
INTRODUCTION
Osteosarcoma is the most common non-haematological primary malignant tumor of the bone, it arises from mesenchymal cells that produce osteoid and immature bone and affects mainly the extremities of adolescents and young adults [1–2].
The 5-years survival rate for patients with osteosarcoma without evidence of metastasis is 60% to 65%, whereas it is only 20% to 28% for osteosarcoma patients with metastases at the time of diagnosis [3]. Treatment of high grade osteosarcoma is based on a multidisciplinary approach that includes neoadjuvant chemotherapy, surgical excision of the primary tumor and metastasis excision; evaluation of response to therapy in the surgical specimen is crucial to eventually schedule a postoperative chemotherapy [4]. Standard therapy regimens often involve the use of high-dose methotrexate, doxorubicin, cisplatin and ifosfamide [4–5]. Evaluation of response to therapy was assessed by percentage area of necrosis in the specimen after surgery; patients who respond to therapy showing tumor necrosis ≥ 90% after surgery and they do not develop metastasis (Responder) while patients that do not respond to neoadjuvant chemotherapy showing tumor necrosis ≤ 90% after surgery and eventually developing metastasis (Non-responder) [6].
Although the survival rate has improved considerably after the introduction of neoadjuvant chemotherapy and surgery, metastatic or recurrent disease still occurs and the survival rate of patients is mainly linked to the resistance to therapy and to the development of metastasis [7]. In patients with metastatic osteosarcoma treated with neoadjuvant therapy, the “Responder” status shows improved survival (82% at 5-years) compared to “Non-Responder” (70% at 5-years) [8–9]. Over the last decade, scientific research has provided essential information about the pathogenic, molecular and biochemical pathway involved in osteosarcoma [10–11]; however, molecular mechanisms related to carcinogenesis, progression and resistance to therapy are still largely unknown. Indeed, even now the osteosarcoma karyotype is considered complex and only mutations of tumor suppressors genes TP53 and RB1 are commonly associated with the development of osteosarcoma [12].
There are many examples of “molecular targeted therapy”, where tailored therapeutic agents have been selected to aim against specific molecules and their downstream effector pathways in each patient [13]. Nonetheless survival rates of patients have not greatly improved [14], as little is known at genetic level about the pathogenesis of osteosarcomas resulting in a lack of more effective and tailored chemotherapy drug regimens [15].
Our hypothesis is that only the analysis of the entire coding genome could lead to a better understanding of the molecular mechanisms underlying the development and progression of osteosarcoma. Next generation sequencing (NGS) technologies can unveil DNA sequences whose study can be applied to characterize both common and rare genomic alterations across cancer types. The NGS approach could clarify the landscape of genetic alteration in osteosarcoma and provide relevant biological data. In particular, the knowledge of genetic alterations in coding exon regions, through WES (Whole Exome Sequencing) approach, may result in an easier discovery of new proteins as molecular target which could be aimed at personalized therapies. Indeed, WES could lead to create algorithms to identify relevant genetic alterations networks relevant to the clinical context of the disease [16–18]. The aim of this study was to perform WES analysis in selected tumors samples of osteosarcoma, both from responder and non-responder patients that developed metastasis. The goal will be to improve the knowledge of osteosarcoma carcinogenesis, progression and resistance to therapy, to identify new strategies for therapeutic approaches and personalized therapy.
RESULTS
Sequence coverage and mutation analysis
This analysis provided a vast new reservoir of data and, after filtering the data, within our discovery set of eight osteosarcomas, 5 responder and 3 non-responder, we identified 18,275 somatic variations in 8,247 genes. The number of coding variations in each sample ranged from 496 to 8,500 with an average of 2,347.5 variations. All the variations were missense and the 98.5% were SNVs (Single Nucleotide Variants) while the 1.5% were MNPs (Multiple Nucleotide Polymorphisms).
The mutation rate in the responder group was slightly higher than the non-responder group (Table 1, Figure 1). Moreover, we found that, as in several other cancers [19], C:G>T:A transitions were the most predominant somatic substitution both in the responder group (43%) and in the non-responder group (34%) and we found that the vast majority of these transitions were identified in the context of 5’-CpG-3’ dinucleotide as the result of a 5’-methylcytosine deamination (data not shown).
Table 1: N° of variations and variation rate (n° of variations/Mb) of responder and non-responder osteosarcoma patients
Sample |
N° of variations |
Variations rate |
Status |
|---|---|---|---|
1 |
1390 |
118.8 |
Responder |
5 |
8500 |
726.5 |
Responder |
6 |
496 |
42.4 |
Responder |
7 |
791 |
67.6 |
Responder |
8 |
802 |
68.5 |
Responder |
2 |
4211 |
359.9 |
Non-Responder |
3 |
1495 |
127.8 |
Non-Responder |
4 |
1095 |
93.6 |
Non-Responder |
We did not find common mutated genes in all eight samples but we found three mutated genes (KIF1B, NEB, KMT2C) in 7/8 (87%) samples. Seventeen genes were mutated in 6 samples (75%, 6/8) and 57 genes showed variations in 5 samples (62.5%, 5/8). We noted a great variability of genes mutated among the samples as shown in Supplementary Table 1. Indeed, taking into consideration common mutated genes in at least 5 samples, we noted that sample n°5 showed variations in 77 genes while the sample n°6 showed variations only in 22 genes (Supplementary Table 1).
These 77 genes that were found mutated in more than half of the samples are involved in important biological processes such as transcription regulation, ion transport, DNA damage and repair, cell adhesion and angiogenesis (Table 2).
