obtained from Abcam (Suzhou, China). The cell culture reagents were obtained from Invitrogen (Shanghai, China). MEK-ERK inhibitors, PD98059, U0126 and MEK-162 [33, 34], were provided by Selleck (Beijing, China). The mRNA primers were designed and synthesized by the Genepharm (Shanghai, China).
Rat HCC tumor model
The detailed protocol of rat tumor model was described in detail in our previous studies [10, 11]. Briefly, buffalo rats (4–5 week-old) were obtained from the Animal Center of Soochow University (Suzhou, China). The MRH HCC cells [10, 11] were injected into the right flanks of the Buffalo rats. Four weeks after the initial injection, xenograft tumors were established. Surgery-isolated fresh HCC tumors were then cut into small piece (2×1×1 mm3), and were transplanted to the rat livers [10], using the described procedures [10]. Tumor extension was then determined by ultrasound on days 7, 14, 21 and 28 after tumor establishment. The animal protocols were approved by Institutional Animal Care and Use Committee (IACUC) and Ethics Board of Anhui Medical University. All surgical procedures were performed with anesthesia. All efforts were made to minimize suffering.
Isolation and culture of HSCs
As described in our previous studies [10, 11], HSCs were derived from the livers of normal Buffalo rats or HCC tissues. The livers were subjected to perfusion with described solution [10, 11]. Afterwards, truncated normal liver tissues and HCC tissues were further digested using the described method [10, 11]. Thereafter, the cell suspensions were established, which were purified by centrifugation through a 8% Nycodenz (Axis-Shield PoC) gradient. The achieved HSCs were then cultured in DMEM medium (GIBCO-BRL) with 10% FBS. Cell viability over 90% was verified via trypan blue exclusion. The desmin immuno-staining assay was performed to determine the purity of quiescent HSCs (qHSCs) and tHSCs, ranging from 90–95% [35].
Primary culture of bone marrow dendritic cells (mDCs)
As described [10, 36], bone marrow were isolated from Buffalo rat femurs. After lysis of the red blood cell, bone marrow cells were cultured in described RPMI-1640 medium [10, 11] with rat recombinant granulocyte-macrophage colony stimulating factor (GM-CSF, 4 ng/mL, Sigma) and interleukin-4, (IL-4, 1,000 U/mL, R&D systems, Shanghai, China). The non-adherent cells were released spontaneously from the proliferating cell clusters, harvested, washed, and resuspended in the described complete medium [36].
mDCs and HSC co-culture
For each well, 2.5 × 105 mDCs were incubated with 1.25 × 104 HSCs (20: 1, mDCs to HSCs) for applied time.
Primary culture of spleen T cells
As described in our previous studies [10, 11], the minced rat spleens were filtered. The splenocytes were then isolated from erythrocytes via centrifugation of the cell suspension on a Ficoll gradient (Histopaque 1083, Sigma) [37]. The cells were layered onto the top of the gradient in a 10-ml Falcon tube, followed by centrifugation at 800×g for 20 min at room temperature without braking. Lymphocytes (mainly T-cells) were collected, and washed twice in PBS-1% FBS (washing buffer). Cells were then cultured.
Co-culture of spleen T cells with mDCs and in vitro T cell function detection
The spleen T cells were utilized as antigen-specific responders and co-cultured with mDCs (T cells/mDCs ratio = 20:1). OVA (323–339) peptide assay was performed to test CTL assay [14]. T cell proliferation was determined by [3H]-TdR DNA incorporation assay (after 96 hours [14]) and BrdU ELISA assay (after 96 hours [38]) using the attached protocols. Histone DNA apoptosis ELISA kit was applied to test cell apoptosis (after 96 hours), and detailed protocol was described previously [39–41]. T cells were also treated with LPS (100 ng/mL, Sigma), supernatants were collected after 24 hours for ELISA detection of IFN-γ ( R&D Systems).
RNA extraction and real-time PCR
RNA was always extracted by the TRIzol reagents (Promega, Shanghai, China) [42]. cDNA was achieved via reverse transcription under the SYBR Green PCR kit (Applied Biosystems, Suzhou, China). Quantitative real time-PCR (qRT-PCR) assay was performed via the ABI Prism 7600 Fast Real-Time PCR system as previously described [43, 44]. Melt curve analysis was applied to calculate product melting temperature. Glyceraldehyde-3-phosphatedehydrogenase (GAPDH) mRNA was chosen as the reference gene, and the 2−∆ ∆Ct method was utilized to quantify targeted mRNA change within samples [43, 44]. DIgR2 mRNA primers were described previously [14]. GAPDH primers were also described [43, 44].