Table 2: Genes mutated in more than half of the osteosarcoma patients and their biological functions
Biological function |
Mutated genes |
|---|---|
Transcription regulation |
ATF2; BPTF; CHD7; DENND4A; HIVEP2; IKBKAP; JMJD1C; KMT2C; KMT2E; MYCBP2; NCOR1; RFX7; SPEN; |
Transport |
ABCB5; ABCD2; ANKH; ATP10D; CACNA2D3; DGKD; HERC1; ITPR1; SLC8A1; SLC9B1; TRPM3; |
Cell Adhesion |
DST; FN1; ITGA6; MPDZ; NRXN2; RELN; |
Immunity |
C5; CFH; PKHD1L1; |
Apoptosis |
BIRC6; ITPR1; KIF1B; |
Endocytosis |
DGKD; LRP2; STAB2; |
Cell cycle |
ALMS1; ATM; BIRC6; KMT2E; |
DNA damage |
ATF2; ATM; DNA2; POLQ; PRDM10; PSME4; |
Angiogenesis |
FN1; THSD7A; |
Ubl conjugation pathway |
BIRC6; HECTD1; HERC1; MYCBP2; |
Wnt signaling pathway |
MACF1; |
Differentiation |
ABCB5; OBSCN; PSME4; THSD7A; |
DNA repair |
DNA2; POLQ; PRDM10; PSME4; |
Muscle development |
DMD; NEB; SYNE1; UTRN; |
Regulation of bone mineralization |
ENPP1; FBN2; |
Cell communication |
FRAS1; |
mRNA processing |
FRG1; |
Nucleotide metabolism |
NT5C2; |
Multicellular organism development |
PRTG; |
Regulation of cell migration |
SYNE2; |
Small GTPase mediated signal trasduction |
DOCK5; |
Regulation of GTPase activity |
AKAP13; DENND4A; PREX2; SYDE2; |
Extracellular matrix organization |
EGFLAM; |
Microtuble-based movement |
DNAH9; DNAH10; |
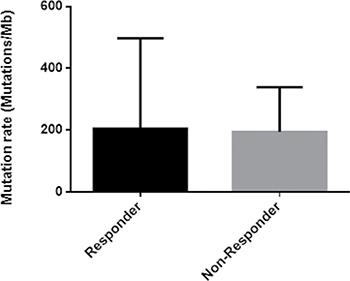
Figure 1: Histogram of the mutation rate in responder and non-responder group.
Despite variations of TP53 and RB1 are associated with development of human osteosarcoma, we found that variations of TP53 were present in only one patient (12.5%, sample n°5) while only two patients showed variations of RB1 gene (25%, sample n°1 and 5).
Genes that may be involved in osteosarcoma tumorigenesis
We found that KIF1B, NEB and KMT2C were the only three genes mutated in 7/8 samples. KIF1B is located on chromosome 1p36, and belongs to the kinesin superfamily of intermediate filaments [20]. NEB, maps to chromosome 2q22, encodes nebulin, a giant protein component of the cytoskeletal matrix [21]. We found 10 and 27 variations respectively in these two genes and they were mutated in all samples except in sample n°7, while sample n°5 had the higher number of variations in both cases (Table 3). Finally, KMT2C is located on chromosome 7q36.1 and it is a member of the mixed-lineage leukemia (MLL) family and it has histone H3 lysine-specific methylation activity. KMT2C is an important component of histone H3 lysine 4 methyltransferase complex, implicated in chromatin remodelling, called ASCOM (ASC-2-and Mll3-containing complex) [22].
Table 3: Somatic variation of KIF1B and NEB in osteosarcoma samples and their genomic and amino acid change
Sample |
Gene |
Genomic change |
Aminoacid |
|
|---|---|---|---|---|
1 |
KIF1B |
c.650C>T |
p.Ser217Phe |
|
1 |
KIF1B |
c.5074G>A |
p.Asp1692Asn |
|
1 |
NEB |
c.21268C>T |
p.Pro7090Ser |
|
1 |
NEB |
c.21197T>A |
p.Leu7066His |
|
2 |
KIF1B |
c.177T>A |
p.His59Gln |
|
2 |
NEB |
c.23803A>G |
p.Arg7935Gly |
|
2 |
NEB |
c.20167C>G |
p.Arg6723Gly |
|
2 |
NEB |
c.20167C>T |
p.Arg6723Trp |
|
2 |
NEB |
c.20165A>G |
p.Tyr6722Cys |
|
2 |
NEB |
c.20165A>T |
p.Tyr6722Phe |
|
2 |
NEB |
c.19249_19250delTAinsCA |
p.Tyr6417His |
|
2 |
NEB |
c.19063T>A |
p.Tyr6355Asn |
|
3 |
KIF1B |
c.2798A>G |
p.Tyr933Cys |
|
3 |
NEB |
c.7325T>A |
p.Ile2442Asn |
|
4 |
KIF1B |
c.5069A>G |
p.Asp1690Gly |
|
4 |
NEB |
c.11109G>C |
p.Lys3703Asn |
|
5 |
KIF1B |
c.538G>A |
p.Asp180Asn |
|
5 |
KIF1B |
c.2332G>A |
p.Val778Met |
|
5 |
KIF1B |
c.2344G>A |
p.Glu782Lys |
|
5 |
NEB |
c.23570A>G |
p.Glu7857Gly |
|
5 |
NEB |
c.22337T>C |
p.Ile7446Thr |
|
5 |
NEB |
c.22333G>C |
p.Asp7445His |
|
5 |
NEB |
c.22021G>C |
p.Ala7341Pro |
|
5 |
NEB |
c.21991G>T |
p.Ala7331Ser |
|
5 |
NEB |
c.21569C>T |
p.Thr7190Ile |
|
5 |
NEB |
c.18996G>C |
p.Gln6332His |
|
5 |
NEB |
c.18448G>C |
p.Asp6150His |
|
5 |
NEB |
c.11026G>A |
p.Asp3676Asn |
|
5 |
NEB |
c.8156A>T |
p.Asn2719Ile |
|
5 |
NEB |
c.8142T>G |
p.Asn2714Lys |
|
5 |
NEB |
c.2633G>C |
p.Ser878Thr |
|
6 |
KIF1B |
c.956C>T |
p.Thr319Ile |
|
6 |
NEB |
c.9705C>A |
p.Asn3235Lys |
|
6 |
NEB |
c.7025G>A |
p.Ser2342Asn |
|
8 |
KIF1B |
c.3814C>T |
p.Arg1272Cys |
|
8 |
NEB |
c.10286C>T |
p.Thr3429Ile |
|
8 |
NEB |
c.9692T>G |
p.Leu3231Trp |
L |
Among these three genes, KMT2C showed the highest number of variations: we found 36 variations, with sample n°5 revealing the highest number of variations, except in sample n°3 (Table 4). Particularly, sample n°3 was a 14-year old boy with diagnosis of high grade chondroblastic osteosarcoma made on femur biopsy (stage IVa), follow-up was 11 years, he did not respond to neoadjuvant chemotherapy and develop metastasis.