Western blotting assay
Cell lysates (30–40 μg each condition) were separated by the 10–12% Sodium Dodecyl Sulphate Poly Acrylamide Gel Electrophoresis (SDS-PAGE) gels, which were transferred to the polyvinylidene difluoride (PVDF) membranes (Millipore, Suzhou, China) [45]. The blots were blocked (with 5% non-fat milk) and incubated with designated primary plus secondary antibodies. Enhanced chemiluminescence (ECL, Roche, Shanghai, China) detection kit was utilized to visualize the targeted band/s under x-ray films. The intensity (total gray) of each band was quantified via ImageJ software, and was normalized to the loading control (β-Tubulin) [40].
Enzyme-linked immunosorbent assay (ELISA) assay
Cytokine production was examined by the corresponding ELISA kit (R&D Systems), and detailed protocol was described previously [46].
DIgR2 shRNA
Three distinct lentiviral DIgR2 shRNAs (named as “Sq1/2/3”) were packed into the GV248-puromycin vector, and were provided by Genepharm (Shanghai, China). The lentiviral shRNA (10 μL/mL per well) was added directly to mDCs for 24 hours. DIgR2 expression in the resulting cells was tested by qRT-PCR assay and Western blotting assay. DIgR2 shRNA “Sq3”, with targeted sequence of AAGAAGAGCTGGTGAACAAC, showed highest efficiency in downregulating DIgR2.
MEK1/2 shRNA
Two lentiviral MEK1/2 shRNAs were also provided by Genepharm (Shanghai, China), which were also added to cultured mDCs for 24 hours. MEK1/2 expression in the resulting cells was tested by Western blotting assay.
Statistical analysis
All statistical analyses were conducted using SPSS 15.0 software. Values were expressed as the mean ± standard deviation (SD). P < 0.05 was considered to indicate a statistically significant result.
CONCLUSIONS
These results indicate that tHSCs directly induces DIgR2 expression in DCs to inhibit T cells.
Authors’ contributions
All authors listed carried out the experiments, participated in the design of the study and performed the statistical analysis, conceived of the study, and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
ACKNOWLEDGMENTS
This work is partly supported by the National Science Foundation of China (No. 81472331).
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interests.
REFERENCES
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017; 67:7–30.
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016; 66:7–30.
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015; 65:5–29.
4. Yang JD, Roberts LR. Hepatocellular carcinoma: A global view. Nat Rev Gastroenterol Hepatol. 2010; 7:448–458.
5. Llovet JM, Hernandez-Gea V. Hepatocellular carcinoma: reasons for phase III failure and novel perspectives on trial design. Clin Cancer Res. 2014; 20:2072–2079.
6. Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer. 2006; 6:674–687.
7. Huang Y, Deng X, Liang J. Modulation of hepatic stellate cells and reversibility of hepatic fibrosis. Exp Cell Res. 2017; 352:420–426.
8. Thompson AI, Conroy KP, Henderson NC. Hepatic stellate cells: central modulators of hepatic carcinogenesis. BMC Gastroenterol. 2015; 15:63.
9. Carloni V, Luong TV, Rombouts K. Hepatic stellate cells and extracellular matrix in hepatocellular carcinoma: more complicated than ever. Liver Int. 2014; 34:834–843.
10. Xia YH, Wang ZM, Chen RX, Ye SL, Sun RX, Xue Q, Huang Y. T-cell apoptosis induced by intratumoral activated hepatic stellate cells is associated with lung metastasis in hepatocellular carcinoma. Oncol Rep. 2013; 30:1175–1184.
11. Xia Y, Chen R, Ye SL, Sun R, Chen J, Zhao Y. Inhibition of T-cell responses by intratumoral hepatic stellate cells contribute to migration and invasion of hepatocellular carcinoma. Clin Exp Metastasis. 2011; 28:661–674.
12. Swiecki M, Colonna M. The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol. 2015; 15:471–485.
13. Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012; 12:265–277.