Table 4: Somatic variations of KMT2C in osteosarcoma samples and their genomic and amino acid change
Sample |
Genomic change |
Aminoacid |
|---|---|---|
1 |
c.2963G>T |
p.Cys988Phe |
1 |
c.8326G>A |
p.Glu2776Lys |
1 |
c.11927C>A |
p.Pro3976Gln |
1 |
c.12014C>T |
p.Ser4005Phe |
1 |
c.14521G>A |
p.Gly4841Arg |
2 |
c.1277C>A |
p.Pro426Gln |
2 |
c.6632G>C |
p.Arg2211Thr |
2 |
c.5459C>G |
p.Ser1820Cys |
2 |
c.8174A>G |
p.Glu2725Gly |
2 |
c.9622T>C |
p.Ser3208Pro |
2 |
c.9617G>C |
p.Arg3206Thr |
4 |
c.10383T>G |
p.Asp3461Glu |
5 |
c.404C>T |
p.Ala135Val |
5 |
c.3155A>T |
p.Lys1052Ile |
5 |
c.3119C>A |
p.Pro1040Gln |
5 |
c.3029G>T |
p.Cys1010Phe |
5 |
c.5669G>C |
p.Arg1890Pro |
5 |
c.7318C>T |
p.Pro2440Ser |
5 |
c.7214A>G |
p.Glu2405Gly |
5 |
c.9182A>G |
p.Gln3061Arg |
5 |
c.9769G>A |
p.Glu3257Lys |
6 |
c.925C>T |
p.Pro309Ser |
6 |
c.1181G>A |
p.Cys394Tyr |
6 |
c.2512G>A |
p.Gly838Ser |
6 |
c.2468T>C |
p.Ile823Thr |
6 |
c.2459C>T |
p.Thr820Ile |
6 |
c.2656C>T |
p.Arg886Cys |
7 |
c.943G>A |
p.Gly315Ser |
7 |
c.925C>T |
p.Pro309Ser |
7 |
c.2512G>A |
p.Gly838Ser |
7 |
c.2468T>C |
p.Ile823Thr |
7 |
c.2963G>T |
p.Cys988Phe |
7 |
c.2917A>G |
p.Arg973Gly |
8 |
c.943G>A |
p.Gly315Ser |
8 |
c.2512G>A |
p.Gly838Ser |
8 |
c.2468T>C |
p.Ile823Thr |
Genes that may be involved in osteosarcoma metastatic progression and resistance to therapy
We found that 0.18% (15/8247) of genes showed variations, all missense (94% SNV, 6% MNP), only in the non-responder group (Table 5). Categorizing them by KEGG pathway analysis and literature review, we found that some of these are involved in important biological processes (Table 6). ERBB4 is located on chromosome 2q34 and it is a receptor tyrosine kinase member of the epidermal growth factor receptor (EGFR) family [23]. ERBB4 is involved in biological process such as signal transduction, transport and catabolism. It is involved in human disease like cancer: it participates in pathways such as ERBB signalling, implicated in tumor migration and invasion, and in other pathway implicated in inhibition of tumor angiogenesis [24]. In signal transduction, transport and catabolism and human disease biological process is involved THBS1 too and moreover is implicated in cell growth and death process. THBS1, thrombospondin 1, is located on chromosome 15q14 [25] and it is involved in RAP1 and TP53 signalling pathways, involved in inhibition of angiogenesis and metastasis, in PI3K-AKT and TGF-beta signalling pathway, involved in the control of cell cycle and apoptosis, and it takes part in focal adhesion and ECM-receptor interaction process [26]. Other genes are enzymes with specific activity like as rRNA and tRNA biogenesis (DIS3, KARS), spliceosome activity (BCLAF1) or charboidrate metabolism (PDHX) and all involved in carcinogenesis [27–32].
Table 5: Variations of genes mutated only in non-responder group and their genomic and amino acid change
Gene |
Genomic change |
Aminoacid |
|---|---|---|
ALDH1L2 |
c.484G>A |
p.Gly162Ser |
ALDH1L2 |
c.1732A>C |
p.Thr578Pro |
ALDH1L2 |
c.2086G>A |
p.Gly696Ser |
BCLAF1 |
c.491A>C |
p.Lys164Thr |
BCLAF1 |
c.1646T>C |
p.Leu549Pro |
BCLAF1 |
c.1742A>T |
p.Lys581Met |
CLCN1 |
c.823_825delGTCinsGGG |
p.Val275Gly |
CLCN1 |
c.1289A>C |
p.Asn430Thr |
CLCN1 |
c.1574C>G |
p.Ala525Gly |
CLCN1 |
c.1073G>A |
p.Cys358Tyr |
CLCN1 |
c.1427C>G |
p.Thr476Ser |
COG3 |
c.943G>C |
p.Glu315Gln |
COG3 |
c.505A>G |
p.Thr169Ala |
COG3 |
c.1411T>A |
p.Tyr471Asn |
DIS3 |
c.2312_2313delTAinsAA |
p.Ile771Lys |
DIS3 |
c.2252C>G |
p.Ala751Gly |
DIS3 |
c.2234C>T |
p.Thr745Ile |
DIS3 |
c.2458C>T |
p.Arg820Trp |
ERBB4 |
c.2761T>A |
p.Tyr921Asn |
ERBB4 |
c.1541G>A |
p.Gly514Glu |
ERBB4 |
c.1841G>A |
p.Cys614Tyr |
KARS |
c.398T>C |
p.Leu133Pro |
KARS |
c.1009G>T |
p.Val337Phe |
KARS |
c.1066G>A |
p.Asp356Asn |
OR52N1 |
c.589A>G |
p.Asn197Asp |
OR52N1 |
c.569C>G |
p.Ser190Cys |
OR52N1 |
c.53G>C |
p.Gly18Ala |
OR52N1 |
c.82T>C |
p.Trp28Arg |
PDE6C |
c.646T>A |
p.Tyr216Asn |
PDE6C |
c.650T>C |
p.Leu217Pro |
PDE6C |
c.1289G>A |
p.Gly430Glu |
PDE6C |
c.1992G>T |
p.Leu664Phe |
PDHX |
c.566G>C |
p.Arg189Pro |
PDHX |
c.626G>A |
p.Gly209Glu |
PDHX |
c.323G>A |
p.Gly108Glu |
SCN8A |
c.2276T>C |
p.Ile759Thr |
SCN8A |
c.4048G>A |
p.Ala1350Thr |
SCN8A |
c.677G>A |
p.Arg226Lys |
SP140L |
c.1425A>C |
p.Lys475Asn |
SP140L |
c.329T>A |
p.Leu110His |
SP140L |
c.1013C>A |
p.Pro338His |
THBS1 |
c.634C>G |
p.Leu212Val |
THBS1 |
c.1699_1700delGAinsGT |
p.Asp567Val |
THBS1 |
c.2365G>A |
p.Asp789Asn |
THBS1 |
c.1532G>C |
p.Gly511Ala |
UBE4A |
c.1130T>A |
p.Ile377Asn |
UBE4A |
c.2209G>A |
p.Glu737Lys |
UBE4A |
c.3136G>T |
p.Asp1046Tyr |
ZNF12 |
c.137C>G |
p.Ser46Cys |
ZNF12 |
c.1265C>T |
p.Ser422Phe |
ZNF12 |
c.2086C>T |
p.Leu696Phe |
Table 6: Mutated genes in non-responder group of osteosarcoma patients and their chromosomal location and biological process
Gene |
Chromosomal location |
Biological process |
|---|---|---|
ALDH1L2 |
12q23.3 |
Metabolism of cofactors and vitamins |
BCLAF1 |
6q23.3 |
Spliceosome |
CLCN1 |
7q34 |
Ion channels |
COG3 |
13q14.13 |
Membrane trafficking |
DIS3 |
13q22.1 |
mRNA biogenesis |
ERBB4 |
2q34 |
Signal trasduction, Transport and catabolism, Human disease |
KARS |
16q23.1 |
tRNA biogenesis |
OR52N1 |
11p15.4 |
Olfactory transduction |
PDE6C |
10q24 |
Sensory transduction |
PDHX |
11p13 |
Carbohydrate metabolism |
SCN8A |
12q13 |
Ion channels |
SP140L |
2q37.1 |
unknown |
THBS1 |
15q15 |
Signal trasduction, Transport and catabolism, Human disease, Cell growth and death |
UBE4A |
11q23.3 |
Ubiquitin mediated proteolysis |
ZNF12 |
7p22.1 |
Transcription factors |
DISCUSSION