14. Shi L, Luo K, Xia D, Chen T, Chen G, Jiang Y, Li N, Cao X. DIgR2, dendritic cell-derived immunoglobulin receptor 2, is one representative of a family of IgSF inhibitory receptors and mediates negative regulation of dendritic cell-initiated antigen-specific T-cell responses. Blood. 2006; 108:2678–2686.
15. Kusters-Vandevelde HV, Willemsen AE, Groenen PJ, Kusters B, Lammens M, Wesseling P, Djafarihamedani M, Rijntjes J, Delye H, Willemsen MA, van Herpen CM, Blokx WA. Experimental treatment of NRAS-mutated neurocutaneous melanocytosis with MEK162, a MEK-inhibitor. Acta Neuropathol Commun. 2014; 2:41.
16. Ascierto PA, Schadendorf D, Berking C, Agarwala SS, van Herpen CM, Queirolo P, Blank CU, Hauschild A, Beck JT, St-Pierre A, Niazi F, Wandel S, Peters M, et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. Lancet Oncol. 2013; 14:249–256.
17. Shurin GV, Tourkova IL, Kaneno R, Shurin MR. Chemotherapeutic agents in noncytotoxic concentrations increase antigen presentation by dendritic cells via an IL-12-dependent mechanism. J Immunol. 2009; 183:137–144.
18. Schnare M, Barton GM, Holt AC, Takeda K, Akira S, Medzhitov R. Toll-like receptors control activation of adaptive immune responses. Nat Immunol. 2001; 2:947–950.
19. Mellman I, Steinman RM. Dendritic cells: specialized and regulated antigen processing machines. Cell. 2001; 106:255–258.
20. Rutella S, Danese S, Leone G. Tolerogenic dendritic cells: cytokine modulation comes of age. Blood. 2006; 108:1435–1440.
21. Kubo N, Araki K, Kuwano H, Shirabe K. Cancer-associated fibroblasts in hepatocellular carcinoma. World J Gastroenterol. 2016; 22:6841–6850.
22. Heindryckx F, Gerwins P. Targeting the tumor stroma in hepatocellular carcinoma. World J Hepatol. 2015; 7:165–176.
23. Rani B, Cao Y, Malfettone A, Tomuleasa C, Fabregat I, Giannelli G. Role of the tissue microenvironment as a therapeutic target in hepatocellular carcinoma. World J Gastroenterol. 2014; 20:4128–4140.
24. Wu Z, Zeng Q, Cao K, Sun Y. Exosomes: small vesicles with big roles in hepatocellular carcinoma. Oncotarget. 2016; 7:60687–60697. https://doi.org/10.18632/oncotarget.10807.
25. Harding JJ, El Dika I, Abou-Alfa GK. Immunotherapy in hepatocellular carcinoma: Primed to make a difference? Cancer. 2016; 122:367–377.
26. Prieto J, Melero I, Sangro B. Immunological landscape and immunotherapy of hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2015; 12:681–700.
27. Fesnak AD, June CH, Levine BL. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer. 2016; 16:566–581.
28. Yu MC, Chen CH, Liang X, Wang L, Gandhi CR, Fung JJ, Lu L, Qian S. Inhibition of T-cell responses by hepatic stellate cells via B7-H1-mediated T-cell apoptosis in mice. Hepatology. 2004; 40:1312–1321.
29. Muhanna N, Horani A, Doron S, Safadi R. Lymphocyte-hepatic stellate cell proximity suggests a direct interaction. Clin Exp Immunol. 2007; 148:338–347.
30. Chen CH, Kuo LM, Chang Y, Wu W, Goldbach C, Ross MA, Stolz DB, Chen L, Fung JJ, Lu L, Qian S. In vivo immune modulatory activity of hepatic stellate cells in mice. Hepatology. 2006; 44:1171–1181.
31. Fu J, Xu D, Liu Z, Shi M, Zhao P, Fu B, Zhang Z, Yang H, Zhang H, Zhou C, Yao J, Jin L, Wang H, et al. Increased regulatory T cells correlate with CD8 T-cell impairment and poor survival in hepatocellular carcinoma patients. Gastroenterology. 2007; 132:2328–2339.
32. Ormandy LA, Hillemann T, Wedemeyer H, Manns MP, Greten TF, Korangy F. Increased populations of regulatory T cells in peripheral blood of patients with hepatocellular carcinoma. Cancer Res. 2005; 65:2457–2464.