Osteosarcoma is a heterogeneous tumor and it is a so-called “orphan cancer” with no known driver oncogenes [33]. It is essential to find useful biomarkers and to detect the potential targets for new drugs, to increase overall survival of these patients. This could be done only if we better understand the complex biology of this tumor and the molecular pathways that lead to the development of metastases and resistance to therapy. At present time, the most innovative approach is represented by NGS, which allows to analyse the entire human genome [18]. In this study, we performed the analysis of exomic sequences of osteosarcomas to advance our understanding of their genetic underpinnings and to correlate the genetic alterations with the clinical and pathological features of each patient. Our aim was to identify clinically relevant variations in the landscape of somatic events in osteosarcoma pathogenesis and so we performed WES analysis on eight high grade osteosarcoma biopsies. Using stringent criteria for the analysis of these presumptive variations, we identified 18,275 somatic variations, all missense, in 8,247 genes. We noted that the mutation rate was similar between responder and non-responder group but overall this rate was higher than previous studies probably due to the small number of our samples [34–36]. We did not find specific structural abnormalities common to all samples, such as specific translocations like other sarcomas (e.g. Ewing sarcoma EWS/FLI1 t(11;22)(q24;q12)), confirming the complexity of osteosarcoma karyotype [15]. Despite TP53 and RB1 genes are considered top driver genes in osteosarcoma cancerogenesis, our study showed lower mutation frequency of TP53 and RB1 than other studies [34,35,37–39]. Indeed, we observed that only one patients (12.5%, 1/8) showed variations of TP53 gene and that only two patients showed variations of RB1 gene (25%, 2/8). Our results could be the effect of the small number of samples, however the range of mutation rate described in osteosarcoma is very large going from 31% to 82% and from 19% to 64% for TP53 and RB1 respectively [41]. Moreover, most TP53 mutations described in some studies, are structural variations in intron 1 [34, 37, 38] that we did not study and the mutation rate outside the intron 1 is low (19–20%) [34, 40]. However, as reported in literature, among all mutations of TP53 reported in previous studies only some mutations are clonal events [37]. Lastly, our data was filtered by Sift and Polyphen-2 bioinformatics software, so we reported only mutations that are considered deleterious for the protein function.
However, beyond TP53 and RB1, between genes considered candidate driver genes in osteosarcoma carcinogenesis [36] we found that ATM, a gene involved in DNA damage control signalling pathway, showed variations in 5/8 osteosarcoma samples. ATM is a tumor suppressor gene that belongs to the family of proteins that respond to DNA damage by phosphorylating key substrates involved in DNA repair and/or cell cycle control [36]. Moreover, we did not find variations in genes associated at inherited cancer predisposition syndromes associated with osteosarcoma as RECQL4, BLM, and WRN [34, 35, 37].
Moreover, we did not find variations common to all osteosarcoma samples but we found three mutated genes in 7/8 samples (87%) never studied in osteosarcoma. Variations in these three genes may be early molecular events and so they may be related to osteosarcoma tumorigenesis. KIF1B is responsible for intracellular vesicular transport [20], a process involved in the production matrix in osteosarcoma [42]. KIF1B has been shown to act as a tumor suppressor in multiple cancers by acting on various inhibitors of cell proliferation and activators of apoptosis [20]. Another interesting NEB, maps to chromosome 2q22, encodes nebulin, a giant protein component of the cytoskeletal matrix [21], that coexists with thick and thin filaments within the sarcomeres of skeletal muscle [21].
Finally, KMT2C catalyses the monomethylation of H3K4 in collaboration with hormone receptors and transcription factors involved in developmental signalling [22, 43]; it is one of the histone modifiers previously implicated in some cancer types [22]. It caught our attention showing more variations than KIF1B and NEB, finally because this gene is found to be inactivated in other tumors causing an alteration of tumor suppressor gene TP53 [43], which is also frequently altered in osteosarcoma [44]. Moreover, recent evidence showed that mutations of KMT2C can modify its cooperation with oestrogen receptor [45], this hormone playing a key role in the development and support of bone remodelling and matrix production [46]. Recently, it has been demonstrated that highly conserved epigenetic regulators are frequently mutated in cancer [47]; indeed, some studies suggest that variations in the coding sequences of regulatory elements, that act on enhancers to recognize specific transcription factors, could be the cause of tumor development [47]. KMT2C acts in collaboration with hormone receptors and transcription factors involved in growth-promoting pathways and it performs its action at transcription enhancer regions [22]. KMT2C is frequently mutated in a broad spectrum of cancers and it has been related to tumorigenesis [48]. Therefore, our hypothesis is that KMT2C could be involved in osteosarcoma carcinogenesis too.
We found that the responder group showed more variations than the non-responder group and we noted that 15 genes showed variations only in the non-responder group which subsequently developed a metastatic disease. We found that some of them are involved in carcinogenesis and tumor progression but not in osteosarcoma. Particularly, ERBB4 is a member of the Tyr protein kinase family and the epidermal growth factor receptor subfamily that regulates cell proliferation and differentiation [23]. It has been shown that ERBB4 is overexpressed in Ewing Sarcoma cell lines derived from chemoresistant or metastatic Ewing sarcoma, a tumor of the bone [24]. ERBB4 leads to activation of PI3K-AKT, focal adhesion kinase (FAK) and the RAC1 GTPase, a mediator of cell migration and invasion [24], pathways that are already been described in osteosarcoma pathogenesis [49–50].
ERBB4 is rarely mutated in human cancers [51] but recently it was shown that ERBB4 can inhibit the tumor suppressor TP53 by regulating the MDMX-MDM2 complex stability, the primary inhibitors of TP53, leading to TP53 inactivation in tumor [52]. Thrombospondin I (THBS1) is an endogenous inhibitor of angiogenesis, which limits blood vessel density in normal tissues and curtails tumor growth. It interacts with a variety of ECM molecules and cell surface receptors and this protein has been shown to play roles in tumorigenesis; particularly, it was showed that THBS1 induced cell migration in several tumor cell lines suggesting that THBS1 is involved in cancer invasion [26]. DIS3 is one of catalytic components of the human RNA processing and degrading exosome it has both endonucleolytic and 3-prime/5-prime exonucleolytic activity [27]; mutations in DIS3 have been observed in several cancer types [28] and in multiple myeloma was observed a shorter survival in patients with DIS3 mutations [29]. BCLAF1 encodes a transcriptional repressor that interacts with several members of the BCL2 family of proteins and ectopical BCLAF1 expression induces apoptosis in various cell types or autophagic cell death in myeloma cells. It was hypothesized that BCLAF1 plays a critical role repressing the transcription of survival genes through TP53 inhibition. Moreover, it was found that tumor cells suppress BCLAF1 expression inducing a cascade of antiapoptotic cellular events that all together determine an increase survival of tumor cells [31]. The KARS gene encodes lysyl-tRNA synthetase, which catalyses the aminoacylation of tRNA-lys in the cytoplasm and mitochondria. It was showed that it was implicated in cancer metastasis indeed the phosphorylation of KARS at the Thr52 residue by p38MAPK, causes the dissociation from the cytosolic multi-tRNA synthetase complex and the following translocation to the plasma membrane, where it associates with a 67 kDa laminin receptor (p67LR) involved in migration and metastasis [30]. The PDHX gene encodes component X of the pyruvate dehydrogenase (PDH) complex that is in the mitochondrial matrix and catalyzes the conversion of pyruvate to acetyl coenzyme A [32]. It was showed that in colorectal cancer cells the glucose metabolism was regulated by miR-26a direct targeting the PDHX, which inhibits the conversion of pyruvate to acetyl coenzyme A in the citric acid cycle to require the glucose uptake to the energy needs of cancer cells [32]. In successive studies, it will be necessary to study whether and how these mutations may modify the expression of these genes and the function of the associated protein to discovery a possible molecular target.
Some of these genes identified in our study are involved in metastatic progression and poor survival in tumors, also of mesenchymal origin, but these have never been studied in osteosarcoma [24, 26, 29, 30]. Our hypothesis is that alterations in these genes could be involved in a mechanism of metastatic progression and drug resistance in osteosarcoma.
We recognize these preliminary results may arise due to the small sample size, which is a limitation of the study, and we believe that it will be necessary first to ascertain our data in a larger cohort of patients.
In conclusion, our WES analysis confirms that the osteosarcoma karyotype is complex, however, we found genes probably involved in osteosarcoma carcinogenesis, as KMT2C, and we identified a group of 15 genes probably involved in metastasis development and drug resistance that could offer the opportunity to identify potential drug targets in order to create a personalized therapy and increase the survival rate among young patients with osteosarcoma.
MATERIALS AND METHODS
Population study
The population of this study included 8 diagnostic biopsies of patients with conventional high grade osteosarcoma (sec. Enneking classification) obtained before neoadjuvant therapy from the files of the Department of Medical and Surgical Sciences and Biotechnologies, Sapienza University of Rome, Polo Pontino, ICOT, Latina, Italy. The WHO 2008 classification of bone tumors was used for classifying and staging the bone lesions. Patients had a mean follow-up time of 9 years (range 1–14 years), 50% of them showed a minimum 5-years overall survival. Evaluation of response to therapy was assessed by percentage area of necrosis in the specimen after surgery; out of these 8 patients, 5 (identified as “responder”) responded to therapy showing tumor necrosis ≥ 90% after surgery and didn’t develop metastasis while 3 patients did not respond to neoadjuvant chemotherapy showing tumor necrosis ≤ 90% after surgery and developed metastasis (identified as “non-responder”). There were not inherited cancer predisposition syndromes associated with osteosarcoma among our samples. The clinical and pathological characteristics of each osteosarcoma patients are described in Table 7. All the tissue samples were reviewed on microscopy examinations by two dedicated pathologists.
Table 7: Clinicopathological characteristics of 8 osteosacoma patients
Sample |
Sex |
Age |
Histological type* |
Grading** |
Staging* |
Site |
Follow-up (Years) |
Status |
|---|---|---|---|---|---|---|---|---|
1 |
F |
29 |
Conventional Osteosarcoma |
High grade |
IIB |
Jaw |
14 |
Responder |
2 |
M |
30 |
Conventional Osteosarcoma |
High grade |
III |
Femur |
3 |
Non-Responder |
3 |
M |
14 |
Chondroblastic osteosarcoma |
High grade |
IVA |
Femur |
11 |
Non-Responder |
4 |
M |
35 |
Conventional Osteosarcoma |
High grade |
III |
Femur |
1 |
Non-Responder |
5 |
M |
21 |
Conventional Osteosarcoma |
High grade |
III |
Femur |
13 |
Responder |
6 |
M |
11 |
Conventional Osteosarcoma |
High grade |
III |
Femur |
5 |
Responder |
7 |
F |
24 |
Conventional Osteosarcoma |
High grade |
IIB |
Mandible |
12 |
Responder |
8 |
M |
31 |
Chondroblastic osteosarcoma |
High grade |
IIA |
Jaw |
13 |
Responder |
*Sec. WHO 2008
** Sec. Enneking grade
Whole-exome sequencing (WES)
Genomic DNA was extracted from FFPE osteosarcoma biopsies using RecoverAll Total Nucleic Acid isolation FFPE kit (Life Technologies, Foster City, CA) according to the manufacturer’s instruction and was used to prepare fragment libraries suitable for massively parallel paired-end sequencing.
DNA was amplified with GenomePlex Single Cell Whole Genome Amplification kit, following the manufacturer’s protocol, because of the degraded material, typical of osteosarcoma samples. We measured DNA concentration of the samples using Qubit 2.0 Fluorometer (Invitrogen, Life Technologies, Grand Island, NY) with the Qubit DNA HS assay kit or Qubit DNA BR Assay kit according to the quantity of the starting material. Moreover, we checked the quality of DNA with 2200 Tapestation Instrument (Agilent Technologies, Santa Clara, CA, USA) using the D1000 screen tape or the High sensitivity D1000 screen tape as appropriate, to see the level of fragmentation of our samples.