33. Zhang J, Hong Y, Shen J. Combination treatment with perifosine and MEK-162 demonstrates synergism against lung cancer cells in vitro and in vivo. Tumour Biol. 2015; 36:5699–5706.
34. Wang K, Fan Y, Chen G, Wang Z, Kong D, Zhang P. MEK-ERK inhibition potentiates WAY-600-induced anti-cancer efficiency in preclinical hepatocellular carcinoma (HCC) models. Biochem Biophys Res Commun. 2016; 474:330–337.
35. Ikeda K, Wakahara T, Wang YQ, Kadoya H, Kawada N, Kaneda K. In vitro migratory potential of rat quiescent hepatic stellate cells and its augmentation by cell activation. Hepatology. 1999; 29:1760–1767.
36. Lu L, McCaslin D, Starzl TE, Thomson AW. Bone marrow-derived dendritic cell progenitors (NLDC 145+, MHC class II+, B7–1dim, B7–2-) induce alloantigen-specific hyporesponsiveness in murine T lymphocytes. Transplantation. 1995; 60:1539–1545.
37. Su H, Kong C, Zhu L, Huang Q, Luo L, Wang H, Xu Y. PPE26 induces TLR2-dependent activation of macrophages and drives Th1-type T-cell immunity by triggering the cross-talk of multiple pathways involved in the host response. Oncotarget. 2015; 6:38517–38537. https://doi.org/10.18632/oncotarget.5956.
38. Zhang YM, Zhang ZQ, Liu YY, Zhou X, Shi XH, Jiang Q, Fan DL, Cao C. Requirement of Galphai1/3-Gab1 signaling complex for keratinocyte growth factor-induced PI3K-AKT-mTORC1 activation. J Invest Dermatol. 2015; 135:181–191.
39. Gong YQ, Huang W, Li KR, Liu YY, Cao GF, Cao C, Jiang Q. SC79 protects retinal pigment epithelium cells from UV radiation via activating Akt-Nrf2 signaling. Oncotarget. 2016; 7:60123–60132. https://doi.org/10.18632/oncotarget.11164.
40. Zhu YR, Zhou XZ, Zhu LQ, Yao C, Fang JF, Zhou F, Deng XW, Zhang YQ. The anti-cancer activity of the mTORC1/2 dual inhibitor XL388 in preclinical osteosarcoma models. Oncotarget. 2016; 7:49527–49538. https://doi.org/10.18632/oncotarget.10389.
41. Chen MB, Zhou ZT, Yang L, Wei MX, Tang M, Ruan TY, Xu JY, Zhou XZ, Chen G, Lu PH. KU-0060648 inhibits hepatocellular carcinoma cells through DNA-PKcs-dependent and DNA-PKcs-independent mechanisms. Oncotarget. 2016; 7:17047–17059. https://doi.org/10.18632/oncotarget.7742.
42. Jia X, Wang F, Han Y, Geng X, Li M, Shi Y, Lu L, Chen Y. miR-137 and miR-491 Negatively Regulate Dopamine Transporter Expression and Function in Neural Cells. Neurosci Bull. 2016; 32:512–522.
43. Dakic A, DiVito K, Fang S, Suprynowicz F, Gaur A, Li X, Palechor-Ceron N, Simic V, Choudhury S, Yu S, Simbulan-Rosenthal CM, Rosenthal D, Schlegel R, et al. ROCK inhibitor reduces Myc-induced apoptosis and mediates immortalization of human keratinocytes. Oncotarget. 2016; 7:66740–66753. https://doi.org/10.18632/oncotarget.11458.
44. Wang L, Zhao Z, Feng W, Ye Z, Dai W, Zhang C, Peng J, Wu K. Long non-coding RNA TUG1 promotes colorectal cancer metastasis via EMT pathway. Oncotarget. 2016; 7:51713–51719. https://doi.org/10.18632/oncotarget.10563.
45. Zhen YF, Li ST, Zhu YR, Wang XD, Zhou XZ, Zhu LQ. Identification of DNA-PKcs as a primary resistance factor of salinomycin in osteosarcoma cells. Oncotarget. 2016; 7:79417–79427. https://doi.org/10.18632/oncotarget.12712.
46. Li P, Wu Y, Li M, Qiu X, Bai X, Zhao X. AS-703026 Inhibits LPS-Induced TNFalpha Production through MEK/ERK Dependent and Independent Mechanisms. PLoS One. 2015; 10:e0137107.