To prepare the DNA library we used Nextera Rapid Capture Expanded Exome Kit (Illumina, San Diego, CA, USA) following the guidelines of the protocol but we used the half of the amount of the fragmentation enzyme because of the high level of fragmentation of all the samples tested with Tapestation. We load a maximum of 5 pooling libraries for each cartridge NextSeq High Output (300 cycles) on NextSeq500 (Illumina, San Diego, CA, USA).
WES data analysis
We sequenced the whole exomes of eight high-grade osteosarcoma tissues using NextSeq550 Illumina platform. The instrument generated approximately an average read length of 120.6 bases. After mapping to the human reference genome GRCh37 (hg19) using Burrows-Wheeler Alignment tool (BWA), we obtained the average depth of each base in the target regions as 17.7x. The average coverage of target regions was 121.7x.
The raw data generated from Illumina NextSeq550 were converted using Bcl2toFastq tools provided by Illumina. The data analysis of exomes was performed by using the SeqMule pipeline1. The final Variant Call Format files (VCFs) were uploaded on the VariantStudio Illumina (Illumina San Diego, CA, USA) software to perform annotation and filtration steps. We filtered genomic data by quality score of 30, by mean read depth of 5, and by GMAF ≤ 1 (Global Minor Allele Frequency: the frequency of the second most frequent allele in the population). Next, the detected mutations were filtered for the variants absent in database of Single Nucleotide Polymorphisms (dbSNP) to distinguish known somatic mutations from germline mutations. We further selected the somatic variations in the coding sequence by excluding those variations in flanking sequences like splicing sites, 5ʹ-UTR, 3ʹUTR, introns and intergenic regions. Moreover, we performed bioinformatics analysis using SIFT (v4.0.3) and PolyPhen-2 (v2.1.0) tools, that predict whether an amino acid substitution affects protein function based on sequence homology. To be sure that the variations to study were only those that caused a protein alteration, we selected only those that were at the same time deleterious for SIFT and probably damaging for PolyPhen-2.
Validation of variations
Genes that showed variation in the 87% of osteosarcoma samples (KIF1B, NEB and KMT2C) were selected for target resequencing and validation by Sanger sequencing. DNAs were amplified using the Gene Amp PCR System 9700 (Life Technologies, Foster City, CA). The PCR products were then purified using Exosap-IT (Affymetrix, Santa Clara, CA) and then sequenced using Big Dye Terminator version 1.1 Cycle Sequencing Kit (Life Technologies, Foster City, CA). Unincorporated primers and dye terminators were removed using the Montage-SEQ96 Sequencing Reaction Cleanup Kit (Merck Millipore, Billerica, MA). Sequencing was performed on an ABI PRISM 3100 Genetic Analyzer (Life Technologies, Foster City, CA) with 3100 Genetic Analyzer Data Collection software version 1.1. The sequencing and each reaction were performed in triplicate.
Abbreviations
NGS: Next Generation Sequencing ; WES: Whole Exome Sequencing; BWA: Burrows-Wheeler Alignment ; SNV: Single Nucleotide Variant; MNP: Multiple Nucleotide Polymorphisms; KIF1B: Kinesin Family member 1B; NEB: Nebulin ; KMT2C: lysine (K)-specific Methyltransferase 2C; TP53: Tumor Protein 53; RB1: Retinoblastoma 1; MLL: Mixed-Lineage Leukemia; ASCOM: ASC-2-and Mll3-containing complex; KEGG: Kyoto Encyclopedia of Genes and Genomes; ERBB4: Erb-b2 receptor tyrosine kinase 4; EGFR: Epidermal Growth Factor Receptor; THBS1: Thrombospondin 1; RAP1: (Ras-proximate-1); PI3K: Phosphoinositide 3-Kinase; AKT: Serine/Threonine Kinase 1; TGF-BETA: transforming growth factor, beta 1; ECM: Extra-Cellular Matrix; DIS3: DIS3 homolog, exosome endoribonuclease and 3’-5’ exoribonuclease; KARS: lysyl-tRNA synthetase; BCLAF1: BCL2 associated transcription factor 1; PDHX: pyruvate dehydrogenase complex component X; EWS: EWS RNA binding protein 1; FLI1: Fli-1 proto-oncogene, ETS transcription factor; FAK: Focal Adhesion Kinase ; RAC1: ras-related C3 botulinum toxin substrate 1; GTP: Guanosine Triphosphate ; MDMX: MDM4, p53 regulator; MDM2: MDM2 proto-oncogene; p38MAPK: p38 Mitogen-Activated Protein Kinases; p67LR: Laminin Receptor; PHD: Pyruvate Dehydrogenase ; FFPE: Formalin-Fixed Paraffin Embedded; VCF: Variant Call Format ; GMAF: Global Minor Allele Frequency; UTR: Untraslated Region.
Authors’ contributions
Study conception and design: Claudio Di Cristofano.Acquisition of data (selection of subjects, bioptic samples, WES): Chiara Puggioni, Francesca Lessi, Massimiliano Mancini, Vincenzo Petrozza, Caterina Chiappetta, Veronica De Gregorio, Prospero Civita, Sara Franceschi. Statistical analysis: Paolo Aretini, Massimiliano Mancini. Interpretation of data: Claudio Di Cristofano, Caterina Chiappetta, Massimiliano Mancini, Chiara Puggioni, Raffaella Carletti. Drafting the article: Caterina Chiappetta. Critical revision of the article: Claudio Di Cristofano, Carlo Della Rocca, Chiara Maria Mazzanti, Massimiliano Mancini, Caterina Chiappetta, Antonio G. Naccarato. Final approval of the version to be submitted: Claudio Di Cristofano, Carlo Della Rocca, Chiara Maria Mazzanti.
ACKNOWLEDGMENTS
The authors thank the “Associazione Serena Talarico”, “Associazione culturale Simone Tetti”, “Fondazione Roma Sapienza,” “AISOS” and “GIOMI S.p.a.” for the precious support to this research.
CONFLICTS OF INTEREST
The authors declare that they do not have anything to disclose regarding founding from industries or conflict of interest with respect to this manuscript.
REFERENCES
1. Raymond AK, Ayala AG, Knuutila S. Conventional osteosarcoma. In: Fletcher CDM, Unni KK, Mertens F, eds. World Health Organization Classification of Tumors Pathology and Genetics of Tumors of Soft Tissue and Bone. Lyon: IARC Press; 2002:264–270.
2. Damron TA, Ward WG, Stewart A. Osteosarcoma chondrosarcoma and Ewing’s sarcoma: National Cancer Data Base report. Clin Orthop Relat Res. 2007; 459:40–47.
3. PosthumaDeBoer J, Witlox MA, Kaspers GJ, van Royen BJ. Molecular alterations as target for therapy in metastatic osteosarcoma: a review of literature. Clin Exp Metastasis. 2011; 28:493–503.
4. Marina NM, Smeland S, Bielack SS, Bernstein M, Jovic G, Krailo MD, Hook JM, Arndt C, van den Berg H, Brennan B, Brichard B, Brown KL, Butterfass-Bahloul T, et al. Comparison of MAPIE versus MAP in patients with a poor response to preoperative chemotherapy for newly diagnosed high-grade osteosarcoma (EURAMOS-1): an open-label, international, randomized controlled trial. Lancet Oncol. 2016; 17:1396–1408.
5. Dorfman HD, Czerniak B. Bone Tumors. Mosby, 1998.
6. Pakos EE, Nearchou AD, Grimer RJ, Koumoullis HD, Abudu A, Bramer JA, Jeys LM, Franchi A, Scoccianti G, Campanacci D, Capanna R, Aparicio J, Tabone MD, et al. Prognostic factors and outcomes for osteosarcoma: an international collaboration. Eur J Cancer. 2009; 45:2367–2375.
7. Baldini N. Multidrug resistance a multiplex phenomenon. Nat Med. 1997; 3:1380–1385.
8. Ren L, Mendoza A, Zhu J, Briggs JW, Halsey C, Hong ES, Burkett SS, Morrow J, Lizardo MM, Osborne T, Li SQ, Luu HH, Meltzer P, et al. Characterization of the metastatic phenotype of a panel of established osteosarcoma cells. Oncotarget. 2015; 6:29469–29481. https://doi.org/10.18632/oncotarget.5177.
9. Wittig JC, Bickels J, Priebat D, Jelinek J, Kellar-Graney K, Shmookler B, Malawer MM. Osteosarcoma: a multidisciplinary approach to diagnosis and treatment. Am Fam Physician. 2002; 65:1123–32.
10. Zhu L, McManus MM, Hughes DP. Understanding the biology of bone sarcoma from early initiating events through late events in metastasis and disease progression. Front Oncol. 2013; 3:230.
11. Broadhead ML, Clark JC, Myers DE, Dass CR, Choong PF. The molecular pathogenesis of Osteosarcoma: a Review. Sarcoma. 2011; 2011:959248.
12. Berman SD, Calo E, Landman AS, Danielian PS, Miller ES, West JC, Fonhoue BD, Caron A, Bronson R, Bouxsein ML, Mukherjee S, Lees JA. Metastatic osteosarcoma induced by inactivation of Rb and p53 in the osteoblast lineage. Proc Natl Acad Sci USA. 2008;105:11851–11856.
13. PosthumaDeBoer J, Witlox MA, Kaspers GJ, van Royen BJ. Molecular alterations as target for therapy in metastatic osteosarcoma: a review of literature. Clin Exp Metastasis. 2011; 28:493–503.
14. Martin JW, Squire JA, Zielenska M. The genetics of osteosarcoma. Sarcoma. 2012; 2012:627254.
15. Batanian JR, Cavalli LR, Aldosari NM, Ma E, Sotelo-Avila C, Ramos MB, Rone JD, Thorpe CM, Haddad BR. Evaluation of pediatric osteosarcomas by classic cytogenetic and CGH analyses. Mol Pathol. 2002; 55:389–393.
16. Rizzo JM, Buck MJ. Key principles and clinical applications of “Next Generation” DNA sequencing. Cancer Prev Res (Phila). 2012; 5:887–900.
17. Wang Z, Liu X, Yang BZ, Gelernter J. The role and challenges of exome sequencing in studies of human diseases. Front Genet. 2013; 4:160.
18. Shyr D, Liu Q. Next generation sequencing in cancer research and clinical application. Biol Proced Online. 2013; 15:4.
19. Parsons DW, Li M, Zhang X, Jones S, Leary RJ, Lin JC, Boca SM, Carter H, Samayoa J, Bettegowda C, Gallia GL, Jallo GI, Binder ZA, et al. The genetic landscape of the childhood cancer medulloblastoma. Science. 2011; 331:435–439.
20. Munirajan AK, Ando K, Mukai A, Takahashi M, Suenaga Y, Ohira M, Koda T, Hirota T, Ozaki T, Nakagawara A. KIF1Bbeta functions as a haploinsufficient tumor suppressor gene mapped to chromosome 1p36.2 by inducing apoptotic cell death. J Biol Chem. 2008; 283:24426–24434.
21. Stedman H, Browning K, Oliver N, Oronzi-Scott M, Fischbeck K, Sarkar S, Sylvester J, Schmickel R, Wang K. Nebulin cDNAs detect a 25-kilobase transcript in skeletal muscle and localize to human chromosome 2. Genomics. 1988; 2: 1–7.
22. Shilatifard A. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem. 2012; 81:65–95.
23. Arteaga CL, Engelman JA. ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer cell. 2014; 25:282–303.
24. Mendoza-Naranjo A, El-Naggar A, Wai DH, Mistry P, Lazic N, Ayala FR, da Cunha IW, Rodriguez-Viciana P, Cheng H, Tavares Guerreiro Fregnani JH, Reynolds P, Arceci RJ, Nicholson A, et al. ERBB4 confers metastatic capacity in Ewing sarcoma. EMBO Mol Med. 2013; 5:1087–1102.
25. Bornstein P. Diversity of function is inherent in matricellular proteins: an appraisal of thrombospondin 1. J Cell Biol. 1995; 130: 503–6.
26. Pal SK, Nguyen CT, Morita KI, Miki Y, Kayamori K, Yamaguchi A, Sakamoto K. THBS1 is induced by TGFB1 in the cancer stroma and promotes invasion of oral squamous cell carcinoma. J Oral Pathol Med. 2016; 45:730–739.
27. Wasmuth EV, Lima CD. Exo- and endoribonucleolytic activities of yeast cytoplasmic and nuclear RNA exosomes are dependent on the noncatalytic core and central channel. Mol Cell. 2012; 48:133–144.
28. Hoskins JW, Ibrahim A, Emmanuel MA, Manmiller SM, Wu Y, O’Neill M, Jia J, Collins I, Zhang M, Thomas JV, Rost LM, Das S, Parikh H, et al. Functional characterization of a chr13q22.1 pancreatic cancer risk locus reveals long-range interaction and allele-specific effects on DIS3 expression. Hum Mol Genet. 2016; 25:4726–4738.
29. Weißbach S, Langer C, Puppe B, Nedeva T, Bach E, Kull M, Bargou R, Einsele H, Rosenwald A, Knop S, Leich E. The molecular spectrum and clinical impact of DIS3 mutations in multiple myeloma. Br J Haematol. 2015; 169:57–70.
30. Nam SH, Kim D, Lee MS, Lee D, Kwak TK, Kang M, Ryu J, Kim HJ, Song HE, Choi J, Lee GH, Kim SY, Park SH, et al. Noncanonical roles of membranous lysyl-tRNA synthetase in transducing cell-substrate signaling for invasive dissemination of colon cancer spheroids in 3D collagen I gels. Oncotarget. 2015; 6:21655–21674. https://doi.org/10.18632/oncotarget.4130.
31. Brown GT, Cash B, Alnabulsi A, Samuel LM, Murray GI. The expression and prognostic significance of bcl-2-associated transcription factor 1 in rectal cancer following neoadjuvant therapy. Histopathology. 2016; 68:556–566.
32. Chen B, Liu Y, Jin X, Lu W, Liu J, Xia Z, Yuan Q, Zhao X, Xu N, Liang S. MicroRNA-26a regulates glucose metabolism by direct targeting PDHX in colorectal cancer cells. BMC Cancer. 2014; 14:443.
33. Reed DE, Shokat KM. Targeting osteosarcoma. Proc Natl Acad Sci U S A. 2014; 111:18100–18101.
34. Chen X, Bahrami A, Pappo A, Easton J, Dalton J, Hedlund E, Ellison D, Shurtleff S, Wu G, Wei L, Parker M, Rusch M, Nagahawatte P, et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep. 2014; 7:104–12.
35. Perry JA, Kiezun A, Tonzi P, Van Allen EM, Carter SL, Baca SC, Cowley GS, Bhatt AS, Rheinbay E, Pedamallu CS, Helman E, Taylor-Weiner A, McKenna A, et al. Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc Natl Acad Sci U S A. 2014; 111: E5564–73.
36. Rickel K, Fang F, Tao J. Molecular genetics of osteosarcoma. Bone. 2016; S8756–3282: 30313–1.
37. Kovac M, Blattmann C, Ribi S, Smida J, Mueller NS, Engert F, Castro-Giner F, Weischenfeldt J, Kovacova M, Krieg A, Andreou D, Tunn PU, Dürr HR, et al. Exome sequencing of osteosarcoma reveals mutation signatures reminiscent of BRCA deficiency. Nat Commun. 2015; 6:8940.
38. Bousquet M, Noirot C, Accadbled F, Sales de Gauzy J, Castex MP, Brousset P, Gomez-Brouchet A. Whole-exome sequencing in osteosarcoma reveals important heterogeneity of genetic alterations. Ann Oncol. 2016; 27:738–44.
39. Joseph CG, Hwang H, Jiao Y, Wood LD, Kinde I, Wu J, Mandahl N, Luo J, Hruban RH, Diaz LA Jr, He TC, Vogelstein B, Kinzler KW, et al. Exomic analysis of myxoid liposarcomas, synovial sarcomas, and osteosarcomas. Genes Chromosomes Cancer. 2014; 53:15–24.
40. Hainaut P, Pfeifer GP. Somatic TP53 Mutations in the Era of Genome Sequencing. Cold Spring Harb Perspect Med. 2016; 6.
41. Gianferante DM, Mirabello L, Savage SA. Germline and somatic genetics of osteosarcoma - connecting aetiology, biology and therapy. Nat Rev Endocrinol. 2017; 13:480–491. https://doi.org/10.1038/nrendo.2017.16.
42. Di Cristofano C, Leopizzi M, Miraglia A, Sardella B, Moretti V, Ferrara A, Petrozza V, Della Rocca C. Phosphorylated ezrin is located in the nucleus of the osteosarcoma cell. Mod Pathol. 2010; 23:1012–1020.
43. Lee J, Kim DH, Lee S, Yang QH, Lee DK, Lee SK, Roeder RG, Lee JW. A tumor suppressive coactivator complex of p53 containing ASC-2 and histone H3 lysine-4 methyltransferase MLL3 or its paralogue MLL4. Proc Natl Acad Sci U S A. 2009; 106:8513–8518.
44. Marina N, Gebhardt M, Teot L, Gorlick R. Biology and therapeutic advances for pediatric osteosarcoma. Oncologist. 2004; 9: 422–441.
45. Ansari KI, Hussain I, Kasiri S, Mandal SS. HOXC10 is overexpressed in breast cancer and transcriptionally regulated by estrogen via involvement of histone methylases MLL3 and MLL4. J Mol Endocrinol. 2012; 48:61–75.
46. Manolagas SC, O’Brien CA, Almeida M. The role of estrogen and androgen receptors in bone health and disease. Nat Rev Endocrinol. 2013; 9:699–712.
47. Herz HM. Enhancer deregulation in cancer and other diseases. Bioessays. 2016; 38:1003–1015.
48. Ford DJ, Dingwall AK. The cancer COMPASS: navigating the functions of MLL complexes in cancer. Cancer Genetics. 2015; 208:178–191.
49. Perry JA, Kiezun A, Tonzi P, Van Allen EM, Carter SL, Baca SC, Cowley GS, Bhatt AS, Rheinbay E, Pedamallu CS, Helman E, Taylor-Weiner A, McKenna A, et al. Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc Natl Acad Sci U S A. 2014; 111:E5564–5573.
50. Chiappetta C, Leopizzi M, Censi F, Puggioni C, Petrozza V, Della Rocca C, Di Cristofano C. Correlation of the Rac1/RhoA pathway with ezrin expression in osteosarcoma. Appl Immunohistochem Mol Morphol. 2014; 22:162–70.
51. Soung YH, Lee JW, Kim SY, Wang YP, Jo KH, Moon SW, Park WS, Nam SW, Lee JY, Yoo NJ, Lee SH. Somatic mutations of the ERBB4 kinase domain in human cancers. Int J Cancer. 2006; 118:1426–1429.
52. Gerarduzzi C, de Polo A, Liu XS, El Kharbili M, Little JB, Yuan ZM. Her4 Suppresses p53 Via Targeting the MDMX-MDM2 Complex: Implication of a Novel MDMX Ser314 Phosphosite. J Biol Chem. 2016; 291:25937–25949.